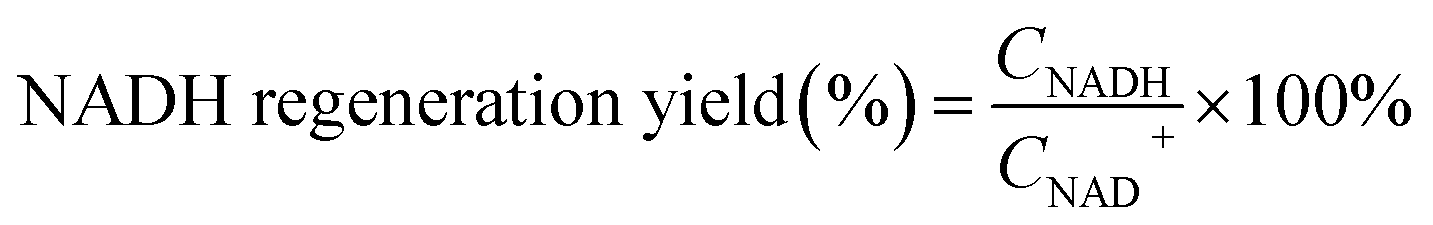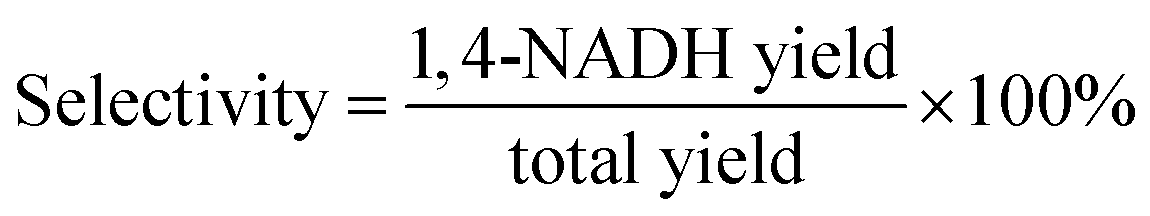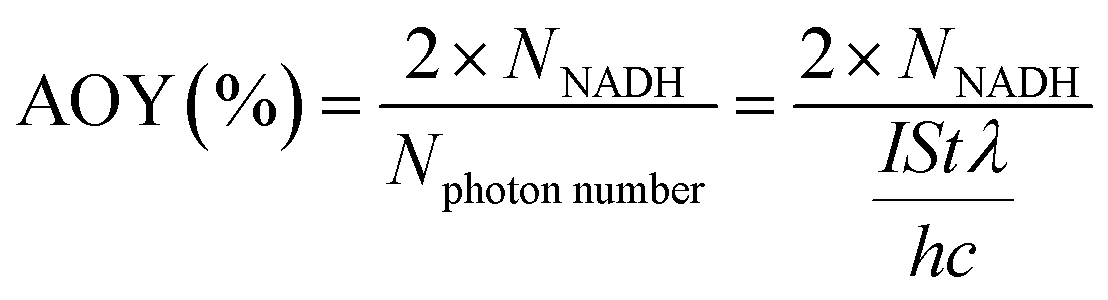DOI:
10.1039/D5TA03036F
(Paper)
J. Mater. Chem. A, 2025, Advance Article
CdS nanofeathers enable efficient electron-mediator-free photocatalytic regeneration of coenzyme NAD(P)H via direct electron–proton coupling†
Received
17th April 2025
, Accepted 7th July 2025
First published on 8th July 2025
Abstract
Photocatalytic regeneration of coenzyme NAD(P)H is essential for energy metabolism and reductive biosynthesis. Traditional systems depend on indirect electron-coupled proton transfer with precious metal-based electron mediators, adding complexity and cost. Here, we demonstrate that CdS nanofeather photocatalysts can achieve visible-light photocatalytic coenzyme NAD(P)H regeneration without electron mediators. Under visible-light irradiation, the NAD+ conversion of the CdS nanofeather photocatalyst reached 66.0% (1 h), showing 70.5% selectivity for the physiologically active product 1,4-NADH. When electron mediators are used, the NAD+ conversion is 72.7% (1 h). Furthermore, the expansion of substrate types indicates that another coenzyme, NADPH, also can be effectively regenerated without the assistance of an electron mediator. The unique morphology facilitates efficient charge separation and rapid migration, satisfying the electron concentration demands for NAD(P)H regeneration. Mechanistic studies show that the process involves stepwise electron–proton–electron transfer characterized by direct electron-coupled proton transfer. NADH is produced via the pathway NAD+ → NAD˙ → enol-NADH˙+ → enol-NADH → 1,4-NADH, fundamentally differing from the indirect electron transfer mechanism that relies on electron mediators. This work introduces visible-light photocatalytic coenzyme NAD(P)H regeneration without electron mediators, achieving competitive conversion.
1. Introduction
Coenzymes NADH and NADPH [abbreviated as NAD(P)H] serve as essential electron donors and carriers of biohydrogen in cellular metabolism, and are widely involved in critical biochemical processes such as energy metabolism, anti-oxidation, and biosynthesis.1–3 For instance, coenzyme NADH, as an electron donor, transfers electrons to oxygen molecules through the electron transport chain during oxidative phosphorylation, generating ATP and providing energy for cells.4 Additionally, coenzyme NAD(P)H participates in various reduction reactions, maintaining the redox balance of cells and protecting them from oxidative stress damage.5 However, due to the low intracellular concentration of coenzyme NAD(P)H and its limited regeneration rate, the efficient regeneration of coenzyme NAD(P)H has become a significant research focus in biomedicine and biochemistry.
In fact, the regeneration of the coenzyme NAD(P)H can be achieved through various methods, including homogeneous, heterogeneous, photocatalytic, and electrocatalytic techniques.6–8 Photocatalytic technology, in particular, utilizes light energy to drive chemical reactions, allowing for the efficient regeneration of coenzyme NAD(P)H under mild conditions while avoiding harsh conditions such as high temperature and high pressure.9,10 Additionally, the photocatalytic process can directly harness sunlight, a renewable energy source, making it both economical and environmentally friendly.11 However, most current photocatalytic coenzyme NAD(P)H regeneration systems depend heavily on the electron mediator [Cp*Rh(bpy)H2O]2+.12 Although electron mediators can improve the efficiency and selectivity of photocatalytic coenzyme NAD(P)H regeneration with physiologically active products, the use of precious metals in these mediators significantly increases the cost. Therefore, the primary challenge, is to enhance the activity and selectivity of photocatalytic coenzyme NAD(P)H regeneration without relying on electron mediators containing precious metals.13–15
Electron mediators, such as [Cp*Rh(bpy)H2O]2+, facilitate NADH regeneration via electron-coupled proton transfer, as illustrated in Fig. 1a. In this process, [Cp*Rh(bpy)H2O]2+ is reduced and protonated by photogenerated electrons to form the intermediate [Cp*Rh(bpy)H]+. NAD+ then interacts with [Cp*Rh(bpy)H]+ to generate a transient [Cp*Rh(bpy)H-NAD]2+ state. Although both NAD+ and [Cp*Rh(bpy)H]+ are cationic, their interaction is driven by the thermodynamic favorability of reducing NAD+ to 1,4-NADH, which renders the hydrogen transfer from [Cp*Rh(bpy)H]+ energetically favorable. Crucially, the amide group of NAD+ coordinates to the Rh metal center via an open coordination site generated through a well-established ring-slip mechanism of the Cp* ligand (η5 to η3). This process facilitates a kinetically favorable six-membered ring transition state, which further drives regioselective hydride transfer to the C4 position of NAD+. The resulting carbonyl coordination may also enhance electrophilicity at the C4 site, promoting hydride attack.12,16 Following this step, 1,4-NADH and [Cp*Rh(bpy)H2O]2+ are formed. Thermodynamically, the stabilization achieved upon reduction favors this reaction. Finally, [Cp*Rh(bpy)H2O]2+ is regenerated by subsequent electron and proton uptake, making it ready for another catalytic cycle.16,17 It can be seen that the primary function of the electron mediator is to facilitate the rapid transfer of electrons and protons to NAD+, thereby ensuring efficient regeneration of NADH. In organisms, the production of coenzymes NADH or NADPH occurs through cellular respiration or photosynthesis, where processes are completed under enzyme catalysis without relying on electron mediators (Fig. 1b). Therefore, biomimetic artificial photosynthesis that does not rely on electron mediators may achieve efficient regeneration of coenzyme NAD(P)H. The main function of the electron mediator is to promote the rapid transfer of electron-coupled protons.18 To achieve high activity and selectivity in the regeneration of coenzyme NAD(P)H without electron mediators, it is essential to ensure that photogenerated electrons and protons are quickly transferred to the reaction substrate NAD(P)+. This requires the photocatalyst to have high charge separation efficiency and a fast migration rate of photogenerated carriers to meet the electron concentration requirements for the regeneration of the coenzyme NAD(P)H. In other words, for the photocatalytic reaction to proceed stably and continuously, the concentration of photogenerated carriers must reach a minimum threshold. Although the exact value is not clear at present, a high concentration of photogenerated carriers is undoubtedly necessary for achieving stable reaction conditions.
 |
| | Fig. 1 (a) The electron mediator [Cp*Rh(bpy)H2O]2+ facilitates electron-coupled proton transfer, promoting efficient photocatalytic regeneration of the coenzyme NADH. (b) Two natural mechanisms for NADH regeneration: photosynthesis and respiration. | |
In this study, we prepared CdS nanofeather photocatalysts using a straightforward one-step hydrothermal method. Owing to their unique morphology, these photocatalysts efficiently regenerated the coenzyme NAD(P)H without the need for electron mediators. Under visible-light irradiation, the NAD+ conversion of the CdS nanofeather photocatalyst reached 66.0%, showing 70.5% selectivity for the physiologically active product 1,4-NADH. In contrast, when electron mediators were employed, the NAD+ conversion increased to 72.7%. Furthermore, the expansion of substrate types indicated that another coenzyme, NADPH, also could be effectively regenerated without the assistance of an electron mediator. The unique nanofeather morphology promoted efficient separation and rapid migration of photogenerated charges, thereby meeting the electron concentration requirements for coenzyme NAD(P)H regeneration. Mechanistic studies revealed that, unlike the electron-coupled proton transfer mechanism assisted by electron mediators, this process involved a stepwise electron–proton–electron transfer allowing for directly incorporating electrons and protons. This work represented the first instance of visible-light photocatalytic regeneration of the coenzyme NAD(P)H without electron mediators, achieving a conversion comparable to that with electron mediators. We believed this advancement would have significant implications for the regeneration of the coenzyme NAD(P)H.
2. Experimental section
2.1. Synthesis of CdS nanofeather photocatalyst
The CdS nanofeather photocatalyst was prepared using a one-step hydrothermal method. To begin, a mixed solution of 40 mL ethylene glycol and deionized water was prepared, to which 1 mmol of CdCl2·2.5H2O and 1.5 mmol of thiourea were sequentially added. After these reagents were fully dissolved, the clear solution was transferred to a 50 mL polytetrafluoroethylene-lined autoclave, which was then sealed in a stainless steel tank for the hydrothermal reaction. The reaction temperature was maintained at 180 °C for 24 hours. After the reaction, the product was collected, rinsed with deionized water, and vacuum-dried at 60 °C. The volume ratios of ethylene glycol to deionized water used were 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, 10:30, 20:20, 30:10, and 40:0, respectively. Accordingly, the resulting catalysts were named CdS-0, CdS-10, CdS-20, CdS-30, and CdS-40.
:40, 10:30, 20:20, 30:10, and 40:0, respectively. Accordingly, the resulting catalysts were named CdS-0, CdS-10, CdS-20, CdS-30, and CdS-40.
2.2. Photocatalytic NAD(P)H regeneration
Photocatalytic NAD(P)H regeneration was performed in a 30 mL quartz test tube reactor. A 300 W xenon lamp equipped with a 420 nm cutoff filter provided visible light irradiation, positioned 15 cm from the reactor. The reactor contained 30 mg of photocatalyst, 1 mL of NAD(P)+ solution (1 mM), 1 mL of triethanolamine (TEOA) solution (15.0 w/v%), and 2 mL of buffer solution (pH 7.4). After stirring the catalyst and reaction solution in the dark for 30 minutes, the light was activated, and 1 mL aliquots of the reaction mixture were collected every 30 minutes. The catalyst in each 1 mL sample was removed using a syringe equipped with a filter membrane to obtain a clear reaction solution. Subsequently, 0.5 mL of the clear solution was transferred to a 1 cm pathlength cuvette and diluted with 2.5 mL of deionized water to ensure optimal absorbance readings within the linear range of the spectrophotometer. NAD(P)H concentrations were measured using a PerkinElmer UV-vis-NIR spectrometer, scanning from 250 to 800 nm at a speed of 8 nm s−1. The yield of NADH was determined using the external standard curve method (Fig. S1†). For the quantitative detection of 1,4-NADH, deuterated water was employed as the solvent, and the post-reaction solution was analyzed using a Jeol ECZ500R NMR spectrometer via 1H NMR spectroscopy. The quantification was based on the integrated peak area of the characteristic proton signal of 1,4-NADH (chemical shift at δ = 6.8 ppm), which exhibited good resolution in the deuterated solvent and was not interfered with by the reaction system. In experiments involving electron mediators, 50 μL of the mediator solution was added following the aforementioned procedure. The NADH regeneration yield and selectivity were calculated according to formulas (1) and (2), respectively. The apparent quantum yield (AQY) was calculated according to formulas (3) and (4).| |
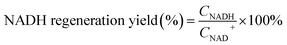 | (1) |
| |
 | (2) |
| |
 | (3) |
| |
 | (4) |
I: light intensity (W m−2), S: illumination area S ≈ 0.015 × 0.01 (m−2), λ: monochromatic light wavelength (m), t: light time (s), h: Planck's constant (6.626 × 10−34), c: speed of light (3 × 108 m s−1).
2.3. Alcohol dehydrogenase (ADH) enzyme-catalyzed reaction
Formaldehyde solution (100 μL) was added to each of two identical cuvettes. Subsequently, 0.5 mL of the solution obtained after the CdS-30 photocatalytic NAD+ reduction was added to both cuvettes. Next, 100 μL of pre-warmed (37 °C) alcohol dehydrogenase (ADH) enzyme solution (188 U) was added to one cuvette (experimental group), while 100 μL of pH 7.8 Tris–HCl buffer (replacing the enzyme solution) was added to the other cuvette (blank control group). Following a 20-minute reaction period at 37 °C for both cuvettes, 2.3 mL of pH 7.8 Tris–HCl buffer was added to each. The absorbance values (Abs340) of the experimental and control groups were then immediately measured at 340 nm using a UV-visible spectrophotometer.
3. Results and discussion
3.1. Characterization of CdS nanofeathers
The CdS nanofeather photocatalyst was successfully prepared using a simple one-step hydrothermal method. The crystal phases of the photocatalysts were characterized by X-ray powder diffraction (XRD), as shown in Fig. 2a. The diffraction peaks of all samples corresponded to the standard diffraction peaks of hexagonal phase CdS (JCPDS No. 41-1049), with no impurity's peaks indicating the absence of impurities.19 This result confirmed the high purity of the photocatalysts. The elemental composition and valence state of the CdS nanofeather photocatalyst were analyzed using X-ray photoelectron spectroscopy (XPS). The full XPS spectrum revealed the presence of Cd, S, C, and O (Fig. S2†). Peaks with binding energies of 161.43 and 162.63 eV were attributed to the S 2p3/2 and S 2p1/2 of the CdS-30 sample, with a spin–orbit splitting component Δ = 1.20 eV (Fig. 2b).20,21 These values corresponded to those typical of metal sulfides. The S spectra of CdS-0 and CdS-40 were consistent with those of CdS, indicating similar chemical environments and valence states among the samples. Similarly, peaks with binding energies of 404.98 and 411.71 eV were assigned to the Cd 3d5/2 and Cd 3d3/2 of the CdS-30 sample, with a spin–orbit splitting component Δ = 6.73 eV, corresponding to Cd2+ (Fig. 2c).22,23 The Cd spectra of CdS-0 and CdS-40 were also consistent with those of CdS, confirming similar chemical environments and valence states. As shown in Fig. S3,† no sulfur defect structures were observed in the photocatalysts by electron paramagnetic resonance (EPR). Transmission electron microscopy (TEM) images showed that CdS-0 and CdS-30 exhibit typical feather-like morphologies, as depicted in Fig. S4† and 2d. The lattice spacing of the CdS-30 sample was 0.358 nm, corresponding to the (1 0 0) crystal plane, as shown in Fig. 2e. Selected area electron diffraction (SAED) further confirmed that the CdS-30 sample had diffraction spots consistent with a single-crystal structure (Fig. 2f). Finally, mapping tests revealed that the S and Cd elements were uniformly distributed on the surface of the CdS nanofeather photocatalyst (Fig. 2g–i). Energy dispersive X-ray spectroscopy (EDS) was employed to confirm the elemental composition and ratio of the sample, as illustrated in Fig. S5.† The EDS spectrum revealed the presence of Cd and S elements, with a Cd/S atomic ratio close to 1:1. These characterizations demonstrated the successful preparation of the CdS nanofeather photocatalyst with a single-crystal structure.
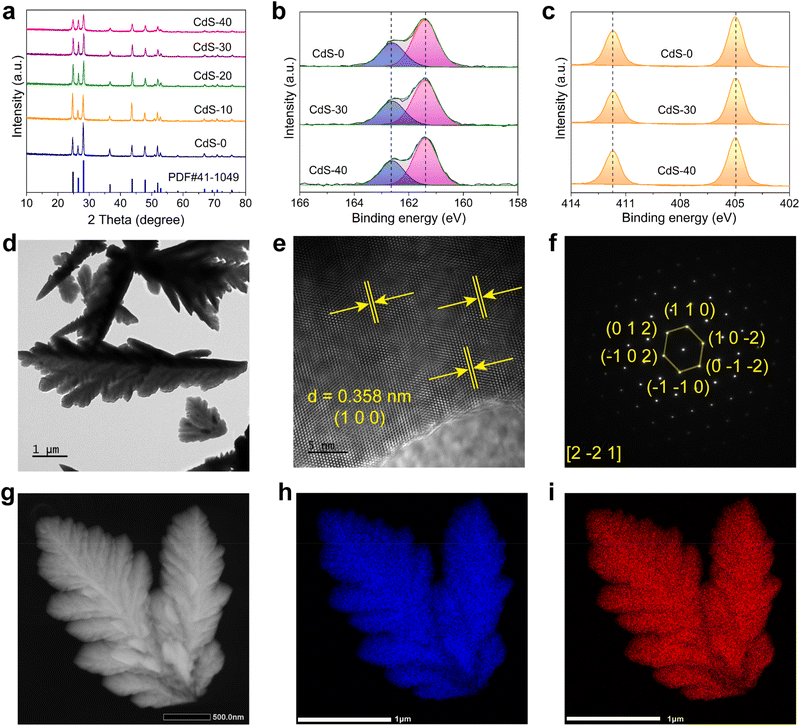 |
| | Fig. 2 (a) XRD patterns of various photocatalysts. High-resolution XPS spectra of CdS: (b) S 2p, (c) Cd 3d. (d) TEM image of the CdS-30 sample. (e) HRTEM image. (f) SAED pattern of the CdS-30 sample, and (g–i) EDS elemental mapping images. | |
3.2. Photocatalytic regeneration of coenzyme NAD(P)H
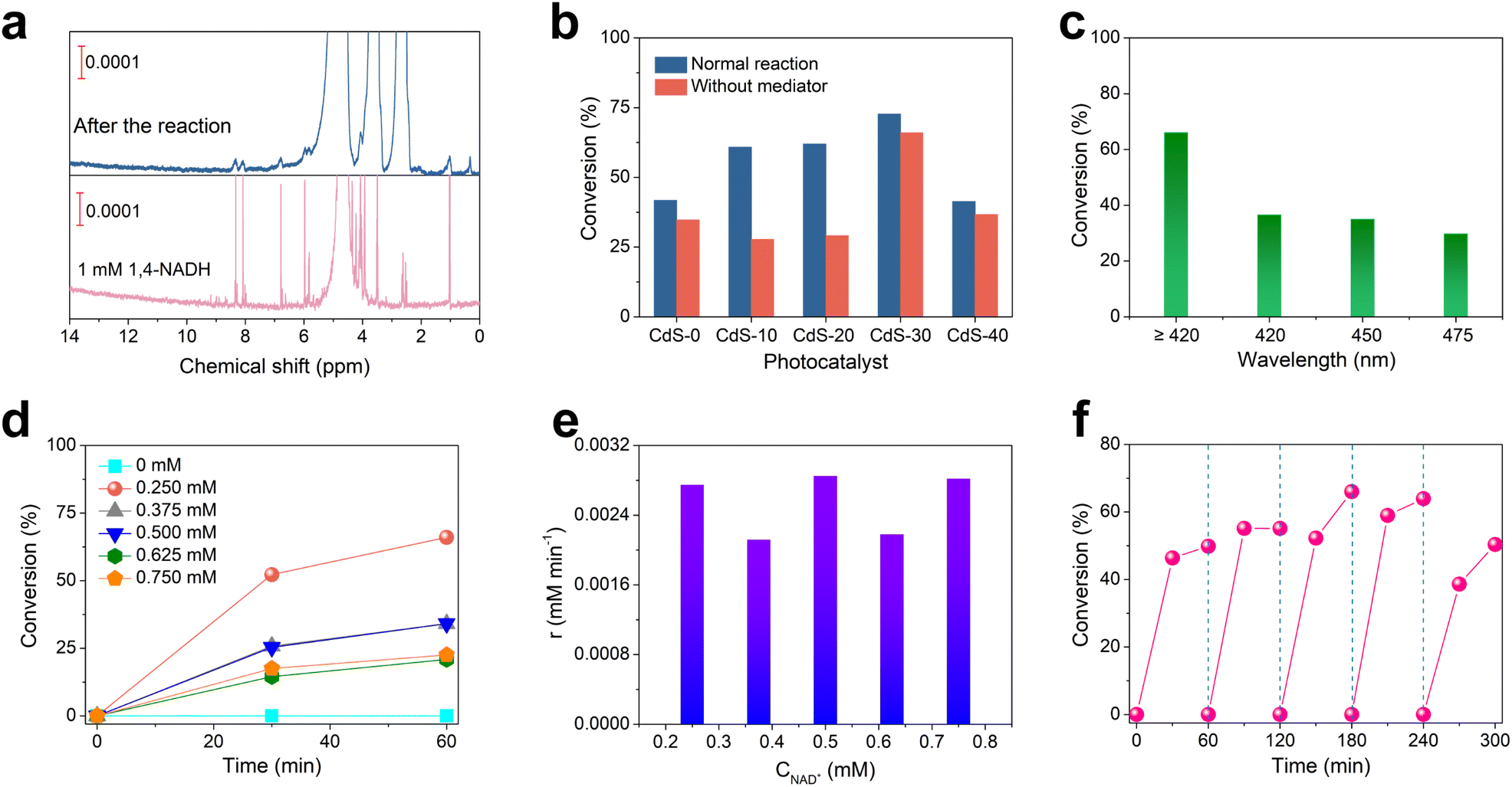
The photocatalytic performance for coenzyme NAD(P)H regeneration was evaluated in a sealed quartz tube under visible light, simulated by a 300 W xenon lamp with a 420 nm cutoff filter. Control experiments demonstrated that NAD+, TEOA, and the photocatalyst were essential for the reaction, as the absence of any of these components resulted in no detectable NADH production (Fig. S6 and S7†). With [Cp*Rh(bpy)H2O]2+ as an electron mediator, the NAD+ conversion reached 72.7% after 1 hour. Surprisingly, even without an electron mediator, CdS-30 was able to photocatalyze the conversion of NAD+ to NADH, achieving a conversion of 66.0% after 1 hour. Thus, the CdS nanofeather photocatalyst enabled direct photocatalytic NADH regeneration without an electron mediator. 1H NMR analysis confirmed the high selectivity of CdS-photocatalyzed NAD+ reduction toward physiologically active 1,4-NADH, with no detectable 1,6-NADH isomers in the absence of an electron mediator (Fig. 3a).24 Reference samples generated via controlled NaBH4 reduction provided definitive spectral signatures: authentic 1,4-NADH showed characteristic proton resonance at δ 6.81 ppm, 1,6-NADH displayed its distinctive signal at δ 7.03 ppm, and the NAD2 dimer exhibited its unique signal at δ 2.71 ppm (Fig. S8†). Comparative analysis revealed the unique identity of the photocatalytic products with the 1,4-NADH standard, in particular the unique 1,4-NADH signal at δ 6.81 ppm observed in both the reference and reaction spectra. The absence of the δ 7.03 ppm signal of 1,6-NADH further confirmed the regioselectivity of the system. Concurrently, 1H NMR quantitative analysis substantiated that the CdS-30 photocatalytic NAD+ reduction achieved a 46.9% conversion to 1,4-NADH in the absence of electron mediators. Consequently, the CdS-30 nanofeather photocatalyst demonstrated 70.5% selectivity toward 1,4-NADH formation. The observed discrepancy in yields between NMR and UV-vis analyses may be attributed to the formation of an additional product, such as NAD2. In addition, 1,4-NADH generation was quantified using ADH, with enzymatic incubation at 37 °C specifically consuming 1,4-NADH and reducing absorbance at 340 nm (Fig. S9†), demonstrating that CdS-30 achieved 29.81% 1,4-NADH conversion and 45.77% selectivity for 1,4-NADH during photocatalytic NAD+ reduction without electron mediators. Fig. 3b, and S10† as well as Fig. S11† displayed the photocatalytic NADH regeneration performance and UV-visible absorption spectra of various photocatalysts with and without electron mediators. The NADH regeneration activity of all samples followed a typical volcano-type catalytic reaction pattern. CdS-30 exhibited the highest NADH regeneration activity, surpassing most reported photocatalysts, especially in the absence of electron mediators (Table S1†). Additionally, the NADH regeneration activity of CdS-30 under different monochromatic light irradiations without electron mediators was evaluated (Fig. 3c and S12†). The catalytic activity decreased as the wavelength increased, with a NAD+ conversion of 36.5% after 1 hour at 420 nm and 29.7% after 1 hour at 475 nm. Monochromatic light intensities measured using a calibrated ELITETECH solar simulator were 4.8 mW cm−2 at 420 nm, 6.8 mW cm−2 at 450 nm, and 9.1 mW cm−2 at 475 nm. Given CdS-30's near-complete (>99%) light absorption at both 420 nm and 450 nm, the incident photon flux was calculated as 5.13 × 1019 at 420 nm and 8.31 × 1019 at 450 nm. Consequently, systematic implementation of formulas (3) and (4) yields apparent quantum yields of 0.86% and 0.51% for these respective wavelengths. Fig. 3d and S13† showed the NADH regeneration activity of CdS-30 at various NAD+ concentrations. Theoretically, within a certain concentration range, as the reaction substrate concentration increased, the substrate's conversion decreased. This occurred because, at higher substrate concentrations, the active sites of the photocatalyst became saturated, leading to a stable reaction rate. This saturation limited the availability of active sites for additional substrate molecules, thereby reducing overall conversion efficiency. When the initial concentration of NAD+ was 0.75 mM, the conversion was only 22.6% after 1 hour, whereas at a concentration of 0.25 mM, the conversion of NAD+ reached 63.9% after 1 hour. Additionally, as the initial concentration of NAD+ increased, the chemical reaction rate remained essentially unchanged, indicating that the reaction followed zero-order kinetics (Fig. 3e).25 This was mainly because the reaction rate was limited by certain non-reactant concentration factors, such as the recombination rate of electron–hole pairs. When the reaction concentration of NAD+ was 0.25 mM, no decrease in activity was observed in the 300-minute cycle test, indicating good cycle stability (Fig. 3f and S14†). XPS analysis before and after the reaction revealed no significant surface structural changes in CdS, confirming the catalyst's good stability (Fig. S15†). Based on these results, we concluded that the CdS nanofeather photocatalyst is an efficient and stable catalyst for photocatalytic coenzyme NADH regeneration, with broad application prospects.
 |
| | Fig. 3 (a) 1H NMR spectrum of products obtained by photocatalytic reduction of NAD+ by CdS-30. (b) Photocatalytic NADH regeneration performance of a series of CdS photocatalysts with and without electron mediators. (c) Photocatalytic performance of coenzyme NADH regeneration on CdS-30 under monochromatic light irradiation. (d) Photocatalytic regeneration performance of CdS-30 for different concentrations of NAD+. (e) Reaction kinetics of NADH regeneration on the CdS-30 catalyst. (f) Photocatalytic NADH regeneration stability test of the CdS-30 sample. | |
Apart from coenzyme NADH, coenzyme NADPH is also a crucial coenzyme extensively involved in cellular energy metabolism and biosynthetic reactions. In cells, coenzyme NADPH primarily functions as a reducing agent, providing the necessary reducing power for the synthesis of fatty acids, nucleotides, and amino acids, while also neutralizing reactive oxygen species within the antioxidant defense system. Consequently, the regeneration of coenzyme NADPH is essential for maintaining normal cellular metabolism and physiological functions. Similar to the coenzyme NADH regeneration mechanism, coenzyme NADPH regeneration occurs through the acceptance of protons and electrons by NADP+. Specifically, NADP+ is converted into NADPH upon acquiring a proton and two electrons. This conversion process involves a series of enzymatic reactions and electron transfer steps within the organism. Notably, the CdS nanofeather photocatalyst directly regenerates the coenzyme NADPH photocatalytically under visible light conditions. Fig. 4a presents the UV-visible absorption spectrum of the CdS-30 nanofeather photocatalyst during the photocatalytic regeneration of NADPH in the presence of the electron mediator [Cp*Rh(bpy)H2O]2+. A distinct UV-visible absorption peak at 340 nm, characteristic of NADPH, is observed.26,27 As the illumination time increases, the absorption peak intensifies, indicating the production of NADPH. From 0 to 60 minutes, the NADPH absorption intensity increases by 0.11. The CdS-30 nanofeather photocatalyst also achieves NADPH regeneration without the addition of an electron mediator. Fig. 4b shows that at 0 minutes of illumination, no significant absorption peak is detected at 340 nm. However, as the illumination time extends to 30 and 60 minutes, a clear UV-visible absorption peak emerges at 340 nm, with its intensity increasing over time. From 0 to 60 minutes, the NADPH absorption intensity rises by 0.072. Similarly, the CdS-0 nanofeather photocatalyst regenerates NADPH, irrespective of the presence of an electron mediator, as illustrated in Fig. S16a and b.† When an electron mediator is used, the NADPH absorption intensity increases by 0.054 from 0 to 60 minutes. Without an electron mediator, the NADPH absorption intensity increases by 0.052 over the same period. It is evident that, regardless of the presence of an electron mediator, CdS-30 exhibits higher photocatalytic activity for coenzyme regeneration than CdS-0. This enhanced activity is attributed to the higher photogenerated charge separation efficiency and the faster migration rate of photogenerated carriers in the CdS-30 nanofeather photocatalyst.
 |
| | Fig. 4 (a and b) Photocatalytic coenzyme NADPH regeneration of CdS-30 with and without electron mediator. | |
3.3. Energy band and photoelectrochemical characterization
The energy band structure determines the light absorption performance and redox ability of the photocatalyst, thereby affecting the photocatalytic activity. The valence band potential of the photocatalyst is characterized by the valence band XPS (VB XPS), as shown in Fig. 5a. The valence band edges of CdS-0, CdS-30, and CdS-40 are estimated by linear extrapolation to be +1.27, +1.26, and +1.26 eV below the Fermi level by the linear extrapolation method, respectively.28,29 The light absorption capacity of the five photocatalysts is tested using solid-state UV-vis diffuse reflectance spectroscopy, as shown in Fig. S17.† The absorption band edges all exceed 420 nm, and with the increase of ethylene glycol dosage during the preparation process, the absorption band edges gradually blue-shift. The absorption cutoff band edges of CdS-0, CdS-10, CdS-20, CdS-30, and CdS-40 are 602 nm, 573 nm, 563 nm, 558 nm, and 549 nm, respectively, and the corresponding optical band gaps are estimated to be 2.06, 2.16, 2.20, 2.22, and 2.26 eV, respectively.30 These band gaps enable good light absorption in the visible light range. Based on the above data, the energy band structures of CdS-0, CdS-30, and CdS-40 are obtained, as shown in Fig. 5b. The conduction band values of the three samples are more negative relative to the reduction potential of the electron mediator ([Cp*Rh(bpy)H2O]2+/[Cp*Rh(bpy)H]+ −0.62 V s NHE) and NAD+ (NAD+/NADH −0.32 V s NHE), while all valence band values exceed the oxidation potential of TEOA.31 This demonstrates that the photogenerated electrons and holes generated by CdS under photoexcitation have sufficient reduction and oxidation capabilities, respectively, to drive the reduction of NAD+ and the oxidation of TEOA.
 |
| | Fig. 5 (a) VB XPS spectra. (b) Energy band data of CdS-0, CdS-30 and CdS-40 photocatalyst. (c) Photoluminescence spectra. (d) Time-resolved photoluminescence of CdS-0, CdS-30 and CdS-40. (e) Photocurrent response and (f) electrochemical impedance spectroscopy of the as-prepared samples. | |
Photoluminescence tests are performed to evaluate the separation and migration of photogenerated charges, as shown in Fig. 5c. Under excitation at 360 nm monochromatic light, all samples exhibit fluorescence emission peaks around 462 nm, with the CdS-30 sample showing the lowest fluorescence emission peak intensity, indicating a lower recombination rate of photogenerated charges.32–34 Additionally, transient fluorescence lifetime tests demonstrate that CdS-30 (0.99 ns) has the shortest excited state lifetime, as the recombination of photogenerated electrons and holes is more limited in the CdS-30 sample compared to other samples, thereby enhancing separation (Fig. 5d).35 Furthermore, photoelectrochemical characterization reveals that CdS-30 exhibits the highest photocurrent density (∼35 μA cm−2) and the smallest electrochemical impedance radius among the catalysts tested, indicating that CdS-30 has the lowest charge migration resistance and the highest photogenerated charge separation efficiency (Fig. 5e and f).36,37 High photogenerated charge separation efficiency and rapid migration rate are essential for improving photocatalytic reactions, particularly in the absence of electron mediators to facilitate electron transfer. Notably, CdS demonstrates high charge separation efficiency and fast migration rate, which enables it to promote the regeneration of coenzyme NAD(P)H through a direct electron-coupled proton transfer process, rather than relying on electron mediators for indirect electron transfer.
3.4. Direct electron-coupled proton transfer mechanism
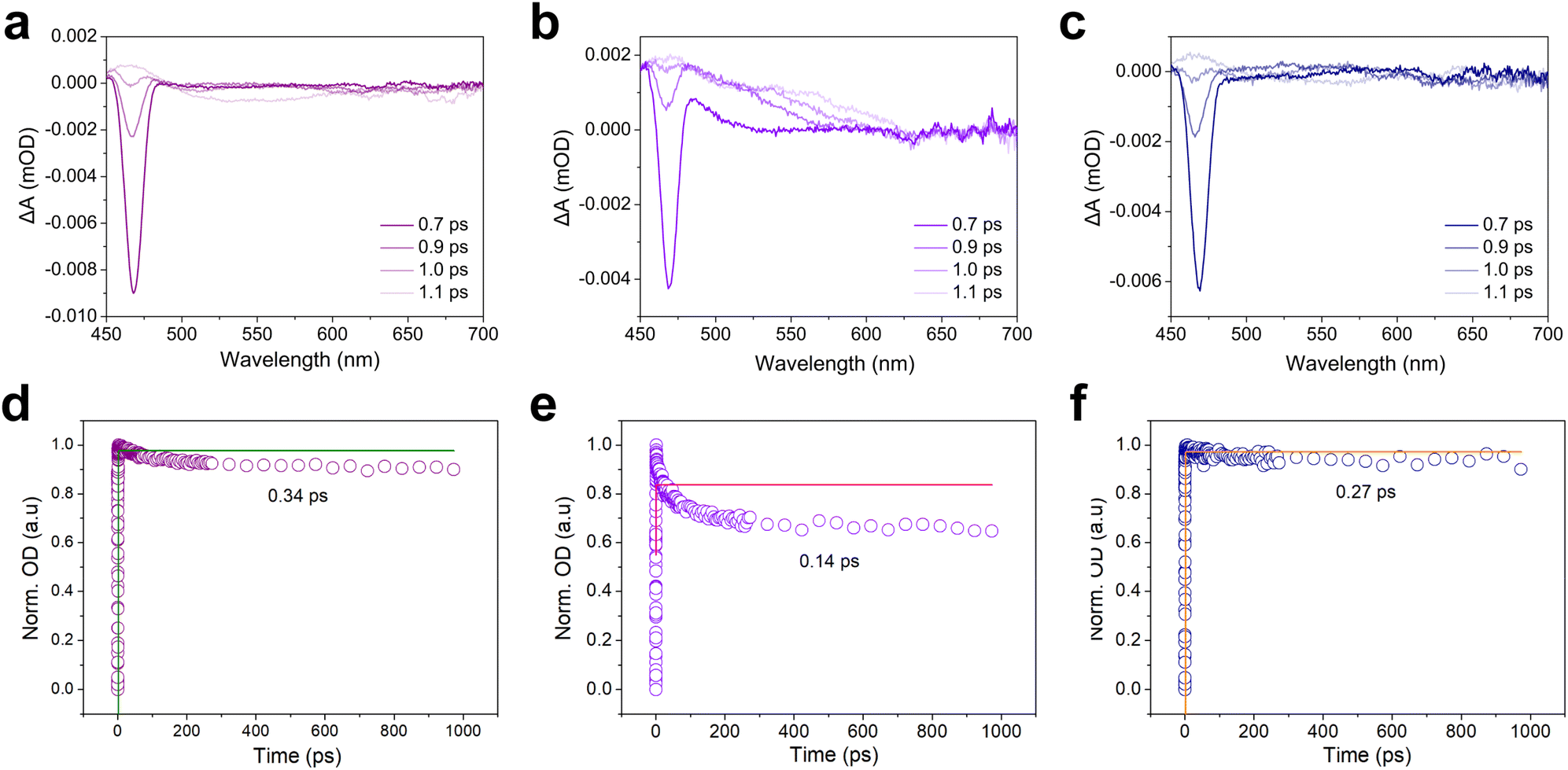
The femtosecond transient absorption (fs-TA) test was used to further study the electron transfer behavior between the electron mediator or the reaction substrate and the CdS. The fs-TA spectrum of CdS-30 showed an obvious broad negative signal, attributed to ground state bleaching (Fig. 6a).38 Generally, the strength of the ground state bleaching peak indicated the number of molecules or atoms in the sample excited to the excited state.39,40 Notably, the addition of electron donor TEOA significantly altered the charge carrier dynamics of CdS-30, as evidenced by a reduction in ground-state bleaching peak intensity and shortened charge carrier lifetime (Fig. S18†). This phenomenon arises from TEOA's dual function: it rapidly scavenges photogenerated holes while simultaneously suppressing electron–hole recombination, thereby promoting electron accumulation. The observed initial signal decay corresponds to sub-picosecond charge separation processes preceding diffusive quenching. Critically, photogenerated electrons undergo sequential kinetic phases: instantaneous charge separation, geminate recombination of Coulombically bound pairs, and ultimately bulk diffusion-mediated trapping or interfacial transfer, with TEOA accelerating the latter pathway by removing hole-induced recombination bottlenecks. After adding the electron mediator [Cp*Rh(bpy)H2O]2+ and the reaction substrate NAD+, the intensity of the ground state bleaching peak of the CdS-30 sample decreased, with a more significant decrease observed after adding electron mediator [Cp*Rh(bpy)H2O]2+ compared to NAD+ (Fig. 6b and c). This indicated that both the electron mediator and the reaction substrate reaction substrate NAD+ could interact with CdS-30 in the excited state, meaning the electrons excited by CdS-30 could interact with both the electron mediator [Cp*Rh(bpy)H2O]2+ and reaction substrate NAD+. For CdS-30, the reduction in the ground state bleaching peak due to the addition of the electron mediator [Cp*Rh(bpy)H2O]2+ was much higher than that due to NAD+, suggesting that the excited state electrons of CdS-30 were more readily transferred to the electron mediator [Cp*Rh(bpy)H2O]2+. However, the addition of NAD+ also led to a decrease in the ground state bleaching peak intensity, indicating that the electrons could directly transfer to NAD+ to achieve coenzyme regeneration via direct electron transfer. The kinetic trajectories under different test conditions were fitted using a single exponential decay function.41 For pure CdS-30 and with the addition of the electron mediator [Cp*Rh(bpy)H2O]2+ and reaction substrate NAD+, the decay lifetimes were 0.34, 0.14, and 0.27 ps, respectively (Fig. 6d–f). The quenching effect of the electron mediator on the excited state was significantly higher than that of NAD+. This indicated that the rate at which the photo-generated electrons excited by CdS-30 were transferred to the electron mediator [Cp*Rh(bpy)H2O]2+ was higher than that to NAD+. However, the addition of NAD+ also led to a decrease in the decay lifetime, demonstrating that the excited electrons could be transferred to NAD+, thereby promoting the photocatalytic reduction regeneration of the coenzyme NADH.
 |
| | Fig. 6 (a) Transient absorption spectra of CdS-30 at various delay times under 400 nm excitation. (b) Transient absorption spectra of CdS-30 at various delay times under 400 nm excitation after adding [Cp*Rh(bpy)H2O]2+. (c) Transient absorption spectra of CdS-30 at various delay times under 400 nm excitation after adding NAD+. (d) Kinetics of CdS-30 probed at 460 nm. (e) Kinetics of CdS-30 probed at 460 nm after adding [Cp*Rh(bpy)H2O]2+. (f) Kinetics of CdS-30 probed at 460 nm after adding NAD+. | |
The regeneration mechanism of the coenzyme NADH is traditionally understood to involve a negative hydrogen transfer. In the presence of an electron mediator, this mechanism primarily relies on the three-step transfer process of [Cp*Rh(bpy)H2O]2+, which includes sequential electron–electron–proton transfers. In contrast, the direct electron transfer mechanism, which does not utilize an electron mediator, transfers through three steps: electron–proton–electron. For the photocatalytic system without an electron mediator, the pathway of NADH generation is illustrated in Fig. 7a. In this system, the NAD+ molecule receives photogenerated electrons from CdS, forming the NAD+ radical. This radical then accepts a proton, leading to the formation of the enol NADH radical cation. Thermodynamically, the enolate radical cation is more favored than its corresponding keto form because the energy required to remove an electron from the π orbital of the enolate is lower than that needed to remove an electron from the n orbital of the ketone.42 After the enolate NADH radical cation accepts another electron, enolate NADH is formed. Finally, the enolate NADH undergoes intramolecular rearrangement to produce 1,4-NADH. To gain a deeper understanding of the photocatalytic NADH regeneration of CdS-30 via indirect and direct electron transfer mechanisms, density functional theory (DFT) calculations elucidate the two pathways involved in the reaction. The CdS (1 0 1) crystal plane serves as the calculation model (Fig. S19†). Fig. 7b and c illustrate the potential energy diagrams for photocatalytic NADH regeneration using CdS nanofeather photocatalysts with and without the electron mediator [Cp*Rh(bpy)H2O]2+, respectively. For the system with the electron mediator, the adsorption of [Cp*Rh(bpy)H2O]2+ on the CdS surface is a spontaneous process with an adsorption energy of −5.979 eV. When CdS is excited by light, photogenerated electrons and holes are generated. The holes oxidize TEOA to generate H+, while the electrons react with [Cp*Rh(bpy)H2O]2+ to generate [Cp*Rh(bpy)]. The formation of [Cp*Rh(bpy)] is a non-spontaneous process that requires overcoming an energy barrier of 2.722 eV. Subsequently, [Cp*Rh(bpy)] acquires an H+ to form [Cp*Rh(bpy)H]+, which is also an endothermic process that absorbs 1.173 eV of energy. The interaction between [Cp*Rh(bpy)H]+ and NAD+ is thermodynamically non-spontaneous and requires overcoming an energy barrier of 2.732 eV. NAD+ coordinates to the Cp*Rh metal center through its amide oxygen atom. It is worth noting that the reduction reaction, after [Cp*Rh(bpy)H]+ interacts with NAD+ to generate the transition state [Cp*Rh(bpy)H-NAD]2+, is a spontaneous process that releases 14.621 eV of energy. This process achieves the reduction of NAD+ and produces NADH. Finally, under the action of H2O, 1,4-NADH desorbs and [Cp*Rh(bpy)H2O]2+ regenerates. However, for the system without an electron mediator, the interaction of NAD+ with photoexcited CdS to generate electrons and form NAD radicals is a spontaneous process, releasing 0.765 eV of energy. The subsequent process of the radical accepting a proton to form the enolate NADH radical cation requires overcoming an energy barrier of 2.406 eV. When the enolate NADH radical cation accepts an additional electron to form the enol form NADH, it is a spontaneous process, releasing 0.383 eV of energy. Finally, the enol form NADH undergoes an intramolecular rearrangement to form 1,4-NADH, which is also a spontaneous process because the formation of the more stable 1,4-NADH product reduces the free energy of the system. Consequently, regardless of whether an electron mediator is used, the occurrence of the reaction depends on the energy changes along the reaction path and the electron transfer characteristics of the reactants. Thus, under the same reaction conditions, the reaction can proceed via different pathways.
 |
| | Fig. 7 (a) The mechanism of photocatalytic regeneration of coenzyme NADH by CdS-30 without an electron mediator. (b and c) Gibbs free energy diagram of photocatalytic regeneration of coenzyme NADH by CdS-30 with and without an electron mediator. | |
4. Conclusions
The CdS nanofeather photocatalyst, prepared by a simple hydrothermal method, enabled the photocatalytic regeneration of the coenzyme NAD(P)H. In the indirect electron transfer reaction using the [Cp*Rh(bpy)H2O]2+ electron mediator, the conversion of NAD+ reached 72.70% (1 h). However, in the direct electron transfer reaction without the [Cp*Rh(bpy)H2O]2+ electron mediator, the NAD+ conversion was 66.0% (1 h), showing 70.5% selectivity for the physiologically active product 1,4-NADH. In the indirect electron transfer reaction using the electron mediator, the [Cp*Rh(bpy)H-NAD]2+ intermediate was generated through an electron-coupled proton process. This intermediate was then hydrolyzed into NADH and [Cp*Rh(bpy)H2O]2+ in the presence of H2O. In contrast, the direct electron transfer process without an electron mediator produced NADH via the pathway of NAD+ → NAD˙ → enol-NADH˙+ → enol-NADH → 1,4-NADH. This work revealed two distinct pathways for the photocatalytic regeneration of NAD(P)H and identified a direct electron transfer system for coenzyme NAD(P)H regeneration that eliminated the need for expensive electron mediators containing precious metals.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
Yao Chai: responsible for experimental design, operation, and manuscript preparation. Leyi Zhao: conducted TA testing and performed data analysis. Zirui Pang: carried out catalyst characterization. Liang Wan: carried out catalyst characterization. Heng Jiang: evaluated photocatalytic performance. Chi Chung Tsoi: evaluated photocatalytic performance. Yu Du: conducted catalyst characterization. Huaping Jia: conducted catalyst characterization. Yujiao Zhu: contributed to writing, review, and editing. Detao Liu: contributed to writing, review, and editing. Mingjie Li: performed TA testing and data analysis. Fengjia Xie: contributed to writing, review, and editing. Xuming Zhang: provided supervision and contributed to writing, review, and editing.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Research Grants Council (RGC) of Hong Kong (15215620, N_PolyU511/20), Innovation and Technology Commission (ITC) of Hong Kong (ITF-MHKJFS MHP/085/22, PiH/236/24) and The Hong Kong Polytechnic University (1-CD4V, 1-CD6U, G-SB6C, 1-CD8U, 1-BBEN, 1-W28S, 1-CD9Q, 1-SBVB, 1-CDJW, 1-CDJ8 and 1-W32A). The technical assistance and facility support of the UMF-Materials Research Centre (MRC) and UMF-Cleanroom (UMF-Cleanroom) of the University Research Facility in Material Characterization and Device Fabrication (UMF), the University Research Facility in Life Sciences (ULS), University Research Facility in 3D Printing (U3DP), and University Research Facility in Chemical and Environmental Analysis (UCEA) of The Hong Kong Polytechnic University are acknowledged.
References
- X. Pan, M. L. Heacock, E. N. Abdulaziz, S. Violante, A. L. Zuckerman, N. Shrestha, C. Yao, R. P. Goodman, J. R. Cross and V. Cracan, Nat. Chem. Biol., 2024, 20, 594–604 CrossRef CAS PubMed.
- W. Xiao, R. S. Wang, D. E. Handy and J. Loscalzo, Antioxid. Redox Signaling, 2018, 28, 251–272 CrossRef CAS.
- C. T. Walsh, B. P. Tu and Y. Tang, Chem. Rev., 2018, 118, 1460–1494 CrossRef CAS.
- X. Yuan, Y. Liu, B. M. Bijonowski, A. C. Tsai, Q. Fu, T. M. Logan, T. Ma and Y. Li, Commun. Biol., 2020, 3, 774 CrossRef CAS PubMed.
- W. Grabowska, E. Sikora and A. Bielak-Zmijewska, Biogerontology, 2017, 18, 447–476 CrossRef CAS PubMed.
- G. Zhao, C. Yang, W. Meng and X. Huang, J. Mater. Chem. A, 2024, 12, 3209–3229 RSC.
- C. Trotta, G. M. Rodriguez, C. Zuccaccia and A. Macchioni, ACS Catal., 2024, 14, 10334–10343 CrossRef CAS.
- L. B. F. Browne, T. Sudmeier, M. A. Landis, C. S. Allen and K. A. Vincent, Angew. Chem., Int. Ed., 2024, 63, e202404024 CrossRef CAS.
- F. Xing, X. Xue, J. Li, J. Liu, W. Wang, W. Dong, H. Yuan and J. Liu, ACS Catal., 2024, 14, 11366–11377 CrossRef CAS.
- L. Zhou, Z. Su, J. Wang, Y. Cai, N. Ding, L. Wang, J. Zhang, Y. Liu and J. Lei, Appl. Catal., B, 2024, 341, 123290 CrossRef CAS.
- Z. Xu, F. Zhou, H. Chen and J. Wang, ACS Catal., 2024, 14, 5868–5878 CrossRef CAS.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19–20, 167–176 CrossRef CAS.
- A. Bianco, M. Zaffagnini and G. Bergamini, Sustainable Energy Fuels, 2022, 6, 4393–4397 RSC.
- N. H. A. Besisa, K. S. Yoon and M. Yamauchi, Chem. Sci., 2024, 15, 3240–3248 RSC.
- N. H. A. Besisa, K. S. Yoon, T. G. Noguchi, H. Kobayashi and M. Yamauchi, ACS Sustainable Chem. Eng., 2024, 12, 9874–9881 CrossRef CAS.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 10 CrossRef.
- F. Xie, H. Jia, C. K. T. Wun, X. Huang, Y. Chai, C. C. Tsoi, Z. Pan, S. Zhu, K. Ren, T. W. B. Lo, Y. Zhu and X. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 11002–11011 CrossRef CAS.
- M. M. Grau, M. Poizat, I. W. C. E. Arends and F. Hollmann, Appl. Organomet. Chem., 2010, 24, 380–385 CrossRef CAS.
- M. Ding, S. Cui, Z. Lin and X. Yang, Appl. Catal., B, 2024, 357, 124333 CrossRef CAS.
- Z. Y. Wang, L. Xu, C. H. Liu, S. J. Han, M. L. Fu and B. Yuan, J. Mater. Chem. A, 2024, 12, 10991–11003 RSC.
- Z. Zhao, X. Wang, M. Feng, R. Cao, Y. Wang, Z. Wu, H. Si and J. Zhang, Appl. Surf. Sci., 2024, 657, 159672 CrossRef CAS.
- S. Lu, Y. Yin, J. Bao, H. Wang, Z. Lei, E. Hu, Q. Xin, Y. Quan, J. Li and Q. Wang, J. Environ. Chem. Eng., 2024, 12, 112989 CrossRef CAS.
- H. Li, R. Li, Y. Jing, B. Liu, Q. Xu, T. Tan, G. Liu, L. Zheng and L. Z. Wu, ACS Catal., 2024, 14, 7308–7320 CrossRef CAS.
- S. Tian, G. Long, P. Zhou, F. Liu, X. Zhang, C. Ding and C. Li, J. Am. Chem. Soc., 2024, 146, 15730–15739 CrossRef CAS.
- D. F. Ollis, J. Phys. Chem. B, 2005, 109, 2439–2444 CrossRef CAS.
- D. Han, B. Ding, P. Zheng, M. Yuan, Y. Bian, H. Chen, M. Wang, M. Chang, A. A. A. Kheraif, P. Ma and J. Lin, Adv. Healthcare Mater., 2024, 13, 2303309 CrossRef CAS.
- O. Bachar, M. M. Meirovich and O. Yehezkeli, Adv. Funct. Mater., 2024, 34, 2314443 CrossRef CAS.
- S. Yan, J. Wang, H. Gao, N. Wang, H. Yu, Z. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2012, 23, 1839–1845 CrossRef.
- Y. Chai, Y. Chen, B. Wang, J. Jiang, Y. Liu, J. Shen, X. Wang and Z. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 8825–8834 CrossRef CAS.
- G. Yang, D. Wang, Y. Wang, W. Hu, S. Hu, J. Jiang, J. Huang and H. L. Jiang, J. Am. Chem. Soc., 2024, 146, 10798–10805 CrossRef CAS PubMed.
- L. Tong, Z. Gong, Y. Wang, J. Luo, S. Huang, R. Gao, G. Chen and G. Ouyang, J. Am. Chem. Soc., 2024, 146, 21025–21033 CrossRef CAS PubMed.
- N. Jin, Y. Sun, W. Shi, P. Wang, Y. Nagaoka, T. Cai, R. Wu, L. Dube, H. N. Nyiera, Y. Liu, T. Mani, X. Wang, J. Zhao and O. Chen, J. Am. Chem. Soc., 2023, 145, 21886–21896 CrossRef CAS.
- K. Wu, Y. Du, H. Tang, Z. Chen and T. Lian, J. Am. Chem. Soc., 2015, 137, 10224–10230 CrossRef CAS PubMed.
- M. Y. Qi, Y. H. Li, F. Zhang, Z. R. Tang, Y. Xiong and Y. J. Xu, ACS Catal., 2020, 10, 3194–3202 CrossRef CAS.
- K. Q. Lu, Y. H. Li, F. Zhang, M. Y. Qi, X. Chen, Z. R. Tang, Y. M. A. Yamada, M. Anpo, M. Conte and Y. J. Xu, Nat. Commun., 2020, 11, 5181 CrossRef CAS PubMed.
- Y. Chai, Y. Kong, M. Lin, W. Lin, J. Shen, J. Long, R. Yuan, W. Dai and X. Wang, Nat. Commun., 2023, 14, 6168 CrossRef CAS.
- Y. Chai, Y. Chen, J. Shen, M. Ni, B. Wang, D. Li, Z. Zhang and X. Wang, ACS Catal., 2021, 11, 11029–11039 CrossRef CAS.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- J. Y. Xu, X. Tong, P. Yu, G. E. Wenya, T. McGrath, M. J. Fong, J. Wu and Z. M. Wang, Adv. Sci., 2018, 5, 1800221 CrossRef PubMed.
- B. A. Chen, G. T. Pang, X. Q. Lan, Z. B. He and R. Chen, Mater. Today Phys., 2020, 14, 100228 CrossRef.
- Y. Liu, D. A. Cullen and T. Lian, J. Am. Chem. Soc., 2021, 143, 20264–20273 CrossRef CAS PubMed.
- J. Gebicki, A. Marcinek and A. Zielonk, Acc. Chem. Res., 2004, 37, 379–386 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Liang Wana,
Heng Jianga,
Chi Chung Tsoia,
Yu Dua,
Huaping Jiaa,
Yujiao Zhu
a,
Liang Wana,
Heng Jianga,
Chi Chung Tsoia,
Yu Dua,
Huaping Jiaa,
Yujiao Zhu