
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Haloboration: scope, mechanism and utility
Sven
Kirschner†
 ,
Kang
Yuan†
and
Michael J.
Ingleson
*
,
Kang
Yuan†
and
Michael J.
Ingleson
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.ingleson@ed.ac.uk
First published on 8th July 2020
Abstract
Haloboration, the addition of B–X (X = Cl, Br, I) across an unsaturated moiety e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Y or C
Y or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Y (Y = C, N, etc.), is dramatically less utilised than the ubiquitous hydroboration reaction. However, haloboration of alkynes in particular is a useful tool to access ambiphilic 1,2-disubstituted alkenes. The stereochemical outcome of the reaction is easily controlled and the resulting products have proven to be valuable building blocks in organic synthesis and materials chemistry. This review aims at providing the reader with a brief summary of the historic development and of the current mechanistic understanding of this transformation. Recent developments are discussed and select examples demonstrating the use of haloboration products are given with a focus on the major areas, specifically, natural product synthesis and the development of boron-doped polycyclic aromatic hydrocarbons (B-PAHs).
Y (Y = C, N, etc.), is dramatically less utilised than the ubiquitous hydroboration reaction. However, haloboration of alkynes in particular is a useful tool to access ambiphilic 1,2-disubstituted alkenes. The stereochemical outcome of the reaction is easily controlled and the resulting products have proven to be valuable building blocks in organic synthesis and materials chemistry. This review aims at providing the reader with a brief summary of the historic development and of the current mechanistic understanding of this transformation. Recent developments are discussed and select examples demonstrating the use of haloboration products are given with a focus on the major areas, specifically, natural product synthesis and the development of boron-doped polycyclic aromatic hydrocarbons (B-PAHs).
 Sven Kirschner | Sven Kirschner studied chemistry at the Goethe-University Frankfurt/Main (Germany), where he received his PhD in 2019 for his contributions to the field of boron-doped polycyclic aromatic hydrocarbons. He joined the group of Michael Ingleson in 2019 as a postdoctoral research associate and is currently working on the development of main-group element mediated C–C coupling reactions. |
 Kang Yuan | Kang Yuan received his BSc degree in 2013 from Sun Yat-sen University under the guidance of Prof. Ming-Liang Tong. Then, he moved to Canada to explore main group chemistry and obtained his PhD degree under the supervision of Prof. Suning Wang at Queen's University. In 2018, he joined the Ingleson group as a postdoctoral research associate. His current research focuses on developing new methods for constructing novel boron containing molecules. |
 Michael J. Ingleson | Mike Ingleson received his PhD. in organometallic chemistry in 2004 working with Prof. Andrew S. Weller. He then pursued postdoctoral studies with Prof. Kenneth G. Caulton at Indiana University and with Professor Matthew J. Rosseinsky at the University of Liverpool. He started his independent career at the University of Manchester in 2008 with a Royal Society University Research Fellowship. At Manchester, he was promoted to Reader in 2012 and Full Professor in 2018. He joined the University of Edinburgh in 2019. His research focuses on main group chemistry, and he has a particular fondness for all things boron. |
1. Introduction
Since the end of the last millennium, the principle of sustainability and atom economy increasingly has impacted the way scientific research is done.1 A chemical transformation that is 100% atom efficient is the addition reaction, in which a reagent is added across a multiple bond (e.g., CC, CC, or CE, E: O, NR). One very important addition reaction is the Nobel Prize winning hydroboration reaction.2 Initially, it was almost exclusively used to access alcohols from alkenes by oxidative B–C bond cleavage. However, organic transformations like the Matteson homologation,3 Petasis variant of the Mannich reaction,4 Chan–Lam coupling,5 and, of course, the Suzuki–Miyaura reaction6 all utilise substrates that can be accessed by hydroboration reactions, rendering it a powerful tool in the synthetic chemist's toolbox. Unsurprisingly, researchers still strive to develop new hydroboration methods, e.g., by expanding the scope or reducing the environmental impact.7 In contrast, haloboration – although discovered at roughly the same time – has remained a niche technique that has gained only a small fraction of the attention even though it adds an additional highly valuable group (a halide) in the same step. The concomitant installation of a boron unit and a halide generates functionality rich molecules, containing a nucleophilic C–B and an electrophilic C–X unit. Thus, it is surprising that this reaction is so under-utilised, despite being potentially useful to many.
In this review, we give a brief discourse of the historic development of the haloboration reaction, from the curiosity driven fundamental research mainly by the group of Lappert, to the usage of Pd catalysed cross-coupling reactions to demonstrate the full potential of haloboration from the group of Suzuki who broadened the scope and deepened the understanding of this reaction.8 We also provide an in-depth discussion of the underlying mechanism and select applications of the products from the haloboration reaction in the field of synthesis. Recently, haloboration has started to gain wider interest through its use in natural-product synthesis to introduce CC double bonds stereoselectively, and in the synthesis of boron-doped polycyclic aromatic hydrocarbons (B-PAHs), thus these are the main applications focused on herein. B-PAHs are a relatively new class of organic materials with interesting optoelectronic properties and haloboration is a fast and convenient way to incorporate a borane unit and a halogen functionality into a PAH at the same time. Since the publication of the last reviews on haloboration, which were in the 1980s to the best of our knowledge,8d the scope and utility of haloboration in synthetic chemistry has increased significantly. We hope that this review, focused on the use of boron electrophiles to haloborate CY and CY nucleophiles (Y = C, N or O based substituents) facilitates the wider application of this useful, yet often overlooked, reaction.
2. Haloboration of simple alkynes using Y2B–X (Y = X or R)
2.1 The early work
Chloroboration of alkynes was first explored by H. R. Arnold in 1946.9 With mercury(I) chloride on activated carbon as the catalyst, chloroboration of acetylene with boron trichloride (BCl3) was achieved to afford 2-chlorovinyldichloroborane at 150–300 °C (Scheme 1a). In this patent, the stereoselectivity of chloroboration was not determined. Subsequently, Jensen et al. repeated the synthesis under similar conditions and measured the dipole moment of the obtained product.10 The experimentally determined dipole moment (1.06(0.05) D) was found to be very close to the predicted value of trans-product 1 (1.05 D). For comparison, the dipole moment of the cis-isomer was predicted to be 3.23 D. Thus, trans-product 1 was believed to be formed. In addition, the trans-chloroboration product 1 was found to be significantly more stable than the cis-isomer by 145 kJ mol−1 in electronic energy based on ab initio calculations.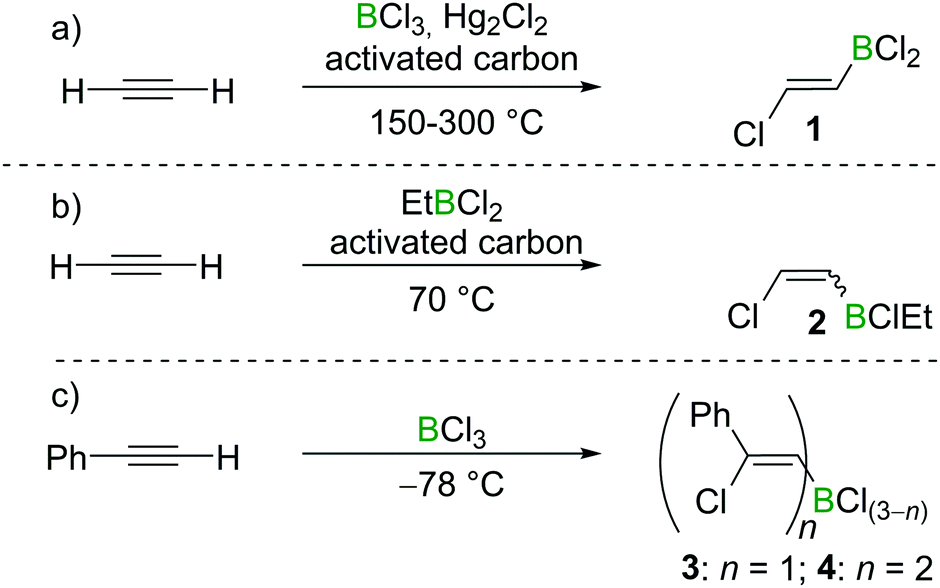 | ||
| Scheme 1 Chloroboration of acetylene/phenylacetylene under various conditions. | ||
Subsequent to Arnold's work, Gipstein et al. found that when EtBCl2 was used, in spite of the reduced Lewis acidity compared to BCl3, the chloroboration of acetylene in the presence of activated carbon could be realised at 70 °C, affording product 2 (configuration not determined) in 90% yield (Scheme 1b).11 Later, Lappert and co-workers studied the chloroboration reaction with a variety of alkynes and boranes.12 For instance, phenylacetylene was reported to undergo chloroboration with one equivalent of BCl3 readily even at −78 °C to afford the syn-addition product 3. The obtained product 3 was shown to react with another equivalent of phenylacetylene in a syn-manner to give compound 4 (Scheme 1c). In the initial report by Lappert and co-workers, the configurations of 3 and 4 were assigned with incomplete evidence. Subsequent studies confirmed that chloroboration of terminal alkynes with BCl3 proceeds in a syn-manner.13 Although the reaction between BCl3 and terminal alkynes such as phenylacetylene occurs promptly, no reactivity was observed when internal alkynes such as diphenylacetylene and BCl3 were mixed at 15 °C. In accordance with the reactivity of BCl3, PhBCl2 readily reacted with two equivalents of phenylacetylene and compound 5 was obtained (Scheme 2a).12 In contrast, when 1-hexyne was treated with half an equivalent of PhBCl2, both chloroboration and carboboration occurred to give the product 6 (Scheme 2b). In a controlled reaction of 1-hexyne with Ph2BCl, carboboration occurred exclusively yielding compound 7 (Scheme 2c).
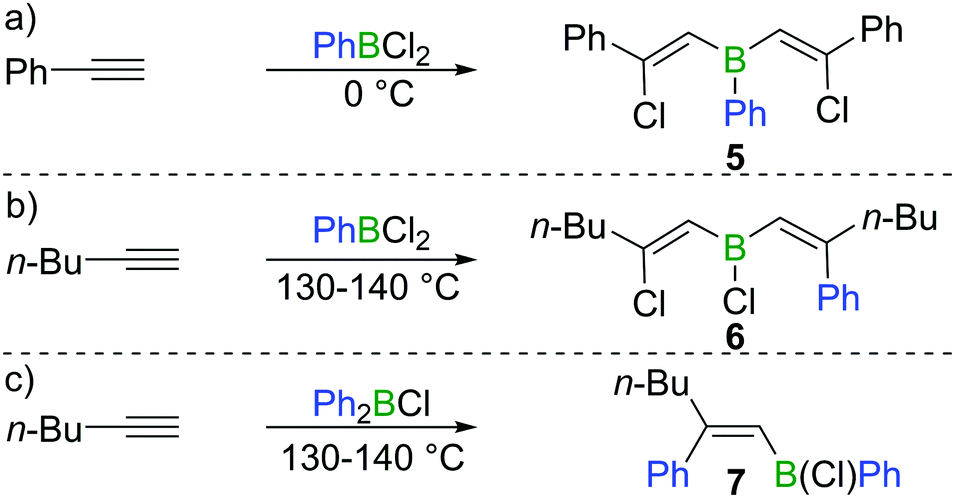 | ||
| Scheme 2 Chloroboration and carboboration of terminal alkynes. | ||
Lappert et al. also investigated the haloboration of acetylene using BBr3. The increased Lewis acidity of BBr3 (relative to BCl3) drastically facilitated the transformation which proceeded at room temperature and the addition product 8 was obtained (Scheme 3a). Alcoholysis of compound 8 with n-butanol afforded the known compound E-9Bu with specified configuration. The configuration of E-9Bu led the authors to assign a trans-configuration to 8. However, a recent study suggested the bromoboration of acetylene typically gives a mixture of E/Z isomers at around 0 °C (for further details and mechanistic discussion, see Sections 2.2 and 2.3). Lappert et al. also found that the addition of pyridine (Py) to 8 resulted in the elimination of Py·BBr3 and regeneration of acetylene, which indicated that the bromoboration of acetylene might be reversible (Scheme 3a). The bromoboration of acetylene with BBr3 followed by esterification with alcohols provides a convenient route to halo-alkenylboronates that serve as versatile building blocks (vide infra).
 | ||
| Scheme 3 Bromoboration reactions to gain insight into the selectivity. | ||
Although BCl3 does not react with internal alkynes, Lappert and co-workers found that bromoboration of diphenylacetylene occurs readily in neat BBr3 at room temperature within one hour. In this case, syn-addition product 10 was formed as confirmed by subsequent protodeboronation reactions with acetic acid (AcOH) to give 11 (Scheme 3b), with protodeboronation known to proceed with retention.12,14a Blackborow performed detailed studies on the bromoboration of 1-hexyne under various conditions (Scheme 3c).14 Generally, the bromoboration of the terminal alkyne proceeds in a Markovnikov fashion. The stereoselectivity, however, was found to be highly dependent on the reaction conditions. For example, when the reaction was performed at −80 °C in petroleum or dichloromethane, syn-addition product 12 was found to be dominant (98%) as determined by analysis post protodeboronation with AcOD. In contrast, when the reaction was performed at −40 °C, the stereoselectivity decreased with the major product being the anti-bromoboration product 13 (64%) while only 30% syn-addition product 12 was observed under these conditions. In addition to E/Z isomers, multiple haloborations to form the respective divinylbromoborane and trivinylborane also were observed. However, as the formation of these multiple borylation products is relatively slow under the reaction conditions and the product distribution was not well defined, these details are not discussed further here.
Subsequent to Lappert's work with BBr3, Eisch and co-workers found that the less Lewis acidic borane PhBBr2 reacts with diphenylacetylene in a reversible manner (Scheme 4a).15 Upon mixing PhBBr2 and diphenylacetylene, cis-bromoboration product 14 was formed rapidly, which was confirmed by protodeboronation with acetic acid. However, prolonged storage of compound 14 in hydrocarbon solvents led to the irreversible formation of carboboration product 15. This suggests that the bromoboration of diphenylacetylene with PhBBr2 is a kinetically favoured but reversible process while the carboboration is a slower and irreversible competing process. Wrackmeyer studied the reaction of 3-hexyne with BBr3 at −78 °C (Scheme 4b).16 When the sample was kept at room temperature for one hour, the syn-addition product 16 was found to be the major product in the reaction mixture (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 = 15:1). However, after several days, the E-isomer 17 became dominant with the ratio of 16:17 switching to 1:7.
:17 = 15:1). However, after several days, the E-isomer 17 became dominant with the ratio of 16:17 switching to 1:7.
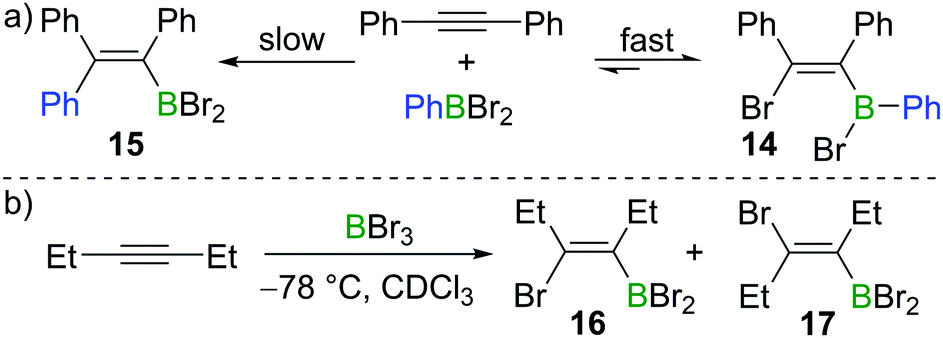 | ||
| Scheme 4 Isomerisation in reactions of internal alkynes with haloboranes. | ||
Eisch also studied the reactivity of MeBI2 with diphenylacetylene. In this case, iodoboration occurred rapidly and compound 18 was obtained (Scheme 5a).17 Siebert found that iodoboration of 3-hexyne with BI3 furnished the syn-addition product 19 rapidly, which, in line with Wrackmeyer's observations, underwent slow isomerisation to form the anti-addition product 20 at room temperature (Scheme 5b).18,19
 | ||
| Scheme 5 Haloboration reactions on various alkynes with different haloboranes. | ||
As discussed above, bromoboration of terminal alkynes with BBr3 does not occur exclusively in a 1:1 stoichiometry due to further reactions of the vinylBBr2 species with additional alkyne. One solution to this problem is to use 9-halo-9-borabicyclo[3.3.1]nonane (9-X–BBN; X = Br, I). Suzuki and co-workers found that 9-Br–BBN could react with one equivalent of 1-octyne to afford 21 in high regio- and stereoselectivity (Scheme 5c).20 They also found that 9-Br–BBN is inert to internal alkynes likely due to its lower Lewis acidity. These early studies clearly demonstrated the viability of alkyne haloboration, albeit complicated in many cases by formation of different haloboration isomers. The origin of Z- and E-configured haloboration products was at the time unclear and required subsequent DFT calculations to provide mechanistic insight.
2.2 Mechanistic studies
The haloboration of alkynes was investigated computationally initially by Uchiyama and co-workers.21Ab initio calculations with second-order Møller–Plesset perturbation theory (MP2) were performed on the haloboration of acetylene and propyne using a dichloromethane continuum solvent model (Scheme 6). The two alkynes first form loose van der Waals complexes A with BX3 (X = Cl, Br, I), which then may transform into π-bonded complexes B if X = Br or I but not Cl. Intermediates A (X = Cl) or B (X = Br, I) then convert into the syn-addition products cis-Pvia four-centred transition state TS. Consistent with the observed reactivity of BX3 with alkynes (BBr3 > BCl3), the reaction energy barriers (energy of TS) decrease in order of BCl3 > BBr3 > BI3 for the syn-addition pathway.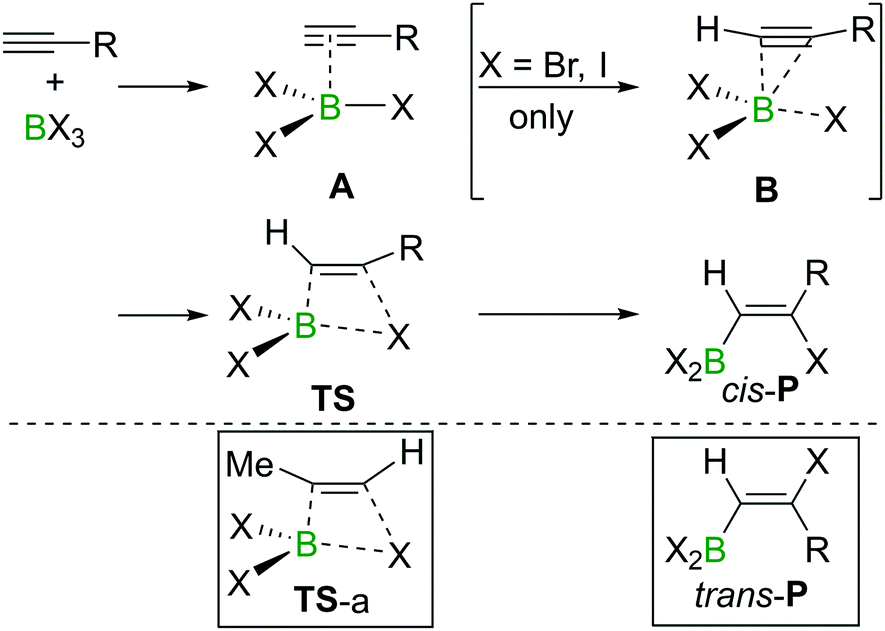 | ||
| Scheme 6 Reaction pathway for cis-haloboration of alkynes. | ||
An energy barrier of 33.8 kcal mol−1 was determined computationally for the cis-chloroboration of acetylene, which agrees with the experimental observations that chloroboration of acetylene required a high reaction temperature and a catalyst. In addition, the haloboration of propyne was found to have a lower energy barrier than acetylene, which is ascribed to hyperconjugation stabilising the developing positive charge at carbon in TS (when R = Me). Furthermore, the anti-Markovnikov pathway for the chloroboration of propyne was also explored computationally. The transition state of anti-Markovnikov pathway TS-a (X = Cl, ΔG‡ = 35.6 kcal mol−1) was found to be much higher than the Markovnikov pathway (X = Cl, ΔG‡ = 15.5 kcal mol−1). The results fit well with the high regioselectivity of terminal alkyne haloboration reactions. For all transformations, the trans-haloboration products trans-P also were computed. For acetylene, they were found to be thermodynamically more stable than the cis-products, which is in agreement with the observation that the trans-chloroboration product was formed exclusively at high temperature in the aforementioned reports (cf. Section 2.1). In contrast, the syn-addition products of propyne haloboration are very close in energy to their anti-addition isomers. Again, this result is consistent with the observation that bromoboration of 1-hexyne at temperatures above −40 °C gave a mixture of both isomers. After the exploration of several different potential reaction pathways, Uchiyama and co-workers proposed that the stereoconversion proceeded via a haloboration/retro-haloboration mechanism of cis-P with BX3 (Scheme 7).
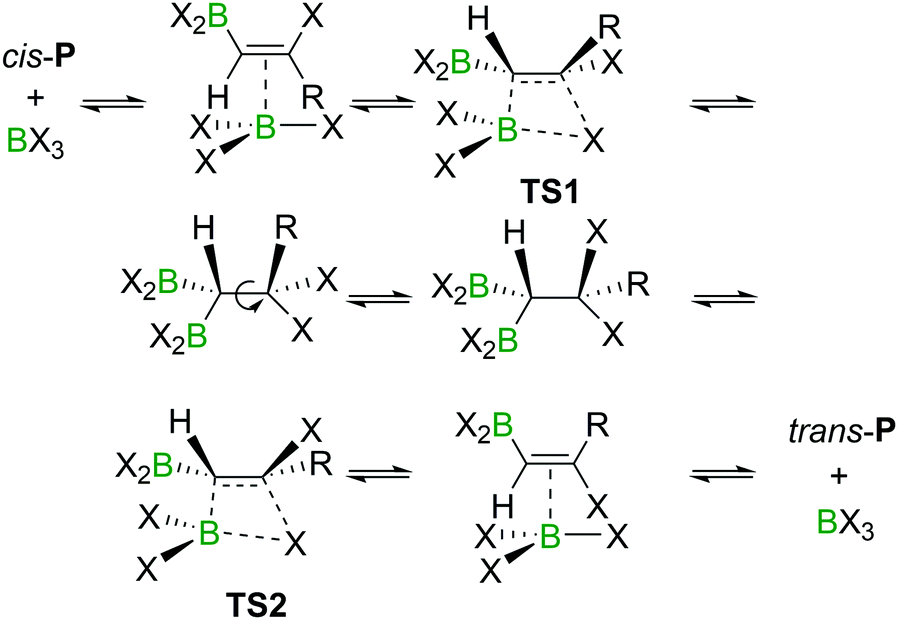 | ||
| Scheme 7 Possible mechanism for stereoconversion mediated by BX3. | ||
The chloroboration of the internal alkyne 2-butyne, and propene was also investigated. For both substrates, the chloroboration was found to be endergonic at 293 K (ΔG = 2.8 kcal mol−1 for 2-butyne; ΔG = 9.4 kcal mol−1 for propene) consistent with that lack of reactivity between BCl3 and internal alkynes and olefins.
2.3 Recent studies into the bromoboration of simple alkynes with BBr3
In Lappert's initial studies, the stereoselectivity for the bromoboration of acetylene was determined by analysis of the post-esterification (conversion of C–BBr2 to C–B(OR)2) products. Recently, Mazal and co-workers carried out similar haloboration experiments at 0 °C and monitored the formation of E/Z vinylbromoborane 8 by NMR spectroscopy (Scheme 8a). With BBr3 distilled from Mg turnings, a mixture of E/Z isomers was obtained (E:Z = 15:85). Interestingly, addition of small amounts of water, NEt3 or [n-Bu4N]Br to the bromoboration reaction facilitated the formation of anti-addition product E-8. It is even more notable that the authors exclusively found the trans-vinylboronate E-9Pin post esterification workup. They proposed radical or polar addition mechanisms involving adventitious HBr to rationalise the observed reactivity.22 By modifying the workup procedures to avoid the formation of HBr, a mixture of E/Z-bromovinylboronate E/Z-9Et could be obtained, which was subjected to transesterification with pinacol, yielding E/Z-9Pin. Selective decomposition of the E-isomer (AcOK, MeOH) enabled the isolation of Z-bromovinylboronate Z-9Pin in useful yields (Scheme 8b).
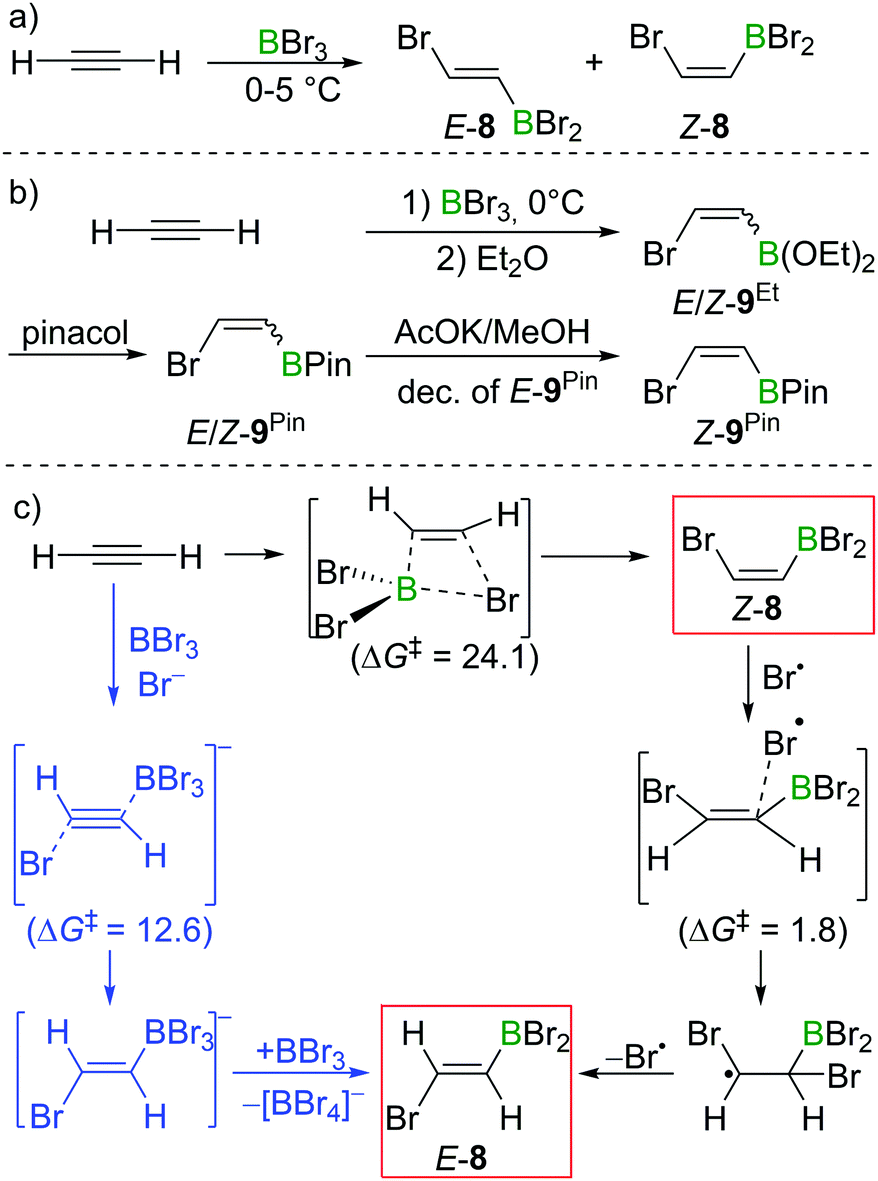 | ||
| Scheme 8 (a) Reaction of acetylene with BBr3. (b) Brønsted-acid-free workup conditions allow for the selective isolation of Z-9Pin. (c) Possible reaction pathways and energies (kcal mol−1) of acetylene bromoboration. | ||
In their proposed radical mechanism, a cis-bromoboration of acetylene occurs first to give Z-8 with an energy barrier of 24.1 kcal mol−1 (MP2/6-31+(d)/PCM(DCM)). Attack of a bromine radical (proposed to be generated from adventitious HBr) on Z-8 proceeds with almost no barrier (1.8 kcal mol−1). Bond rotation followed by bromine radical elimination was then assessed to afford the trans-bromoboration product E-8. An anti-addition of BBr3 and Br− to acetylene was also suggested for the formation of E-8. Acetylene was calculated to first form a π-bonded complex with BBr3, which was then attacked by Br− to form a borate intermediate. Abstraction of Br− with BBr3 from the intermediate gave E-8. The energy barrier for this process was determined to be 12.6 kcal mol−1.
2.4 1,1-Bromoboration of internal alkynes
Recently, the group of Ingleson have revisited the reaction of internal alkynes with BBr3. Consistent with Lappert's observations, they found that in non-polar solvents such as heptane, the reaction of diphenylacetylene with BBr3 at room temperature afforded the cis-1,2-addition product 10 within a few minutes.23 However, upon heating to 60 °C or prolonged standing at room temperature, the 1,1-addition product 22 was observed (Scheme 9a). When a polar solvent such as dichloromethane was used in the haloboration reactions, both 10 and 22 were formed within a few minutes with 22 being the major product (Scheme 9b). No trans-1,2-bromoboration product was observed by in situ NMR spectroscopy at any stage of the reaction. These observations suggest that 1,1-bromoboration proceeds through a polar transition state, likely a vinyl cation type intermediate(s) which is more stabilised by the more polar solvent dichloromethane. Upon reaction with pinacol and NEt3, compound 22 was converted into the corresponding pinacol ester 23 while 10 was converted back into diphenylacetylene. The 1,1-bromoboration could be extended to diarylalkynes and arylalkylalkynes, providing a convenient way to 1-bromo-2,2-diaryl substituted vinylboronate esters.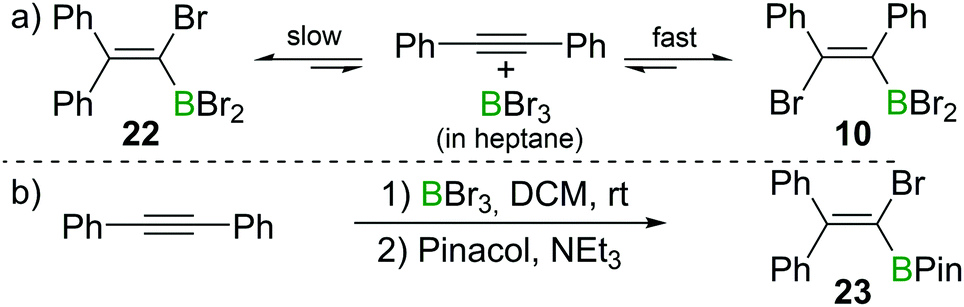 | ||
| Scheme 9 1,1-Bromoboration of diphenylacetylene yields mixtures of the syn-1,2-addition product 10 and the 1,1-addition product 22. | ||
This is a further example that the reactions of alkynes and BX3 can give conditions dependent outcomes. Nevertheless, the use of the appropriate conditions and work-up can lead to isolation of a single haloboration product in useful yield. This diversity in outcome (1,1-, syn-1,2- or anti-1,2-haloboration products being accessible) arguably increases the power of the haloboration transformation.
3. Other alkyne haloboration reactions
This section covers recent synthetic reports that use other (i.e., not Y2B–X, Y = X or R) boron electrophiles or provides alkyne haloboration products that are distinct to those reported in the work discussed in Section 2.3.1 Borocation mediated chloroboration of alkynes
As discussed in Section 2.2, Uchiyama and co-workers found the chloroboration of internal alkynes with BCl3 to be thermodynamically uphill. However, more electrophilic boranes such as BBr3 and BI3 react with internal alkynes readily. Therefore, Ingleson et al. envisioned the use of more electrophilic chloroborane species, such as borocations, to enable internal alkyne chloroboration.24 They prepared the boronium (tetracoordinate at B mono-cation) salt [Cl2B(2-DMAP)][AlCl4] (2-DMAP = 2-dimethylaminopyridine) by sequential addition of 2-DMAP and AlCl3 to BCl3 (Scheme 10a). Due to the strain within the four-membered boracycle, this complex showed a low energy barrier to ring opening and reacted as a masked borenium (= tricoordinate B monocation) ion. [Cl2B(2-DMAP)][AlCl4] reacted with one equivalent of a terminal alkyne at room temperature with high regio- and stereoselectivity. In contrast to the haloboration of alkynes with neutral BX3 which proceeded via a four-membered transition state with concerted formation of B–C and Cl–B bonds, chloroboration of phenylacetylene with [Cl2B(2-DMAP)][AlCl4] was calculated to proceed via a vinyl-cation intermediate, 24a. Intramolecular chloride transfer in 24a occurs with an energy barrier of 24.9 kcal mol−1 to give 24b (at the M06-2X/6-311G(d,p)/PCM(DCM) level of theory), which then rearranges to afford the chelated compound 24. Although the reaction between the boronium complex and terminal alkynes was facile, no reactivity was observed between [Cl2B(2-DMAP)][AlCl4] and internal alkynes. This is presumably due to the requirement to open the four-membered boracycle within [Cl2B(2-DMAP)]+ prior to the haloboration reaction, coupled with the significant N → B π donation (as shown in 24b) in the borenium isomer of [Cl2B(2-DMAP)]+ (reducing the Lewis acidity at B) leading to unfavourable energetics.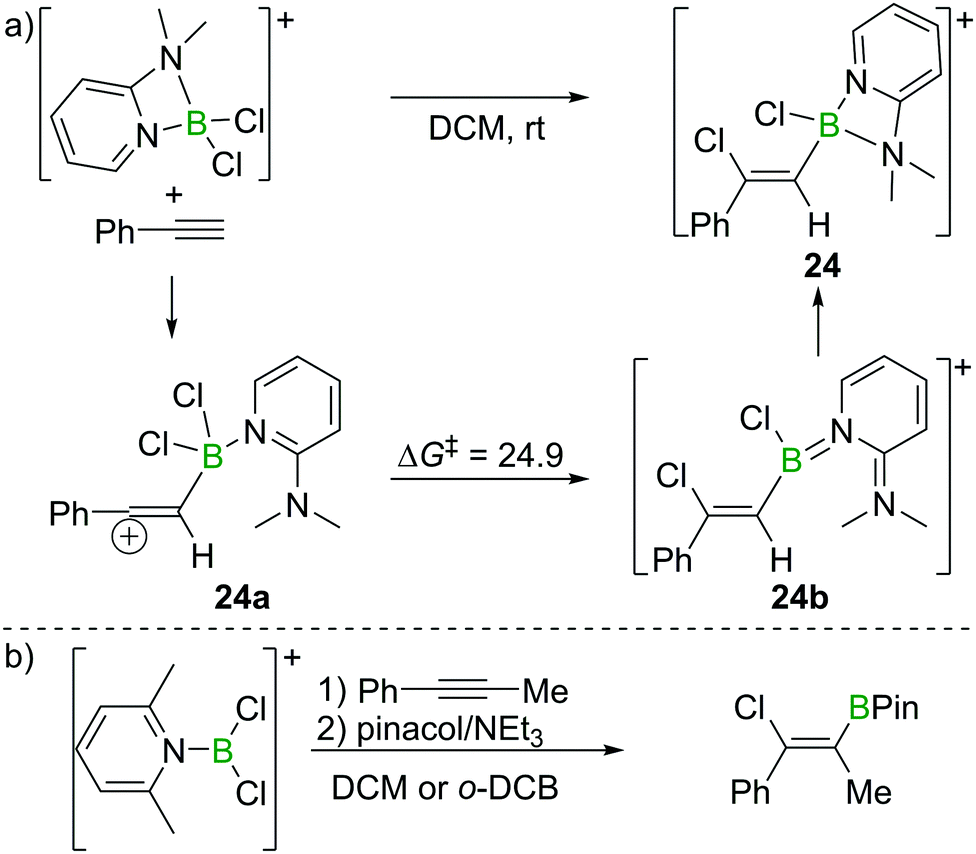 | ||
| Scheme 10 Reaction route and calculated energies (kcal mol−1) for chloroboration of phenylacetylene with boronium/borenium complexes. | ||
Ingleson and co-workers also reported that when the borenium salt [Cl2B(Lut)][AlCl4] (Lut = 2,6-lutidine) was used, syn-addition of the B–Cl bond across both terminal and internal alkynes occurred (Scheme 10b), presumably due to the enhanced electrophilicity at B in this borocation relative to the 2-DMAP analogue. Good chloroboration stereoselectivity was achieved for dialkyl, diaryl and arylalkylalkynes. Remarkably, for arylalkylalkynes, regioselective chloroboration could also be readily realised. All the products could be converted into the corresponding pinacol boronate esters by subsequent esterification with no loss in stereo-/regioisomeric purity.
3.2 1,2-trans-Chloroboration of alkynes
Chloroboration of alkynes typically proceeds in a syn-manner. The group of Ingleson found that treatment of 2-dimethylaminotolan with BCl3 gave the unusual anti-addition product 25 (Scheme 11a).25 Subsequently, Pei and co-workers reported that 2-aminotolan also reacted with PhBCl2 to give the trans-chloroboration product 26 (Scheme 11b).26 The reaction was proposed to be initiated through the activation of the triple bond by the boron moiety, which was then followed by a nucleophilic attack of the alkyne with chloride. This method serves as a convenient way to prepare B,N-fused polycyclic aromatic hydrocarbons. The disparity between the formation of five membered 25 and six membered 26 is notable. This can be attributed to the all sp2-containing 26 having a strong preference for forming six membered boracycles via electrophilic borylation, while the incorporation of a single tetrahedral centre (as in 25) leads to five membered boracycles from electrophilic borylation being the favoured products.27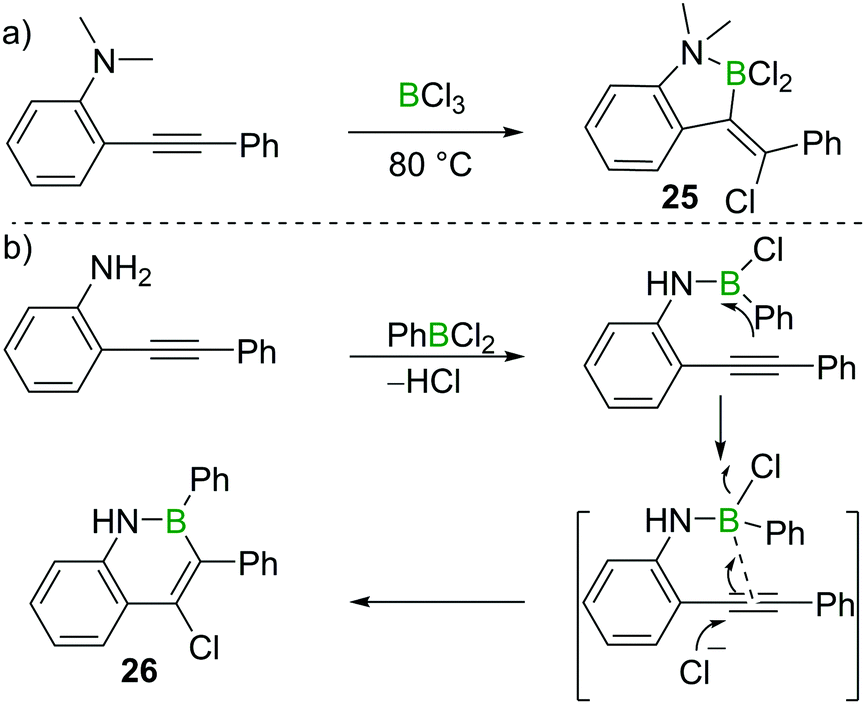 | ||
| Scheme 11 Amino group directed trans-chloroboration of internal alkynes. | ||
3.3 1,3-Chloroboration of alkynes
In 2018, Melen et al. reported a unique 1,3-chloroboration of propargyl esters.28 By treating propargyl benzoate with 1 equivalent of PhBCl2, an intermediate dioxolanonium ion was proposed to be formed via boron promoted cyclisation. The intermediate was assumed to subsequently undergo ring-opening and chloride migration to furnish the corresponding product 27 in high yield at room temperature (Scheme 12a). Interestingly, for a related propargyl ester with two methyl groups at the propargylic position, 1,1-carboboration occurred when PhBCl2 was added to give 27a (Scheme 12b), presumably due to dimethyl substituents leading to a higher barrier for the 1,4-chloride transfer.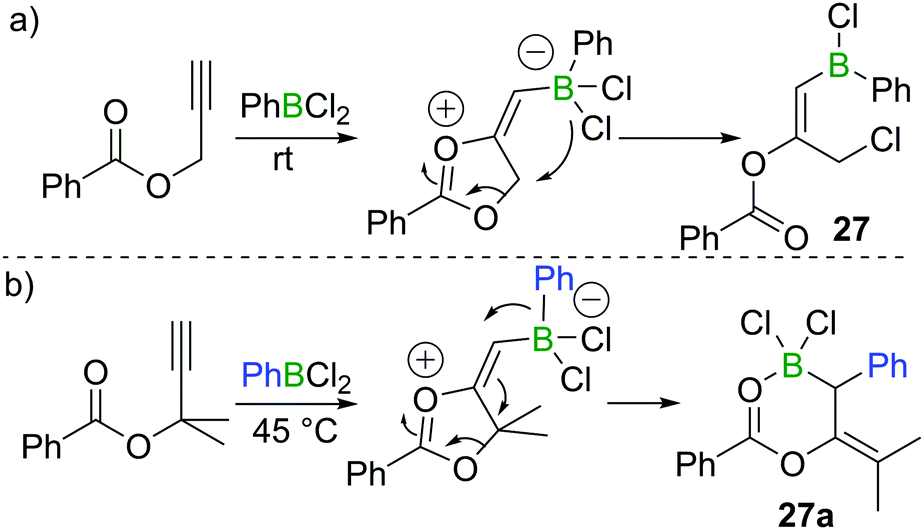 | ||
| Scheme 12 1,3-Chloroboration/1,1-carboboration of propargyl esters. | ||
3.4 Chloroboration of diynes
Ingleson and co-workers found that 1,6-heptadiyne reacts rapidly with BCl3 at room temperature to afford a chlorinated cyclohexene featuring an exocyclic vinylBCl2 moiety.29 The corresponding pinacol protected compound 28 could be isolated in high yield post esterification with pinacol/NEt3 (Scheme 13a). In this case terminal alkyne 1,2-haloboration must have a higher barrier than intramolecular reaction of the alkyne–BCl3 adduct with the second alkyne. Furthermore, the reaction outcome was highly solvent dependent, with dichloromethane and dichloroethane affording 28 in good yield, whereas the use of chloroarenes resulted in very low yields of 28, suggesting the vinyl chloride is made via a carbocationic intermediate, which can engage in side-reactions with aromatic solvents.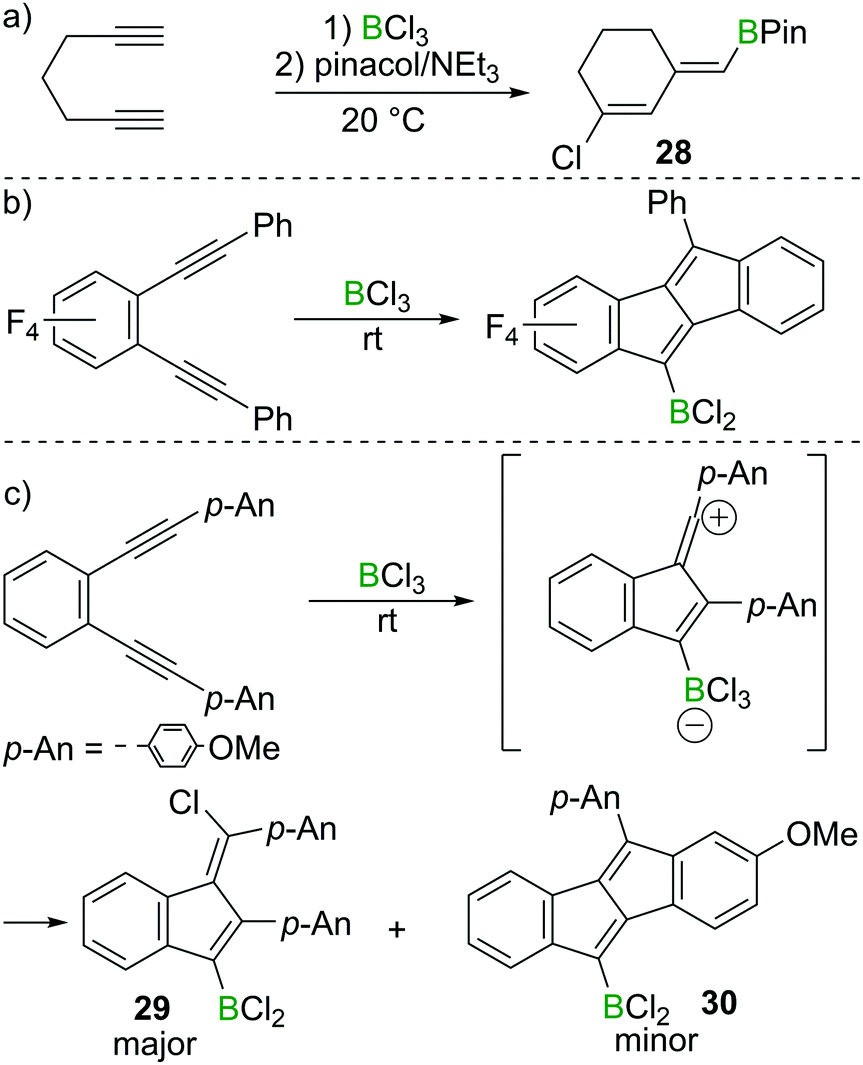 | ||
| Scheme 13 Borylative cyclisation/chloroboration of diynes. | ||
Although BCl3 shows no reactivity towards internal alkynes, facile transformations between BCl3 and 1,2-dialkynyl benzenes have been reported by the groups of Erker, Yamaguchi and subsequently Ingleson.30,31 Presumably these proceed from the adduct between the internal alkyne and BCl3. In these reports, dibenzopentalenes or analogues were the major products in most cases (Scheme 13b). However, Ingleson et al. found that by introduction of electron donating groups such as p-methoxyphenyl (p-An) to 1,2-dialkynyl benzene substrates, the major product is the benzofulvene 29via a formal 1,4-chloroboration reaction. In this case the dibenzopentalene 30 is only observed as the minor product (Scheme 13c).31
Interestingly, the formation of 1,4-chloroboration products becomes favoured in other cases by the addition of an exogenous chloride donor such as [BCl4]− (Scheme 14). For example, 1,2-bis(p-tolylethynyl)benzene reacts with BCl3 affording the corresponding benzofulvene 31 and dibenzopentalene 32 in a 1:2 ratio. However, in the presence of three extra equivalents of a [BCl4]− salt, the ratio of 31 and 32 switched to 1.5:1, indicating the role of [BCl4]− in promoting the formal chloroboration reaction potentially by transferring chloride to the vinyl cation zwitterionic intermediate. Furthermore, precluding the presence of [BCl4]− or [RBCl3]− species in the reaction mixture by using boronium salt [Cl2B(2-DMAP)][AlCl4] significantly reduced the amount of the 1,4-chloroboration product observed. These studies therefore reveal that in the haloboration of diynes the halide source must be considered carefully to ensure a successful reaction outcome.
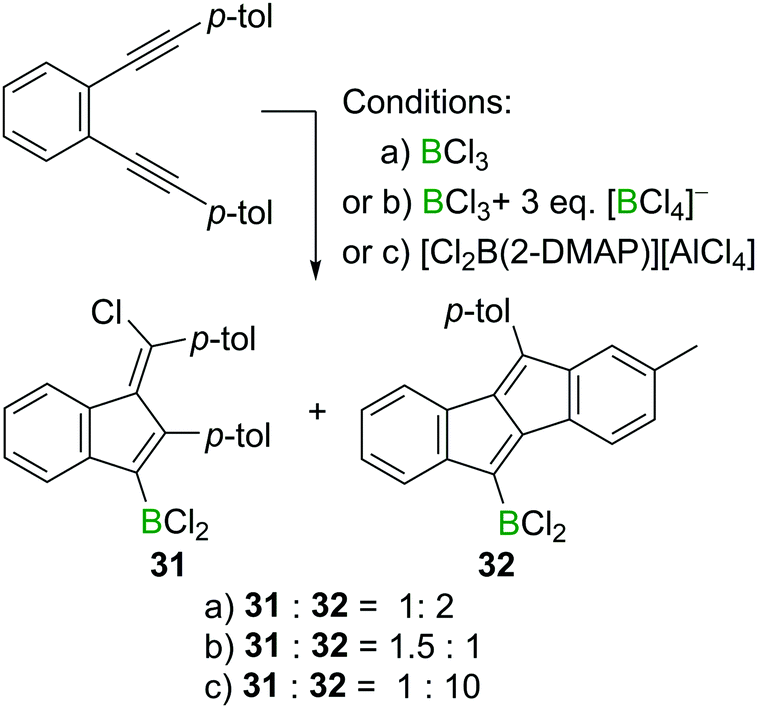 | ||
| Scheme 14 Change in product ratio on altering the boron electrophile or by the addition of an exogenous chloride source. | ||
3.5 Directed trans-bromoboration of alkynes
In addition to the aforementioned trans-bromoboration of acetylene and terminal alkynes, there are a limited number of other examples of internal alkyne trans-bromoboration reactions. Yamato and co-workers reported a BBr3 induced transformation of a o,o′-dimethoxy-substituted tolan derivative (Scheme 15).32 The highly Lewis acidic BBr3 induced a twofold ether cleavage and intramolecular trans-bromoboration, yielding benzofurochromene derivative 33. | ||
| Scheme 15 Directed trans-bromoboration of internal alkynes. | ||
Pei and co-workers also reported a directed trans-bromoboration similar to their trans-chloroboration reaction discussed above (cf. Scheme 11).26 Due to the similarities the details are not discussed again herein.
Sections 2 and 3 show the utility of alkyne haloboration, and this is by far the most developed reaction. The application of the haloboration reaction to other π systems is much less developed and the limited examples reported currently to the best of our knowledge are discussed in Section 4.
4. Haloboration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C double bonds
C double bonds
In their attempted hydroboration of vinyl chloride with B2H6 at −80 °C, DuPont et al. observed decomposition of the putative tris(2-chlorovinyl)borane upon warming to room temperature, whereas allyl chloride underwent hydroboration smoothly even at ambient conditions.33 Those findings were confirmed by Brown and Köster, who investigated the selectivity of allyl chloride hydroboration. Hydroboration resulted in a formal 6:4 anti-Markovnikov (34)/Markovnikov (35) selectivity. However, the Markovnikov product underwent rapid elimination of the vicinal BH2 and Cl groups to form H2BCl and propene (Scheme 16a).34,35 These observations foreshadowed the subsequent reports that found formation of olefin 1,2-haloboration products to be an energetically unfavourable process. Haloboration experiments on cyclohexene by Lappert et al. supported olefin haloboration being energetically uphill: only a mixture of products could be identified, with no α-haloalkyl borane (the primary product from haloboration) observed, indicating that additional reactivity has to take place to lead to an overall exergonic process (Scheme 16b).36
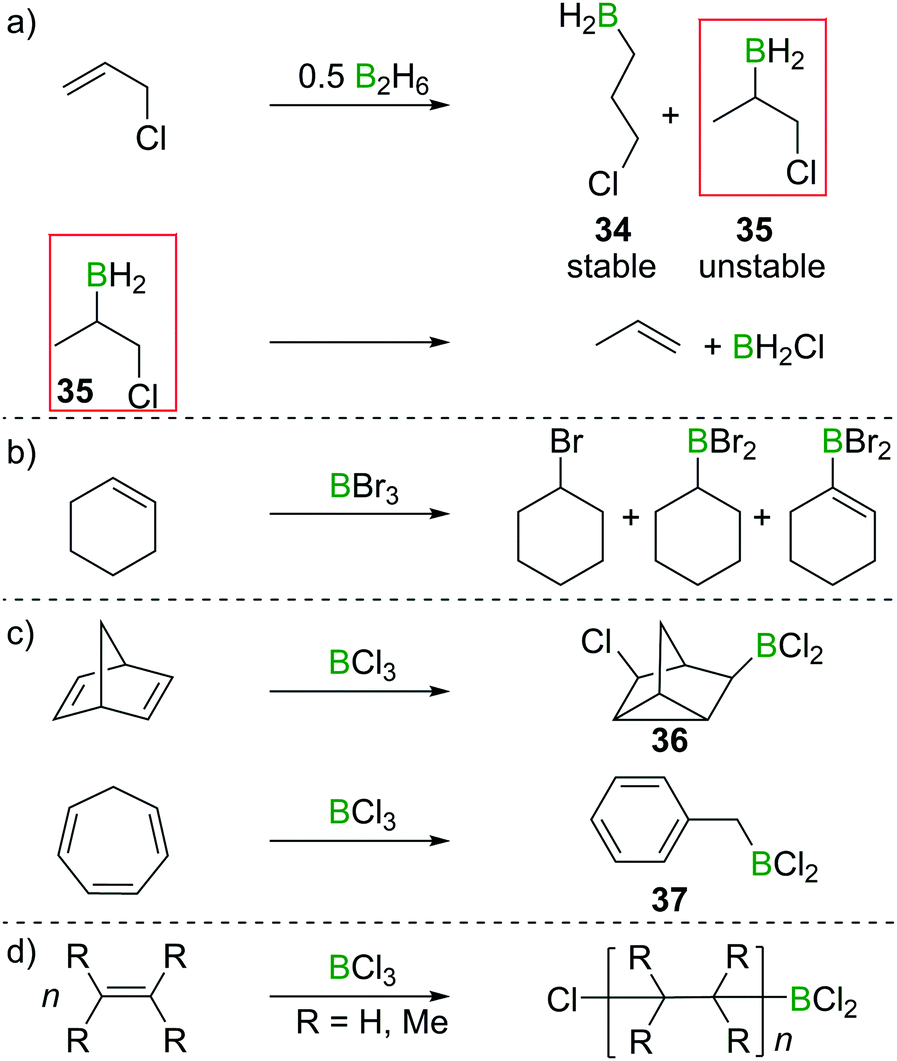 | ||
| Scheme 16 (a) Hydroboration of allyl chloride gives 3-chloropropylborane (34) and 1-chloro-2-propylborane (35); the latter decomposes under elimination of BH2Cl to give propene. (b) Reaction of cyclohexene with BBr3 yields a mixture of products. (c) and (d) Select examples of BCl3 initiated cationic rearrangements or polymerisation of alkenes. | ||
Rearrangements also can be used to trap initial olefin haloboration products, for example the reaction of BCl3 with suitable olefins such as norbornadiene or cycloheptatriene, furnishes tricyclene 36 or BnBCl2 (37, Scheme 16c).36,37
These observation are consistent with the ability of boron trihalides to function as initiators in alkene polymerisation (Scheme 16d), instead of resulting in simple olefin haloboration.38 These experimental findings were corroborated through the earlier discussed calculations by Uchiyama et al., who showed that haloboration of alkenes is thermodynamically unfavourable.21 This is further supported by the experimental finding that alkynyl substituted alkene 38 reacts selectively with BBr3via the alkyne moiety to yield 39 (Scheme 17a).39 Thus haloboration of olefins is an uncommon route to β-haloalkylboranes with only one inter- and one intramolecular synthesis reported to our knowledge.40,41 Other methods are preferred to prepare this versatile structural motif.42
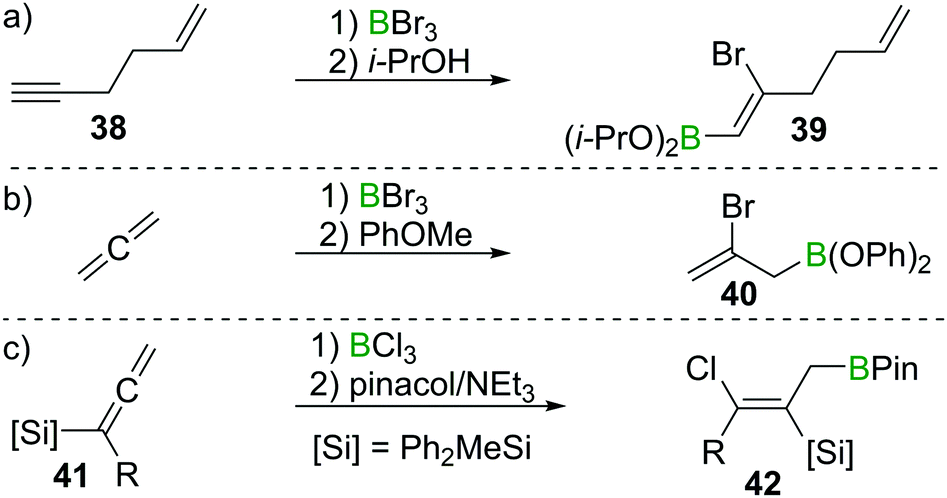 | ||
| Scheme 17 Select examples of selectivity of alkyne haloboration over alkene haloboration and of haloboration reactions on CC double bonds. | ||
While isolated olefins do not undergo haloboration, Lappert et al. showed that allenes reacted smoothly at −20 °C with BBr3 to give the respective 1,2 adduct. Isolation of the bromoborane, however, was problematic as polymerisation of the remaining alkene function occurred at elevated temperatures during attempted distillation.36 By esterification of the BBr2 group through reaction with anisole, polymerisation was prevented and the adduct could be isolated (as 40), thus providing stereoselective access to allylboronic esters (Scheme 17b).43 Iodoboration of terminal allenes with 9-I–BBN also can be a useful route to 2-iodoalkenes, if post haloboration the 9-BBN moiety is removed via acetolysis.44 Notably, if acetolysis is omitted, this reaction can furnish a sought after allylboronate without the need for allyl-metal species which are prone to 1,3 metallotropic shifts.45 If silylated allenes such as 41 are employed, the outcome of the reaction changes. Instead of the 1,2 haloboration product, the formal 1,3 haloboration product 42 is obtained (Scheme 17c). Quantum chemical calculations suggest silyl migration followed by sterically driven 1,3 boryl shifts to be responsible for this special stereoselectivity.46
5. Haloboration of EC bonds (EO, NR)
Lappert et al. investigated the reactivity of aldehydes towards BX3 and suggested haloboration across CO to be a first step in a cascade of reactions that, depending on the nature of the aldehyde, eventually lead to borate esterification, formation of alkylhalides, haloalkylethers, or enolisation with concomitant polymerisation of the resulting vinylboric ester or alkenylether (Scheme 18a).47 The reaction of acetone with BCl3, however, only yielded an ill-defined mixture of products upon release of HCl, which precluded characterisation.48 Remarkably, when perhalogenated ketones (Hal = F, Cl, Br) were employed, usually haloboration of the CO bond occurred,49 but in some instances, simple halide exchange is favoured, giving a different boron halide and the respective perhaloketone.50 In the case of isocyanates and isothiocyanates (43), multiple CE double bonds exist. In these cases, haloboration proceeds stepwise via an R–NCE → BX3 (E = O, S) adduct 44, followed by 1,3 migration of X to C. The higher stability of a CO bond compared to a CN bond then leads to a B shift, giving aminoborane derivatives 45 (Scheme 18b).
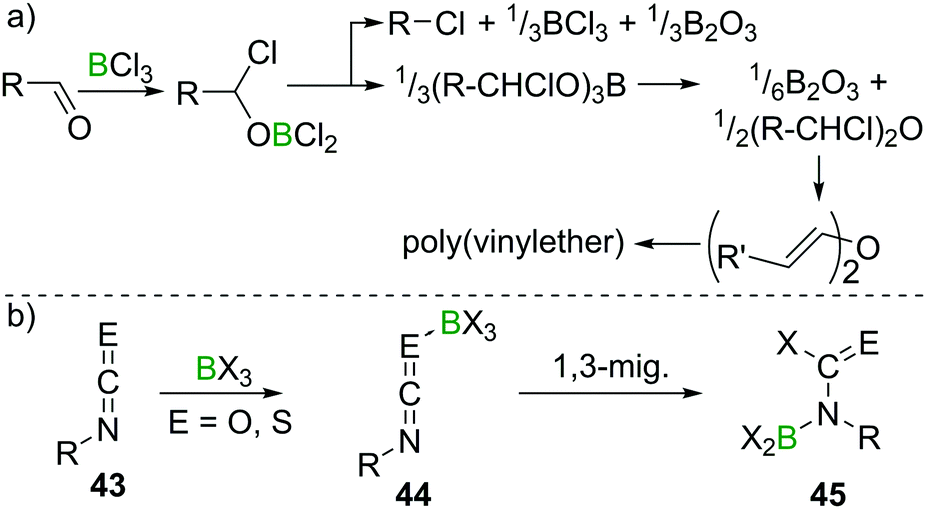 | ||
| Scheme 18 Reaction of aldehydes with BCl3 leading to complex mixtures, depending on the nature of the aldehyde. With iso(thio)cyanates, well defined products are obtained. | ||
Nitriles, which are isostructural and isoelectronic to alkynes can react with BX3 in a fashion related to their CC analogues. However, due to the lone pair at the N atom, the resulting aminoboranes 46 exist in an equilibrium with their cyclic dimers (47, Scheme 19a).51,52 In aminobenzonitriles, reactivity depends on the position of the two groups relative to each other. For para- and meta-substituted aminobenzonitrile, simple adducts of the type R–NH2 → BX3 form. In the case of ortho-aminobenzonitrile and PhBCl2, haloboration of the CN triple bond occurs, furnishing aminoborane 48 which at elevated temperatures cyclises to give the heterocyclic product 49 (Scheme 19b).53 In contrast to the meta- and para-substituted benzonitriles, ambidentate cyanamide reacts exclusively via haloboration of the CN triple bond giving dimeric iminoboranes, due to the strong and immediate mesomeric effect of the NH2 group on the CN triple bond and the thereby increased basicity of the cyano N-atom.54 Isonitriles form stable adducts 50 with the higher haloboranes BX3 (X = Cl, Br, I; for X = F, polymerisation occurs). If heated, these compounds undergo 1,1-haloboration and dimerisation to form 2,5-dihydrodiborapyrazines (51, Scheme 19c).55
 | ||
| Scheme 19 Select examples of EC haloboration reactions. | ||
Finally, reports on the reaction of BX3 with CNR double bonds are, just like in the case of CC double bonds, scarce, and limited to either highly electron deficient perfluorinated imines56,57 or chelating 1,4 diazabutadienes such as 52.58 The latter is an early example of a simple route to 1,3,2-diazaborolidines (53, Scheme 20), which play a crucial role in accessing nucleophilic boranes.59
 | ||
| Scheme 20 Chloroboration of 1,4-diazabutadienes to access 1,3,2-diazaborolidines. | ||
6. Application of alkyne haloboration products in organic synthesis
As described in Sections 2 and 3, haloboration of terminal alkynes is a versatile tool to access selectively 1,1- or 1,2-difunctionalised alkenes. The stereoselectivity can be controlled and the resulting vinylboranes show ambiphilic reactivity: the halogenated C atom is a potential electrophile, whereas the boron-bonded C atom behaves as a nucleophile. In this section we focus on applications that take advantage of both these groups or use the electrophilic vinylBX2 intermediate (particularly in reactions other than esterification). The applications highlighted are distinct to the plethora of reports on forming and utilising vinyl-boronate esters, and the reader is directed to the excellent recent reviews on these topics.60,61Haloboration initially was used to stereoselectively produce singly or doubly halogenated terminal alkenes by reacting either terminal alkynes or haloalkynes with e.g., 9-Br–BBN13,14,62 to give the intermediate alkenylborane 54 followed by acetolysis (55, Scheme 21a).63–65 1,2-Dihaloalkenes can also be accessed from terminal alkynes if post haloboration the boron moiety is transformed into an R–BF3K salt and then treated with an electrophilic halogenating agent.66 Suzuki et al. first employed vinylBBr2 derivative 56 in a one-pot two-steps Negishi/Suzuki–Miyaura cross-coupling sequence to access 1,2-disubstituted alkenes (57) selectively (Scheme 21b).67,68 Although yields were good, the β-bromoalkenyl dibromoboranes 56 were found to be prone to retro-haloboration in the presence of Pd complexes, making further derivatisation to boronic esters expedient to lower the Lewis acidity of the B atom.69–71 During their fundamental studies, Lappert et al. used esterification of R–BX2 with catechol or alkanols to transform their products into stable and conveniently analysed derivatives.12,37 Esterification by ether cleavage72 or by reaction with the respective alcohol73 gave boronic esters with prolonged shelf-life.25 Other functionalisation includes formation of R–B(dan) (dan = 1,8-diaminonaphthalen-N,N′-diyl),74 R–B(MIDA) complexes (MIDA = N-Methyliminodiacetate),75 or R–BF3K salts.66,76 As expected, these classes of compounds are versatile reagents in transition-metal catalysed transformations such as Suzuki–Miyaura cross-coupling or Rh-catalysed [2 + 2 + 2] cycloadditions owing to their ambiphilic nature (electrophilic C–X/nucleophilic C–B).39,77–80
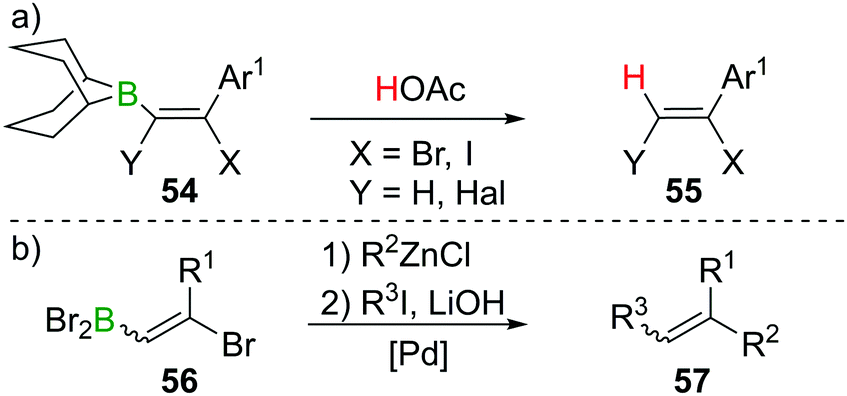 | ||
| Scheme 21 Early examples of functionalisation via B terminus. | ||
The high B-centred Lewis acidity confers the alkenyl dihaloboranes a unique reactivity. The electrophilicity of the B atom paired with the nucleophilicity of the adjacent C atom enables the carboboration of alkynes. For example, the borocation [LutBCl2]+ (Lut = 2,6-lutidine) first reacts with R–CCH (R = alkyl, alkenyl, aryl) under 1,2-syn-haloboration to form compounds of general formula 58. Upon addition of Me–CC–SiMe3, 1,2-syn-carboboration takes place, furnishing a borylated butadiene 59 which can be transformed into the corresponding pinacol ester by reaction with pinacol/NEt3 (60, Scheme 22a).81 Similar reactivity was observed for (F5C6)2BCl.82 If, however, (F5C6)2BX (X = Cl, Br) is reacted with enynes or cyclopropylacetylene, oligomerisation of the alkyne moiety to give 61 without interference of the alkene part of the molecule occurs. In this process, the newly formed C–B bond is added across the CC triple bond in a 1,2 carboboration reaction (Scheme 22b).83 Experiments by Eisch et al. suggest that haloboration is kinetically favoured over carboboration, but due to its irreversibility the latter is often the observed reaction outcome.15 Taking advantage of boron's intrinsically high oxophilicity, haloboration can be exploited to form 1,4-dienes from alkynes, BX3 and aldehydes under deoxygenative conditions in a one-pot process without isolation of the haloboration product 62 that is the intermediate in this process. Remarkably, the stereoselectivity is dependent on the halide employed with BCl3 furnishing exclusively the (E,Z) diastereomer and BBr3 the respective (Z,Z) isomer of 63 (Scheme 22c).84,85
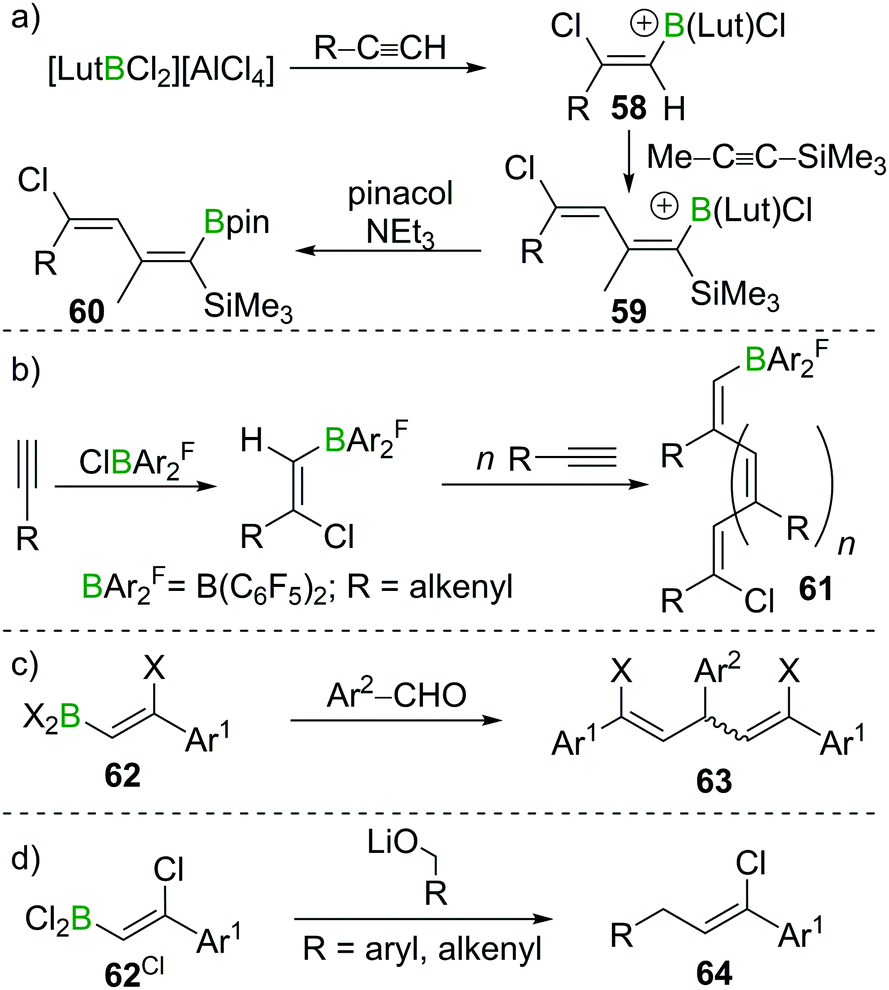 | ||
| Scheme 22 Select examples of functionalisation of haloborated alkynes via the B terminus. | ||
In a similar fashion, propargyl,86 benzyl,87 or allyl88 alcohols can be used to access propargyl-, benzyl-, or allyl-substituted styrenes 64 (Scheme 22d). If instead of BX3 9-I–BBN is used, haloboration of ethoxyethyne yields a haloboration product which reacts with aldehydes via a formal CO carboboration. The resulting secondary alkylborinic esters are readily hydrolysed to α,β unsaturated carbonic esters.89
The electron withdrawing nature of a BX2 group also renders alkenes like E-8 electron poor, making them good substrates in Diels–Alder reactions. Due to the instability of α-halogenated alkylboranes (cf. Section 4), BBr3 elimination transforms the intermediate cyclohexene 65 into a 1,4-cyclohexadiene 66 (Scheme 23).90
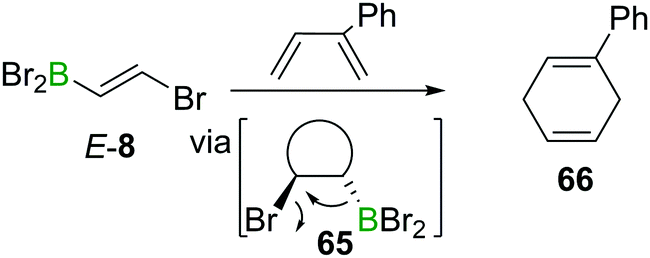 | ||
| Scheme 23 Diels–Alder reactions of haloboration products yield 1,4-cyclohexadienes. | ||
6.1 Haloboration as a tool in natural product synthesis
Polyenes are common substructures in natural products (Scheme 24).91,92 Usually, the double bonds are formed via Wittig-type olefinations,93 and thus can suffer from forming mixtures of both E and Z isomers. Using the appropriate conditions the haloboration of terminal alkynes, as discussed above, proceeds stereoselectively (usually >98%70,94,95) and furnishes ambiphilic halogenated vinyl boronic acid derivatives post workup.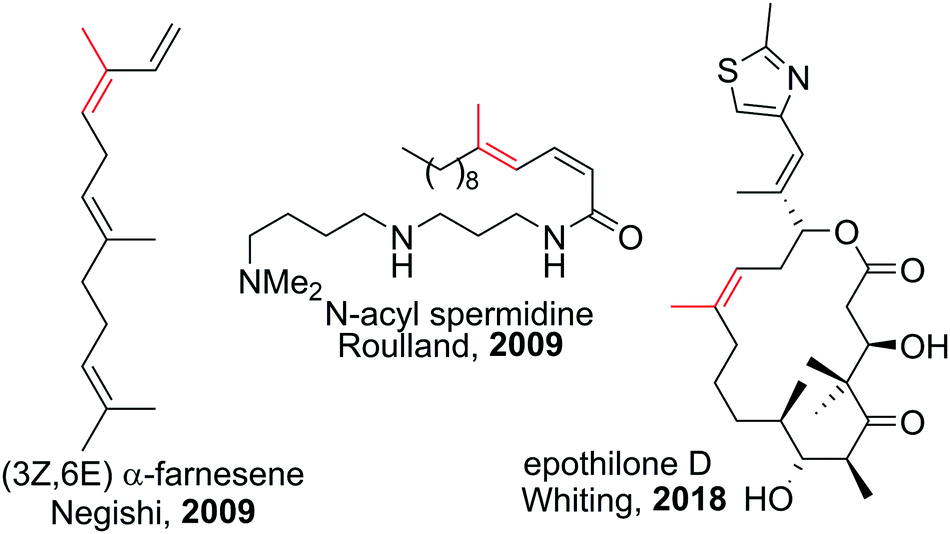 | ||
| Scheme 24 Select natural products synthesised via haloboration/cross-coupling sequences. The fragment incorporated in such manner is coloured in red. | ||
Under the mild conditions of Pd catalysed cross-coupling reactions, the stereo information of the double bond is conserved.94,95 Thus, haloboration/cross-coupling sequences can be applied in the synthesis of olefinic natural products (Scheme 25).96 The inherent instability of polyene boronic acids can be circumvented by either using them directly without further purification or using the MIDA boronate derivatives. Indeed, only 12 different MIDA boronates derived inter alia from the haloboration product of acetylene are necessary to build most polyene natural products via consecutive Suzuki–Miyaura reactions.75,92
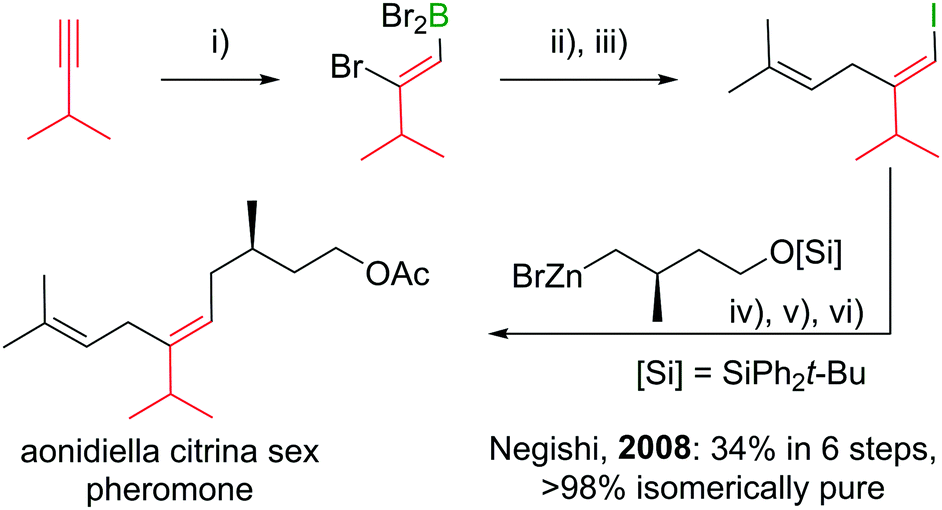 | ||
| Scheme 25 Example of a natural product synthesis sequence based on haloboration. Reagents and conditions: (i) BBr3, DCM, −78 °C to 23 °C, 1 h; (ii) Me2CCH–CH2ZnBr, Pd(PPh3)2Cl2 (1 mol%), THF, 23 °C, 2 h; (iii) I2, NaOAc, THF/H2O, 23 °C, 1 h; (iv) zinc organyl, Pd(PPh3)4 (1 mol%), THF, 23 °C, 5 h; (v) [n-Bu4N]F, THF, 23 °C, 1 h; (vi) Ac2O, pyridine, DCM, 23 °C, 12 h. | ||
6.2 Haloboration in the synthesis of B-doped PAHs
Besides providing a useful route to form β-functionalised boronic-acid derivatives, haloboration is an expedient tool to form boron-doped polycyclic aromatic hydrocarbons (B-PAHs), which are promising candidate materials in the field of organic electronics as they are electron deficient aromatics with a comparatively low LUMO energy.23,97 Haloboration of 1-alkynylnaphthalenes 67 can lead selectively (under appropriate conditions) to either trans-1,2-haloboration products (R = H, alkyl) or 1,1-haloboration products 68 (R = aryl, Scheme 26), which then undergo bora Friedel–Crafts reactions furnishing 1-boraphenalenes (69, Scheme 26a).97 If pyrene instead of naphthalene is used as the hydrocarbon core (70), singly or doubly (71) B-doped PAHs with very low LUMO energies are accessible.23 The utility of bromoboration is then demonstrated by usage of the vinyl-Br unit in a subsequent Negishi coupling enabling donor–acceptor–donor complex 72 to be formed readily (Scheme 26b). In other materials synthesis applications the high Lewis acidity of BBr3 allows for a one-shot double cyclisation of o,o′-dimethoxy-substituted tolan derivatives by ether cleavage and concomitant trans-haloboration, yielding benzofurochromene derivatives (cf.Scheme 15, Section 3).32 In a similar fashion, N-protected propargylamines underwent intramolecular N–B bond formation post haloboration.98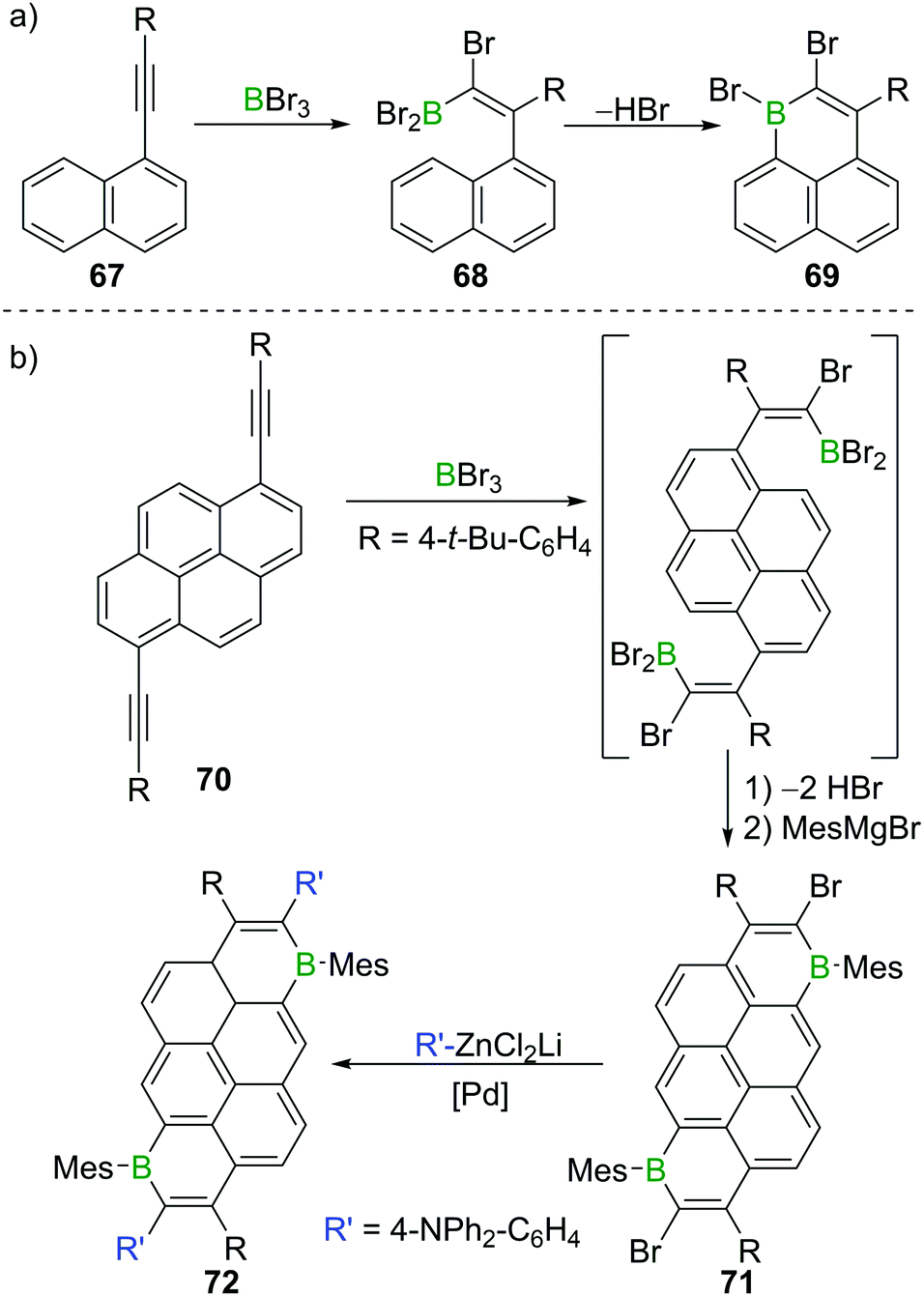 | ||
| Scheme 26 (a) Synthesis of 1-boraphenalene 69via haloboration/bora Friedel–Crafts reaction sequence. (b) Extension to pyrene derivatives. | ||
A related reaction sequence was used to construct even larger B,N doped PAHs.99 Besides being useful to incorporate boron into PAHs, haloboration has also been demonstrated to be a valuable tool to construct B-incorporated polymers. This was achieved by subjecting diynes such as 73 to multiple haloboration reactions using either BBr3 or R2BBr as the B source to yield polymers such as 74 (Scheme 27).100
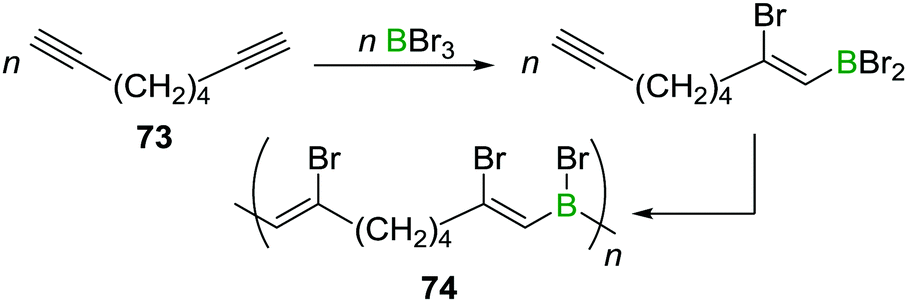 | ||
| Scheme 27 Haloboration of diynes can be used to construct B incorporating PAHs or to access poly(vinylboranes). | ||
7. Conclusion
Since its discovery almost 80 years ago, the haloboration of alkynes has developed into a reliable way to form bifunctional alkenes with excellent control of stereoselectivity possible. The haloboration of other substrates is more limited and further work is required to develop these into broadly useful transformations. Regarding alkyne haloboration, although some mechanistic details are still subject to debate, experimental evidence clearly demonstrates that the stereoselectivity of haloboration can be controlled. The addition of BX3 (X = Cl, Br, I) to a terminal alkyne proceeds via syn-addition of a B–X bond across the CC triple bond and is usually very fast, even at low temperatures. Thus, low-temperature quenching furnishes the Z-adduct selectively. At higher temperatures, isomerisation to the E-adduct can occur. The initial addition as well as the subsequent isomerisation are highly efficient, allowing access to the respective adduct in high yields with stereoselectivities often >98%. Haloboranes such as PhBBr2 or 9-Br–BBN can be employed in haloboration as well. For internal alkynes, stronger electrophiles than BCl3 are needed to effect haloboration, and most simply BBr3 can be used. The lower reactivity of internal CC triple bonds allows for the isolation of the syn-addition product even at room temperature within minutes. Yet, prolonged storage in solution or elevated temperatures (or more polar solvents) can lead to rearrangement of the 1,2-syn addition product to the 1,1-haloboration product in a reversible process. Thus, the addition of BBr3 to internal alkynes yields mixtures of both 1,1- and 1,2-adducts. Notably both the 1,2-adduct and the 1,1 adduct can be isolated selectively using the appropriate conditions.
The combination of halide and boron moieties in the addition product allows for broad diversification of the obtained olefin. For example, haloboration outperforms most other common CC bond formation reactions in terms of selectivity and it has proved its worth in the field of polyene natural product synthesis. Notably, the primary products from haloboration contain vinylBX2 units which often have distinct reactivity compared to vinylB(OR)2 analogues due to the stronger electron withdrawing nature of the BX2 unit. An emerging application of the haloboration reaction is in the incorporation of B atoms into large delocalised π systems. This may either be achieved by using strategically positioned heteroatoms (e.g., O, N) to direct alkyne haloboration, or by alkyne haloboration followed by a bora Friedel–Crafts reaction on alkyne-substituted PAHs. These are attractive as the formed B-doped PAHs are halogenated enabling facile subsequent diversification. We hope that further applications are forthcoming that help bring haloboration out of the shadow of the ubiquitous hydroboration reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made possible by financial support from the Horizon 2020 Research and Innovation Programme (Grant No. 769599). Dr Jürgen Pahl and Saqib Iqbal (both University of Edinburgh) are thanked for useful comments.Notes and references
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- H. C. Brown and B. C. Subba Rao, J. Am. Chem. Soc., 1956, 78, 5694–5695 CrossRef CAS.
- D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS.
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1992, 34, 583–586 CrossRef.
- (a) D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863–3865 CrossRef CAS; (b) P. Y. S. Lam, G. Vincent, D. Bonne and C. G. Clark, Tetrahedron Lett., 2003, 44, 4927–4931 CrossRef CAS.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- For recent reviews on hydroboration of CE bonds, cf.
(a) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS;
(b) A. M. Carroll, T. P. O’Sullivan and P. J. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS;
(c) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS;
(d) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed;
(e) M. L. Shegavi and S. K. Bose, Catal. Sci. Technol., 2019, 9, 3307–3336 RSC.
- Reviews on the haloboration reaction were published by Suzuki and Hara, but the articles were either in Japanese or the journal they were published in ceased to exist: (a) S. Hara, J. Synth. Org. Chem., Jpn., 1990, 48, 1125–1137 CrossRef CAS; (b) A. Suzuki and S. Hara, J. Synth. Org. Chem., Jpn., 1985, 43, 100–111 CrossRef CAS; (c) A. Suzuki, Rev. Heteroat. Chem., 1997, 17, 271–314 CAS . To the best of our knowledge, the last broadly accessible review is: ; (d) A. Suzuki, Pure Appl. Chem., 1986, 58, 629–638 CAS.
- H. R. Arnold, New Organoboron Compounds, US Pat., 2402589, E. I. DuPont de Nemours & Company, 1946 Search PubMed.
- R. J. Jensen, J. K. Hayes, C. L. Cluff and J. M. Thorne, IEEE J. Quantum Electron., 1980, 16, 1352–1356 CrossRef.
- E. Gipstein, P. R. Kippur, M. A. Higgins and B. F. Clark, J. Org. Chem., 1961, 26, 943 CrossRef CAS.
- M. F. Lappert and B. Prokai, J. Organomet. Chem., 1964, 1, 384–400 CrossRef CAS.
- N. Zhou, Q. Wang, A. J. Lough and H. Yan, Can. J. Chem., 2012, 90, 625–630 CrossRef CAS.
- (a) J. R. Blackborow, J. Chem. Soc., Perkin Trans. 2, 1989, 1973 Search PubMed; (b) J. R. Blackborow, J. Organomet. Chem., 1977, 128, 161–166 CrossRef CAS.
- J. J. Eisch and L. J. Gonsior, J. Organomet. Chem., 1967, 8, 53–64 CrossRef CAS.
- B. Wrackmeyer, Polyhedron, 1986, 5, 1709–1721 CrossRef CAS.
- J. J. Eisch and H. P. Becker, J. Organomet. Chem., 1979, 171, 141–153 CrossRef CAS.
- W. Siebert, R. Full, J. Edwin and K. Kinberger, Chem. Ber., 1978, 111, 823–831 CrossRef CAS.
- Y. Nie, S. Schwiegk, H. Pritzkow and W. Siebert, Eur. J. Inorg. Chem., 2004, 1630–1638 CrossRef CAS.
- S. Hara, Y. Satoh, H. Ishiguro and A. Suzuki, Tetrahedron Lett., 1983, 24, 735–738 CrossRef CAS.
- C. Wang and M. Uchiyama, Eur. J. Org. Chem., 2012, 6548–6554 CAS.
- J. Polášek, J. Paciorek, J. Stošek, H. Semrád, M. Munzarová and C. Mazal, J. Org. Chem., 2020, 85, 6992–7000 CrossRef PubMed.
- K. Yuan, R. J. Kahan, C. Si, A. Williams, S. Kirschner, M. Uzelac, E. Zysman-Colman and M. J. Ingleson, Chem. Sci., 2020, 11, 3258–3267 RSC.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518–7522 CrossRef CAS PubMed.
- A. J. Warner, A. Churn, J. S. McGough and M. J. Ingleson, Angew. Chem., Int. Ed., 2017, 56, 354–358 CrossRef CAS PubMed.
- F. D. Zhuang, J. M. Han, S. Tang, J. H. Yang, Q. R. Chen, J. Y. Wang and J. Pei, Organometallics, 2017, 36, 2479–2482 CrossRef CAS.
- S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- L. C. Wilkins, Y. Soltani, J. R. Lawson, B. Slater and R. L. Melen, Chem. – Eur. J., 2018, 24, 7364–7368 CrossRef CAS PubMed.
- A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 11245–11249 CrossRef CAS PubMed.
- C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992–5996 CrossRef CAS PubMed.
- A. J. Warner, K. M. Enright, J. M. Cole, K. Yuan, J. S. McGough and M. J. Ingleson, Org. Biomol. Chem., 2019, 17, 5520–5525 RSC.
- Y. Uchikawa, K. Tazoe, S. Tanaka, X. Feng, T. Matsumoto, J. Tanaka and T. Yamato, Can. J. Chem., 2012, 90, 441–449 CrossRef CAS.
- M. F. Hawthorne and J. A. DuPont, J. Am. Chem. Soc., 1958, 80, 5830–5832 CrossRef CAS.
- H. C. Brown and K. A. Keblys, J. Am. Chem. Soc., 1964, 86, 1791–1795 CrossRef CAS.
- R. Köster, G. Griasnow, W. Larbig and P. Binger, Justus Liebigs Ann. Chem., 1964, 672, 1–34 CrossRef.
- F. Joy, M. F. Lappert and B. Prokai, J. Organomet. Chem., 1966, 5, 506–519 CrossRef CAS.
- F. Joy and M. F. Lappert, Proc. Chem. Soc., 1960, 1, 353–354 Search PubMed.
- For examples of BX3 used as a catalyst in cationic polymerisation, cf. (a) E. L. Muetterties, J. Am. Chem. Soc., 1960, 82, 4163–4166 CrossRef CAS; (b) L. Balogh, L. Wang and R. Faust, Macromolecules, 1994, 27, 3453–3458 CrossRef CAS; (c) L. Balogh, Z. Fodor, T. Kelen and R. Faust, Macromolecules, 1994, 27, 4648–4651 CrossRef CAS.
- K. K. Wang, Z. Wang, A. Tarli and P. Gannett, J. Am. Chem. Soc., 1996, 118, 10783–10791 CrossRef CAS.
- B. M. Mikhailov and M. E. Nikolaeva, Russ. Chem. Bull., 1963, 12, 1246–1250 CrossRef.
- Y. N. Bubnov, N. Y. Kuznetsov, F. V. Pastukhov and V. V. Kublitsky, Eur. J. Org. Chem., 2005, 4633–4639 CrossRef CAS.
- H. Abu Ali, V. M. Dembitsky and M. Srebnik, in Science of Synthesis 6: Category 1, Organometallics, ed. D. E. Kaufmann, D. S. Matteson, E. Schaumann and M. Regitz, Georg Thieme Verlag, Stuttgart, 1st edn, 2005, pp. 867–879 Search PubMed.
- S. Hara and A. Suzuki, Tetrahedron Lett., 1991, 32, 6749–6752 CrossRef CAS.
- S. Hara, S. Takinami, S. Hyuga and A. Suzuki, Chem. Lett., 1984, 345–348 CrossRef CAS.
- C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2–14 CrossRef CAS PubMed.
- Z. Yang, T. Liu, X. Chen, R. Wan, Y. Li, X. Wang, C. H. Yang and J. Chang, Org. Lett., 2019, 21, 9541–9544 CrossRef CAS PubMed.
- M. J. Frazer, W. Gerrard and M. F. Lappert, J. Chem. Soc., 1957, 739 RSC.
- A. G. Massey, J. Chem. Soc., 1961, 1103–1105 RSC.
- (a) G. W. Parshall, Inorg. Chem., 1965, 4, 52–54 CrossRef CAS; (b) E. W. Abel, D. J. Walker and J. N. Wingfield, Inorg. Nucl. Chem. Lett., 1969, 5, 139–140 CrossRef CAS; (c) E. W. Abel, N. Giles, D. J. Walker and J. N. Wingfield, J. Chem. Soc. A, 1971, 1991–1993 RSC.
- E. W. Abel, D. J. Walker and J. N. Wingfield, Inorg. Nucl. Chem. Lett., 1969, 5, 139–140 CrossRef CAS.
- A. Meller and W. Maringgele, Monatsh. Chem., 1968, 99, 2504–2513 CrossRef CAS.
- A. Meller and A. Ossko, Monatsh. Chem., 1969, 100, 1187–1194 CrossRef CAS.
- A. Meller, W. Maringgele and G. Maresch, Monatsh. Chem., 1974, 105, 637–647 CrossRef CAS.
- B. Bonnetot, B. Frange, F. Guilhon, H. Mongeot and W. Einholz, Polyhedron, 1994, 13, 2211–2216 CrossRef CAS.
- A. Meller and H. Batka, Monatsh. Chem., 1969, 100, 1823–1828 CrossRef CAS.
- K. Niedenzu, K. E. Blick and C. D. Miller, Inorg. Chem., 1970, 9, 975–977 CrossRef CAS.
- C. D. Miller and K. Medenzu, Synth. React. Inorg. Met.-Org. Chem., 1972, 2, 217–222 CrossRef CAS.
- F. S. Mair, R. Manning, R. G. Pritchard and J. E. Warren, Chem. Commun., 2001, 1136–1137 RSC.
- (a) M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS PubMed; (b) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed; (c) A. Yagi, H. Kisu and M. Yamashita, Dalton Trans., 2019, 48, 5496–5499 RSC.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- J. Carreras, A. Caballero and P. J. Pérez, Chem. – Asian J., 2019, 14, 329–343 CrossRef CAS PubMed.
- M. J. Bayer, H. Pritzkow and W. Siebert, Z. Naturforsch. B, 2002, 57, 295–300 CAS.
- S. Hara, H. Dojo, S. Takinami and A. Suzuki, Tetrahedron Lett., 1983, 24, 731–734 CrossRef CAS.
- S. Hara, T. Kato and A. Suzuki, Synthesis, 1983, 1005–1006 CrossRef CAS.
- R. V. Edwankar, C. R. Edwankar, J. R. Deschamps and J. M. Cook, J. Org. Chem., 2014, 79, 10030–10048 CrossRef CAS PubMed.
- (a) N. Zhou, Q. Wang, A. J. Lough and H. Yan, Can. J. Chem., 2012, 90, 625–630 CrossRef CAS; (b) M. L. Yao, G. W. Kabalka, D. W. Blevins, M. S. Reddy and L. Yong, Tetrahedron, 2012, 68, 3738–3743 CrossRef CAS.
- S. Hyuga, Y. Chiba, N. Yamashina, S. Hara and A. Suzuki, Chem. Lett., 1987, 1757–1760 CrossRef CAS.
- Y. Satoh, H. Serizawa, N. Miyaura, S. Hara and A. Suzuki, Tetrahedron Lett., 1988, 29, 1811–1814 CrossRef CAS.
- M. L. Yao, M. S. Reddy, W. Zeng, K. Hall, I. Walflsh and G. W. Kabalka, J. Org. Chem., 2009, 74, 1385–1387 CrossRef CAS PubMed.
- C. Wang, Z. Xu, T. Tobrman and E. I. Negishi, Adv. Synth. Catal., 2010, 352, 627–631 CrossRef CAS PubMed.
- C. Mazal and M. Vaultier, Tetrahedron Lett., 1994, 35, 3089–3090 CrossRef CAS.
- N. Yamashina, S. Hyuga, S. Hara and A. Suzuki, Tetrahedron Lett., 1989, 30, 6555–6558 CrossRef CAS.
- S. Hyuga, N. Yamashina, S. Hara and A. Suzuki, Chem. Lett., 1988, 809–812 CrossRef CAS.
- Y. Sumida, R. Harada, T. Kato-Sumida, K. Johmoto, H. Uekusa and T. Hosoya, Org. Lett., 2014, 16, 6240–6243 CrossRef CAS PubMed.
- S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS PubMed.
- X. Fang, J. Sun and X. Tong, Chem. Commun., 2010, 46, 3800–3802 RSC.
- K. K. Wang and Z. Wang, Tetrahedron Lett., 1994, 35, 1829–1832 CrossRef CAS.
- A. Tarli and K. K. Wang, J. Org. Chem., 1997, 62, 8841–8847 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2002, 124, 5025–5036 CrossRef CAS PubMed.
- M. Sato, Y. Yamamoto, S. Hara and A. Suzuki, Tetrahedron Lett., 1993, 34, 7071–7074 CrossRef CAS.
- J. R. Lawson, V. Fasano, J. Cid, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 6060–6070 RSC.
- K. Škoch, C. Pauly, C. G. Daniliuc, K. Bergander, G. Kehr and G. Erker, Dalton Trans., 2019, 48, 4837–4845 RSC.
- A. Ueno, J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem. – Eur. J., 2018, 24, 10044–10048 CrossRef CAS PubMed.
- G. W. Kabalka, Z. Wu and Y. Ju, Org. Lett., 2002, 4, 1491–1493 CrossRef CAS PubMed.
- G. W. Kabalka, M. L. Yao, S. Borella, Z. Wu, Y. H. Ju and T. Quick, J. Org. Chem., 2008, 73, 2668–2673 CrossRef CAS PubMed.
- G. W. Kabalka, Z. Wu and Y. Ju, Org. Lett., 2004, 6, 3929–3931 CrossRef CAS PubMed.
- G. W. Kabalka, M. L. Yao, S. Borella and Z. Z. Wu, Org. Lett., 2005, 7, 2865–2867 CrossRef CAS PubMed.
- G. W. Kabalka, M. L. Yao, S. Borella and Z. Wu, Chem. Commun., 2005, 2492–2494 RSC.
- Y. Satoh, T. Tayano, S. Hara and A. Suzuki, Tetrahedron Lett., 1989, 30, 5153–5156 CrossRef CAS.
- D. A. Singleton and S. W. Leung, J. Organomet. Chem., 1997, 544, 157–161 CrossRef CAS.
- C. Thirsk and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2002, 999–1023 RSC.
- E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS PubMed.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- S. Xu, C. T. Lee, H. Rao and E. I. Negishi, Adv. Synth. Catal., 2011, 353, 2981–2987 CrossRef CAS PubMed.
- E. I. Negishi, T. Tobrman, H. Rao, S. Xu and C. T. Lee, Isr. J. Chem., 2010, 50, 696–701 CrossRef CAS PubMed.
- (a) N. Miyaura, Y. Satoh, S. Hara and A. Suzuki, Bull. Chem. Soc. Jpn., 1986, 59, 2029–2031 CrossRef CAS; (b) A. B. Smith, G. K. Friestad, J. J. W. Duan, J. Barbosa, K. G. Hull, M. Iwashima, Y. Qiu, P. Grant Spoors, E. Bertounesque and B. A. Salvatore, J. Org. Chem., 1998, 63, 7596–7597 CrossRef CAS; (c) M. D. Chappell, S. J. Stachel, C. B. Lee and S. J. Danishefsky, Org. Lett., 2000, 2, 1633–1636 CrossRef CAS PubMed; (d) Z. Xu and E. I. Negishi, Org. Lett., 2008, 10, 4311–4314 CrossRef CAS PubMed; (e) X. Guinchard, X. Bugaut, C. Cook and E. Roulland, Chem. – Eur. J., 2009, 15, 5793–5798 CrossRef CAS PubMed; (f) C. Wang, T. Tobrman, Z. Xu and E. I. Negishi, Org. Lett., 2009, 11, 4092–4095 CrossRef CAS PubMed; (g) A. M. Haydl and B. Breit, Chem. – Eur. J., 2017, 23, 541–545 CrossRef CAS PubMed; (h) K. S. Madden, B. Laroche, S. David, A. S. Batsanov, D. Thompson, J. P. Knowles and A. Whiting, Eur. J. Org. Chem., 2018, 5312–5322 CrossRef CAS.
- R. J. Kahan, D. L. Crossley, J. Cid, J. E. Radcliffe and M. J. Ingleson, Angew. Chem., Int. Ed., 2018, 57, 8084–8088 CrossRef CAS PubMed.
- A. Meller, F. J. Hirninger, M. N. W. Maringgele, A. Meller, F. J. Hirninger and M. Noltemeyer, Chem. Ber., 1981, 114, 2519–2535 CrossRef.
- F. D. Zhuang, Z. H. Sun, Z. F. Yao, Q. R. Chen, Z. Huang, J. H. Yang, J. Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2019, 58, 10708–10712 CrossRef CAS PubMed.
- (a) Y. Chujo, I. Tomita and T. Saegusa, Macromolecules, 1990, 3, 687–689 CrossRef; (b) Y. Chujo, I. Tomita and T. Saegusa, Makromol. Chem., Macromol. Symp., 1993, 70–71, 47–56 CrossRef CAS; (c) N. Matsumi and Y. Chujo, Polym. Bull., 1997, 39, 295–302 CrossRef CAS; (d) M. Miyata, N. Matsumi and Y. Chujo, Polym. Bull., 1999, 42, 505–510 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |