
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective vinylogous-Mukaiyama-type dearomatisation by anion-binding catalysis†
Kirandeep
Kaur‡
a,
Jorge
Humbrías-Martín‡
b,
Leon
Hoppmann
a,
Jose A.
Fernández-Salas
 bc,
Constantin G.
Daniliuc
a,
José
Alemán
*bc and
Olga García
Mancheño
*a
bc,
Constantin G.
Daniliuc
a,
José
Alemán
*bc and
Olga García
Mancheño
*a
aOrganic Chemistry Institute, University of Münster, 48149 Münster, Germany. E-mail: olga.garcia@uni-muenster.de
bOrganic Chemistry Department (Module 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 – Madrid, Spain. E-mail: jose.aleman@uam.es; Web: http://www.uam.es/jose.aleman
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 – Madrid, Spain
First published on 20th August 2021
Abstract
The first enantioselective vinylogous Mukaiyama-type dearomatisation of heteroarenes under anion-binding catalysis is presented. A recyclable tetrakistriazole catalyst was used for the enantiocontrol of the remote vinylogous active position of silyl dienol ethers. This approach provided chiral heterocycles bearing α,β-unsaturated chains with complete regioselectivity and excellent enantioselectivities (up to 97.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 e.r.).
:2.5 e.r.).
Asymmetric vinylogous-Mukaiyama reactions have become a powerful tool for the generation of multifunctional chiral scaffolds commonly occurring in natural products bearing easily derivatisable unsaturated carbon chains, which allow the construction of more complex structures.1 Based on Fuson's principle of vinylogy,2 the incorporation of an additional adjacent double bond to silyl enol ethers extends and strengthens their nucleophilicity at the γ-position, which provides a different, appealing connectivity in the products. As a result, several enantioselective approaches for vinylogous Mukaiyama-type reactions have been developed with silyl dienolates.1 Among them, multiple organocatalytic systems based on different activation modes, including hydrogen-bonding, Lewis-base and ion-pairing catalysis, have been described (eqn (A), Scheme 1).3 However, vinylogous Mukaiyama reactions under anion-binding organocatalysis4 has long been prevented, most probably as a consequence of the long-range enantioinduction that the silyl dienolate would require.5
 | ||
| Scheme 1 Previous organocatalytic vinylogous Mukaiyama strategies (eqn (A)) and the present work (eqn (B)). | ||
Inspired by the power of dearomatisation methods6 and the anion-binding catalysed asymmetric functionalisation of activated N-heteroarenes such as (iso)quinolines and pyridines by nucleophilic addition,4,7,8 we tackled this deficiency in the vinylogous-Mukaiyama technology. To the best of our knowledge, the use of silyl dienol ether derivatives as vinylogous nucleophiles in this type of asymmetric dearomatisation process has never been accomplished. This approach will allow straightforward access to highly functionalised enantiopure N-heterocyclic derivatives, which are abundant structural motifs in high demand for drug discovery processes,9–11 in a single operation from readily available heteroaromatic platforms.12
Based on previous studies,8d,13 we envisioned that multidentate foldameric triazole-based catalysts, able to form a chiral contact ion-pair by recognition of the counterion of the in situ generated N-acyl salt as well as bridge both the cationic substrate and nucleophile, would be an excellent platform to test this vinylogous reaction. Hence, in this work, we present the asymmetric dearomatisation of heteroarenes such as quinazolines with silyl dienol ethers, enabling the first vinylogous Mukaiyama, Mannich-type dearomatization reaction under anion-binding catalysis (eqn (B), Scheme 1).
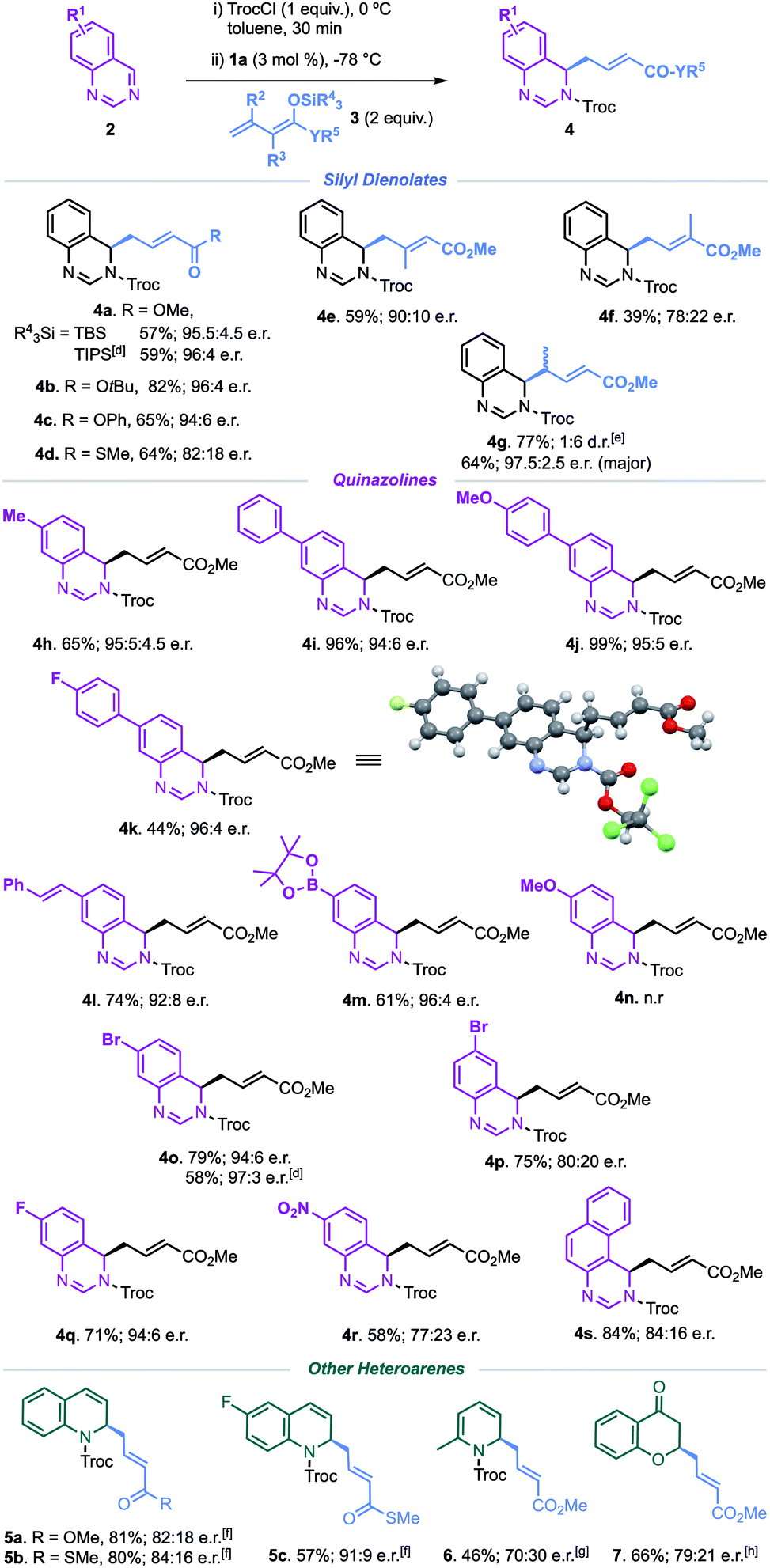
We began our investigations by studying the reaction of quinazoline (2a) as the model substrate with tert-butyldimethylsilyl (TBS)-substituted methylcrotonate nucleophile 3a (Table 1, see the ESI† for full screening). The reaction was performed in the presence of different hydrogen-bond (HB) donor organocatalysts 1 and 2,2,2-trichloroethoxycarbonyl chloride (TrocCl) as the acylating agent for the in situ formation of the quinazolinium salt. At first, all catalysts were examined in MTBE at –78 °C (entries 1–5), as a strong uncatalysed background reaction to form a regioisomeric mixture of the vinylogous products 4a and 4a′ was always observed (1.7:1 ratio, entry 6).14 Interestingly, catalytic reactions with the HB-donors 1 only delivered the C4-addition product 4a. The tetrakistriazoles 1a and 1b showed superior efficiency regarding both yield and enantioselectivity (entries 1 and 2), while the more powerful HB-donor bisthiourea 1e displayed almost no stereocontrol (entry 5). After selecting 1a as the optimal catalyst, the catalytic loading was considered (entries 7 and 8), the best enantioselectivity was shown when employing 3 mol% of 1a (entry 7). Finally, changing the solvent to toluene led to the 3,4-dihydroquinazoline 4a in an enhanced excellent enantioselectivity of 95.5:4.5 e.r. (entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | 4a/4a′b | Yield 4ac (%) | e.r.d |
| a Conditions: (i) 2a (0.1 mmol, 1 equiv.) and TrocCl (1 equiv.) in the appropriate solvent at 0 °C, 30 min; (ii) at −78 °C, catalyst 1 (x mol%) and 3a (2 equiv.) were added and the reaction was stirred for 18 h. b 4a/4a′ ratio determined using 1H NMR of the reaction mixture. c Yield of the isolated product. d The enantiomeric ratios (e.r.) were measured using chiral-phase SFC. e In the uncatalysed reaction, 4a was formed in 31% yield and the C2-addition regioisomer 4a′ in 18% yield (see the ESI). | |||||
| 1 | 1a (10) | MTBE | >25:1 |
69 | 84:16 |
| 2 | 1b (10) | MTBE | >25:1 |
69 | 82:18 |
| 3 | 1c (10) | MTBE | >25:1 |
46 | 77:23 |
| 4 | 1d (10) | MTBE | >25:1 |
58 | 75:25 |
| 5 | 1e (10) | MTBE | >25:1 |
48 | 52:48 |
| 6 | — | MTBE | 1.7:1 |
49e | — |
| 7 | 1a (3) | MTBE | >25:1 |
73 | 90:10 |
| 8 | 1a (1) | MTBE | >25:1 |
63 | 88:12 |
| 9 | 1a (3) | Et2O | >25:1 |
54 | 87:13 |
| 10 | 1a (3) | Toluene |
>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_i_char_2009.gif) :1 :1
|
57 |
95.5:4.5
|

With the optimised conditions in hand (entry 10, Table 1), the scope of the reaction was studied (Table 2). Different silyl-dienol derivatives were initially tested (Table 2, top). While the tert-butyl and phenyl dienol ethers performed very efficiently, giving rise to the desired products (4b and 4c) in excellent yields and enantioselectivities, a thioester substitution led to inferior enantiocontrol (4d). It should be highlighted that the more sterically hindered triisopropylsilyl (TIPS) substitution at the dienol derivative provided a slightly improved enantioselectivity, furnishing 4a in 96:4 e.r. Moreover, substitution on the diene-unit was also tolerated. The 2-methyl dienol 3e gave rise to the β,β-disubstituted product 4e in a 90:10 e.r., whereas substitution at the C-3 position led to a decrease in both the yield and the enantioselectivity (4f). Remarkably, the γ-methyl derivative built the product 4g as a 1:6 diasteromeric mixture and at an excellent 97.5:2.5 e.r. (major isomer).
|
a Reaction conditions: (i) 2 (0.1 mmol, 1.0 equiv.) and TrocCl (1.0 equiv.) in toluene 30 min at 0 °C; (ii) after cooling down to −78 °C, 1a (3 mol%) and TBS-3 (2 equiv.) were added and the reaction was stirred for 18–24 h.
b Yields of the isolated products.
c Enantiomeric ratios determined using chiral-phase SFC.
d Reaction performed with TIPS-dienolate 3b.
e Determined using 1HNMR.
f Reaction using 1d (10 mol%) in Et2O:C6F6 (3:1) at −30 °C.
g Reaction using 1d (10 mol%) in C6F6 at 6 °C.
h Reaction using 1a (5 mol%) in toluene at −78 °C.
|
|---|
|
Next, the scope of the quinazoline was examined (Table 2, middle). The method tolerated a variety of groups, proceeding remarkably smoothly with the 7-alkyl and aryl substituted quinazolines. Thus, the dearomatised products 4h and 4i–k were obtained with excellent enantiocontrol, no matter the electronic nature of the substituent. An olefin conjugated system or the versatile pinacolatoboron (Bpin) moiety were likewise well-suited, building the desired products 4l and 4m with excellent enantioselectivities. Unexpectedly, even when full conversion of 7-methoxyquinazoline into its stabilized N-acyl iminium ion was observed, 4n was not formed and the starting quinazoline was mostly recovered after workup. Conversely, reaction with halogenated 7-bromo and 7-fluor quinazolines led to the corresponding substituted adducts 4o and 4q with very good enantioselectivities (up to 97:3 e.r.), while the nitro group showed significantly lower enantiocontrol (4r). Moreover, substitution at positions 6 (4p) and 5 (4s) was also compatible. Moreover, in order to extend the applicability of the method, other heteroarenes such as quinoline, pyridine and 4-chromenone15 derivatives were also enrolled (see the ESI† for more details), providing the corresponding products 5–7 in moderate to good enantioselectivities. The absolute configuration of the newly formed stereocentre was unequivocally assigned as (R) using X-ray crystallographic analysis of the quinazoline product 4k (see Table 2 and the ESI†).16 The same stereochemical outcome was assumed for all the compounds of this series.
To our delight, upscaling the model reaction of quinazoline (2a) 10 times (1 mmol vs. 0.1 mmol) led to a remarkable enhancement in the efficiency of the process (Scheme 2, top). In this case, the TIPS-dienolate 3b that led to the best enantioselectivity was employed, providing the desired product in a significantly higher yield (83% vs. 59%), while no erosion of the enantioselectivity of the process was observed. In addition, to further prove the utility and applicability of the method, we showed the possibility of recovering the catalyst from the reaction mixture (in 90% yield) and re-using it (Scheme 2, middle). Hence, a subsequent reaction with the recovered catalyst gave similar results in terms of both yield and enantioselectivity (see entry 10, Table 1). We next investigated the synthetic value of the obtained multifunctional chiral quinazoline derivatives with respect to the presence of the α,β-unsaturated system introduced through the vinylogous addition. Thus, the N-deprotection of 4a (0.5 mmol, 96:4 e.r.) with Zn followed by selective hydrogenation of the conjugated double bond led to the interesting chiral lactam derivative 8 after in situ cyclisation without significant erosion of the enantiopurity of the final product (Scheme 2, bottom).
 | ||
| Scheme 2 Upscaling of the reaction, catalyst recovery/reusability and derivatisation towards ring-fused systems. | ||
Finally, evolving from earlier reports,4,13 a plausible mechanism that explains the observed enantioselectivity outcome of this vinylogous asymmetric anion-binding catalytic reaction is proposed (Scheme 3). After the in situ generation of the substrate of the reaction, the quinazolium salt I, upon treatment of quinazoline with TrocCl, a chiral contact ion pair II between catalyst 1a and I by anion-binding to its counteranion is formed. A plausible transition state (III) presenting multiple interactions of the chiral contact ion pair complex II with both reactants can then be envisioned. Thus, the cationic substrate I is fixed via coulomb attractions with the anionic 1a–Cl complex and π–π stacking with one arm of the catalyst. In addition, the TBS-dienolate 3a shows hydrogen-bonding with the chloride counteranion, orientating its distal δ-carbon towards the C4 position of the substrate17 and allowing for the selective vinylogous nucleophilic attack of the more accessible Re-face of the quinazolinium moiety. Hence, (R)-4a is obtained as the major product, with concomitant formation of TBSCl.
 | ||
| Scheme 3 Proposed mechanism. | ||
In conclusion, we have described the first enantioselective organocatalysed vinylogous Mukaiyama-type reaction for the dearomatisation of heteroarenes under anion binding catalysis. The process takes place with high enantiocontrol (up to 97.5:2.5 e.r.) and with a range of differently substituted quinazolines, which could be extended to other types of N- and O-heterocycles. In addition, different experiments and studies such as catalyst recovery/reusability and the derivatisation of the final products have been performed in order to further illustrate the utility and applicability of the presented method.
The European Research Council (ERC-CoG-FRICatANIONS, No. 724695; ERCCoG-UNBICAT, No. 647550) and the Deutsche Forschungsgemeinschaft (DFG) within the SFB858 are gratefully acknowledged for generous support. We are also grateful to the Spanish Government (RTI2018-095038-B-I00) and the “Comunidad de Madrid” and European Structural Funds (S2018/NMT-4367). J. A. F.-S. thanks the Spanish Government for a Ramón y Cajal Contract.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Kalesse and J. Hassfeld, in Asymmetric Synthesis—The Essentials, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, 2008, 2nd edn, pp. 112–116. See also Search PubMed; (b) Y. Kim, R. A. Singer and E. M. Carreira, Angew. Chem., Int. Ed., 1998, 37, 1261–1263 CrossRef CAS; (c) M. Christmann, U. Bhatt, M. Quitschalle, E. Claus and M. Kalesse, Angew. Chem., Int. Ed., 2000, 39, 4364–4366 CrossRef CAS; (d) D. A. Evans, D. M. Fitch, T. E. Smith and V. J. Cee, J. Am. Chem. Soc., 2000, 122, 10033–10046 CrossRef CAS; (e) J. Hassfeld, M. Christmann and M. Kalesse, Org. Lett., 2001, 3, 3561–3564 CrossRef CAS PubMed; (f) J. Hassfeld and M. Kalesse, Synlett, 2002, 2007–2010 CAS; (g) I. Paterson, R. D. M. Davies, A. C. Heimann, R. Marquez and A. Meyer, Org. Lett., 2003, 5, 4477–4480 CrossRef CAS PubMed; (h) F. Liesener and M. Kalesse, Synlett, 2005, 2236–2238 CAS; (i) S. E. Denmark and S. Fujimori, J. Am. Chem. Soc., 2005, 127, 8971–8973 CrossRef CAS PubMed; (j) E. H. Sessions and P. A. Jacobi, Org. Lett., 2006, 8, 4125–4128 CrossRef CAS PubMed; (k) M. Yamaoka, Y. Fukatsu, A. Nakazaki and S. Kobayashi, Tetrahedron Lett., 2009, 50, 3849–3852 CrossRef CAS.
- R. C. Fuson, Chem. Rev., 1935, 16, 1–27 CrossRef CAS.
- (a) M. Frías, W. Cieslik, A. Fraile, A. Rosado-Abón, A. F. Garrido-Castro, F. Yuste and J. Alemán, Chem. – Eur. J., 2018, 24, 10906–10933 CrossRef PubMed; (b) C. Curti, L. Battistini, A. Sartori and F. Zanardi, Chem. Rev., 2020, 120, 2448–2612 CrossRef CAS PubMed . Selected vinylogous Mannich-type examples: ; (c) M. Sickert and C. Schneider, Angew. Chem., Int. Ed., 2008, 47, 3631–3634 CrossRef CAS PubMed; (d) D. S. Giera, M. Sickert and C. Schneider, Org. Lett., 2008, 10, 4259–4262 CrossRef CAS PubMed; (e) M. Sickert, F. Abels, M. Lang, J. Sieler, C. Birkemeyer and C. Schneider, Chem. – Eur. J., 2010, 16, 2806–2818 CrossRef CAS PubMed; (f) Q. Wang, M. van Gemmeren and B. List, Angew. Chem., Int. Ed., 2014, 53, 13592–13595 CrossRef CAS PubMed.
- Selected reviews: (a) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (c) S. Beckendorf, S. Asmus and O. García Mancheño, ChemCatChem, 2012, 4, 926–936 CrossRef CAS; (d) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed; (e) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed; (f) D. Seidel, Synlett, 2014, 783–794 CrossRef CAS; (g) M. D. Visco, J. Attard, Y. Guan and A. E. Mattson, Tetrahedron Lett., 2017, 58, 2623–2684 CrossRef CAS.
- Example of vinylogous addition to allenoates: V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203–11205 RSC.
- Selected reviews: (a) C. Tsukano and Y. Takemoto, Dearomatisation Reactions of Electron-Deficient Aromatic Rings in Asymmetric Dearomatisation Reactions, ed. S.-L. You, Wiley-VCH, 2016, pp. 247–278 Search PubMed; (b) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed . For selected examples, see: ; (c) M. Takamura, K. Funabashi, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 6327–6328 CrossRef CAS; (d) K. Funabashi, H. Ratni, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 10784–10785 CrossRef CAS PubMed; (e) E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808–11809 CrossRef CAS PubMed.
- Pioneering work: M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700–6704 CrossRef CAS PubMed.
- Selected examples: (a) Y. Yamaoka, H. Miyabe and Y. Takemoto, J. Am. Chem. Soc., 2007, 129, 6686–6687 CrossRef CAS PubMed; (b) C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed; (c) A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321–11324 CrossRef CAS PubMed; (d) M. Zurro, S. Asmus, S. Beckendorf, C. Mück-Lichtenfeld and O. García Mancheño, J. Am. Chem. Soc., 2014, 136, 13999–14002 CrossRef CAS PubMed; (e) O. García Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823–8827 CrossRef PubMed; (f) A. R. Choudhury and S. Mukherjee, Chem. Sci., 2016, 7, 6940–6945 RSC; (g) J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047–3051 RSC; (h) T. Fischer, Q.-N. Duong and O. García Mancheño, Chem. – Eur. J., 2017, 23, 5983–5987 CrossRef CAS PubMed; (i) M. Gómez-Martínez, M. C. Pérez-Aguilar, D. G. Piekarski, C. G. Daniliuc and O. García Mancheño, Angew. Chem., Int. Ed., 2021, 60, 5102–5107 CrossRef PubMed.
- (a) E. C. Taylor and J. E. Saxton, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1983/1994 Search PubMed; (b) J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 2000 Search PubMed; (c) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co, Weinheim, 2nd edn, 2003 CrossRef; (d) N. Saracoglu, Top. Heterocycl. Chem., 2007, 11, 145–178 CrossRef.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- Asymmetric Synthesis of Nitrogen Heterocycles, ed. J. Royer, Wiley-VCH Verlag, Weinheim, 2009 Search PubMed.
- (a) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807–4815 RSC; (b) A. M. M. M. Faisca Phillips,Organocatalytic Enantioselective Dearomatisation Reactions for the Synthesis of Nitrogen Heterocycles In Synthetic Approaches to Nonaromatic Nitrogen Heterocycles, ed. A. M. M. M. Faisca Phillips, Wiley-VCH, 2020, pp. 273–323 CrossRef; (c) C. Zhuo, W. Zhang and S. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed; (d) C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432–444 CrossRef CAS PubMed.
- D. G. Piekarski, P. Steinforth, M. Gómez-Martínez, J. Bamberger, F. Ostler, M. Schönhoff and O. García Mancheño, Chem. – Eur. J., 2020, 26, 17598–17603 CrossRef CAS PubMed.
- The catalyst provokes a notable reaction acceleration, reaching completion after only 1 h (see the ESI† for details).
- (a) A. M. Hardman-Baldwin, M. D. Visco, J. M. Wieting, C. Stern, S.-i. Kondo and A. E. Mattson, Org. Lett., 2016, 18, 3766 CrossRef CAS PubMed; (b) T. Fischer, J. Bamberger, M. Gómez-Martínez, D. G. Piekarski and O. García Mancheño, Angew. Chem., Int. Ed., 2019, 58, 3217–3221 CrossRef CAS PubMed.
- The crystallographic data of 4k (CCDC 2069596†).
- This pre-orientation seems crucial for achieving the observed selectivity towards 4a, suggesting a less efficient ion-pair recognition and reaction of the other possible 1N-Troc salt.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical data of new compounds, SFC and HPLC chromatograms, kinetics, X-ray analysis of 4k and NMR collection (PDF). CCDC 2069596. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc03514b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |