
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA divergent synthetic route to functional copolymer libraries via modular polymers
Rachel H.
Bianculli†
 ,
Connor M. B.
Gallagher†
,
Zhen
Shi
,
Rhone B.
Jenkins
,
Timothy D.
Ermolaev
and
Michael D.
Schulz
*
,
Connor M. B.
Gallagher†
,
Zhen
Shi
,
Rhone B.
Jenkins
,
Timothy D.
Ermolaev
and
Michael D.
Schulz
*
Virginia Tech, Department of Chemistry and Macromolecules Innovation Institute, USA. E-mail: mdschulz@vt.edu
First published on 17th February 2026
Abstract
High-throughput polymer synthesis enables rapid exploration of chemical space but remains limited by batch-to-batch inconsistencies that can obscure structure–property relationship trends. To address this challenge, we developed a synthetic approach to produce multifunctional copolymers using post-polymerization modification of activated ester modular polymers with commercially available amines. Easily derivitized parent polymers—poly(tetrafluorophenyl acrylate) and poly(tetrafluorophenyl styrene sulfonate)—were synthesized by RAFT polymerization to yield single polymer batches containing highly reactive tetrafluorophenyl esters or sulfonate esters on each repeat unit. Tuning post-polymerization modification reaction conditions enabled the addition of sub-stoichiometric amounts of amines (relative to the repeat unit) to yield partially functionalized intermediates that could then be further derivatized. Reaction monitoring by 19F NMR spectroscopy confirmed good control over these sequential post-polymerization modifications. This synthetic route produced a variety of copolymers with defined comonomer ratios while preserving the underlying polymer structure (degree of polymerization, dispersity, tacticity) for both the acrylate and styrene sulfonate backbones. We further applied this approach in a divergent manner to create a small library of structurally distinct copolymers from a single parent batch in three synthetic steps. This modular, divergent synthesis demonstrates a general route to structurally consistent copolymer libraries that enable systematic studies of structure–property relationships and can accelerate functional materials discovery.
Introduction
As high-throughput screening methods have advanced, high-throughput synthetic approaches have developed in parallel. High-throughput methods have been used in the pharmaceutical industry for decades,1–4 and have more recently begun being employed in polymer chemistry.5–9 In the context of polymer synthesis, these approaches have enabled the rapid production of large libraries of polymers with varying compositions to quickly discover new structures, establish structure–property relationships, and optimize polymerization conditions.Ideally, the creation of these polymer libraries accelerates the discovery of new materials by enabling researchers to rapidly explore a wide range of chemical space and identify materials with optimal properties for a specific application. For example, copolymer libraries have been synthesized and screened for antibacterial activity, ultimately revealing structure–activity relationships that can guide the future development of antibacterial materials.10–13 Additionally, high-throughput synthetic techniques have been applied to stimuli-responsive polymers to optimize synthetic methods and generate libraries of polymers with tuneable properties.14–19 Despite these successes, many polymer structures remain inaccessible by direct (co)polymerization due to incompatibility in polymerization conditions, synthetically challenging monomers, or differences in comonomer reactivity ratios.20,21 Furthermore, even the most well-controlled syntheses produce mixtures of polymer chains with slight (or not so slight) differences in degree of polymerization (DP), tacticity, and dispersity. While robotic platforms can often keep these variations to a minimum, structural variables are not perfectly controlled among different samples.22–26 In applications where these minor structural changes can produce large effects, a synthetic approach capable of holding these variables constant—while still enabling rapid synthesis of many structurally related materials—is desirable.
Modular polymers, that is, polymers that can be easily modified post-polymerization,27,28 offer several key advantages that streamline and enhance materials discovery. With a modular approach, a single parent material may be synthesized and subsequently derivatized to produce a library of materials that share key structural features, e.g., DP, dispersity, and tacticity. While many modular polymer strategies exist,29–31 modular polymers featuring activated esters, specifically perfluorophenyl esters, are particularly effective because of their facile functionalization by nucleophilic acyl substitution. This approach has been well-developed to produce homopolymer libraries,32–34 and to a more limited extent, copolymers.35,36 For example, Hawker, Bates, and coworkers recently demonstrated a high-throughput combined chromatographic and robotic method to generate a block copolymer library with varied block length and composition, based on a modular polymer block. Though limited to a series of homo-di-block copolymers, this work highlights the opportunity to extend modular polymer strategies to more compositionally complex copolymers.37 As high-throughput synthesis evolves, the full potential of copolymer synthesis by a modular approach invites further exploration.
Here, we demonstrate a divergent synthetic strategy to copolymer libraries by sub-stoichiometric functionalization in a modular polymer-based approach. This strategy enables synthesis of copolymer libraries with high degrees of uniformity in composition, DP, and dispersity. This approach also enables synthesis of diverse copolymers while eliminating the variability inherent in other synthetic approaches and enabling access to copolymer structures inaccessible by direct polymerization.
Results and discussion
Synthesis and post-polymerization modification of poly(tetrafluorophenyl acrylate) (PTFPA)
We chose poly(tetrafluorophenyl acrylate) (PTFPA) as a model for this study because activated esters are a well-known class of modular polymer, functionalization of the polyacrylate backbone enables access to a variety of polyacrylates and polyacrylamides, and the para-proton allows for reaction monitoring by both 1H and 19F NMR spectroscopy.30,32,38 Monomeric tetrafluorophenyl acrylate (TFPA) was synthesized in one step by the reaction of tetrafluorophenol with acryloyl chloride (Scheme 1 and Fig. S1, S2, SI). TFPA was then polymerized by reversible addition–fragmentation chain-transfer (RAFT) polymerization using 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT; Fig. S3 and S4, SI) as a chain-transfer agent to control the molecular weight and dispersity. Size exclusion chromatography (SEC) and NMR confirmed a well-defined polymer structure (Fig. S5–S7, SI). | ||
| Scheme 1 Synthesis of PTFPA by RAFT polymerization. Final polymer Mn (SEC) = 24.3 kDa, Đ = 1.58. | ||
Activated-ester polymers react quantitatively with nucleophiles (typically primary amines or, in the presence of a catalyst, alcohols32) to yield the corresponding functionalized polymer (polyacrylamides or polyacrylates, respectively). In this study, we used unhindered, primary amines due to their facile reactivity. To test our hypothesis that addition of sub-stoichiometric amounts (relative to the repeat unit) of nucleophile would produce isolable polymers with retained modular tetrafluorophenyl functionality, we reacted PTFPA with 0.25 molar equivalents of benzylamine in DMF (Fig. S8, SI). After reacting for 15 hours, we observed ∼43% conversion by 19F NMR, higher than what would be expected for only 0.25 equivalents of nucleophile. Similar to previous mechanistic studies,32 we concluded that the solvent, DMF, may be displacing the activated ester moiety. This solvent displacement would not impact the polymer structure once fully functionalized; however, due to our desire to easily track functionalization by 19F NMR, we screened a series of common organic solvents to determine which would not displace the activated ester (Fig. S9 and Table S1, SI). Gratifyingly, ethyl acetate did not appear to displace the activated ester moiety, even after extensive heating or in the presence of another nucleophile (benzylamine) (Fig. S10, SI). Therefore, we continued to use ethyl acetate for the modification of PTFPA. Addition of 0.5 equivalents of benzylamine in ethyl acetate yielded a functionalization of ∼53% after 2 hours. Subsequent addition of 0.5 equivalents of n-hexylamine functionalized the remaining tetrafluorophenyl acrylate to form a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 poly(benzylacrylamide-co-hexylacrylamide) copolymer (Fig. 1).
:50 poly(benzylacrylamide-co-hexylacrylamide) copolymer (Fig. 1).
 | ||
| Fig. 1 (A) Scheme for functionalization of PTFPA with benzyl- and hexylamine to yield a 50:50 random copolymer. (B) Post-polymerization modification was monitored by 19F NMR (375 MHz, DMSO-d6) with an internal standard and quantified by the ratio of polymeric fluorine (pink circles) to tetrafluorophenoxide (blue triangles) in the reaction solution. Error is calculated by propagating the standard deviation between the two sets of fluorines (ortho and meta to the oxygen). | ||
Synthesis and post-polymerization modification of poly(tetrafluorophenyl styrene sulfonate) (PTFPSS)
Activated-ester moieties can be appended to other polymer backbones in addition to polyacrylates. To demonstrate this versatility, we synthesized tetrafluorophenyl styrene sulfonate (TFPSS) to access styrenic backbones. The tetrafluorophenyl styrene sulfonate monomer was synthesized in two steps, first converting sodium styrene sulfonate to the corresponding sulfonyl chloride with thionyl chloride followed by substitution of chloride with tetrafluorophenol (Scheme 2 and Fig. S11–S15, SI). The TFPSS monomer was then polymerized via RAFT polymerization resulting in a similar DP to the PTFPA batch (Scheme 2). Molecular weight of the final polymer was confirmed by size-exclusion chromatography (SEC) and matching NMR end-group analysis (Fig. S16–S18, SI). This polymer batch was then used for all subsequent post-polymerization modifications using PTFPSS.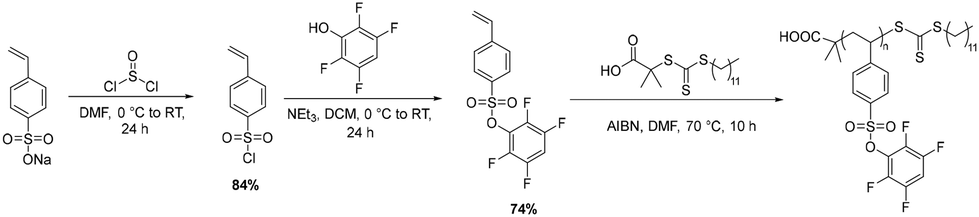 | ||
| Scheme 2 Synthesis of TFPSS monomer and subsequent RAFT Polymerization. Final polymer Mn (SEC) = 33 kDa, Đ = 1.40. | ||
Unlike PTFPA, PTFPSS was significantly less susceptible to unwanted displacement by solvent and these post-polymerization reactions could be conducted in DMF (Fig. 2A). To form a 50:50 poly((benzylsulfonamido)styrene-co-(hexylsulfonamido)styrene) copolymer analogous to the polyacrylate shown in Fig. 1A, 0.52 equivalents of benzylamine were added to the PTFPSS. This addition resulted in an apparent 60% conversion of polymeric tetrafluorophenyl groups to tetrafluorophenoxide (Fig. 2B). We attributed this slight discrepancy to hydrolysis by residual water present in DMF. A subsequent addition of 0.54 equivalents of hexylamine displaced the remaining tetrafluorophenyl units, yielding the desired 50:50 copolymer.
 | ||
| Fig. 2 (A) Scheme for functionalization of PTFPSS with benzyl- and hexylamine to yield a 50:50 random copolymer. (B) Post-polymerization modification was monitored by 19F NMR (375 MHz, DMSO-d6) with an internal standard and quantified by the ratio of polymeric fluorine (pink circles) to tetrafluorophenoxide (blue triangles) in the reaction solution. Error is calculated by propagating the standard deviation between the two sets of fluorines (ortho and meta to the oxygen). | ||
Polymer library generation by a divergent synthetic approach
To further evaluate the scope of the post-polymerization modification of PTFPA and PTFPSS, we added 0.1 equivalents of benzylamine to the parent polymers in ten sequential portions. We used a sequential addition of amines in this reaction and in subsequent experiments to show that a controlled amount of the polymeric tetrafluorophenyl functionalization was retained after each post-polymerization modification. These reactions were quantified after each addition (0.1 mol equiv. benzylamine, 2 h reaction time) using 19F NMR spectroscopy. Interestingly, the PTFPA modification displayed limited control with all the tetrafluorophenyl groups being cleaved by the sixth addition (Fig. S19, SI). We attributed this excess displacement to a combination of the reaction solvent, ethyl acetate, displacing the activated ester after long exposure periods and the NMR solvent, DMSO-d6, displacing the ester during the NMR analysis. Closer monitoring of the reaction progress by 19F NMR spectroscopy revealed that functionalization was complete after only 45 minutes and at a lower temperature of 45 °C (Fig. S20 and Table S2, SI). Excess displacement was avoided by decreasing the reaction time and temperature for future reactions. Additionally, to prevent displacement during NMR spectroscopic characterization, we carried out NMR experiments in acetone-d6 rather than DMSO-d6. Under these conditions, the reaction behaved as expected with each addition of 0.1 equivalents of nucleophile corresponding to an approximately 10% increase in the amount of free leaving group (tetrafluorophenoxide) present in solution (Fig. 3, left, blue). In contrast to the PTFPA, the PTFPSS system immediately behaved as expected by cleaving ∼10% of the tetrafluorophenyl groups with each 0.10 mol equiv. of benzylamine after 2 h of reaction (Fig. 3, right, pink). | ||
| Fig. 3 Synthesis of benzylamine homopolymers with PTFPA (left, blue) or PTFPSS (right, pink) parent polymer through 10, sequential 0.1 mol equivalent additions of benzylamine with near quantitative functionalization tracked through 19F NMR. | ||
To access more complex multifunctional copolymers, we used a divergent synthetic approach to generate a small library of four structurally different polymers in three synthetic steps (Fig. 4). First, 0.25 equivalents of benzylamine were added and allowed to react, then the reaction solution was split into two equal portions. To the first portion, 0.5 equivalents of n-hexylamine were added while 0.5 equivalents of furfurylamine were added to the second and both were allowed to react. The reaction solutions for this second generation were then split into two equal portions again and the remaining 0.25 equivalents of tetrafluorophenyl ester were functionalized by allylamine or either n-hexylamine or furfurylamine depending on what was used in the second generation. Following each reaction period, the reaction solution was characterized by 19F NMR (Tables S3 and S4, SI) to ensure full substitution of each individual functionality, as these amines exhibit different intrinsic reactivities toward the tetrafluorophenyl ester and a single-portion addition would obscure control over the extent of functionalization. While the PTFPA behaved as expected, it should be noted that for the divergent modification of the PTFPSS targeting a (1) 25% benzylamine, (2) 50% hexylamine, (3) 25% furfurylamine composition, the expected 1H NMR integration ratio would be 1:2:1; however, the observed ratio was benzylamine:hexylamine:furfurylamine = 1:2:0.5. When the order of addition was changed to (1) 25% benzylamine, (2) 25% furfurylamine, (3) 50% hexylamine, the observed ratio improved to 1:1.7:1. We attribute the incomplete incorporation of amines in this series to steric hindrance arising from the combined use of three relatively bulky amines. This dependence on the order of addition indicates nearby repeat units interact during substitution, which in turn could affect the distribution of different substituents along the polymer chain. Although full analysis of the substituent distribution is beyond the scope of this study, we observed no anomalous behaviors indicative of block- or gradient-like distributions. However, consideration of these interactions in reaction design is critical to maintain uniform functionalization.
 | ||
| Fig. 4 Divergent route to a polymer library with varying benzyl-, hexyl-, furfuryl-, and allylamine functionalization. | ||
While we did not isolate the first- or second-generation materials in this work, this divergent synthetic approach could yield a total of seven distinct polymers in three synthetic steps. The number of distinct, final polymers could be increased by increasing the number of divisions per generation, thereby enabling rapid production of large polymer libraries. The final four polymers synthesized in this work varied in comonomer identity and amount (Fig. S21–S28, SI). The two final benzyl acrylamide-co-hexyl acrylamide-co-furfuryl acrylamide copolymers had the same comonomer identities but different ratios of functionalization. Although four polymers were produced using this divergent approach, we demonstrated control over ten sequential additions, in theory potentially generating over a thousand different materials even with only two divisions per generation. Additionally, while we used relatively simple, commercially available amines for this study to avoid complex characterization, this method could be expanded to incorporate a wide range of functionality, including orthogonal functionality that could be further derivatized by subsequent post-polymerization modification (e.g., thiol–ene click chemistry at the allylacrylamides). Thus, this divergent method enables the synthesis of copolymers with controlled structural variations in few synthetic steps while holding parent polymer parameters (DP and dispersity) consistent.
Conclusions
In conclusion, we demonstrated a divergent synthetic strategy to generate a library of multi-functional copolymers using a modular polymer approach. This strategy was effective for polystyrene (PTFPSS) and polyacrylate (PTFPA) backbones, though conditions differed due to displacement by solvent in the reaction and NMR experiments, or steric effects. We showed access to a variety of polymer compositions using commonly available amines, but we believe this approach could be more generalizable to other nucleophiles, allowing for diverse functionalities to be incorporated. This strategy will enable synthesis of copolymer libraries with high degrees of uniformity in composition, DP, and dispersity, offering a route to improving existing high-throughput polymer synthesis methods.Author contributions
RHB: conceptualization, investigation, methodology, visualization, writing-original draft. CMBG: conceptualization, investigation, methodology, visualization, writing-original draft. ZS: investigation, methodology, visualization, writing-review & editing. RBJ: investigation. TDE: investigation. MDS: conceptualization, funding acquisition, project administration, supervision, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: detailed experimental procedures and full NMR spectra. See DOI: https://doi.org/10.1039/d5py01156f.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0023035. The PTFPSS portion of this work was supported by GlycoMIP, an NSF Materials Innovation Platform funded through Cooperative Agreement DMR-1933525. The authors would like to thank Jonathan Mase for synthesizing the DDMAT and for helpful comments on the manuscript. The authors would also like to thank Prof. Paul Deck for helpful comments on the manuscript.References
- J. Götz, M. K. Jackl, C. Jindakun, A. N. Marziale, J. André, D. J. Gosling, C. Springer, M. Palmieri, M. Reck, A. Luneau, C. E. Brocklehurst and J. W. Bode, High-Throughput Synthesis Provides Data for Predicting Molecular Properties and Reaction Success, Sci. Adv., 2023, 9(43), eadj2314, DOI:10.1126/sciadv.adj2314.
- B. Mahjour, R. Zhang, Y. Shen, A. McGrath, R. Zhao, O. G. Mohamed, Y. Lin, Z. Zhang, J. L. Douthwaite, A. Tripathi and T. Cernak, Rapid Planning and Analysis of High-Throughput Experiment Arrays for Reaction Discovery, Nat. Commun., 2023, 14(1), 3924, DOI:10.1038/s41467-023-39531-0.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future, Org. Process Res. Dev., 2019, 23(6), 1213–1242, DOI:10.1021/acs.oprd.9b00140.
- S.-H. Hou, F.-F. Zhou, Y.-H. Sun and Q.-Z. Li, Deconstructive and Divergent Synthesis of Bioactive Natural Products, Molecules, 2023, 28(17), 6193, DOI:10.3390/molecules28176193.
- E. C. Day, S. S. Chittari, M. P. Bogen and A. S. Knight, Navigating the Expansive Landscapes of Soft Materials: A User Guide for High-Throughput Workflows, ACS Polym. Au, 2023, 3(6), 406–427, DOI:10.1021/acspolymersau.3c00025.
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Living in the Fast Lane—High Throughput Controlled/Living Radical Polymerization, Macromolecules, 2019, 52(1), 3–23, DOI:10.1021/acs.macromol.8b01864.
- A. P. Grimm, S. T. Knox, C. Y. P. Wilding, H. A. Jones, B. Schmidt, O. Piskljonow, D. Voll, C. W. Schmitt, N. J. Warren and P. Théato, A Versatile Flow Reactor Platform for Machine Learning Guided RAFT Synthesis, Amidation of Poly(Pentafluorophenyl Acrylate), Macromol. Rapid Commun., 2025, 2500264, DOI:10.1002/marc.202500264.
- E. C. Day, S. S. Chittari, K. C. Cunha, R. J. Zhao, J. N. Dodds, D. C. Davis, E. S. Baker, R. B. Berlow, J.-E. Shea, R. U. Kulkarni and A. S. Knight, A High-Throughput Workflow to Analyze Sequence-Conformation Relationships and Explore Hydrophobic Patterning in Disordered Peptoids, Chem, 2024, 10(11), 3444–3458, DOI:10.1016/j.chempr.2024.07.025.
- S. Baudis and M. Behl, High-Throughput and Combinatorial Approaches for the Development of Multifunctional Polymers, Macromol. Rapid Commun., 2022, 43(12), 2100400, DOI:10.1002/marc.202100400.
- P. Pham, S. Oliver, E. H. H. Wong and C. Boyer, Effect of Hydrophilic Groups on the Bioactivity of Antimicrobial Polymers, Polym. Chem., 2021, 12(39), 5689–5703, 10.1039/d1py01075a.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Block versus Random Amphiphilic Copolymers as Antibacterial Agents, Biomacromolecules, 2011, 12(10), 3581–3591, DOI:10.1021/bm200780r.
- P. R. Judzewitsch, T. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Towards Sequence–Controlled Antimicrobial Polymers: Effect of Polymer Block Order on Antimicrobial Activity, Angew. Chem., Int. Ed., 2018, 57(17), 4559–4564, DOI:10.1002/anie.201713036.
- S. Schaefer, T. T. P. Pham, S. Brunke, B. Hube, K. Jung, M. D. Lenardon and C. Boyer, Rational Design of an Antifungal Polyacrylamide Library with Reduced Host-Cell Toxicity, ACS Appl. Mater. Interfaces, 2021, 13(23), 27430–27444, DOI:10.1021/acsami.1c05020.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Libraries of Statistical Hydroxypropyl Acrylate Containing Copolymers with LCST Properties Prepared by NMP, Macromolecules, 2008, 41(14), 5132–5140, DOI:10.1021/ma800469p.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Tunable pH- and Temperature-Sensitive Copolymer Libraries by Reversible Addition−Fragmentation Chain Transfer Copolymerizations of Methacrylates, Macromolecules, 2007, 40(4), 915–920, DOI:10.1021/ma062199r.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, Libraries of Methacrylic Acid and Oligo(Ethylene Glycol) Methacrylate Copolymers with LCST Behavior, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(21), 7138–7147, DOI:10.1002/pola.23018.
- B. Luan, B. W. Muir, J. Zhu and X. Hao, A RAFT Copolymerization of NIPAM and HPMA and Evaluation of Thermo-Responsive Properties of Poly(NIPAM-Co-HPMA), RSC Adv., 2016, 6(92), 89925–89933, 10.1039/C6RA22722H.
- R. Yañez-Macias, I. Kulai, J. Ulbrich, T. Yildirim, P. Sungur, S. Hoeppener, R. Guerrero-Santos, U. S. Schubert, M. Destarac, C. Guerrero-Sanchez and S. Harrisson, Thermosensitive Spontaneous Gradient Copolymers with Block- and Gradient-like Features, Polym. Chem., 2017, 8(34), 5023–5032, 10.1039/C7PY00495H.
- T. Yildirim, I. Yildirim, R. Yañez-Macias, S. Stumpf, C. Fritzsche, S. Hoeppener, C. Guerrero-Sanchez, S. Schubert and U. S. Schubert, Dual pH and Ultrasound Responsive Nanoparticles with pH Triggered Surface Charge-Conversional Properties, Polym. Chem., 2017, 8(8), 1328–1340, 10.1039/C6PY01927G.
- K. A. Günay, P. Theato and H. A. Klock, History of Post–Polymerization Modification, in Functional Polymers by Post–Polymerization Modification, ed. P. Theato and H. A. Klock, Wiley, 2012, pp. 1–44. DOI:10.1002/9783527655427.ch1.
- K. A. Günay, P. Theato and H.-A. Klok, Standing on the Shoulders of Hermann Staudinger: Post-Polymerization Modification from Past to Present, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(1), 1–28, DOI:10.1002/pola.26333.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Scalable Synthesis of Sequence-Defined, Unimolecular Macromolecules by Flow-IEG, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(34), 10617–10622, DOI:10.1073/pnas.1508599112.
- Y. Zhou, Y. Gu, K. Jiang and M. Chen, Droplet-Flow Photopolymerization Aided by Computer: Overcoming the Challenges of Viscosity and Facilitating the Generation of Copolymer Libraries, Macromolecules, 2019, 52(15), 5611–5617, DOI:10.1021/acs.macromol.9b00846.
- B. Lin, J. L. Hedrick, N. H. Park and R. M. Waymouth, Programmable High-Throughput Platform for the Rapid and Scalable Synthesis of Polyester and Polycarbonate Libraries, J. Am. Chem. Soc., 2019, 141(22), 8921–8927, DOI:10.1021/jacs.9b02450.
- J. Lee, P. Mulay, M. J. Tamasi, J. Yeow, M. M. Stevens and A. J. Gormley, A Fully Automated Platform for Photoinitiated RAFT Polymerization, Digital Discovery, 2023, 2(1), 219–233, 10.1039/D2DD00100D.
- C. Ramirez, E. Ahmed, E. D. Mare, M. Pineiro-Goncalves, A. Maroulis, P. Mulay, D. C. Radford and A. J. Gormley, Automation-Assisted Photoinduced Atom Transfer Radical Polymerization, ACS Polym. Au, 2025 DOI:10.1021/acspolymersau.5c00067.
- J. Lutz, Polymerization of Oligo(Ethylene Glycol) (Meth)Acrylates: Toward New Generations of Smart Biocompatible Materials, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(11), 3459–3470, DOI:10.1002/pola.22706.
- R. Kakuchi and P. Theato, Sequential Post-Polymerization Modification Reactions of Poly(Pentafluorophenyl 4-Vinylbenzenesulfonate), Polym. Chem., 2014, 5(7), 2320–2325, 10.1039/C3PY01604H.
- R. Kakuchi and P. Theato, Post–Polymerization Modifications via Active Esters, in Functional Polymers by Post–Polymerization Modification, ed. P. Theato and H. A. Klok, Wiley, 2012, pp. 45–64. DOI:10.1002/9783527655427.ch2.
- A. Das and P. Theato, Activated Ester Containing Polymers: Opportunities and Challenges for the Design of Functional Macromolecules, Chem. Rev., 2016, 116(3), 1434–1495, DOI:10.1021/acs.chemrev.5b00291.
- K. Kasai, H. Amii and R. Kakuchi, Reversible Aminolysis: A New Concept for Postpolymerization Modification, Polym. Chem., 2024, 4622 RSC.
- A. Das and P. Theato, Multifaceted Synthetic Route to Functional Polyacrylates by Transesterification of Poly(Pentafluorophenyl Acrylates), Macromolecules, 2015, 48(24), 8695–8707, DOI:10.1021/acs.macromol.5b02293.
- K. Nilles and P. Theato, Polymerization of an Activated Ester Monomer Based on 4-Vinylsulfonic Acid and Its Polymer Analogous Reaction, Polym. Chem., 2011, 2(2), 376–384, 10.1039/c0py00261e.
- E. Molle, H. Mutlu and P. Theato, Synthesis and Post–Polymerization Modification of Poly(N,-(4-Vinylphenyl)Sulfonamide)s, Macromol. Rapid Commun., 2021, 42(10), 2100063, DOI:10.1002/marc.202100063.
- W. Xue, H. Mutlu and P. Theato, Post-Polymerization Modification of Polymeric Active Esters towards TEMPO Containing Polymers: A Systematic Study, Eur. Polym. J., 2020, 130, 109660, DOI:10.1016/j.eurpolymj.2020.109660.
- H. Zhao and P. Theato, Copolymers Featuring Pentafluorophenyl Ester and Photolabile Amine Units: Synthesis and Application as Reactive Photopatterns, Polym. Chem., 2013, 4(4), 891, 10.1039/c2py21050a.
- E. A. Murphy, K. G. Roth, M. W. Bates, M. C. Murphy, J. Edmund, C. M. Bates and C. J. Hawker, High-Throughput Generation of Block Copolymer Libraries via Click Chemistry and Automated Chromatography, Macromolecules, 2025, 58(15), 8369–8376, DOI:10.1021/acs.macromol.5c01024.
- W. R. Archer, C. M. B. Gallagher, V. Vaissier Welborn and M. D. Schulz, Exploring the Role of Polymer Hydrophobicity in Polymer–Metal Binding Thermodynamics, Phys. Chem. Chem. Phys., 2022, 24, 3579–3585, 10.1039/D1CP05263B.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |