
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrically conductive charge-segregated pseudo-polymorphs comprising highly planar expanded π-electronic cations†
Yohei
Haketa
 a,
Ryoya
Nakajima
a,
Yuto
Maruyama
a,
Hiroki
Tanaka
a,
Wookjin
Choi
b,
Shu
Seki
b,
Shunsuke
Sato
c,
Hitomi
Baba
c,
Yoshiki
Ishii
d,
Go
Watanabe
cd,
Kirill
Bulgarevich
e,
Kazuo
Takimiya
efg,
Kenzo
Deguchi
h,
Shinobu
Ohki
h,
Kenjiro
Hashi
i,
Takashi
Nakanishi
j,
Yukihide
Ishibashi
k,
Tsuyoshi
Asahi
k,
Kazuchika
Ohta
l and
Hiromitsu
Maeda
*a
a,
Ryoya
Nakajima
a,
Yuto
Maruyama
a,
Hiroki
Tanaka
a,
Wookjin
Choi
b,
Shu
Seki
b,
Shunsuke
Sato
c,
Hitomi
Baba
c,
Yoshiki
Ishii
d,
Go
Watanabe
cd,
Kirill
Bulgarevich
e,
Kazuo
Takimiya
efg,
Kenzo
Deguchi
h,
Shinobu
Ohki
h,
Kenjiro
Hashi
i,
Takashi
Nakanishi
j,
Yukihide
Ishibashi
k,
Tsuyoshi
Asahi
k,
Kazuchika
Ohta
l and
Hiromitsu
Maeda
*a
aDepartment of Applied Chemistry, College of Life Sciences, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: maedahir@ph.ritsumei.ac.jp
bDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan
cDepartment of Physics, Graduate School of Science, Kitasato University, Sagamihara 252-0373, Japan
dDepartment of Data Science, School of Frontier Engineering, Kitasato University, Sagamihara 252-0373, Japan
eCenter for Emergent Matter Science (CEMS), RIKEN, Wako 351-0198, Japan
fDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan
gAdvanced Institute for Materials Research (AIMR), Tohoku University, Sendai 980-8577, Japan
hResearch Network and Facility Services Division, National Institute for Materials Science (NIMS), Tsukuba 305-0003, Japan
iCenter for Basic Research on Materials, National Institute for Materials Science (NIMS), Tsukuba 305-0003, Japan
jResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Tsukuba 305-0044, Japan
kDepartment of Applied Chemistry, Graduate School of Science and Engineering, Ehime University, Matsuyama 790-8577, Japan
lInterdisciplinary Graduate School of Science and Technology, Shinshu University, Ueda 386-8567, Japan
First published on 19th February 2025
Abstract
Independently stacked positively and negatively charged π-electronic systems in charge-segregated columnar structures are desired for electronic properties derived from their electron-deficient and -rich assembling states, respectively. An expanded π-electronic cation, benzoporphyrin AuIII complex, was synthesized as the component of ion pairs in combination with counteranions. In contrast to benzoporphyrin, which is known for its insolubility in organic solvents, the ion pairs with bulky anions in this study are soluble in common organic solvents. The ion pairs formed charge-segregated assemblies as two pseudo-polymorphs of single-crystal and less-crystalline (LeC) states based on the stacking of the benzoporphyrin AuIII complex. XRD and solid-state NMR measurements, along with molecular dynamics (MD) simulation, revealed that the LeC states were formed by a less-ordered arrangement of constituting ions induced by bulky counteranions. The electric conductivity properties were observed in the single-crystal and LeC charge-segregated assemblies.
Introduction
The ordered arrangement of π-electronic systems is crucial for charge-carrier transport properties.1 Expanded π-planes are adequate for achieving high performance in organic semiconductive materials.1g Since substituents affect the electronic states of molecules and their arrangement, π-electronic systems that have no substituents are in great demand.1e However, such systems have low solubility (high crystallinity), making it difficult to arrange the constituents to form assembled structures (Fig. 1a top left). A promising strategy is the preparation of ion pairs of charged π-electronic systems by combining them with counterions that improve solubility (Fig. 1a top right).2 An appropriate combination of charged constituents enables facile handling of π-electronic systems to form counterion-dependent assemblies for applications (Fig. 1a bottom). In particular, independently stacked positively and negatively charged π-electronic systems are fascinating because of their ability to form electron-deficient and -rich assembling states, which can function as n- and p-type semiconductive pathways, respectively, in charge-segregated columnar structures.3 Expanded π-systems contribute to influential dispersion forces that overcome electrostatic repulsion in charged columns. The potential positively charged π-systems are porphyrin AuIII complexes, which have been included in various ion-pairing assemblies in the form of crystals, supramolecular gels and liquid crystals depending on the substituents.4,5 Expansion of π-electronic systems can be achieved by modifications at the pyrrole β-positions (Fig. 1b(i)). Numerous porphyrin derivatives that have been synthesized to date offered the choice to use benzoporphyrin61 (Fig. 1b(ii)) as a framework to provide highly planar charged expanded π-electronic systems. Large planes of benzoporphyrin-based cations are suitable for stacking to form charge-segregated assemblies, whose packing structures can be controlled by coexisting counteranions. The arrangement of substituent-free planar cations and bulky anions can be modulated by crystallization conditions. This study shows π-expanded cation-based ion-pairing assemblies in single-crystal and pseudo-polymorph less-crystalline (LeC) states and their electric conductivity properties derived from the charge-segregated assemblies. | ||
| Fig. 1 (a) Expanded π-electronic systems that have no peripheral substituents (top left) and their charged and ion-pairing states (top right, represented as cations), forming a charge-segregated assembly by stacking (bottom) and (b) (i) π-expanded porphyrin AuIII complexes as expanded π-electronic cations that can be used in (a) and (ii) benzoporphyrin 1. The positive signs were omitted in the charge-segregated assembly in (a). | ||
Results and discussion
Synthesis and characterization of expanded π-electronic cations
Benzoporphyrins, including metal complexes, can be synthesized from bicyclo[2.2.2]octadiene precursors via retro-Diels–Alder reactions.6f In this study, AuIII complexation was conducted for bicycloporphyrin 2 to afford the AuIII complex 2au+ mainly as a triflate (OTf−) ion pair by treatment with KAuCl4 in the presence of AgOTf and NaOAc (Fig. 2 top). The ion pair 2au+-OTf− was converted to the Cl− ion pair 2au+-Cl− in 28% yield (two steps) using an ion-exchange resin (Amberlite: IRA402BL Cl). As the selection of the counteranions was crucial in this study, the ion-pair metathesis of 2au+-Cl− with AgPF6, LiB(C6F5)4 (LiFABA), NaB(3,5-(CF3)2C6H3)4 (NaBArF) and NaPCCp (PCCp−: pentacyanocyclopentadienide)7 afforded the corresponding ion pairs 2au+-X− (X− = PF6−, FABA−, BArF− and PCCp−) in 66–76% yields. The obtained ion pairs were characterized by 1H, 13C and 19F NMR and ESI-TOF-MS. DMSO solutions of 2au+ ion pairs (4 μM) exhibited Soret and Q-bands at 392 and 507/542 nm, respectively, indicating that 2au+ exists as a monomeric state with a negligible counteranion effect on the electronic properties of 2au+ (Fig. S12†).82au+-FABA− and 2au+-BArF− were also characterized by X-ray analysis for single crystals prepared by vapour diffusion from CH3CN/water (Fig. 3, S18 and S19†). In the solid-state structures, the cation 2au+, refined as one of the stereoisomers, formed stacked dimers with stacking/Au⋯Au distances of 3.56/3.41 and 3.48/3.37 Å, respectively, and a rotation of ∼45° owing to dispersion forces at the core planes and β-bicyclo units (Fig. 3a and b(i)). The stacking of 2au+ is visualized by Hirshfeld surface analysis, which shows a bow-tie arrangement of red and blue triangles in the shape-index property and flat regions in the curvedness property (Fig. S24 and S25†).9 Mean-plane deviations of 0.052/0.068 and 0.036 Å for stacking 2au+ planes (core 25 atoms), respectively, indicate slightly curved 2au+ planes, owing to the Au⋯Au distances being less than the stacking distances. Such remarkably close Au⋯Au distances are also ascribable to the orbital interactions arising from overlap of the 5dz2 and 6pz orbitals of the adjacent AuIII.10 The stacked 2au+ dimers in ion pairs are aligned along the c-axis with an offset, which is smaller in 2au+-BArF− with a longer distance between the stacked dimers by including two CH3CN molecules (Fig. 3a and b(ii)). In either case, counteranions are located at the side of the stacked 2au+ dimers. In particular, BArF− anions are located proximally at the side of the stacked 2au+ dimers, forming a pseudohexagonally arranged packing structure along the c-axis (Fig. S19a†). | ||
| Fig. 2 Synthesis of benzoporphyrin AuIII complex ion pairs 1au+-X− (X− = PF6−, FABA−, BArF− and PCCp−) via the corresponding bicycloporphyrin AuIII complex ion pairs 2au+-X−. | ||
 | ||
| Fig. 3 Single-crystal X-ray structures of (a) 2au+-FABA− and (b) 2au+-BArF−: (i) side and top views of stacked dimers with stacking and Au⋯Au (italic) distances and (ii) packing structures with the yellow parts corresponding to the stacked dimers in (i). Atom colour codes in (i): brown, pink, blue and orange refer to carbon, hydrogen, nitrogen and gold, respectively. Colour codes in (ii): cyan and magenta refer to cation and anion parts, respectively. Solvent molecules are omitted in (a), whereas CH3CN molecules are shown in (b). | ||
Stacking of 2au+ by overcoming electrostatic repulsion was also observed in the solution state. In CD3CN, the 1H NMR of 2au+-FABA− showed broad signals at 9.96, 8.37/6.94, 5.98 and 3.49–1.30 ppm ascribable to meso-CH, bicyclo-sp2-CH, bicyclo-bridged methine-CH and bicyclo-sp3-CH, respectively, for the stacked dimer.11 Concentration-dependent UV/vis absorption spectra of 2au+-FABA− in CH3CN exhibited a blue shift of λmax from 389 to 374 nm when increasing the concentration from 1.0 × 10−6 to 1.0 × 10−5 M, suggesting the formation of an H-like stacked dimer at the higher concentration (Fig. S71†). The transition dipole moments of 2au+ in the optimized structure of the stacked dimer 2au+2 based on PCM-GD3BJ-B3LYP/6-31+G(d,p) with LanL2DZ for Au (CH3CN) are arranged at ∼45°, suggesting that the H-like stacking mode induces a blue shift (Fig. S61a†).12 In addition, TD-DFT calculation of the optimized 2au+2 revealed an absorption at 375 nm, which is blue-shifted by 16 nm compared to the monomer state (Fig. S46†).
According to the synthetic procedure for benzoporphyrin 1,6f2au+-X− (X− = PF6−, FABA−, BArF− and PCCp−) were quantitatively transformed to the corresponding benzoporphyrin ion pairs 1au+-X− by heating at 180 °C for 20–60 min in the absence of solvent (Fig. 2 bottom). In contrast to 1,6f which is insoluble in most organic solvents, the obtained 1au+-X− showed enhanced solubility. For example, 1au+-FABA− was soluble in acetone, DMF, CH3CN and DMSO. In contrast, another ion pair 1au+-Cl−, which was synthesized from 2au+-Cl−, was not soluble in these solvents.13 It is noteworthy that 1au+ is soluble with facile handling in the form of the ion pairs with appropriate counteranions, although the optimized structure of 1au+ estimated at B3LYP/6-31+G(d,p) with LanL2DZ for Au12 exhibits completely planar geometry with a mean-plane deviation of 0.00 Å (Fig. S29†). 1H NMR of 1au+-FABA− in DMSO-d6 (1.0 mM), as an example, at r.t. exhibited broad signals at 10.21, 9.18 and 8.11 ppm, suggesting soluble but aggregated structures as also indicated by concentration-dependent 1H NMR (Fig. S74†).14 Such 1H NMR signals in the downfield region suggested the aromatic ring current effect of 1au+, which was further supported by nucleus-independent chemical shift (NICS) and the anisotropy of the current induced density (ACID) calculations (Fig. S54 and S55†). Interestingly, 19F NMR in the same solvent shows sharp signals derived from FABA− in the dispersed state. The UV/vis absorption spectrum of 1au+-FABA− in DMSO (4 μM), as a monomer state, exhibits Soret and Q bands of 408 and 564/616 nm, respectively (Fig. 4a), which are more red-shifted than those of 2au+-FABA−. The TD-DFT-based UV/vis absorption stick spectrum of 1au+ in DMSO shows that these absorptions are mainly derived from the HOMO−1-to-LUMO+1 and HOMO-to-LUMO+1 transitions, respectively (Fig. 4a inset, S49†).
 | ||
| Fig. 4 (a) UV/vis absorption spectrum of 1au+-FABA− in DMSO (4 μM) (inset: photograph of the DMSO solution (4 μM)) and TD-DFT-based UV/vis absorption stick spectrum (grey bar) of 1au+ at PCM-B3LYP-GD3BJ/6-31+G(d,p) with LanL2DZ for Au (DMSO) and (b) UV/vis absorption spectra of 1au+-FABA− in CH3CN according to (i) concentrations (solid line: 0.18 μM and broken line: 20 μM) and (ii) temperatures (solid line: 70 °C and broken line: −40 °C). | ||
The 1H NMR of 1au+-FABA− in CD3CN (1.0 mM) shows broader signals than those in DMSO-d6, suggesting more aggregated structures in the less polar solvent (Fig. S9†). Similar to 2au+-FABA−, 1au+-FABA− shows a λmax blue-shift from 402 to 384 nm upon increasing the concentration from 1.8 × 10−7 to 2.0 × 10−5 M in CH3CN, suggesting the formation of H-like stacked structures (Fig. 4b(i) and S72†). Such a blue shift of the λmax is also observed in variable-temperature (VT)-UV/vis absorption spectra at lower temperatures (Fig. 4b(ii)). TD-DFT calculation of the optimized structure for the stacked dimer 1au+2 at PCM-B3LYP-GD3BJ/6-31+G(d,p) with LanL2DZ for Au (CH3CN) suggests a slightly blue-shifted Soret band compared to that of the monomeric state (Fig. S51†). The dimerization constant (Kdim) of 1au+ for 1au+-FABA− is estimated to be 5 × 106 M−1 in CH3CN at r.t. from concentration-dependent UV/vis absorption spectral changes (Fig. S72†). π-Expansion of positively charged π-electronic systems induces influential dispersion forces, which enable the stacking of identically charged π-electronic systems.
Single-crystal-state charge-segregated assemblies
Prism-shaped single crystals of the ion pairs 1au+-X− (X− = FABA−, BArF− and PCCp−), obtained from CH3CN/CHCl3,15 1,1,1-trichloroethane/n-heptane and DMF/o-dichlorobenzene, respectively, were suitable for X-ray analysis, revealing the exact structures of the ion pairs and their assembled structures (Fig. 5). In these structures, the cation 1au+, showing planar geometry with mean-plane deviations (core 25 atoms) of 0.016, 0.031/0.011 and 0.023 Å, respectively, forms closely stacked columnar structures with stacking distances of 3.29, 3.36/3.44 and 3.37 Å, respectively, in the charge-segregated mode (Fig. 5a–c(i)). The stacked parts of 1au+ are clearly shown by Hirshfeld surface analysis, exhibiting a bow-tie arrangement of red and blue triangles in the shape-index property and flat regions in the curvedness property (Fig. S26–S28†).9 The Au⋯Au distances are 4.76, 3.93/4.59 and 3.81 Å, respectively, suggesting that the offset stacking of the cations is larger for 1au+-FABA−. The angles of 44.1°, 48.4°/60.1° and 62.1° are estimated, respectively, for the lines passing through two Au atoms of stacked 1au+ to the corresponding 41-atom mean planes. In these ion pairs, counteranions FABA−, BArF− and PCCp− are located at the side of the columnar structures (Fig. 5a–c(ii) and (iii)). The ion-pair crystals 1au+-FABA−, 1au+-BArF− and 1au+-PCCp− formed orthorhombic, monoclinic and orthorhombic packing, respectively, with the columns of stacked 1au+ aligned along the a-, c- and a-axes, respectively, which are the long axes of the prism crystals (Fig. S23†). Notably, the stacked 1au+ in the columns are tilted, with angles of 30.4°, 2.9°/3.0° and 21.2°, respectively, relative to the stacking axis (Fig. 5a–c(ii)). Interestingly, in the crystal packing of 1au+-FABA−, an ion-pair framework composed of columnar cation structures and counteranions forms two tubular spaces per unit cell, with a volume of 5.56 nm3, containing disordered solvent molecules. The solvent molecules in the single crystal are not removed after heating at 100 °C under vacuum, as revealed by the X-ray analysis. | ||
| Fig. 5 Single-crystal X-ray structures of (a) 1au+-FABA−, (b) 1au+-BArF− and (c) 1au+-PCCp−: (i) side views of the columnar structures of stacked 1au+ with stacking and Au⋯Au (italic) distances and top views along with Hirshfeld surface mapped over the shape-index property of the stacked dimer, (ii) packing structures of columns of stacked 1au+ and counteranions and (iii) packing structures with the yellow parts corresponding to the packing structures in (ii). In (b), the different types of stacking arrangements are indicated by A and B (stacking arrangement A is shown as a representative). Atom colour codes in (i): brown, pink, blue and orange refer to carbon, hydrogen, nitrogen and gold, respectively. Colour codes in (ii and iii): cyan and magenta refer to cation and anion parts, respectively. | ||
To evaluate the stacking behaviour of 1au+ in the crystal structures, energy decomposition analysis (EDA)16,17 was performed using the fragment molecular orbital (FMO) method (FMO2-MP2) with mixed basis sets including NOSeC-V-DZP with MCP with NOSeC-V-TZP with MCP for Au.18,19 The EDA calculations using FMO yielded Ees, Edisp, Ect and Eex (energies for electrostatic, dispersion, charge-transfer forces and exchange repulsion, respectively) and Etot (total energy). In the columnar structure of 1au+ in 1au+-FABA−, an Etot of −164.2 kcal mol−1 is observed, whereas Edisp and Ees are −214.0 and 6.5 kcal mol−1, respectively, indicating that Edisp is a major force in the stacking structure (Fig. 6 and S58†). EDA calculations for 1au+-BArF− and 1au+-PCCp− also elucidated similar energy balances for neighbouring π-electronic ions (Fig. S59 and S60†).
 | ||
| Fig. 6 Decomposition of the total intermolecular interaction energies (Etot) of 1au+-FABA− for (a) the single-crystal X-ray structure and (b) estimated interaction energies (kcal mol−1) according to EDA based on the FMO2-MP2 method using a basis set of NOSeC-V-DZP with MCP with NOSeC-V-TZP with MCP for Au (see Table S4† for the complete data list). Colour codes in (i): brown, pink, blue, yellow, green and orange refer to carbon, hydrogen, nitrogen, boron, fluorine and gold, respectively. | ||
Crystal-state absorptions of the ion pairs 1au+-FABA−, 1au+-BArF− and 1au+-PCCp− were evaluated via optical microscopy for spectroscopic examination (Fig. S75 and S76†). In 1au+-FABA−, the absorptions at 587 and 623 nm, which are slightly blue- and red-shifted, respectively, compared to those of the monomer in DMSO (616 nm), are ascribable to the exciton coupling in a predominantly J-like arrangement and also very weak coupling for orthogonally arranged transition dipole moments (Fig. 7 and S62†). These behaviours are derived from the D4h geometry of 1au+. Similar to 1au+-FABA−, the crystal-state absorption of 1au+-PCCp− shows absorptions at 588 and 623 nm, which are ascribable to the exciton coupling of stacked 1au+. On the other hand, 1au+-BArF− mainly exhibits a blue-shifted broad absorption at 585 nm with a shoulder at 654 nm. The blue-shifted absorption can be attributed to the larger contribution of the H-like arrangement of transition dipoles in the stacked 1au+ (Fig. S62†).
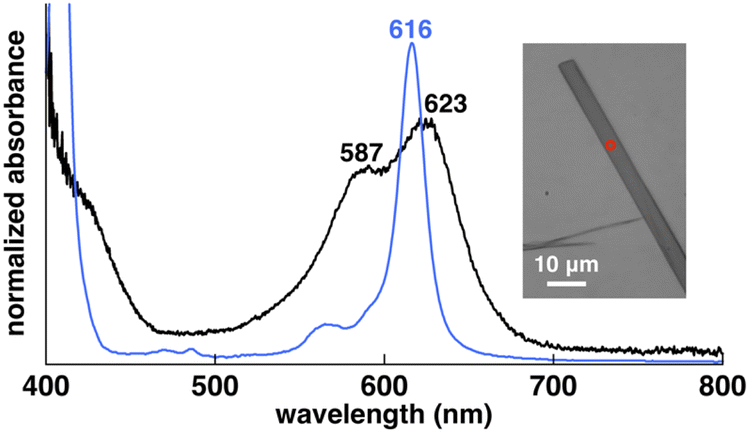 | ||
| Fig. 7 Solid-state UV/vis absorption spectra of 1au+-FABA− in the single crystal (black line) and in DMSO (4 μM) (blue line) as a reference (inset: photograph of the single crystal (red circle indicates the position of measurement)). | ||
Charge-segregated assemblies of π-electronic ion pairs show fascinating electric conductivity properties. The electric conductivity properties of stacked 1au+ in the ion pairs (1au+-FABA−, 1au+-BArF− and 1au+-PCCp−) were evaluated by flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements (Fig. 8a and S91†).20 Electric conductivity (ϕΣμ) values of 4.6 × 10−9, 9.2 × 10−9 and 2.9 × 10−9 m2 V−1 s−1 were observed for the longer axes of the respective single crystals. Clear anisotropic electric conductivity was shown in 1au+-BArF− (7.9 × 10−9 m2 V−1 s−1 for the shorter axis). These values are comparable to and greater than those of previously reported charge-segregated assemblies.3 The order of the ϕΣμ values, 1au+-BArF− > 1au+-FABA− > 1au+-PCCp−, is consistent with the theoretically estimated transfer integrals t at PW91/TZP for the stacked 1au+ units in the crystal structures, showing hole transfer integrals |t|h of 118.5/50.5, 21.3 and 9.8 meV, respectively (Fig. 8b and S63†).21 Furthermore, theoretically estimated HOMO band dispersions using the tight-binding approximation22 for the stacked 1au+ in the single-crystal structures of 1au+-FABA−, 1au+-BArF− and 1au+-PCCp− exhibited one-dimensional band structures, which are consistent with the stacking structures of 1au+ (Fig. S64†). The Fermi levels lie in the middle of the band gaps, suggesting that charge-segregated assemblies comprising stacked 1au+ exhibit semiconductive behaviours. In the discussed ion pairs, decreased on-site Coulomb repulsion between stacked π-electronic cations with expanded π-electronic systems would induce hole transport rather than electron transport. The conductivity transients recorded under an SF6 atmosphere with negligible quenching of charge carriers suggest a major contribution from holes photo-injected into the assemblies (Fig. S92†).23
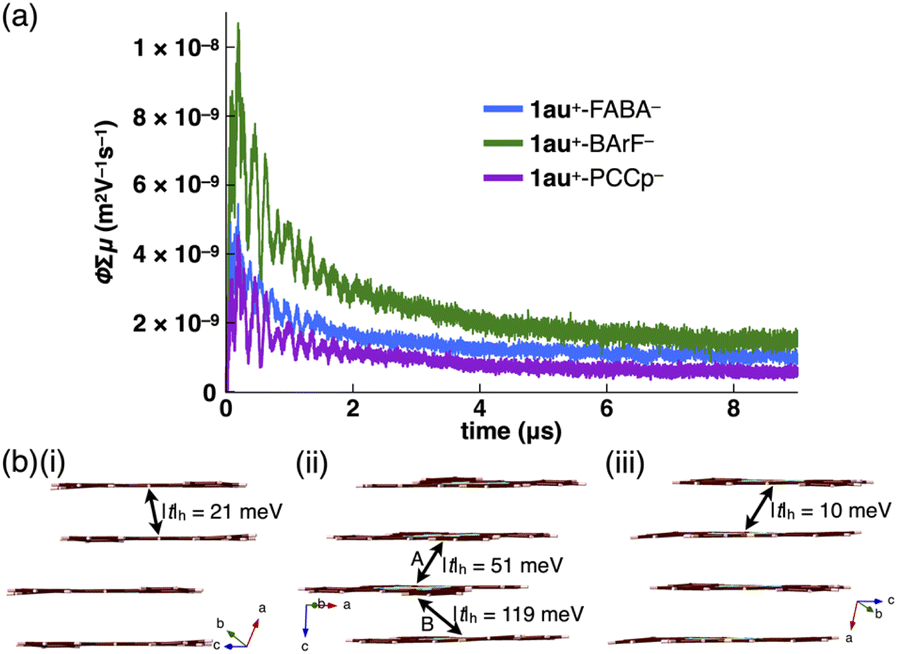 | ||
| Fig. 8 (a) Photoconductivity transients observed upon excitation at 355 nm, 9.1 × 1015 photons per cm2 per pulse, for the long axis of the single crystals of 1au+-FABA− (blue), 1au+-BArF− (green) and 1au+-PCCp− (purple) and (b) stacked 1au+ in the single crystal structures of (i) 1au+-FABA−, (ii) 1au+-BArF− and (iii) 1au+-PCCp− and hole transfer integrals (|t|h) estimated at PW91/TZP. Stacking modes A and B in (ii) correspond to Fig. 5b(i). | ||
Charge-segregated assemblies in pseudo-polymorph less-crystalline states
Substituent-free benzoporphyrins in the forms of free base and metal complexes have been known to show highly crystalline states after heating the corresponding bicyclo[2.2.2]octadiene precursors in the film state.6h In contrast, 1au+ as ion pairs can form bulk materials that are not single crystals via recrystallization from appropriate solvents. Precipitation of 1au+-FABA− from acetone/n-hexane provided a material (labelled as 1au+-FABA−P) that appeared to differ from the single crystals (Fig. 9a inset).24 The synchrotron XRD of 1au+-FABA−P at 25 °C exhibited broad peaks instead of crystalline diffraction, suggesting the formation of an LeC state for the obtained precipitates. The diffraction peaks of 1.81, 1.04, 0.90, 0.68, 0.60, 0.52, 0.50, 0.45, 0.41 and 0.34 nm, showing Debye–Scherrer rings, were assigned to the hkl parameters derived from a hexagonal pattern (100, 110, 200, 210, 300, 220, 310, 400 and 320) as a = 2.09 nm (Fig. 9a and S81†).25 The intense peak at 0.34 nm was assigned to 001 as the stacking distance of 1au+ in the hexagonal columnar (Colh) structure (Z = 1 for ρ = 1.79 g cm−3) (Fig. 9b). Interestingly, heating the powder sample of 2au+-FABA−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 at 190 °C (labelled as 1au+-FABA−H) also formed a Colh structure identical to that of 1au+-FABA−P (Fig. S79–S82†). In contrast to the tilted 1au+ plane along the a-axis in the single crystal of 1au+-FABA−, the 1au+ plane in 1au+-FABA−P should be arranged perpendicularly to the stacking axis, as indicated by the intracolumnar stacking period of 0.34 nm. On the basis of the Colh structure with a = 2.09 nm and the sizes of 1au+ and FABA−, columnar structures comprising less-ordered stacking of 1au+, as indicated by cyan circles, are located close to FABA−, as indicated by magenta circles (Fig. 9b(ii)).
26 at 190 °C (labelled as 1au+-FABA−H) also formed a Colh structure identical to that of 1au+-FABA−P (Fig. S79–S82†). In contrast to the tilted 1au+ plane along the a-axis in the single crystal of 1au+-FABA−, the 1au+ plane in 1au+-FABA−P should be arranged perpendicularly to the stacking axis, as indicated by the intracolumnar stacking period of 0.34 nm. On the basis of the Colh structure with a = 2.09 nm and the sizes of 1au+ and FABA−, columnar structures comprising less-ordered stacking of 1au+, as indicated by cyan circles, are located close to FABA−, as indicated by magenta circles (Fig. 9b(ii)).
 | ||
| Fig. 9 (a) Synchrotron XRD pattern of 1au+-FABA−P at 25 °C for the sample obtained by precipitation from acetone/n-hexane (inset: photograph of the precipitates), (b) schematic representation for the Colh packing structure: (i) packing diagram and (ii) top view of Colh (1au+ and FABA− are represented in cyan and magenta, respectively), (c) top and side views of the optimized structure of 1au+-FABA− as a dimer using B3LYP-GD3BJ/6-31+G(d,p) with LanL2DZ for Au and (d) snapshot of the MD simulation result for 1au+-FABA−P after 100 ns of the equilibration run at 25 °C showing (i) the top view of the packing diagram and (ii) the side view of 1au+ columns extracted from the packing diagram (1au+ and FABA− are represented in cyan and magenta, respectively). In (b) (ii), magenta circles suggest the possible locations of FABA− with a 50% occupancy rate on average in a cross section, whereas cyan circles show the diameter of 1au+ columns in slipped stacking. | ||
The proximal location of 1au+ and FABA− is also suggested by the optimized structure of dimeric 1au+-FABA− using B3LYP-GD3BJ/6-31+G(d,p) with LanL2DZ for Au (Fig. 9c). FABA− should be paired with several 1au+ in the proximal location, although the XRD pattern suggests that the location of FABA− is less clear, probably indicating an amorphous-like state. In light of the stoichiometry of the constituents and their contrasting planar and bulky shapes, FABA− would be observed in three sites on average among the 1au+ columns, and, in another cross section according to the 1au+ planes, the anions should be located in the other three sites. As a result, FABA− can be hexagonally arranged around the 1au+ columns. The Colh structure suggested by the XRD pattern is clearly demonstrated by all-atom molecular dynamics (MD) simulations at 25 °C after 100 ns of the equilibration run (Fig. 9d and S66†). Notably, as the initial structure for the MD simulation, the columns comprising tilted 1au+ units, as observed in the single-crystal structure, are transformed to a structure with barely tilted 1au+. The combination of planar 1au+, suitable for stacking, and bulky FABA−, with a less-ordered arrangement via noncovalent interactions induces the LeC state.27,28 The less-ordered FABA− interferes with the ordered packing of 1au+ for crystallization. 1au+-FABA−P maintains the Colh structure up to 195 °C and is converted to a complicated crystalline state at higher temperatures, with decomposition at 355 °C. This behaviour is also supported by differential scanning calorimetry (DSC) analysis (Fig. S78†). The appearance of 1au+-FABA−P as a dark blue powder in polarized optical microscopy (POM) was maintained through the heating process. The condition-dependent assembly (single-crystal and LeC states) as pseudo-polymorphism29 is fascinating for tuneable properties according to the arrangement of building blocks. The Colh LeC state, in the absence of aliphatic chains, is rare28 but can be achieved by pairing the planar π-electronic cation with a bulky counteranion.30
The details of the structures of 1au+-FABA− were investigated by solid-state NMR (SSNMR) spectroscopy. 13C NMR was performed using cross-polarization magic-angle spinning (CPMAS) (Fig. 10 and S86†). Dipolar dephasing experiments were conducted to support the signal assignment for the ion pair (Fig. S86 and S88†). The broad signals at 132.2, 120.8, 93.4 and 89.3 ppm were assigned to 1au+, whereas those at 149.7, 137.7 and 126.0 ppm were assigned to FABA− (Fig. 10a). 13C CPMAS NMR of 1au+-FABA−H showed slightly broader signals than 1au+-FABA−P, suggesting the formation of a more disordered arrangement of constituting ions (Fig. 10b). In contrast, 13C CPMAS NMR for 1au+-FABA− as single crystals showed narrower signals than those of 1au+-FABA−P and 1au+-FABA−H (Fig. 10c), suggesting that the broader signals of 1au+-FABA−P and 1au+-FABA−H indicated a less-ordered arrangement of ions that form Colh structures. Similar signal broadening was observed for 19F and 11B MAS NMR of 1au+-FABA−P and 1au+-FABA−H (Fig. S89 and S90†). Moreover, the characteristic up-field split signals at 93.4 and 89.3 ppm in 1au+-FABA−P are derived from unsubstituted meso-carbons,31 as also suggested by theoretically estimated NMR signals for 1au+ using B3LYP/6-311+G(d,p) with SDD for Au (Fig. S65†).12 Such signal splitting is also observed in the 13C CPMAS NMR of the single crystals due to slipped stacking of 1au+ observed in the crystal structure of 1au+-FABA− (Fig. 5a(i)).32 These observations, along with the XRD analysis and MD simulation, support a less-ordered slipped stacking structure for 1au+ in a column of Colh LeC states, wherein the 1au+ planes are more perpendicularly arranged to the column compared to those in the single crystal.33
 | ||
| Fig. 10 Solid-state 13C CPMAS NMR spectra of (a) 1au+-FABA−P, (b) 1au+-FABA−H and (c) 1au+-FABA− as single crystals recorded at 20 kHz MAS spinning frequency at r.t. with the corresponding packing diagrams. | ||
The formation of such an LeC state was also observed in the precipitate of 1au+-BArF−P prepared from acetone/n-hexane.34 Similar to 1au+-FABA−P, 1au+-BArF−P shows a Colh LeC state (a = 2.28 nm, c = 0.34 nm, and Z = 1 for ρ = 1.71 g cm−3) (Fig. S84 and S85†), also exhibiting condition-dependent pseudo-polymorphs. Colh LeC states in the precipitates were observed for 1au+-FABA− and 1au+-BArF−, which formed orthorhombic and monoclinic single-crystal packing structures, respectively. Such pseudo-polymorphic phenomena are fascinating because stacking of 1au+ can be easily controlled by the assembly conditions. The electric conductivity (ϕΣμ) values of LeC-state 1au+-FABA−P, 1au+-FABA−H and 1au+-BArF−P were estimated to be 1.2 × 10−9, 1.0 × 10−9 and 3.4 × 10−9 m2 V−1 s−1, respectively, suggesting that electrically conductive pathways also exist in the columnar structures of LeC materials, although the values are smaller than those of the corresponding single crystals (Fig. S93†).
Conclusions
Ion-pairing assemblies in charge-segregated modes were constructed from a highly planar expanded π-electronic cation in combination with counteranions. Charge-segregated assemblies were formed with both planar and bulky counteranions by means of stable stacked structures of the expanded π-electronic cation. The stacking arrangement and resulting absorption spectra in the single crystals were modulated by coexisting anions. Depending on crystallizing solvent conditions, ion pairs with bulky borate anions also provided LeC states as pseudo-polymorphs of their single crystals. Both single crystals and LeC states exhibited electric conductive properties due to stacking of the π-expanded porphyrin AuIII complex.35 It is noteworthy that the discussed planar π-electronic cation, benzoporphyrin AuIII complex, is soluble in organic solvents in the ion-pairing states. Ion pairing is an effective strategy to increase the solubility of planar π-electronic systems for their facile handling and the fabrication of assembled structures with ordered arrangements. Further modifications of charged π-electronic systems would lead to ion-pairing assemblies that can be applied for functional electronic materials and devices.Data availability
Data supporting the work in this publication are available via the ESI and associated crystallographic data.Author contributions
H. M. designed and conducted the project. Y. H., R. N., Y. M. and H. T. carried out the synthesis, characterization and property examinations. W. C. and S. Se. evaluated the electric conductivities. S. Sa., H. B., Y. I. and G. W. conducted the MD calculations. K. B. and K. T. conducted the transfer integral calculations. K. D., S. O., K. H. and T. N. evaluated the SSNMR. Y. I. and T. A. recorded the solid-state absorption spectra. K. O. supported the discussion on the assemblies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP18H01968, JP22H02067 and JP23K23335 for Scientific Research (B), JP19K05444 and JP24K08389 for Scientific Research (C), JP20J22745 for JSPS Fellows, JP23K17951 for Challenging Research (Exploratory) and JP20H05863 for Transformative Research Areas (A) “Condensed Conjugation”, JST SPRING, Grant Number JPMJSP2101 and Ritsumeikan Global Innovation Research Organization (R-GIRO) project (2017–22 and 2022–27). Theoretical calculations were partially performed using the Research Center for Computational Science, Okazaki, Japan (Projects: 20-IMS-C079, 21-IMS-C077, 22-IMS-C077, 23-IMS-C069 and 24-IMS-C067). Synchrotron-radiation analysis was performed at BL40XU (2021B1703 and 2022B1149), BL02B1 (2024A1644) and BL40B2 (2021B1828, 2022A1689, 2022B1546, 2023A1331, 2023B1409 and 2024A1463) of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). A part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We thank Dr Noboru Ohta, JASRI/SPring-8, for synchrotron-XRD measurements, Dr Nobuhiro Yasuda, JASRI/SPring-8 and Prof. Takuji Hatakeyama, Kyoto University, for single-crystal synchrotron-X-ray analysis, Prof. Osamu Tsutsumi, Ritsumeikan University, for single-crystal X-ray analysis, Prof. Yasuteru Shigeta, University of Tsukuba, for theoretical study and Prof. Hitoshi Tamiaki, Ritsumeikan University, for various measurements.Notes and references
- Selected books and reviews on supramolecular assemblies for electronic materials:
(a)
Functional Supramolecular Architectures: For Organic Electronics and Nanotechnology, ed. P. Samorì and F. Cacialli, Wiley, Weinheim, 2011 Search PubMed
; (b) Supramolecular Materials for Opto-Electronics, ed. N. Koch, RSC, Cambridge, 2015 Search PubMed
- Y. Haketa, K. Yamasumi and H. Maeda, Chem. Soc. Rev., 2023, 52, 7170–7196 Search PubMed
-
(a) K. Yamasumi, K. Ueda, Y. Haketa, Y. Hattori, M. Suda, S. Seki, H. Sakai, T. Hasobe, R. Ikemura, Y. Imai, Y. Ishibashi, T. Asahi, K. Nakamura and H. Maeda, Angew. Chem., Int. Ed., 2023, 62, e202216013 CrossRef CAS PubMed
- Selected reports of porphyrin AuIII complexes:
(a) E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373–376 CrossRef CAS
-
(a) Y. Haketa, Y. Bando, Y. Sasano, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, iScience, 2019, 14, 241–256 CrossRef CAS PubMed
- A review and selected reports of benzoporphyrins:
(a) C. M. B. Carvalho, T. J. Brocksom and K. T. de Oliveira, Chem. Soc. Rev., 2013, 42, 3302–3317 RSC
-
(a) O. W. Webster, J. Am. Chem. Soc., 1965, 87, 1820–1821 CrossRef CAS
- 2au + can be obtained as a single isomer prepared from the separated isomer of 2. Details on the synthetic procedures and properties will be discussed elsewhere.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed
- W. Lu, K. T. Chan, S.-X. Xu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752–755 RSC
- The dimerization constant (Kdim) for 2au+-FABA− was estimated to be 1.1 × 105 M−1 from the monomer/dimer ratio of 1H NMR signals in CD2Cl2 (5 × 10−5 M) at 20 °C (Fig. S73†).
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- 1au + with BArF− was also soluble in CH2Cl2 and MeOH, whereas the PF6− ion pair showed less solubility in the solvents that were available for 1au+-FABA−.
- K dim of 1au+ for 1au+-FABA− in DMSO-d6 was estimated to be 400 M−1.
- Single crystals of 1au+-FABA− prepared from acetone/1,2-dichloroethane also afforded a similar packing structure.
- Articles for GAMESS:
(a) M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS
- M. J. S. Phipps, T. Fox, C. S. Tautermann and C.-K. Skylaris, Chem. Soc. Rev., 2015, 44, 3177–3211 RSC
- Report for FMO: K. Kitaura, E. Ikeo, T. Asada, T. Nakano and M. Uebayasi, Chem. Phys. Lett., 1999, 313, 701–706 Search PubMed
- Report for pair interaction energy decomposition analysis (PIEDA): D. G. Fedorov and K. Kitaura, J. Comput. Chem., 2007, 28, 222–237 CrossRef CAS PubMed
-
(a) A. Acharya, S. Seki, A. Saeki, Y. Koizumi and S. Tagawa, Chem. Phys. Lett., 2005, 404, 356–360 CrossRef CAS
- Program and report for calculation of transfer integrals:
(a)
ADF (2024.1), SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, 2024 Search PubMed
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627–633 CrossRef CAS
- Hole transport can be acceptable in light of the electronic properties of 1au+. Theoretical studies revealed a higher HOMO of 1au+ than that of 2au+, suggesting that π-expanded 1au+, which is positively charged, would be suitable for hole transport (Fig. S35 and S40†). In addition, the HOMO of 1au+ is comparable to those of typical hole-transporting materials (ref. 1d).
- Other precipitation conditions such as rapid precipitation using CH3CN/CHCl3 afforded crystalline precipitates with a packing structure similar to the single-crystal structure (Fig. S83†).
- Small diffraction peaks would be derived from the unidentified packing structure of a minor species formed in the precipitation process.
- The powder sample of 2au+-FABA− was prepared by precipitating from CH2Cl2/n-hexane.
-
K. Ohta, Physics and Chemistry of Molecular Assemblies, World Scientific, 2020 Search PubMed
- Selected examples of liquid crystals and plastic crystals based on π-electronic molecules that have no aliphatic chains:
(a) S. Basurto, S. García, A. G. Neo, T. Torroba, C. F. Marcos, D. Miguel, J. Barberá, M. B. Ros and M. R. de la Fuente, Chem.–Eur. J., 2005, 11, 5362–5376 CrossRef CAS PubMed
- B. Dario and G. Fabrizia, Chem. Soc. Rev., 2000, 29, 229–238 RSC
- The LeC states in this study might be considered as liquid crystals. The details will be discussed elsewhere.
- M. Okazaki and C. A. McDowell, J. Am. Chem. Soc., 1984, 106, 3185–3190 CrossRef CAS
- A. P. M. Kentgens, B. A. Markies, J. F. van der Pol and R. J. M. Nolte, J. Am. Chem. Soc., 1990, 112, 8800–8806 CrossRef CAS
- Although the current evaluations for the bulk states suggest frozen-state properties at r.t. that are similar to those of single crystals, investigations according to thermal conditions would reveal more detailed information on dynamic thermal motion and less-ordered states.
- Higher crystallinity compared to 1au+-FABA−P was suggested by the XRD analysis.
- Ion-pairing assemblies in this study, exhibiting unconventional properties derived from charged π-electronic systems, are completely dissimilar from charge-transfer (CT) complexes that are obtained from electronically neutral donor and acceptor molecules. A review of CT complexes: M. Baharfar, A. C. Hillier and G. Mao, Adv. Mater., 2024, 36, 2406083 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical data, computational details and CIF files for the single-crystal X-ray structural analysis. CCDC 2387040–2387044. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07576e |
| This journal is © The Royal Society of Chemistry 2025 |