
Polymeric nanoparticle synthesis for biomedical applications: advancing from wet chemistry methods to dry plasma technologies
Elmer S.
Austria
Jr.
ab and
Behnam
Akhavan
 *abcd
*abcd
aSchool of Biomedical Engineering, Faculty of Engineering, University of Sydney, Sydney, NSW 2006, Australia. E-mail: Behnam.Akhavan@Newcastle.edu.au
bSydney Nano Institute, University of Sydney, Sydney, NSW 2006, Australia
cSchool of Engineering, College of Engineering, Science and Environment, University of Newcastle, Callaghan, NSW 2308, Australia
dHunter Medical Research Institute (HMRI), Precision Medicine Program, New Lambton Heights, NSW 2305, Australia
First published on 20th May 2025
Abstract
Nanotechnology has introduced a transformative leap in healthcare over recent decades, particularly through nanoparticle-based drug delivery systems. Among these, polymeric nanoparticles (NPs) have gained significant attention due to their tuneable physicochemical properties for overcoming biological barriers. Their surfaces can be engineered with chemical functional groups and biomolecules for a wide range of biomedical applications, ranging from drug delivery to diagnostics. However, despite these advancements, the clinical translation and large-scale commercialization of polymeric NPs face significant challenges. This review uncovers these challenges by examining the interplay between structural design and payload interaction mode. It provides a critical evaluation of the current synthesis methods, beginning with conventional wet chemical techniques, and progressing to emerging dry plasma technologies, such as plasma polymerization. Special attention is given to plasma polymerized nanoparticles (PPNs), highlighting their potential as paradigm-shifting platforms for biomedical applications while identifying key areas for improvement. The review concludes with a forward-looking discussion on strategies to address key challenges, such as achieving regulatory approval and advancing clinical translation of polymeric NP-based therapies, offering unprecedented opportunities for next-generation nanomedicine.
 Behnam Akhavan | AProf Behnam Akhavan is an Australian Research Council (ARC) DECRA Fellow and an Associate Professor of Biomedical Engineering at the University of Newcastle, Australia. He leads the Plasma Bio-engineering Laboratory at the School of Engineering and the Hunter Medical Research Institute (HMRI). Since obtaining his PhD in Advanced Manufacturing from the University of South Australia in 2015, he has held postdoctoral and academic positions at the Max Planck Institute for Polymer Research and Fraunhofer Institute of Microtechnology in Germany, and the University of Sydney. AProf Akhavan's pioneering work in plasma bio-engineering, published in over 90 journal articles, has led to innovative applications in healthcare and beyond. He is recognised by Engineers Australia as one of the nation's Most Innovative Engineers. |
1. Introduction
Nanomedicine has become a focal point in healthcare, offering innovative solutions for diagnosing and treating diverse diseases through nanoparticle-based technologies.1,2 This specialized field of nanotechnology focuses on the development of nanoscale platforms designed to deliver drugs and diagnostic agents with enhanced precision and efficacy.3 Among the various nanomaterial platforms available, nanoparticles (NPs), such as inorganic, lipid, and polymeric NPs, have demonstrated tremendous potential as drug delivery systems capable of addressing the limitations linked with free drug formulations.4 The payload can also be conjugated onto the surface or encapsulated within the NP core, which provides protection, while facilitating sustained and controlled drug release.5 Targeting ligands can also be co-functionalized with the payload, allowing for precise drug delivery to the intended target locations.6Polymeric NPs, nanoscale particles primarily composed of macromolecular polymers, have garnered significant interest due to their unique physicochemical properties, enabling their applications in diverse fields such as drug delivery,7,8 imaging,9,10 biosensing,11 catalysis,12,13 energy storage,14–16 environmental remediation,17–19 and electronics20,21 (Fig. 1). For nanomedicine, polymeric NPs have been of particular interest as suitable drug carriers due to their advantages over other types of nanoscale platforms for transporting and delivering drug payloads into the body. The nature of polymer growth and synthesis processes allows for a high degree of control of the polymeric NP size,22 shape and morphology,23,24 and surface chemistry.25 We highlighted in a recent review how precise engineering of these properties enables the drug payload to overcome biological barriers in the body.26 The surfaces of polymeric NPs can also be functionalized with chemical or biological moieties that respond to specific stimuli (i.e., pH, temperature, enzymatic activity).27–29 This responsiveness enhances their ability to penetrate tumours, escape the endosomal compartments, and facilitate the delivery of therapeutic payloads into the cytoplasm or nucleus. Controlled release of the encapsulated and loaded drug payload, which could be hydrophilic or hydrophobic, is also achievable by optimizing the chemical properties of the polymeric core, matrix and/or surface.30–32 These distinctive advantages of polymeric NPs translate to excellent results during the discovery and development phases and in preclinical evaluations.
 | ||
| Fig. 1 Schematic representation of the diverse applications of polymeric NPs, including drug delivery, bioimaging, biosensing, catalysis, energy storage, environmental remediation, and optoelectronics. Polymeric NPs can encapsulate anticancer therapeutics such as doxorubicin (DOX) and curcumin (Cur) to inhibit tumour progression. Surface functionalization with fluorophores enables deep-tissue imaging, while stimuli-responsive smart polymers, sensitive to pH, reactive oxygen species (ROS), and temperature, facilitate biosensing applications. Incorporation of metal and metal oxide NPs imparts catalytic properties to polymeric nanostructures, reducing reaction activation energy. Surface engineering with polymers such as polyimide (PI), poly(arylene ether urea) (PEEU), and polyetherimide (PEI) enhances discharged energy density across broad temperature ranges, a critical feature for dielectric materials in electric vehicles and renewable energy systems. Amphiphilic polyurethane (APU) NPs demonstrate efficacy in remediating soil and water contaminated with polynuclear aromatic hydrocarbons (PAHs). Polymeric NPs composed of π-conjugated block copolymers facilitate efficient charge carrier mobility through π-orbital overlap, leading to superior electronic and optical properties compared to bulk materials. | ||
The crux of the matter lies in the significantly lower number of market-approved polymeric NPs compared to other NP types, such as liposome and lipid NPs.26 Several general factors contribute to this disparity, including a limited understanding of how polymeric NPs overcome biological delivery barriers, the translational gap between animal and human studies, and the inherent heterogeneity across populations.4 Beyond these overarching challenges, there are specific limitations associated with the structural and mechanistic design of polymeric NP subclasses. These include an overreliance on chemical linkers and/or weak electrostatic interactions for payload incorporation,33,34 premature disassembly of polymeric nanostructure in the bloodstream leading to burst payload release,35 and difficulties in achieving high encapsulation and loading efficiencies, which results in a higher proportion of excipients relative to the therapeutic agent.36 The current predominant approach of synthesizing polymeric NPs through wet chemical methods is also laden with pitfalls, such as the excessive dependence on solvents and starting reactants that require meticulous purification, as well as multi-step wet chemical processes that are labour-intensive and complex. These material- and process-related challenges collectively impede commercial scalability and hinder the clinical translation of polymeric NPs.
The challenges in the production of polymeric NPs via wet chemistry approaches can potentially be bypassed through alternative, dry methods which utilize physical processes rather than solvent-based techniques. However, the physical processes may lack the intricate control necessary in tuning the physicochemical properties of polymeric NPs and their subsequent functionalization with the drug payload. Limitations in their production efficiency and the energy-intensive processes may also hinder the scalability of such dry methods. As such, there is a demand for precise, reproducible, and scalable dry methods to produce polymeric NPs that retain the advantages of wet chemical techniques while eliminating their solvent-related and time-intensive challenges.
In this review, we first provide a comprehensive analysis of polymeric NPs, detailing their structural diversity, interaction mechanisms with payloads, and the inherent design challenges that limit their clinical translation. We critically examine conventional wet chemical synthesis methods, evaluating their advantages and drawbacks, before introducing a unique comparative analysis with emerging dry plasma technologies, particularly plasma polymerization. A key focus is placed on plasma polymerized nanoparticles (PPNs), emphasizing their potential as versatile, multi-functional nanoplatforms. The review concludes with a forward-looking discussion, outlining the material and process-related challenges that both wet chemical and plasma polymerization methods have addressed while identifying remaining barriers that must be overcome to enable large-scale manufacturing, regulatory approval, and clinical translation. Special emphasis is placed on the scalability and economic feasibility of plasma polymerization, highlighting how advancements in reactor design, cost-effective processing, and reproducibility strategies could position PPNs as a next-generation platform for biomedical applications.
2. Polymeric nanoparticles
The tuneable physicochemical properties and adaptable structures of polymeric NPs make them ideal candidates for drug delivery in biomedical applications. As we have recently reviewed, the size, shape and surface chemistry of polymeric NPs can be modified using a number of methods which endow capability to load the payload and cross biological barriers inside the body.26 Payloads could be in a variety of forms in terms of charge (i.e., anionic, cationic), polarity (i.e., hydrophobic, hydrophilic), and molecular weight. They can be incorporated to the drug delivery system either through encapsulation into the core, entrapment at the polymer matrix, or binding onto the polymeric surface through electrostatic or covalent interactions (Fig. 2). The mode of incorporation largely depends on the structural organisation of the polymeric NPs, which can be classified as nanocapsules, polymeric micelles, dendrimers, polymersomes, polyplexes, and solid nanospheres (Fig. 2).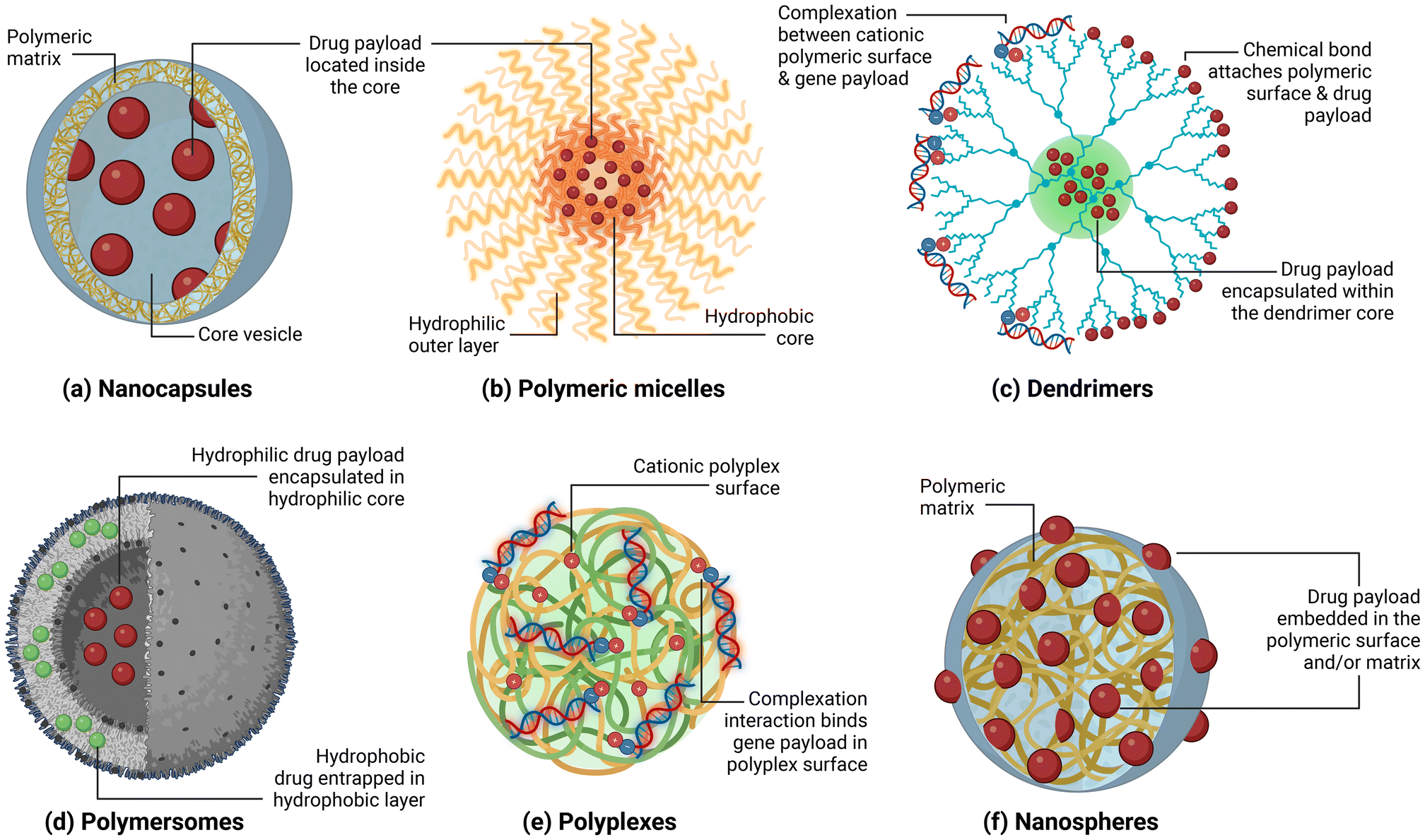 | ||
| Fig. 2 Various classes of polymeric NPs and the nanomaterial-drug interactions; (a) nanocapsules, (b) polymeric micelles, (c) dendrimers, (d) polymersomes, (e) polyplexes, and (f) nanospheres. | ||
This section discusses the different classes of polymeric NPs based on their structure and how each of them interacts with the payload. The advantages of each type of polymeric NP are highlighted, along with examples of how they are applied in preclinical trials, specifically for cancer drug delivery applications. Finally, the limitations of each polymeric NP type in terms of payload protection, controlled release, encapsulation efficiency, stability and requirement for chemical modification are discussed. Table 1 provides a summary of various polymeric NPs and their classifications, which have been assessed in preclinical and clinical studies.
| Polymeric NP | Classification | Payload | Application/findings | Ref. |
|---|---|---|---|---|
| Cation-free disulfide bond-crosslinked polymer-siRNA nanocapsule T-SS(−) | Nanocapsules | siRNA-PLK1 | Reduced growth of PC-3 prostate cancer tumour in mice samples | 37 |
| PLGA-VRL-RES-CAN | Nanocapsules | Vinorelbine bitartrate and resveratrol | Reduced growth of 4T1 triple negative breast cancer tumour in mice samples | 38 |
| Genexol®-PM | Micelles | Paclitaxel | Commercially available for breast and non-small cell lung cancer treatment | 39 and 40 |
| NK105 | Micelles | Paclitaxel | Breast, colon and gastric cancer treatment | 41–43 |
| DEP® docetaxel | Dendrimers | Docetaxel | Phase II clinical trials for lung and prostate cancer treatment | 44 |
| AZD0466 | Dendrimers | AZD4320 | Tumour growth inhibition of RS4;11 lymphoblast cells in mice samples; Phase I clinical trials for blood cancer | 45 and 46 |
| Tf@TBP-Ps-Dox | Polymersomes | Doxorubicin | Inhibited growth of HCT116 colorectal cancer tumours in mice samples | 47 |
| APT-DOX-QD-NPs | Polymersomes | Gd-based quantum dots and doxorubicin | Treatment and MR/fluorescence imaging of triple negative breast cancer | 48 |
| PPLC/siCCR7 | Polyplexes | siCCR7 | Inhibited lung metastasis in 4T1 triple negative breast cancer cells and reduced tumour cell migration to lymph nodes in mice samples | 49 |
| PONI-Guan-Zwit/siRNA | Polyplexes | siRNA targeting STAT3 (si_STAT3) | Successfully knocked down STAT3 and caused complete tumour inhibition in 4T1 triple negative breast cancer tumour-bearing mice samples | 50 |
| BIND-014 | Solid nanospheres | Docetaxel | Prostate and non-small cell lung cancer treatment | 51 |
| Plasma-polymerized NPs (PPNs) | Solid nanospheres | Recombinant green fluorescent protein | Bioconjugation with fluorescent protein enables bioimaging | 52 |
| PPNs | Solid nanospheres | Nile blue | Fluorescence imaging of breast cancer cells | 53 |
| PPNs | Solid nanospheres | Paclitaxel, IgG-Cy7, and IgG-Cy-5 | Cellular uptake studies of triple-functionalized PPNs into MCF7 breast cancer cells | 54 |
| PPNs | Solid nanospheres | n/a | Comprehensive cytotoxic evaluation highlighting the biosafety of PPNs at elevated dosages | 55 |
| PPNs | Solid nanospheres | Platelet-derived growth factor AB | Feasibility study showing PPNs are safe nanoplatforms for delivering growth factor payload into heart | 56 |
| PPNs | Solid nanospheres | Arginylglycylaspartic acid | Bioactivation of inert substrates for cell growth and differentiation | 57 |
2.1. Nanocapsules
Nanocapsules are reservoir systems consisting of a polymeric membrane or shell that trap the payload within the core vesicle cavity (Fig. 2a).58 Polymeric shell has been a preferred option as a nanocapsule shell because it offers increased drug loading efficiencies, protects the payload from degradation, and can be engineered in ways that improves overall biocompatibility.59 The core vesicle, on the other hand, can be solid, hollow, or liquid phase. The liquid core of the polymeric nanocapsules can either be aqueous (hydrophilic) or oil-based (lipophilic) in nature. Aqueous core is utilized for trapping hydrophilic payloads such as proteins60 and other water-soluble therapeutics.61,62 In contrast, oil-based cores are used to encapsulate lipophilic and hydrophobic payloads like latanoprost63 and curcumin.64The payload can be released from the core though a variety of mechanisms such as rupturing of the shell, or through diffusion of the payload to the surface. The buckling and rupture dynamics of polymeric nanocapsules, leading to payload release, were observed using in situ liquid-phase transmission electron microscopy (TEM).65 The polymeric nanocapsules synthesized from butyl methacrylate (BMA), tert-butyl methacrylate (t-BMA) and ethylene glycol dimethacrylate (EGDMA) have demonstrated that the release of the aqueous payload is due to the buckling and subsequent collapse of the polymeric walls. Moreover, the excessive wrinkling of the walls is a by-product of their elevated Föppl–von Kármán number γ. The authors were able to identify that this “buckling and collapse” mechanism can be controlled through careful engineering of the capsule radius (R), shell wall thickness (h), and collapse rate.65 In particular, when h > R/5, polymeric nanocapsules maintain their spherical morphology without buckling and collapsing. Conversely, when h < R/5, the morphology depends heavily on size. Nanocapsules with R < 100 nm and h < 10 nm become crumpled, exhibiting numerous indentations. In contrast, larger nanocapsules (100 nm < R < 10 μm) with thicker shells (h > 50 nm) show single indentations, a design that likely reduces morphological deformation and improves controlled release.
Diffusion of the payload from the core outwards to the surface can also be a mode of release for polymeric nanocapsules. In a separate work, polymeric nanocapsules loaded with calcitriol prepared through nanoprecipitation displayed a classic biphasic drug release profile wherein a burst release phase during the first hour is observed, followed by a plateauing of the payload release.66 The initial spike in the release can be attributed to the calcitriol adsorbed directly to the nanocapsule surface. The authors implied that the subsequent constant release is a result of multiple factors, including the equilibrium exchange between the core/shell structure of the polymeric nanocapsule and the solvent media, the volume of each phase, and the surfactant concentration present in the release media.66 Moreover, diffusion of the payload can also be manipulated by tuning the thickness of the polymeric shell through the polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oil ratio.67 The control over the shell thickness thus translates to an increase or decrease of the diffusion flux of the payload while limiting the effects of the burst release.
:oil ratio.67 The control over the shell thickness thus translates to an increase or decrease of the diffusion flux of the payload while limiting the effects of the burst release.
In general, polymeric nanocapsules are preferred structures whenever a sensitive payload needs protection from degradation, or when controlled drug delivery and enhanced drug stability are required. Small interfering RNAs (siRNAs), for instance, can easily be degraded by serum nuclease and cause immunogenicity if delivered as an attachment to polymeric NPs. However, encapsulating siRNA into polymeric nanocapsules based on cationic triblock copolymer cRGD-PEG-PAsp(MEA)-PAsp(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-DETA) resulted in improved stability in serum while maintaining its characteristic cancer cell targeting through the cRGD functionalization and GSH-triggered payload release (Fig. 3a).37 This T-SS(-) nanocapsule formulation encapsulated with siRNA-PLK1 resulted in reduced tumour growth while having an enhanced survival rate, wherein 83% of the mice survived longer than 60 days.37 Furthermore, since the payload is tucked within its core, controlled release is one of the main advantages of polymeric nanocapsules, especially for drug delivery applications against cancer. For example, a nanocapsule formulation based on polylactic-co-glycolic acid (PLGA) with folic acid and lactobionic acid surface decorations loaded with pterostilbene (PTN) was able to demonstrate a controlled release for 2 days.68 This sustained release from the polymeric nanocapsule led to a significant decrease in the IC50 values (5.9 ± 0.8 μg mL−1) compared to free PTN (121.3 ± 9.4 μg mL−1) upon testing on HepG2 liver cancer cells.
N-DETA) resulted in improved stability in serum while maintaining its characteristic cancer cell targeting through the cRGD functionalization and GSH-triggered payload release (Fig. 3a).37 This T-SS(-) nanocapsule formulation encapsulated with siRNA-PLK1 resulted in reduced tumour growth while having an enhanced survival rate, wherein 83% of the mice survived longer than 60 days.37 Furthermore, since the payload is tucked within its core, controlled release is one of the main advantages of polymeric nanocapsules, especially for drug delivery applications against cancer. For example, a nanocapsule formulation based on polylactic-co-glycolic acid (PLGA) with folic acid and lactobionic acid surface decorations loaded with pterostilbene (PTN) was able to demonstrate a controlled release for 2 days.68 This sustained release from the polymeric nanocapsule led to a significant decrease in the IC50 values (5.9 ± 0.8 μg mL−1) compared to free PTN (121.3 ± 9.4 μg mL−1) upon testing on HepG2 liver cancer cells.
 | ||
| Fig. 3 Polymeric nanostructures serve as transport vehicles in various biomedical applications. (a) Cation-free T-SS(−) nanocapsules exhibit optimal size, shape and surface charge, enabling enzyme-sensitive release of FITC-siRNA at physiological pH, reproduced with permission from ref. 37. Copyright © 2023, Elsevier. (b) Hybrid micelles (D&R-HM-MCA) facilitate targeted delivery of DOX and R848 into glioblastoma (GBM) cells, reproduced with permission from ref. 69. Copyright © 2024, American Chemical Society. (c) Tf@TBP-PS-Dox polymersomes precisely target transferrin receptors, which are overexpressed in HCT-116 colorectal cancer cells, reproduced with permission from ref. 47. Copyright © 2020, Elsevier. (d) Polyplexes loaded with PPLC/siCCR7 evade lysosomes within 5 hours, primarily due to the amino group in PPLC, while siCCR7 effectively silences the CCR7, reducing the cellular response rate, reproduced with permission from ref. 49. Copyright © 2022, John Wiley and Sons. | ||
A significant challenge in employing polymeric nanocapsules as drug delivery systems lies in achieving high encapsulation efficiency, which often necessitates assembling the nanocapsule around the therapeutic payload. However, this strategy can be detrimental, as it may expose the sensitive drug molecules to physical agitation, sonication, toxic chemicals, or elevated temperatures, leading to potential chemical modifications. Such alterations can compromise the drug's inherent efficacy against its target. Therefore, the development of milder reaction conditions and, ideally, dry synthesis and encapsulation methods, is crucial to mitigate these issues and enhance the stability and functionality of polymeric nanocapsules.
2.2. Polymeric micelles
Polymeric micelles consist of di- or tri-block amphiphilic components that undergo self-assembly in aqueous environments due to hydrophobic–hydrophilic interactions.70 The typical structure of a polymeric micelle features a hydrophobic core surrounded by a hydrophilic outer layer (Fig. 2b). Hydrophobic molecules within the core trap the payload, while the hydrophilic surface plays a key role in the nano–bio interface of polymeric micelles. The payload encapsulated within the core of polymeric NPs can be released in response to various changes in endogenous stimuli, such as pH or enzymes, which alter the micellar structure. For instance, NC-6300, a polymeric micelle formulation comprising of hydrazone-functionalized PEG-b-PAsp conjugated to epirubicin, enables triggered drug release in acidic environments characteristic of tumour tissues.71 The pH gradient between the normal tissues and blood (pH 7.4) and the tumour microenvironment (pH < 6.5) have been utilized in designing such pH-responsive polymeric NPs.27 NC-6300 is currently in Phase 1b/2 clinical trials (NCT03168061) to determine the maximum tolerated dose and dose-limiting toxicities in patients with advanced solid tumours or soft tissue sarcoma.71Beyond pH sensitivity, the elevated levels of enzymes like cathepsin B in glioblastoma can be leveraged to cleave peptides integrated into polymeric micellar structures, thereby triggering the release of encapsulated payload. For instance, hybrid polymeric micelles composed of poly(lactic-co-glycolic acid)–poly(ethylene glycol)-p-aminophenyl-α-D-mannopyranoside (PLGA–PEG2k–MAN) and PLGA-FRRG-PEG5k-angiopep-2 block copolymers, co-loaded with doxorubicin (DOX) and immunomodulator R848, have been developed as a monodisperse and brain-targeted drug delivery system aimed at both tumour-associated macrophages (TAMs) and glioblastoma cells (Fig. 3b).69 Glioblastoma is one of the most lethal and treatment-resistant malignancies of the central nervous system (CNS).72 Once the polymeric micelles reach the glioblastoma microenvironment, the high concentration of cathepsin B cleaves the FRRG peptides, resulting in the release of the angiopep-2 moiety. Such cleavage reduces abluminal low-density lipoprotein receptor-associated protein 1-mediated efflux, enhancing drug retention within the brain parenchyma. The released DOX induces cytotoxicity on glioblastoma cells, while R848 reprograms TAMS from an immunosuppressive M2 phenotype to a pro-inflammatory M1 phenotype, thereby strengthening the anti-glioblastoma immune response.69
The payload in polymeric micelles is encapsulated within its core, providing protection from premature degradation. This core is shielded by an outer hydrophilic surface layer, which imparts “stealth” properties to the micelles. These stealth characteristics enable the particles to evade rapid recognition and clearance by the immune system, thereby extending their circulation time in vivo. Prolonged systemic circulation enhances the likelihood of the drug cargo being delivered to target sites, such as solid tumours and cancer cells, thereby improving therapeutic efficacy. For instance, Genexol®-PM, a polymeric micelle formulation, features a hydrophilic shell that imparts stealth properties, enabling it to evade clearance by the mononuclear phagocyte system (MPS). This formulation consists of polyethylene glycol (PEG) and poly(D,L-lactic acid) blocks, which encapsulate paclitaxel (PTX).39 Genexol®-PM is currently approved for use in South Korea and is also accessible in other markets such as Hungary and Bulgaria for the treatment of metastatic breast cancer and advanced lung cancer. It is also undergoing Phase II clinical trials in the United States.40 Clinical studies have demonstrated that Genexol®-PM exhibits superior antitumour efficacy compared to the FDA-approved Taxol®, which has been attributed to a higher concentration of PTX within tumours.73 The higher efficacy of the Genexol®-PM was attributed by the authors to the higher PTX tumour concentration in contrast to Taxol®.73 This enhanced efficacy is largely due to the PEG-based outer shell, which provides stealth capabilities, allowing for extended systemic circulation and increased tumour accumulation of the micelles.
Another example is NK105, a polymeric micelle currently in the late stages of clinical trials. NK105 is formulated through the self-assembly of PEG and polyaspartate in an aqueous phase.74 This micellar NP system is designed for the delivery of PTX to treat breast,41 colon,42 and gastric cancers.43 Notably, NK105 demonstrated a 20-fold longer half-life compared to free PTX, primarily due to its PEG-based hydrophilic coating.75 However, NK105 failed to reach the primary endpoint in its Phase III clinical trial (NCT01644890), which could be attributed to the lower enhanced permeability and retention (EPR) effects in the patients, and a lower administered dose of NK105 (65 mg m−2) compared to standard PTX (80 mg m−2). Despite these challenges, the encapsulation of PTX in polymeric micelle formulations like Genexol®-PM and NK105 offers protection against premature drug release and clearance, thereby prolonging in vivo circulation and improving drug delivery to tumour sites.
The micellar system has been one of the most successful types of polymeric drug delivery systems in the past 10 years. However, several limitations are still hindering polymeric micelles in becoming the preferred nanoplatform for drug delivery. These challenges include stability issues from premature dissociation and disassembly, drug loading limitations, immunogenicity of PEG-based coatings and scalability issues. Since the formation of polymeric micelles is a function of the critical micelle concentration,76 instability and disruptions caused by changes in the environmental conditions (i.e., temperature, pH, ionic strength) may cause the micelles to prematurely dissociate, which results in leakage, non-specific delivery, and/or denaturation of the loaded drug. PEGylation, which has been the primary method of providing stealth properties to avoid clearance, is known to cause accelerated blood clearance (ABC) and induce immunogenicity in the polymeric micelles.77 Scalability issues beyond laboratory and pilot scales are also critical in commercializing the polymeric micelle technology at a reasonable cost.78
2.3. Dendrimers
Dendrimers are highly-ordered and well-defined branched polymeric NPs with multiple repeating units originating from their core.44 Like other core–shell nanostructures, the dendrimer architecture consists of a central core, an interior branched layer referred to as the “generation”, which radiates outward from the core, and terminal functional groups. These multiple generation and branching, which originate from repeated growth reactions, lead to the forming of a 3D spherical structure (Fig. 2c). The drug payload can be incorporated into the dendrimer structure in three different ways. First, the payload can be chemically conjugated into the dendrimer surface, wherein the drug can be released passively through hydrolysis of ester or amide moieties, or actively via bond cleavage by the presence of various stimuli (e.g., glutathione,79 reactive oxygen species,80 and pH81). Second, hydrophobic drugs can be entrapped into dendrimers, which consist of a hydrophobic core and a hydrophilic surface, much like that of a polymeric micelle. Lastly, electrostatic complexation between cationic dendrimers and gene payloads such as siRNA82 enables dendrimers to act as non-viral gene carriers.Dendrimers, owing to their unique generation-based structure, have several innate advantages over other types of polymeric NPs. Dendrimer surface is multivalent, which allows further modification or conjugation with different types of payloads (e.g., drugs,83 targeting groups,84 and genes82). As an extension to this multi-modality, the dendrimer surface can covalently attach solubility-improving moieties such as PEG to improve the overall efficacy of a NP formulation. For example, a PEGylated poly(L-lysine) (PLL) dendrimer with a docetaxel load, is presently in its Phase II clinical trials for lung and prostate cancer.44 The water-soluble DEP® docetaxel (DEP DTX) formulation displayed improved efficacy towards various cancer types and a lower toxicity profile compared to the FDA-approved chemotherapeutic drug Taxotere®. The DEP® formulation developed by Starpharma does not include polysorbate 80, a detergent typically used to improve hydrophilicity to drug compounds (e.g., Taxotere® and Jevtana®), which ironically can trigger side effects such as anaphylaxis and neutropenia.85
Encapsulation of hydrophobic payload into its intramolecular cavity can also be achieved using dendrimers. This pseudo-encapsulation capability is owed to its hydrophobic-hydrophilic core–shell structure which can be akin to a unimolecular micelle. For instance, cabazitaxel, a hydrophobic taxane mitotic inhibitor, was incorporated in a separate DEP®-based nanoformulation.86 Preclinical trials in pancreatic cancer models in mice showed that the DEP® cabazitaxel (DEP CTX) completely inhibited tumour growth. In the same study, FDA-approved Nab paclitaxel (Abraxane®) only inhibited 85% of the tumour growth after day 37.86 Completion of the enrolment and treatment stage of Phase II clinical trials for DEP CTX has been recently announced, along with observed tumour size reduction and biomarker improvement across various cancer types, including prostate cancer, ovarian cancer, gastroesophageal cancer, cholangiocarcinoma, and head and neck cancer.87
Despite the advantages of dendrimers as drug delivery vehicles, dendrimers face several challenges that directly impact their efficacy and commercial viability. Unlike polymeric micelles, which typically form through simple self-assembly dynamics, dendrimers require multi-step synthetic processes to achieve the desired physicochemical properties. Tuning the size of the dendrimer is based on the number of generations during synthesis, and has its own complexities in terms of design, ease of production, and cost. Specifically, dendrimers with generations of 5 and below produce smaller-sized polymeric NPs (4 to 6 nm), which can be excreted through the urine. In contrast, dendrimers formed from six generations and above are dependent on hepatic clearance.88,89 To add to these challenges, the larger polymeric dendrimers (i.e., generation 6 and above) are expensive and time-consuming to produce,88 which further translates to scalability and large-scale production issues. These higher-generation dendrimers also face biocompatibility and toxicity issues primarily due to their cationic surfaces, particularly for NH2-functionalized terminal groups.90 Moreover, chemical modification is needed to attach the drug to the terminal functional groups of the dendrimer. Dendrimers also encounter some of the hurdles encountered by polymeric micelles, such as limited drug loading efficiencies and the risk of structural degradation leading to pre-emptive drug release.91
2.4. Polymersomes
Polymersomes are self-assembled nanostructures comprising a bilayer architecture that closely resembles the structure of liposomes.92 However, unlike liposomes, which are formed from phospholipid bilayers, polymersomes consist of bilayers constructed from amphiphilic copolymers. These copolymers can be arranged as a block, dendronized, graft, or alkylated moieties.93 This polymeric bilayer membrane consists of hydrated hydrophilic coronas on both the inner and outer surfaces, surrounding the hydrophobic core of the membrane (Fig. 2d). This configuration serves to isolate and protect the fluidic core from the external environment. In turn, this aqueous core is utilized to trap and encapsulate the drug payload.94Although no clinical trials involving polymersomes have been reported to date, preclinical studies on functionalized polymersomes have shown their potential as a promising DDS. For example, transferrin-binding peptides (TBP) were surface functionalized into polymersome NPs through co-assembly of poly(ethylene glycol)-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) (mPEG-P(TMC-DTC)) with TBP-PEG-P(TMC-DTC).47 Such polymersome NPs (Tf@TBP-Ps-Dox) facilitated DOX delivery into HCT-116 cancer cells in mice, inhibiting colorectal tumour growth. The circulation half-life was also higher for Tf@TBP-Ps-Dox compared to the non-peptide functionalized polymersome formulation (Ps-Dox) with 9.5 and 8.9 h, respectively. The potency of the Tf@TBP-Ps-Dox NPs in HCT-116 cells was 2.5- and 4.5-fold higher compared to Ps-Dox and the commercially available Lipo-Dox formulation, respectively. The in vitro results corresponded with the in vivo studies wherein Tf@TBP-Ps-Dox NPs formulations garnered higher tumour inhibition rates after 20 days of dosing compared to Ps-Dox and Lipo-Dox (Fig. 3c). These findings highlight the importance of the transferrin-targeting moiety of the polymeric NP.
Several other preclinical trials for polymersomes have been reported over the past few years for the treatment of various cancer types, such as breast cancer,95 non-small cell lung cancer,96 peritoneal cancer,97 and head and neck cancer.98 Moreover, unlike its liposome analogues, which have been extensively translated into clinical applications, polymersomes have not yet entered clinical trials due to several obstacles. These challenges include lower biocompatibility compared to liposomes,99 slower drug release due to thicker membranes (∼50 nm),100 and potential disintegration because of non-covalent interactions during the self-assembly that leads to premature release of payload.101 In addition, wet chemical methods used to prepare polymersomes are through post-polymerization self-assembly, that are hard to scale-up, expensive, and not eco-friendly.102
2.5. Polyplexes
Polyplexes are polymeric NP systems wherein the therapeutic payload, typically a gene or siRNA, is loaded through electrostatic and/or hydrophobic interactions between the cationic polymeric components and the anionic nucleic acid (Fig. 2e).103 The polyplexes prevent the enzymatic degradation of the nucleic acids while ensuring targeted release at appropriate sites.The primary advantage of incorporating therapeutic payloads into polyplexes via complexation with polymers lies in the elimination of the need for chemical modification of the therapeutic molecule. This complexation approach preserves the delicate intermolecular interactions within the payload, minimizing the disruption caused by external chemical alterations.104 Consequently, the intrinsic bioactivity of the drug is retained post-complexation, offering a significant improvement over approaches involving polymer functionalization.
Another advantage of polyplexes is their enhanced transfection efficiency, driven by the cationic surface charge, which promotes stronger interactions with the negatively charged cell membrane. For example, the interaction between CC chemokine receptor 7 (CCR7) overexpressed in tumour cells, and CC chemokine ligand 21 (CCL21) in lymph nodes were selectively blocked by CCR7-targeting small interfering RNA (siCCR7) functionalized in mPEG-poly-(lysine)-based polyplex (PPLC/siCCR7).49 Cell uptake studies showed that the cationic charge of the polyplex enhanced its cellular internalization, while the amino groups from the PPLC aided the lysosomal escape of siCCR7 without altering its functionality (Fig. 3d). Optimization of the polyplex design resulted to the inhibition of about 92% of lung metastasis in 4T1 breast tumour mice models while reducing tumour cell migration in lymph nodes by 80%.49 The PPLC/siCCR7 designed in the polyplex formulation demonstrated enhanced knockdown of CCR7 in 4T1 tumour cells based on western blot results (Fig. 3d), which resulted in a lower response rate on CCL21 and LN tropism. Overall, the site-specific knockdown improved the efficacy of the polyplex nanoplatforms for both in vitro and in vivo conditions.
The design of the polyplex plays a critical role in its viability towards non-viral gene therapy. For example, linear and branched polyethylenimine (PEI)-based polyplexes can be used as vectors to deliver plasmid DNA (pDNA) which encodes small hairpin RNAs (shRNA) that blocks oncogene responsible for triple-negative breast cancer (TNBC) cell proliferation.105 Results showed that the branched PEI polyplexes were more cytotoxic to the 4T1 mouse TNBC cells than the linear ones. Moreover, increasing the polymer:pDNA ratio also resulted in increased cytotoxicity towards the same cell line.
The use of polyplexes in drug delivery, however, is subject to several limitations. For instance, the assembly of polyplexes with certain proteins and polypeptides can be difficult because these biomolecules may possess moderate or even neutral charges under physiological conditions which are all dependent on their respective isoelectric points.106 Since the polyplex–payload interaction is mostly a complexation reaction and not a covalent attachment, the payload can be cleaved and detached under physiological conditions. The number of moieties with appropriate charges within a protein structure is limited and varies from protein to protein,107 limiting the formation mechanism of polyplexes into proteins. Instability under physiological environments also plagues the systemic delivery of polyplexes as salts and protein corona formation can cause unwanted early payload release and/or disassembly of the structure. Premature clearance is also problematic since immune cells and negatively charged serum proteins can interact with the cationic-dominated polyplex formulation.34
2.6. Solid nanospheres
Solid polymeric nanospheres, one of the most commonly applied polymeric NPs, are single-phased solid matrix systems assembled through dense packing of polymeric groups.108 The drug payload is either encapsulated within the matrix, or attached to the surface through physical or covalent interactions (Fig. 2f). A main advantage of polymeric nanospheres is the ease of incorporating “smart” polymers into the surface. For example, a pH-responsive drug delivery system formed from hydroxypropyl methacrylamide (HPMA)-based polymer was developed to deliver doxorubicin (DOX) into breast cancer cells.109 The conjugation of DOX into pH-sensitive hydrazone functional groups facilitated faster drug release at intracellular (pH 5.5) and intratumoral pH (pH 6.5) compared to physiological levels (pH 7.4), suggesting the sensitivity of the final nanoplatform to changes in pH.109One advantage of polymeric nanospheres is their stability as nanoplatforms, which arises from the hydrophobic interactions within the polymer matrix. An example of this type of polymeric NPs is BIND-014, which is made up of a hydrophobic polylactic acid (PLA) core coated with hydrophilic PEG and Prostate-Specific Membrane Antigen (PSMA) targeting ligands.51 Overexpression of PSMA is seen in prostate adenocarcinoma and other solid malignancies.110,111 The active anti-cancer compound of BIND-014 is docetaxel, and it is encapsulated within this nanocarrier system. BIND-014 has successfully passed preclinical and Phase 1 trials where it showed notable activity towards multiple tumour types with manageable levels of toxicity.51 Phase 2 trials for BIND-014, on the other hand, have shown mixed results for various cancer types. Positive outcomes were observed in the clinical trials for prostate cancer (NCT01812746) and non-small-cell lung cancer (NCT02283320; NCT01792479). However, Phase 2 trials for advanced cervical or head and neck cancer (NCT02479178) using BIND-014 were terminated due to adverse side effects, including neutropenia, fatigue, nausea, and mucositis.112,113
A significant challenge associated with nanospheres is the efficient encapsulation and/or loading of active payloads. Two primary strategies are employed to incorporate payloads into nanospheres. First, the drug payload can be loaded into the nanospheres post-synthesis. However, low diffusion coefficients (<10−6 cm2 s−1) of large biomacromolecules in aqueous solutions hinder efficient incorporation using this method.114 Furthermore, the use of chemical linkers to covalently attach the payload to the surface introduces additional complexity to the synthesis and purification processes. This approach also increases costs and carries the risk of chemically modifying the payload, potentially affecting its functionality. Alternatively, higher loading efficiency can be achieved by forming the nanospheres directly around the payload. However, this approach often involves harsh formulation conditions, including ultrasonication, exposure to organic solvents, or reaction conditions, such as elevated temperature and UV irradiation. Similarly, these conditions can compromise the structural integrity of sensitive biomolecules, potentially diminishing the effectiveness of the therapeutic payload.
Polymeric NPs face several challenges as drug delivery systems associated with the nanostructure design and its interaction with the payload. These include the need for chemical linkers to attach payloads, difficulties in achieving high loading efficiencies, and the requirement for chemical modification of payloads to integrate with polymeric platforms. Additional limitations include the restriction to charged payloads in polyplexes and dendrimers, reliance on unpredictable electrostatic interactions under physiological conditions, and premature drug release caused by structural disassembly. Moreover, the high cost and complexity of large-scale production further hinder their application.
Emerging as a promising alternative, plasma polymerized nanoparticles (PPNs) offer potential solutions to many of these limitations. PPNs are synthesized through plasma polymerization, a dry and environmentally friendly process. In this process, the PPNs are formed through plasma-assisted fragmentation of monomer precursors, followed by their recombination into polymeric NPs.25,54 PPNs feature radical-rich structures, enabling single-step, linker-free covalent conjugation of multiple molecules (Fig. 4). Moreover, the radical-based covalent attachment is universal and multimodal, which means it can be used to anchor a wide range of molecules. Their solid, carbonaceous nanosphere structure is compact and resistant to premature disintegration during circulation. The production process is scalable and considered ‘green’ due to the minimal use of harmful solvents and the low generation of waste.52 Toxicological evaluations have demonstrated that PPNs are well-tolerated in both cellular and animal models.55 Collectively, PPNs and the plasma polymerization technique offer a promising solution to addressing current challenges in polymeric NP design, potentially bridging the gap to successful clinical translation. A comprehensive discussion on the formation mechanisms and fabrication processes of PPNs is presented in Section 3.
 | ||
| Fig. 4 Plasma polymerized nanoparticles containing long-lived radicals facilitate covalent attachment of multiple payloads using a single-step, linker-free approach. | ||
3. Approaches to synthesizing polymeric nanoparticles
Polymeric NP synthesis can be broadly classified into wet chemical methods and dry technologies. Traditional synthesis techniques often involve either the breakdown of larger, preformed polymers or the in situ polymerization of monomer precursors,115,116 relying heavily on aqueous or lipophilic reactants, solvents, and linkers. In this review, methods such as emulsification – solvent evaporation, emulsification – solvent diffusion, emulsification – reverse salting-out, and nanoprecipitation, as well as controlled radical polymerization (CRP) of monomeric precursors are collectively categorized as wet chemical approaches (Fig. 5). | ||
| Fig. 5 Wet chemical methods of synthesizing polymeric NPs include emulsification – solvent evaporation, emulsification – solvent diffusion, emulsification – reverse salting-out, nanoprecipitation and controlled radical polymerization. | ||
In contrast to wet chemical methods, dry synthesis methods, such as inert gas condensation, laser ablation, physical vapour deposition, laser pyrolysis, high-energy ball milling, and plasma polymerization, are emerging as promising alternatives to address the limitations of wet chemical techniques.52,117 Among these, plasma polymerization stands out for its ability to overcome key challenges associated with wet chemical synthesis, offering greater control over particle composition, size, and surface functionality. This section provides an overview of each synthesis method, discussing their respective advantages and drawbacks, as summarized in Table 2.
| Method | Classification of synthesis method | Advantages | Limitations | Types of polymeric NPs that can be fabricated |
|---|---|---|---|---|
| Emulsification – solvent evaporation | Wet approach | • Versatile and scalable | • Prone to aggregation | • Nanospheres118 |
| • High encapsulation efficiencies for lipophilic drugs | • Requires heating mechanism for evaporation | • Micelles119 | ||
| • Requires source of mechanical stress to generate nanoemulsions | • Dendrimers120 | |||
| • Polymersomes121 | ||||
| • Polyplexes122 | ||||
| Emulsification – solvent diffusion | Wet approach | • Does not require homogenizer | • Scaling up requires efficient solvent extraction process | • Nanospheres123 |
| • High yield and encapsulation efficiencies | • Possibility of leakage for hydrophilic cargo | • Micelles124 | ||
| • Can utilize reusable organic solvents that are pharmaceutically acceptable | • Polyplex125 | |||
| Emulsification – reverse salting-out | Wet approach | • Encapsulation of lipophilic drugs without using harmful chlorinated solvents | • Not applicable to all drug cargos | • Nanospheres126 |
| • Requires salting agents in purification step which could be incompatible with some drugs | ||||
| • Wider size distribution range | ||||
| • Scalability issue | ||||
| Nanoprecipitation | Wet approach | • Robust, cost-effective, and reproducible method | • Hydrophilic cargo has low encapsulation efficiencies due to aqueous phase diffusion | • Nanospheres126 |
| • Microfluidic platforms have improved the mixing of solvent/non-solvent | • Slow mixing leads to low drug encapsulation due to difference in solubility | • Micelles127 | ||
| • Dendrimers83 | ||||
| • Polymersomes128 | ||||
| • Polyplexes129 | ||||
| Controlled radical polymerization –ATRP | Wet approach | • Tunability of the number of monomer units | • Removal of transition-metal catalyst is necessary | • Nanospheres130 |
| • No homopolymer contaminations during the addition of subsequent monomers | • High temperature conditions | • Micelles131 | ||
| • Polymersomes132 | ||||
| • Polyplexes133 | ||||
| Controlled radical polymerization –RAFT | Wet approach | • Compatible to a wide range of monomers | • Collapsed hollow NPs and unwanted solids can be formed | • Nanospheres134 |
| • Mild reaction conditions | • High homopolymer contamination | • Micelle135 | ||
| • Heavy metals, which are hard to remove, are not needed | • Dendrimers136 | |||
| • Chain extension is quicker and less solvent-sensitive than ATRP | • Polymersome135 | |||
| • Polyplexes136,137 | ||||
| Plasma polymerization | Dry approach | • Can be generated using gaseous precursors only | • Higher start-up cost to build vacuum chamber set-up | • Nanospheres52,138 |
| • No toxic by-product during fabrication process | ||||
| • Fast NP formation | ||||
| • NP surface contains radicals that covalently bond with any molecule |
3.1. Wet chemical approaches
The emulsification–solvent evaporation method can be used to control the different physicochemical properties of polymeric NPs. For example, the optimization of fabricating PLGA NPs loaded with docetaxel through emulsification–solvent evaporation was performed by ranking the different factors vital to the synthetic process.118 The ratio of organic and aqueous phase, MW of the polymer, terminal end functional group, lactide:glycolide ratio, poly(vinyl alcohol) (PVA) ratio, and drug concentration were used as primary determinants in optimizing the size, polydispersity index (PDI), zeta potential and drug-loading efficiency of the PLGA NPs. Importantly, the factorial design of the emulsification – solvent evaporation parameters enabled the identification of an optimized method yielding controllable particle size and surface charge, low polydispersity index (PDI < 0.3) suggesting monodispersity, and high docetaxel loading efficiency.118
Although the solvent evaporation method is considered straightforward and versatile, it has several drawbacks. Solvent evaporation is mostly usable only for liposoluble cargo, and NP aggregation may occur during the evaporation process.115 Extensive discussion of the preparative variables, conditions, and mechanisms of formation of the emulsion-evaporation method is provided elsewhere.140
An efficient solvent removal/retrieval system is necessary for scaling up the emulsification – solvent diffusion. For example, a tangential flow filtration system via membrane module was used in purifying and concentrating the o/w emulsion to form disulfram-loaded methoxy poly(ethylene glycol)-b-poly(lactide-co-glycolide)/poly(ε-caprolactone) (mPEG5k-b-PLGA2k/PCL3.4k) micelles.53 The filtration system removed the organic solvents added to the water phase while also concentrating the final polymeric micelle product. The polymeric micelles were then filtered using a 0.45 μm membrane filter as the final purification step, underscoring the critical dependence of the emulsification-solvent diffusion method on efficient solvent removal for its applicability in in vivo studies.
The solvent diffusion method has a relatively high yield, reproducibility, and encapsulation efficiency. The use of high-pressure homogenizers is also avoided.141 However, hydrophilic drugs are prone to leak during the emulsification process, and high volumes of water are needed to be removed from the suspension.115
The crucial role of the colloidal stabilizer in controlling particle size in the reverse salting-out method was highlighted in an earlier work.143 PVA was used as the emulsifying agent in fabricating methacrylic acid copolymer-based NPs. The PVA chains present in the bulk solution increased the viscosity of the external phase, which eventually led to enhanced hydrodynamic stabilization. Moreover, at the droplet interface, the polymeric chains promote mechanical and steric stability apart from their function in reducing the interfacial tension. In addition, the composition of both organic and aqueous phases also led to size variations in this study. A narrower size distribution was observed for the salting-out method (123 to 710 nm) compared to the solvent diffusion method (108 to 715 nm), although both methods have wider size distribution ranges when compared to nanoprecipitation (147 to 245 nm) from the same work.143
The reverse salting-out method is an excellent approach to encapsulating lipophilic drugs into nanospheres without using chlorinated solvents.108 Nonetheless, the method is not applicable to all cargo molecules, and the use of salting-out agents is not compatible with all therapeutics, thereby entailing intensive purification steps for the NPs.
Nanoprecipitation is a simple, rapid, and reproducible method of producing polymeric NPs which are more favourable in terms of size, size distribution, and entrapment efficiency than the products of emulsion-based procedures.146,147 The main challenge of this technique is finding the appropriate polymer and drug in a suitable solvent/non-solvent system.115 Encapsulating hydrophilic drugs can also be problematic because of the diffusion of the mixture into the aqueous phase during the precipitation stage. The difference in solubility leads to low drug encapsulation efficiencies, especially when the mixing process is slow.
Recent approaches, such as the use of microfluidics and flash nanoprecipitation, have been incorporated to address the issues of traditional nanoprecipitation methods by improving the co-precipitation of both drug and polymeric NPs. For example, a two-phase microfluidic reactor was used to co-encapsulate 7-ethyl-10-hydroxycamtothecin (SN-38) and curcumin into PCL-b-PEG block copolymer.148 Both SN-38 and curcumin have shown tremendous anti-cancer properties but are quite limited in clinical use because of their low water solubility and bioavailability. Evaluation of the size (37 to 47 nm) and PDI (0.18 to 0.27) showed that increasing the flow rate of the microfluidic reactor (0 to 400 μL min−1) has negligible effects on both NP properties. Results from the same study showed that the microfluidic method yielded better encapsulation efficiency of SN-38 compared to the traditional nanoprecipitation technique.148 In a separate work, a flash nanoprecipitation technique to combine poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-b-PLA) and hydrophobic zein yielded polymeric micelles useable for PTX delivery.127 Overall, it can be concluded that flash nanoprecipitation is a simple and scalable modification of the traditional nanoprecipitation method wherein a rapid micromixer promotes increased supersaturation, yielding to the precipitation and entrapment of the therapeutic cargo inside the polymeric NPs.149 The flash nanoprecipitation technology in the same study was facilitated by injecting the polymer and PTX dissolved in a water-miscible solvent into a jet/multi-inlet vortex mixer.127 The hydrophobic PTX formed nuclei and was entrapped within the amphiphilic polymer-based outer layer, which resulted in enhanced NP stability, along with improved encapsulation efficiency and sustained release of the PTX cargo. The use of microfluidics and flash nanoprecipitation technology may need an indirect use of homogenizers similar with some of the emulsification methods. Nonetheless, both modifications improve the feasibility and upscaling potential of the nanoprecipitation method in producing polymeric NPs loaded with drug cargo.
The ATRP technique operates through an equilibrium between active growing radicals and dormant species, primarily in the form of alkyl halides.150 The process is initiated by a redox reaction involving an alkyl halide initiator and a transition metal complex, typically a copper-based catalyst.151 This reaction facilitates the homolytic cleavage of the covalent bond in the alkyl halide, followed by the complexation of the halogen atom with the catalyst. The resulting radical site serves as the initiation point for monomer addition, enabling chain propagation and the formation of the polymer. The polymer growth can be terminated with rapid exchange with the catalyst, which has a dynamic equilibrium between active and dormant polymeric chains.150 In comparison to FRP, the molecular weight of the final product of ATRP can be precisely controlled by modulating the ratio of the concentrations of the monomer and initiator.116,151 Another advantage of ATRP is the potential synthesis of block copolymers through the chain extension of the initiator. Thus, a second monomer could be added to the chain without homopolymer contamination. The main drawback of ATRP is the use of a heavy metal/ligand complex, such as CuBr/1,10-phenanthroline or CuCl/1,1,4,7,10,10-hexamethyl triethylene tetramine, to drive the reaction forward.152,153 The presence of such metal-based complexes requires an additional purification step (e.g., precipitation and adsorption on gel columns) to avoid polymer contamination.
The RAFT polymerization, in contrast, begins analogously to FRP, in which the free radicals are produced via homolytic cleavage of the initiator. This cleavage can be induced by thermal, photochemical, or redox reactions.116 The resulting radicals initiate monomer addition, generating an active polymer chain capable of reacting with a chain transfer agent (CTA). The CTA is a hallmark of RAFT polymerization, which is typically a thiocarbonyl compound such as 2-cyano-2-propyl dodecyl trithiocarbonate154 and (S)-2-(ethyl propionate)-(O-ethyl xanthate).155 The CTA facilitates the degenerative transfer of radicals, thereby mediating the polymerization process.156 The process is considered degenerate, as it involves only the exchange of functionality, with the primary difference between the two sides of the equilibrium being their degree of polymerization.157 Moreover, the homolytic leaving group or the polymer chain itself is subsequently released after the reversible addition–fragmentation step between the CTA and the polymer. The resulting radical species can then reinitiate polymerization with a second monomer, leading to the formation of a new polymer chain. This process may continue through additional addition–fragmentation or equilibrium steps.158
The continuous equilibrium in the addition–fragmentation step controls the reaction between the active polymer chain and the dormant CTA. The molecular weight of RAFT polymer products, in parallel with ATRP, is modulated by the ratio of monomer and CTA concentration. As previously pointed out, RAFT does not require heavy metal compounds in the process. A wider range of monomers can also be utilized in RAFT while having milder reaction conditions compared to ATRP. The main disadvantage of the method is high homopolymer contamination when di-block copolymers are synthesized. In particular, RAFT is challenging when two radical species having similar leaving group abilities are used. Furthermore, the cost of the CTA presents an additional challenge, as functionalized and custom-synthesized CTAs containing reactive groups are often associated with high production and procurement expenses.
Throughout the years, wet chemical methods discussed in this section have been the favoured synthetic approach for producing polymeric NPs because of the distinct advantages that these methods offer, such as the precise control of physicochemical properties, streamlined protocols for incorporating either hydrophilic or hydrophobic drug payloads, and to some extent, robust and reproducible techniques of polymeric NP fabrication. However, these methods are also limited by their complex, multi-step and time-intensive processes, along with the innate requirement of using solvents and reagents that add unwanted toxicity, waste issues, and cost inflation, thus impeding their commercial scalability and clinical translation as preferred drug delivery systems.
3.2. Dry plasma technologies
Dry nanoparticle synthesis methods eliminate the reliance on solvents, offering an environmentally conscious and scalable alternative for NP production. As discussed, traditional wet chemical methods often rely on expensive and potentially toxic solvents and linkers, requiring extensive purification steps, especially when the resulting nanoplatforms are intended for biomedical applications.159 The complexity of these conventional synthesis methods increases both cost and time, leading to scalability and regulatory approval challenges.In the broader field of nanomaterials, dry synthesis approaches have found success in fabricating metallic,160 metal oxide,161 quantum dots162 and carbon-based NPs,163 as examples. These methods use physical processes such as inert gas condensation,164 laser ablation,165 physical vapour deposition,166 laser pyrolysis,167 and high-energy ball milling.168 While applicable to non-polymeric NPs, such physical methods often fail to deliver the level of control required for polymeric systems, especially regarding surface chemistry and functionalisation. Moreover, limitations such as low production efficiency and energy-intensive operations further reduce their suitability for polymeric NP synthesis. Thus, there is an increasing demand for reliable, nontoxic, high-yield, and environmentally sustainable techniques to produce polymeric NPs that also offer a comparable level of precision and control to traditional wet chemical synthesis methods. Plasma polymerization, as discussed in the following subsections, stands out as a transformative technology in this domain.
The plasma polymerization process utilizes a precursor monomer that is excited into the plasma state, where it undergoes fragmentation and subsequent polymerization, forming plasma polymers in the form of thin films171–175 and/or NPs.52,53,176–178 Unlike conventional polymers, plasma polymers form a dense, highly cross-linked, and heterogeneous network rather than a structure based on repetitive monomer units (Fig. 6a).
 | ||
| Fig. 6 (a) Schematic illustration comparing a plasma polymer and a conventional polymer derived from the same monomer. Dependence of the formation mechanism of (b) ethylene180 and (c) acetylene181 on plasma polymerization parameters such as pressure and flow rate of monomer. (b) and (c) were reproduced from ref. 180. Copyright © 2003, John Wiley and Sons, and ref. 181. Copyright © 1974, American Chemical Society, respectively. | ||
The formation of PPN powders and the deposition of polymeric films during plasma polymerization are closely interconnected. PPN formation is characterized by the concentrated production of polymeric species within a localized plasma region, and the proportion of PPNs incorporated into a deposited film is influenced by the film's formation rate.179 Early studies by Kobayashi et al. in 1973 demonstrated that operational parameters, such as working pressure and flow rate of monomer applied during the plasma polymerization of ethylene, determine whether the process yields a film or a combination of film and powder species (Fig. 6b).180 Using plasma polymerization to synthesize NPs thus necessitates a judicious choice of process parameters.
Nanoparticle production using plasma polymerization can be performed either under low-pressure conditions using vacuum chambers or at atmospheric pressure. The following two sections provide further details on these approaches.
3.2.1.1. Low-pressure plasma polymerization. Most plasma polymerization studies reported in the literature are conducted under low-pressure conditions. The mechanism of PPN formation in low-pressure environments is strongly influenced by the reactor design. In his seminal book on plasma polymerization, Yasuda highlighted that PPN production depends heavily on synthesis parameters, such as precursor flow rate and working pressure – which can vary significantly across different reactor designs.179 For instance, PPN powder formation from acetylene was observed exclusively in a bell-jar reactor (Fig. 6c), whereas acetylene-based plasma polymerized films dominated when an inductively coupled reactor was used.181 Thus, achieving controlled PPN synthesis through plasma polymerization necessitates not only the optimisation of operating parameters but also the careful selection and design of the reactor.
The production of PPNs under low-pressure conditions can be achieved either with or without using a gas aggregation source (GAS) within a vacuum chamber. A typical GAS setup comprises a tubular vacuum chamber equipped with a DC or RF electrode, or a magnetron, to generate a plasma discharge for polymerization (Fig. 7a). The vacuum chamber is typically designed with a relatively high aspect ratio, which promotes the homogeneous nucleation and formation of nano- and micro-clusters along an extended aggregation zone.182 The GAS configuration facilitates coaxial gas flow, resulting in the efficient transport of PPNs from the discharge region via an orifice into a secondary deposition chamber for collection.183
 | ||
| Fig. 7 Schematic diagram of various plasma-based systems used for synthesizing PPNs. (a) A typical GAS set-up, reproduced with permission from ref. 183. Copyright © 2017, Beilstein-Institut; (b) an RF-powered magnetron-based GAS set-up, adapted from ref. 184 with permission from the authors, Copyright © 2019; (c) a non-GAS vacuum chamber system, adapted with permission from ref. 52. Copyright © 2022, American Chemical Society; and (d) atmospheric pressure DBD plasma system, reproduced with permission from ref. 185. Copyright © 2014, John Wiley and Sons. | ||
Magnetron sputtering, a well-established method that is used in various modes for thin film deposition,186–188 can also be used as a GAS-assisted technique for producing PPNs at sub-atmospheric pressures (∼100 Pa). For instance, Kylián et al. reported a planar RF magnetron set-up consisting of a Nylon 6,6 target (Fig. 7b) to synthesize, on separate occasions, carbonaceous PPNs and PPN-metal nanocomposites.184 Changing the working gas from Ar to N2 resulted in nitrogen-rich PPNs, which have different morphology and surface chemistry than those PPNs produced with Ar.
Another avenue the same group explored is the production of heterogeneous PPN/metal nanocomposites. They reported the production of Cu/Nylon-based PPN composites by adding a copper strip to the Nylon 6,6 at relatively high power (80 W) and pressure (>100 Pa) conditions. This metal/polymer sputtering method yielded Cu cores enclosed within a shell of plasma polymer.184 This technology provides a suitable fabrication template for the layered shell of plasma polymers onto inorganic (i.e., metal) NP cores, which could be of interest for applications such as theranostic,189 post-translational modification (PTM) enrichment,190 and environmental remediation.191 We clarify that the fundamental operation of magnetron sputtering for the synthesis of PPNs using solid targets reported by Kylián et al. is quite different from a typical plasma polymerization process where organic monomers in the form of gas/vapour are excited into plasma to form polymers.
Since the early 1970s, low-pressure systems without the GAS modality have been used for the plasma polymerization of NPs. Examples include the pioneering works of Kobayashi on ethylene-180 and acetylene-based PPNs.181 More recently, Santos et al. increased the collection rate of PPNs inside a low-pressure non-GAS system by replacing the two-dimensional sample holder (stainless steel), commonly used in thin-film deposition, with a three-dimensional tissue culture plate.192 Scanning electron microscopy (SEM) micrographs showed that less than 1% of the stainless-steel surface was covered with NPs, and that the majority of the substrate was coated with plasma polymer. In contrast, powder-like brown NPs were collected using a 24-well polystyrene tissue culture plate without a noticeable presence of the plasma polymer coating on the stainless steel holder. The authors attributed the increased yield of PPN collection in the tissue culture plate to two factors. First, the positive plasma potential can expand locally in each individual well, which enables entrapment of the PPN. In addition, plasma sheath forms around the wells, resulting in an electric field that localizes the particles towards the centre of the well. The authors also demonstrated that doubling the RF input power from 50 W to 100 W resulted in an increase in PPN yield from 31 mg h−1 to 88 mg h−1 under a constant working pressure of 0.15 Torr.192
We have recently reported that the collection yield of PPNs in a similar plasma polymerization system (Fig. 7c) can be significantly enhanced by controlling the inflow sequence of the precursor gas mixture.52 A 2.5-fold increase in terms of the number of PPNs/unit area was observed by simply delaying the introduction of acetylene by 90 seconds after the plasma ignition of Ar/N2 gas mixture.52 Through this strategy, a collection efficiency of approximately 70–88% is achieved, which is markedly higher than the 27–37% obtained when all gases are inserted simultaneously. This work demonstrates that the yield of collected PPNs can be enhanced while minimising losses due to deposition on chamber walls or removal through the vacuum system. These types of optimization studies are significant, especially for upscaling PPN production without compromising energy efficiency and economics, which have been some of the existing challenges in implementing plasma processes at the industrial level.193
The formation dynamics of PPNs in Ar/C2H2 plasma follow a periodic pattern and can be divided into three distinct phases; nucleation, coagulation, and accretion52,194 (Fig. 7c). The nucleation phase is characterized by the formation of stable, nanosized clusters, known as protoparticles, though radical/ion plasma polymerization. In the coagulation phase, the protoparticles collide and grow while increasingly collecting negative charges. During the accretion phase, the particle size increases nearly linearly over time, as radicals and ions from the plasma are transferred onto the PPNs. Toward the end of the accretion phase, PPNs reach a critical size and exit the plasma discharge as regulated by an interplay of gravitational, electrostatic and ion drag forces, initiating a new growth cycle. The resulting PPNs are monodispersed in water, with PDI of less than 0.2.192 Such monodispersity is a result of plasma polymerisation-driven self-limiting growth mechanisms. During the coagulation phase, protoparticles accumulate negative charges as electrons, being more mobile than ions, preferentially diffuse to their surfaces. This charge buildup induces coulombic repulsion, preventing uncontrolled agglomeration and ensuring a narrow size distribution. Consequently, electrostatic stabilization restricts growth to particles within a specific size range, minimizing excessive aggregation. The monodispersed nature of PPNs in aqueous solution enhances their suitability for biomedical applications, as discussed in Section 4.
3.2.1.2. Atmospheric pressure plasma polymerization. Plasma polymerization has primarily been carried out in low-pressure systems over the past three decades.195 Atmospheric pressure plasma polymerization, however, has emerged as a potentially lower-cost alternative to low-pressure systems, requiring less complex equipment.196–198 Whether incorporating GAS or not, low-pressure plasma polymerization processes require vacuum chambers and vacuum pumps, which involve significant initial investment. In contrast, atmospheric pressure plasma polymerization processes do not rely on vacuum systems and costly chambers. Instead, plasmas typically take the form of dielectric barrier discharges, where an insulating dielectric material is positioned between electrodes.199 However, atmospheric pressure plasma processes often require high volumes of inert gases (e.g., He or Ar) along with monomeric precursors, which can increase the overall costs in the long term.
Atmospheric pressure plasma polymerization can produce PPNs with core–shell morphology.185 A dielectric barrier plasma reactor operating in atmospheric conditions (Fig. 7d) and powered by an RF source was used to fabricate polypyrrole-based PPNs.185 Two different types of hollow polypyrrole PPNs were produced based on their core cavity. The first type has a single spherical core, while the other has multiple bubble-like cores. These results contrast the PPNs produced in low-pressure environments wherein solid NPs, without core cavities, are typically produced due to homogeneous nucleation, crosslinking and particle growth.200 The authors proposed that small droplets of the oligomeric material coalesce and form a single drop with a larger surface area (Fig. 8).185 Polymerization happens at the surface and an outward diffusion of the liquid phase inside results in single-cored hollow polymeric NPs. In contrast, the small, bubble-like cores were hypothesized to be produced due to higher concentration and monomer reactivity.185 This mechanism leads to the agglomeration of the oligomeric precursor, which is carried into the core of PPNs post-polymerization.
 | ||
| Fig. 8 Monomer concentration and reactivity influence the formation of diverse hollow NPs, featuring either spherical cores or bubble-derived cores through atmospheric plasma polymerization, reproduced with permission from ref. 185. Copyright © 2014, John Wiley and Sons. | ||
3.2.1.3. Chemically functionalized plasma polymerized nanoparticles. The versatility of plasma polymerization is exemplified by its ability to utilize organic precursors in either gaseous or liquid form. Gaseous precursors, such as hydrocarbons (e.g., acetylene, ethylene, and methane),201,202 can be precisely delivered into the plasma polymerization system using high-precision flow controllers. Liquid precursors like acrylic acid,203,204 aniline,205 hexamethyldisiloxane,206 octadiene,207–209 and thiophene210,211 can be aerosolized,212 atomized,213 or evaporated214 before being transported to the plasma discharge. The plasma polymers produced using these monomers are functionalized with the corresponding chemical groups, including hydrocarbons, amines, and carboxylic acids. Also, PPNs can contain high concentrations of long-lived reactive radicals that facilitate covalent attachment of molecules,53,54 as further discussed in Section 4.
Alkane and alkene groups can be incorporated into PPNs by using monomeric hydrocarbons such as acetylene. For example, plasma polymerization of C2H2/Ar mixture at low pressure (150 mTorr) yielded solid, carbonaceous PPNs (Fig. 9a) containing hydrocarbon groups.54 Functional group analysis using Fourier transform infrared (FTIR) spectroscopy revealed the presence of multiple C–H stretching (2876, 2934, and 2957 cm−1) and C–H bending (1377 and 1454 cm−1) groups (Fig. 9b), indicating the retention of carbon-based moieties from the acetylene precursor.54 In addition, alkene peak (CC) identified at the 1631 cm−1 region implies that the triple bonds from the acetylene were cleaved, forming single and double carbon-to-carbon bonds. Nitrogen-based moieties can also be incorporated into the final PPN structure by adding a tertiary gas into the vacuum chamber during the plasma polymerization process. For example, in the same work,54 using a mixture of Ar/N2/C2H2 (3:10:6 sccm) resulted in the evolution of peaks corresponding to nitrogen-containing functional groups such as amine (1550, 3315, and 3340 cm−1) and nitrile (1250 cm−1) (Fig. 9b). The formation of such functional groups suggests that incorporating a non-polymerizable gas, such as N2, can be used as an effective strategy to control of the surface chemistry of PPNs.
 | ||
| Fig. 9 Tailorable surface chemistry of PPNs achieved by control of plasma polymerization process parameters. (a) Acetylene-based PPNs exhibit a compact, solid, and amorphous morphology, with surface functional groups adjustable by (b) modifying the precursor gas mixture to C2H2/Ar/N2, reproduced with permission from ref. 54. Copyright © 2018, American Chemical Society. (c and d) Aniline-based PPNs form spherical particles that retain nitrogen-containing functional groups from their precursors, reproduced with permission from ref. 215. Copyright © 2018, AIP Publishing. (e and f) Carboxyl-functionalized PPNs, derived from acrylic acid, display tuneable size and O–CO bond abundance by adjusting the plasma duty cycle during polymerization, reproduced with permission from ref. 176. Copyright © 2018, American Chemical Society. (g and h) PPNs feature radical-rich surfaces governed by temperature-dependent kinetics, reproduced with permission from ref. 54. Copyright © 2018, American Chemical Society. | ||
Aside from reacting N2 with other precursors like C2H2, amine functionalization in PPNs can be achieved through plasma polymerization of aniline. Amine-functionalized PPNs with aromatic moieties were produced by plasma polymerizing aniline with argon gas under low pressure.215 Similar to the other PPNs, the aniline-based PPNs are spherical-shaped with an estimated particle size of ∼451 nm (Fig. 9c). In situ FTIR measurements revealed that the spectral profile of the aniline-based PPNs was similar to polyaniline thin films synthesized in the literature (Fig. 9d). Nonetheless, the authors have acknowledged that there are differences between the PPNs and polyaniline thin films found in the literature. First, the PPNs partially lost their aromatic characteristics, demonstrated by the evolution of a shoulder peak between 1600 and 1750 cm−1, which can be attributed to either carbonyl stretching or alkene stretching from non-aromatic alkenes. Moreover, the authors credit the cross-linking of the polymeric structure to the Ar plasma interaction. The structure of plasma polymers is irregular due to fragmentation, random poly-recombination, and crosslinking mechanisms, which occur during the synthesis process (Fig. 6a).216 Thus, it is not surprising that the polyaniline films in the literature would have differences with the PPNs synthesized in this work.
Carboxyl-functionalized PPNs can be produced using acrylic acid as a precursor monomer.176,177 For example, carboxyl-bearing PPNs, which are highly stable in aqueous media, were formed by plasma polymerization of acrylic acid in a cylindrical chamber with a horizontal RF electrode.176 Particle size and concentration of carboxyl groups retained in the PPNs were shown to be tuneable by varying the RF-generated plasma duty cycle via changing the time on (ton) and time off (toff). Lowering the duty cycle from 100% to 32% decreased the size of the PPNs from 93 ± 14 nm to 31 ± 5 nm, while also increasing the abundance of carboxyl groups from 9.0 atom % to 12.0 atom %, respectively (Fig. 9e).176 The authors concluded that two different regimes could be classified based on the duty cycle (Fig. 9f). At higher duty cycles (ton > 30 μs and toff < 20 μs), the growth of the carboxyl-based PPNs was hindered by the bombardment of positive ions yielding the removal of the carboxyl functional groups. In contrast, at lower duty cycles (ton < 30 μs and toff > 20 μs), the PPN growth is based on radical-induced chain propagation wherein intact acrylic acid molecules are preserved, hence the higher O–CO abundance. Thus, pulsing the plasma enables control over the size and surface chemistry of carboxyl-based PPNs.
The presence of reactive radicals in PPNs has been previously reported.53,54 Electron paramagnetic resonance (EPR) spectroscopy spectrum of the PPNs shows that they are permeated by radicals (Fig. 9g). The EPR spectrum shows a single resonance peak, centred at 348 mT (g-value of 2.003),54 corresponding to unpaired electrons associated with radical-containing compounds.53 The radicals have shown temperature-dependent behaviour, with a slower decay rate at 24 °C compared to 4 °C (Fig. 9h).54 Notably, approximately 57% of the initial radical concentration was retained 10 days after synthesizing the PPN batch. The EPR findings highlight the stability and longevity of these radicals, which can be harnessed to achieve covalent attachment of biomolecules, as discussed in Section 4.
The dry approach of plasma polymerization for producing PPNs demonstrates significant potential, offering the advantages of a solvent-free process while enabling precise control over their physicochemical properties. The production process is faster and potentially more cost-effective for commercialization compared to conventional wet chemical methods. Plasma polymerization stands out as an ecologically benign, “green” technique, creating minimum waste and almost no effluents, thereby removing the need for expensive environmental remediation operations often needed in other technologies. This renders it highly attractive for industrial applications, where sustainability and regulatory adherence are paramount. Furthermore, the method utilizes minimal precursor materials, thus lowering operating expenses, and its compatibility with continuous production systems improves scalability.169,217 Notably, plasma polymerization can be harnessed to generate PPNs with radical-rich surfaces, providing conjugation sites capable of covalently attaching a wide range of molecules. In the following section, we highlight the versatility and efficacy of these PPNs across various biomedical applications.
4. Biomedical applications of plasma polymerized nanoparticles
Research on PPNs has primarily focused on understanding the mechanisms of their formation and growth in reactive dusty plasmas.218,219 Earlier works aimed to reduce and eliminate particle formation, as these particles were considered contaminants in various industrial applications, including thin film deposition, lithography, and semiconductor manufacturing.220–222 However, since 2018, several studies have been published demonstrating the potential of PPNs for real-world applications. This section reviews these applications, with a focus on their biomedical uses.4.1. Biomedical applications of plasma polymerized nanoparticles
Surface-active binding sites in PPNs enable robust attachment of a wide range of molecules, making it an excellent nanoplatform for biomedical applications. One of the earliest studies demonstrating the utility of PPNs as nanocarriers was conducted in 2018 by Santos et al., who conjugated various payloads, including an antibody (IgG), enzyme (luciferase), small interfering RNA (siRNA), drug (PTX), and a stable free radical molecule (2,2-diphenyl-1-picrylhydrazyl, DPPH), onto PPNs (Fig. 10a).54 The streamlined conjugation procedure involves incubation of the PPNs with various molecules in water at 4 °C for 1 hour. This one-pot functionalization of PPNs avoids the challenges of typical wet chemistry-based procedures, such as the use of chemical linkers and solvents, along with tedious purification steps. Cytocompatibility results of the PPNs with endothelial (hCECs), epithelial (MCF-10A) and human breast adenocarcinoma (MCF-7) cells after 72 and 120 hours of incubation indicated no significant decrease in cell viabilities across all conditions. Furthermore, confocal images from cell uptake experiments suggest that the PPNs conjugated with PTX and IgG with two different fluorescent labels (Cy5 and Cy7) are all co-localized inside the cell membrane of MCF-7 breast cancer cells (Fig. 10b). The authors implied that the multiple molecular cargos successfully underwent cell uptake and accumulated in the cytoplasm, where the molecules remained attached to the PPN after cell entry.54 The biocompatibility of these PPNs, even at elevated concentrations (∼109 PPN per ml), combined with their capability of intracellular delivery, makes them attractive for drug delivery and theranostic applications. | ||
| Fig. 10 PPNs are capable of cell internalization and endosomal escape. (a) SEM imaging demonstrates multifunctionalization of PPNs with 40, 20, and 10 nm IgG gold labels which enable labelling and tracking for cell imaging applications; (b) co-localization of PPNs conjugated with a therapeutic payload (PTX) and two fluorescent labelling agents (Cy5 and Cy7) into MCF-7 breast cancer cells, reproduced with permission from ref. 54. Copyright © 2018, American Chemical Society. (c) PPN conjugated with a fluorophore (NB) shows cell uptake in MCF-7 breast cancer cells, displaying significant difference compared to untreated and PPN-treated cells, reproduced from ref. 53 with permission from the Royal Society of Chemistry, Copyright © 2025. (d) Confocal microscopy imaging shows rapid cell internalization and lysosomal escape of PPNs from human coronary endothelial cells (hCAEC); (e) co-localization studies indicate that the Cy5-stained siVEGF is within the endosomal/lysosomal compartments at various time points, and (f) release profile of siRNA from the PPNs at pH 5 and pH 7.2, reproduced with permission from ref. 138. Copyright © 2020, Springer Nature. | ||
The direct and linker-free attachment of PPNs with molecules has been demonstrated to be through covalent interaction. In our recent work, we provided evidence for the formation of covalent bonds between fluorescent molecules (fluorescein isothiocyanate (FITC) and Nile Blue (NB)) and PPN surfaces.53 The stability of the attachment was demonstrated through detergent washing, using sodium dodecyl sulfate (SDS), with the molecules remaining on the NP surfaces as confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. 11a and b), fluorescence spectroscopy, flow cytometry, and time-of-flight secondary ion mass spectrometry (ToF-SIMS) data (Fig. 11c). SDS removes physisorbed molecules from surfaces while retaining those covalently attached intact.169 We also demonstrated that the PPNs attached to the NB have shown to be internalized into MCF-7 human breast cancer cells (Fig. 10c), demonstrating the potential of PPNs as nanoplatforms for biomedical imaging applications.
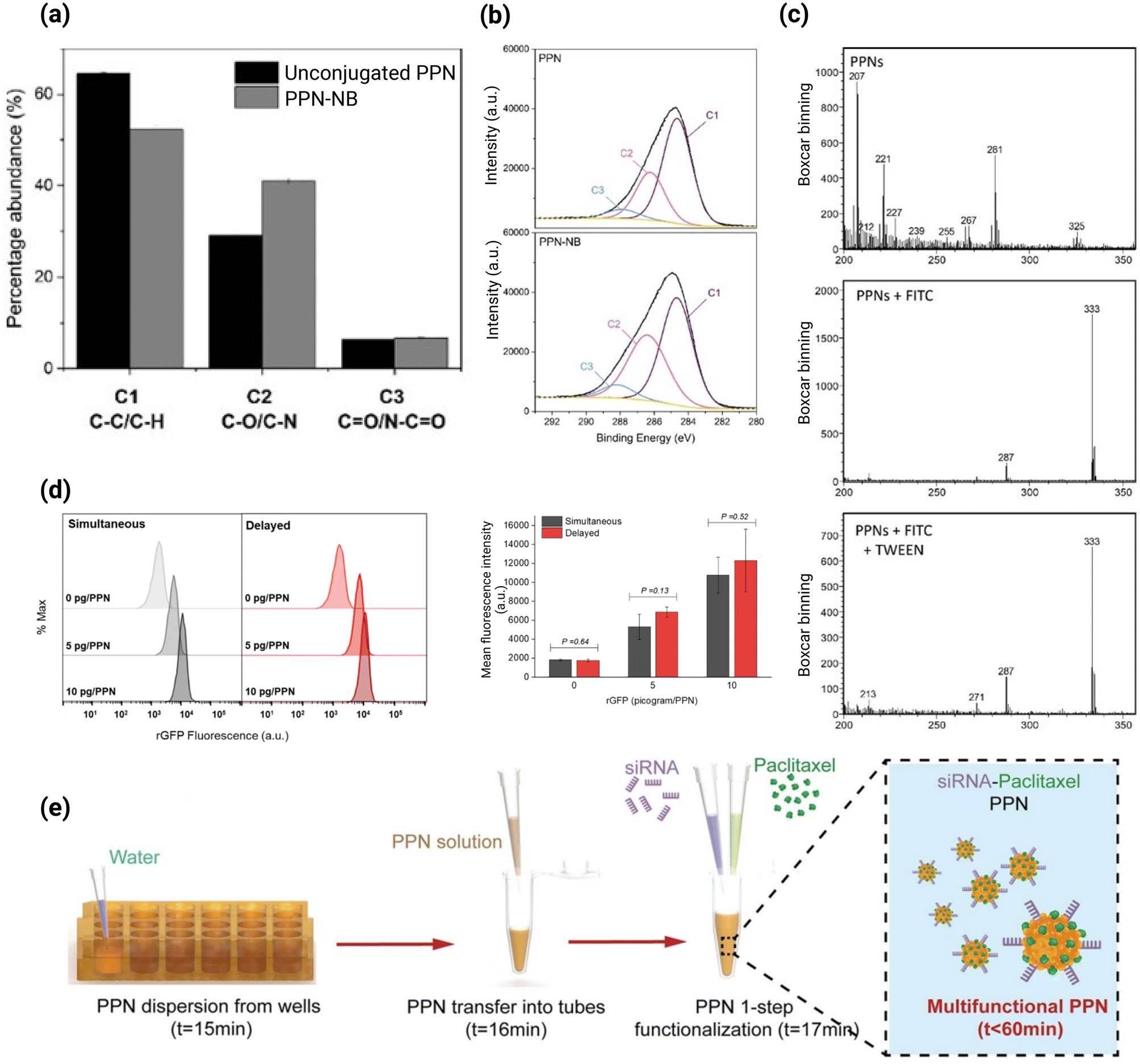 | ||
| Fig. 11 PPNs covalently conjugate payloads onto its surface. (a) Relative abundance of C 1s bond obtained from (b) high-resolution peak-fitted XPS of PPN and PPN-NB; (c) ToF-SIMS spectra of PPN, PPN-FITC, and PPN + FITC washed with Tween-20, reproduced with permission from ref. 53 with permission from the Royal Society of Chemistry, Copyright © 2025. (d) Flow cytometry results of two collection strategies, delayed and simultaneous, in terms of the mean fluorescence intensity of rGFP, reproduced with permission from ref. 52. Copyright © 2022, American Chemical Society; and (e) schematic diagram of the single-step, linker-free covalent conjugation of multiple payloads, reproduced with permission from ref. 138. Copyright © 2020, Springer Nature. | ||
Fluorescent molecules used for tracking, imaging, and theranostic applications can also be attached to PPNs. For example, we conjugated a model fluorophore, recombinant green fluorescent protein (rGFP) obtained from jellyfish Aequorea Victoria, onto the PPNs made from C2H2/N2/Ar plasma. The PPNs were incubated with different concentrations of rGFP (0, 5, and 10 pg per PPN) at room temperature for 30 minutes, resulting in a proportional increase in mean fluorescence intensity, as quantified by flow cytometry52 (Fig. 11d). The flow cytometry results suggest that attachment onto the PPN surface did not compromise the biological activity of rGFP. The strong autofluorescence properties of the PPN-rGFP indicate that the β-barrel tertiary structure responsible for GFP fluorescence remained intact after conjugation. The streamlined conjugation method's simplicity and robustness, preserving cargo activity, highlight PPNs’ versatility for biomedical applications.
The PPNs have demonstrated excellent potential as drug delivery vehicles against cancer therapy. Michael et al. successfully conjugated standard-of-care drug PTX onto the PPNs to form monodispersed PPN-PTX using a similar single-step, rapid incubation attachment procedure to treat breast cancer cells.138 The ensuing PTX attachment resulted in a slight increase in hydrodynamic size from 122 nm to 164 nm for the unconjugated PPN and PPN-PTX, respectively, while maintaining low polydispersity index (PDI) after conjugation (PDI < 0.1). As a proof-of-concept application, the PPN-PTX product was used against MCF-7 breast cancer cells wherein it showed significant inhibition of cell proliferation, and a higher percentage of apoptosis compared to the untreated cells after 24 and 72 hours. These cell study results demonstrate the potential of PPNs to serve as nanoplatforms for delivering therapeutic cargo in in vitro models.
The highly reactive surface of the PPNs can bind multiple functional groups onto their surfaces. Such highly reactive nature of the PPN surfaces was exemplified in the same study when an siRNA against vascular endothelial growth factor (siVEGF) was co-conjugated with the PTX molecule forming a dual functionalized PPN (PPN-Dual) (Fig. 11e).138 The combination of standard-of-care PTX and various siRNA has been explored for cancer therapy in other literature.223,224 Rapid internalization of Cy5-labeled PPN-siRNA was observed in hCAEC cells, with lysosomal co-localization occurring within 5 minutes of incubation (Fig. 10d and e). After 30 minutes, the PPN-siRNA (shown in green) was detected outside the lysosomes (Fig. 10f), implying successful endosomal/lysosomal escape.
The efficiency of the PPN-Dual containing PTX and siVEGF was tested against an orthotopically grown MCF-7 breast cancer tumour in a mice model.138 Results from this in vivo experiment showed that the tumour volume and weight regressed by 40% and 43%, respectively, after three doses of PPN-Dual in 14 days. Furthermore, the overall size of the tumour decreased by three-fold when exposed to the PPN-Dual compared to the saline control condition. Quantitative reverse transcription polymerase chain reaction (RT-PCR) results have also exhibited significantly increased levels of TUBB2A, a gene known to be involved in the regulation of cancer cell proliferation,225,226 post-incubation with PPN-Dual. The effective co-delivery of two therapeutic cargos on a single PPN nanoplatform demonstrated in this work can potentially be applied to other combinatory therapies. With careful optimization of the functionalization protocol (i.e., ratio of each molecule, incubation time, temperature), delivering three or more therapeutic cargos that provide synergistic effects together anchored on a single PPN can possibly be achieved in future works.
PPNs can also be used for on-demand bioactivation of inert substrates, which can be challenging to coat using traditional thin film coatings. In a recent work, porous 3D-printed polyethylene glycol diacrylate (PEGDA) hydrogel gyroids were surface functionalized with arginylglycylaspartic acid (RGD) peptide-functionalized PPNs.57 RGD is a short peptide sequence that serves as a recognition motif for integrins, mediating cell adhesion to the extracellular matrix.227 The PPN-RGD-functionalized hydrogel gyroids were used as substrates to anchor human cardiac fibroblasts. Fluorescence imaging showed enhanced fibroblast viability (Fig. 12a), cell density, and structural coverage on the PPN-RGD-functionalized hydrogels compared to those functionalized with either PPN or RGD alone (Fig. 12b). Furthermore, PEGDA hydrogels functionalized with PPN-RGD promoted fibroblast differentiation into myofibroblast in the presence of TGF-β (Fig. 12c), suggesting that such surface modified substrates provide environments conducive to cellular differentiation. This work highlights the excellent potential of PPNs as a versatile alternative to traditional thin-film coatings for surface biofunctionalization of biomaterials.
 | ||
| Fig. 12 Bioactivation of inert surfaces using PPN formulations. (a) Assay showing low cell growth on the unfunctionalized PEGDA substrate, with significant improvement in cell viability upon bioactivation with PPN and PPN-RGD. (b) Passivation of PEGDA hydrogel with RGD shows non-significant cell viability to untreated hydrogel, whereas high cell density of human cardiac fibroblasts was observed in PPN and PPN-RGD-coated substrate. (c) Hydrogel gyroids functionalized with PPN-RGD induced differentiation of human cardiac fibroblasts. Reproduced from ref. 57 with permission from the authors. Copyright © 2024, John Wiley and Sons. | ||
4.2. Biosafety of plasma polymerized nanoparticles
Translation to clinical-level applications requires the PPN nanoplatform to be biocompatible, monodispersed, stable in aqueous solution, and non-immunogenic, ensuring they do not elicit cytotoxic responses in vivo. Transport and accumulation of the PPNs into the sensitive organs in the body is possible. Therefore, it is crucial to ensure that any PPN-based formulations do not cause harmful toxicological side effects, except when the cargo itself is intended to be cytotoxic, such as a chemotherapeutic drug like PTX.Toxicological assessment of the PPN nanoplatform has been previously performed using various cell lines such as human dermal fibroblasts (hFB), A549 adenocarcinomic human alveolar basal epithelial cells, coronary artery cells (hCAECs), HEK293 embryonic kidney cells, malignant epithelial cells (HeLa), HEPG2 liver cancer cells, MCF7 breast cancer cells, human vascular smooth muscle cells (hSMCs), and induced pluripotent stem cell-derived endothelial cells (IPSC-ECs).52,55 The viability of the cells in the presence of PPNs has been assessed primarily based on proliferation, cellular integrity, mitochondrial membrane potential, and other related parameters. Traditional in vitro cytotoxicity assays such as MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and resazurin (commercially known as AlamarBlue™) assays serve as quantitative methods for determining the number of viable and functionally active cells in a population.228,229 In some cases, cellular integrity following PPN treatment is also evaluated by measuring the amount of lactate dehydrogenase (LDH) released from cells, which is indicative of membrane damage caused by PPNs.230 The mitochondrial membrane potential (MMP), which is directly correlated to the levels of mitochondrial function, can also be quantified via fluorescent probes which can penetrate the cell membrane like tetramethylrhodamine ethyl ester (TMRE), tetramethylrhodamine methyl ester (TMRM), rhodamine 123 (Rhod 123), and 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)).231 In addition, the stability of the PPNs post-conjugation is characterized by its monodispersity, which is measured by the polydispersity index (PDI), and the zeta potential, both of which are obtainable using dynamic light scattering (DLS) analysis. Monodispersity, or the uniformity of size in the dispersed phase, in NPs is characterized by PDI values of less than 0.1.232 However, other literature suggests that a PDI of 0.3 or below is acceptable for NPs used in drug delivery applications.233 The surface charge, in terms of zeta potential, impacts the stability of the NP formulation due to electrostatic interaction between individual particles. Colloidal NPs with zeta potential between −30 mV and +30 mV are typically considered unstable and prone to aggregation in solution.234–236 Overall, these quantitative assays and physicochemical properties serve as indicators of the cytotoxicity, or lack thereof, induced by PPNs at the in vitro scale.
Comprehensive in vitro and in vivo evaluation of PPN cytotoxicity has been reported by Michael et al.55 The cytotoxicity of two PPN sizes (128 and 234 nm, respectively) was assessed based on cell proliferation, cell membrane integrity, and cell apoptosis and necrosis.55 Compared to a commercial lipid-based NP (Lipofectamine RNAiMax), both primary (IPSC-ECs, hCAECs, hSMCs, hFBs) and cancer cell lines (A549, MCF7, HeLa, HEPG2) showed excellent tolerance to PPNs. The cells had high cell proliferation (80%), maintained cellular membrane integrity (<30% LDH release), and caused minimal apoptosis and necrosis (<25% events).55 These in vitro results indicate that PPNs are non-toxic to both healthy and cancerous cells. The toxicological evaluation was expanded to examine the physiological safety of PPNs in BALB/c mice through both acute and repeated-dose studies.55 The PPN formulation was tolerated remarkably well by the mice samples, wherein it exhibited no behavioural differences and loss of weight. The study found no haematological toxicity and adverse effects on kidney and liver functions (Fig. 13a), with parameters remaining within normal limits or comparable to saline controls. Histological analysis showed no organ damage or abnormalities from intravenous PPN treatment after repeated dosage for 14 days, although deposition at higher PPN concentrations was observed in the liver and spleen (Fig. 13b). Even at the highest doses (6000 mg kg−1 for acute studies and 48000 mg kg−1 for repeated injections), no premature deaths occurred, indicating that PPNs are safe for in vivo cargo delivery.
 | ||
| Fig. 13 Biocompatibility of PPNs. (a) Assessment of liver and kidney functions following repeated dosing of PPNs, showing no significant adverse effects; (b) histopathological analysis of mouse organs after 14 days at specific PPN concentrations. Red arrows indicate PPN deposition in the liver and spleen at elevated dosages, with no major structural abnormalities observed in the organs, reproduced from ref. 55 with permission from the authors, Copyright © 2021. (c) Exposure of PPNs (prepared through simultaneous or delayed protocol) did not affect the morphology of the cytoskeleton and nuclei of human dermal fibroblasts regardless of concentration. Evaluation of PPN cytotoxicity via (d) LDH release and (e) cell proliferation assays. Reproduced with permission from ref. 52. Copyright © 2022, American Chemical Society. | ||
Cellular morphology and nuclei size are not affected by the presence of the PPNs. In a separate work, exposure of PPNs fabricated from two varying procedures based on how acetylene is introduced into the N2- and Ar-filled vacuum chamber, namely simultaneous and delayed protocol, onto human dermal fibroblasts (GM3348) yielded negligible differences in terms of the morphology and nuclei size after 3 and 7 days.52 Actin filament and nuclei staining showed that the fibroblast cells remained unaffected by PPNs prepared using either the simultaneous or delayed protocol, even at concentrations ranging from 101 to 109 PPN per mL (Fig. 13c). The cytotoxic effects of the PPNs on fibroblasts were further assessed using LDH proliferation assay wherein basal LDH release percentages were observed for either of the two protocols (Fig. 13d).52 This result indicates the plasma membranes of fibroblasts remain intact after being incubated with the same concentrations of PPNs for 7 days. These findings were further supported by cell proliferation data, which showed no significant changes in fluorescence intensity compared to the positive control after 7 days (Fig. 13e).52 Collectively, it can be concluded that PPNs do not alter cellular properties or cause cytotoxic damage to normal fibroblasts.
The non-cytotoxic effects of the PPN-based nanoplatform have also been demonstrated in cardiomyocytes. In a recent work, the PPNs formed from the plasma polymerization of C2H2/N2/Ar were conjugated with a platelet-derived growth factor AB (PDGF-AB) before delivering the cargo-loaded PPNs into multiple cardiac muscle cells for in vitro assay studies.56 PDGFs are known to promote mitosis and are considered as chemotactic factors for mesenchymal cells.237 In addition, a recent study showed that PDGF-AB enhances cardiac function by stimulating post-myocardial infarction wound repair, resulting in an improved survival rate in the porcine model.238 Cell viability and mitochondrial membrane potential assessments using the MTT assay and TMRE assay demonstrated that both PPN and PPN-PDGF-AB formulations exhibit no significant cytotoxicity in neonatal rat ventricular myocytes (NRVMs) and human coronary artery vascular smooth muscle cells (HCASMCs).56 The lack of cytotoxicity was consistent across all three tested PPN doses (2.5 × 107 PPN, 5 × 107 PPN, and 1 × 108 PPN) and at all evaluated time points (1, 4, 7, 14, and 21 days). No evidence of deteriorating cardiomyocyte contractility was found in pluripotent stem cell derived cardiomyocytes (PSC-CMs) treated with PPN or its PDGF-AB-conjugated counterpart after 4 and 7 days. The in vitro results reported in this work support the findings from the earlier studies that the PPN platform, at least at a biologically relevant dosage, is not harmful at the cellular level.
5. Conclusions and future directions
This review comprehensively analyzed the diverse classes of polymeric NPs, along with the currently available synthesis methods of production for biomedical applications. The review focused on the interaction modes with payloads, discussing their respective advantages and limitations, which shed light on the barriers to the clinical and commercial translation of polymeric NPs.The diverse classes of polymeric NPs – nanocapsules, polymeric micelles, dendrimers, polymersomes, polyplexes, and solid nanospheres – offer unique advantages as polymer-based drug delivery systems. However, their clinical application faces significant challenges related to structural and mechanistic design. These include the need for chemical linkers to attach drug payloads, chemical modifications of the payload to integrate with the polymeric NPs, reliance on relatively weak and unpredictable electrostatic complexation reactions in biological conditions, premature release caused by the collapse and dissociation of nanostructures, and limitations in drug loading and encapsulation efficiencies. Furthermore, the production of most polymeric NPs relies heavily on wet chemical synthesis, which introduces issues of scalability, toxicity, and production complexity that impede their broader application (Fig. 14a).
 | ||
| Fig. 14 (a) Comparative infographic summarizing wet chemical and dry synthesis approaches for polymeric NP production, highlighting the key characteristics and advantages of plasma polymerization. (b) Proposed roadmap illustrating the future trajectory of PPNs toward commercialization and clinical applications. | ||
In contrast, dry, plasma polymerization technology emerges as a transformative, dry, and environmentally sustainable approach to produce PPNs that may overcome these challenges. We detailed the plasma polymerization synthesis process, laboratory setups, and biomedical utility of PPNs, highlighting their tuneable physicochemical properties, as well as their promising performance in drug delivery applications.
As a polymeric NP, PPNs stand out as it addresses the shortcomings of other classes. Their surface radicals enable covalent conjugation of molecules, facilitating single-step, reagent-free immobilization under mild conditions, broadening the potential for diverse biomedical applications. Structurally, PPNs are solid, carbonaceous nanospheres that resist disintegration and dissociation under biological conditions. Their high surface area-to-volume ratio enhances binding efficiencies, making them highly effective for drug delivery. Furthermore, PPNs have demonstrated excellent biocompatibility across various cell lines and animal models, reinforcing their potential for various biomedical applications.
In terms of its viability as a method, the plasma polymerization technique is a dry process that circumvents the challenges associated with conventional wet chemical methods. Unlike traditional approaches, this method does not require solvents or chemical reagents, instead utilizing relatively safe and cost-effective gaseous mixtures such as acetylene, nitrogen, and argon. The production rate of PPNs varies depending on the laboratory setup but typically achieves a throughput of approximately 1011 PPNs per minute. Purification of these NPs is efficiently performed using established washing and centrifugation protocols, which takes approximately one hour – significantly faster than traditional dialysis steps that require at least 4 to 72 hours. These inherent advantages of plasma polymerization method make it a promising, scalable, and efficient method of commercially producing PPNs for clinical applications.
Despite recent advancements in PPNs, several critical challenges related to their design, mechanism of action, and biocompatibility must be addressed to enable clinical translation. Precise size control is a key priority, with an optimal size range of 20–100 nm to ensure prolonged circulation and effective tumour penetration via the EPR effect. Advancements in the field could potentially enable shape modulation to produce geometries beyond spheres, opening new avenues for improving cellular uptake and biodistribution. Surface chemistry remains a particularly versatile feature of PPNs, offering opportunities to co-conjugating ligands that specifically target overexpressed or abundant receptors in cancer cells (e.g., folate receptors in breast cancer) with cytotoxic drugs (e.g., paclitaxel) can transform PPNs into precise, multimodal platforms for cancer therapy. Another potential of particular interest is that incorporating pH-sensitive linkers can allow for charge reversal, enhancing circulation time under physiological conditions while improving cellular uptake in the acidic tumour microenvironment. These advancements could significantly enhance the therapeutic potential of PPNs in cancer treatment.
The design of payload-conjugated PPNs for biomedical applications is quite analogous to polymer–drug conjugates. This design ensures that the payload remains covalently attached, preventing premature release and enabling the entire PPN-payload formulation to reach the target site. Future research could explore the integration of stimuli-responsive polymer–drug conjugates capable of responding to environmental cues such as pH, temperature, or enzymatic activity. These systems can allow controlled disassembly or shedding of the polymer, facilitating localized and sustained drug release at the target site. Comprehensive pharmacokinetic studies are crucial for understanding the interactions of PPNs with biological systems, as these interactions significantly affect safety, efficacy, and dosing regimens. Optimization of PPN design, particularly surface charge and chemistry, should be correlated with blood clearance half-life to refine their performance as drug delivery platforms. Determining optimal storage conditions is critical for commercialization. Research studies should evaluate whether PPNs are more stable in solid or dispersed phases, identify suitable buffer systems, and establish timeframes before aggregation, surface instability, or increased polydispersity occurs.
Last but not the least, scaling up plasma polymerization processes for industrial production will be essential for translating PPNs into clinically viable products (Fig. 14b). Developing cost-effective and reproducible protocols is key to ensuring regulatory compliance and facilitating widespread adoption. Moreover, plasma polymerization offers significant economic advantages as an environmentally sustainable process. Unlike wet chemical methods, which often involve hazardous solvents and generate substantial effluent requiring extensive treatment, plasma polymerization minimizes material usage and virtually eliminates hazardous byproducts. This “green” processing approach not only reduces environmental remediation costs but also enhances the overall economic feasibility of industrial-scale production. Optimizing reactor designs for continuous operation – for example, through hybrid systems that integrate plasma polymerization with existing manufacturing processes – holds significant potential to further improve throughput and reduce operating costs.
As an emerging field of great interest for biomedical applications, PPN research has already demonstrated compelling evidence of their potential as scalable, sustainable, and innovative platforms for drug delivery. By tackling the challenges highlighted here, PPNs have the potential to revolutionize polymeric NP technology, ushering in a new era for nanomedicine and its applications.
Abbreviations
| ABC | Accelerated blood clearance |
| ATRP | Atom transfer radical polymerization |
| BMA | Butyl methacrylate |
| CCR7 | CC chemokine receptor 7 |
| CNS | Central nervous system |
| CRP | Controlled radical polymerization |
| DEP CTX | DEP® cabazitaxel |
| DEP DTX | DEP® docetaxel |
| DiOC6(3) | 3,3′-Dihexyloxacarbocyanine iodide |
| DLS | Dynamic light scattering |
| DOX | Doxorubicin |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| DTX | Docetaxel |
| EGDMA | Ethylene glycol dimethacrylate |
| EPR | Enhanced permeability and retention |
| FITC | Fluorescein isothiocyanate |
| FRP | Free radical polymerization |
| GBM | Glioblastoma |
| hFB | Human dermal fibroblasts |
| HPMA | Hydroxypropyl methacrylamide |
| hSMCs | Human vascular smooth muscle cells |
| IPSC-ECs | Induced pluripotent stem cell-derived endothelial cells |
| LDH | Lactate dehydrogenase |
| Mg(CH3COO)2 | Magnesium acetate |
| MgCl2 | Magnesium chloride |
| mPEG-P(TMC-DTC) | Poly(ethylene glycol)-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) |
| MPS | Mononuclear phagocytic system |
| NB | Nile blue |
| NP | Nanoparticle |
| PDI | Polydispersity index |
| pDNA | Plasmid DNA |
| PEG-b-PLA | Poly(ethylene glycol)-b-poly(D,L-lactide) |
| PEGDA | Polyethylene glycol diacrylate |
| PEI | Polyethylenimine |
| PLGA | Poly(lactide-co-glycolide) |
| PLGA–PEG2k–MAN | Poly(lactic-co-glycolic acid)–poly(ethylene glycol)-p-aminophenyl-α-D-mannopyranoside |
| PNPs | Polymeric nanoparticles |
| PSMA | Prostate-specific membrane antigen |
| PTN | Pterostilbene |
| RF | Radiofrequency |
| rGFP | Recombinant green fluorescent protein |
| shRNA | Small hairpin RNAs |
| siRNA | Small interfering RNA |
| SN-38 | 7-Ethyl-10-hydroxycamtothecin |
| XPS | X-ray photoelectron spectroscopy |
| ABC | Accelerated blood clearance |
| ATRP | Atom transfer radical polymerization |
| BMA | Butyl methacrylate |
| CaCl2 | Calcium chloride |
| CCR7 | CC chemokine receptor 7 |
| CNS | Central nervous system |
| CRP | Controlled radical polymerization |
| CTA | Chain transfer agent |
| DC | Direct current |
| DEP CTX | DEP® cabazitaxel |
| DEP DTX | DEP® docetaxel |
| DiOC6(3) | 3,3′-Dihexyloxacarbocyanine iodide |
| DLS | Dynamic light scattering |
| DOX | Doxorubicin |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| DTX | Docetaxel |
| EGDMA | Ethylene glycol dimethacrylate |
| FITC | Fluorescein isothiocyanate |
| FRP | Free radical polymerization |
| FTIR | Fourier transform infrared |
| GAS | Gas aggregation source |
| GBM | Glioblastoma |
| hFB | Human dermal fibroblasts |
Data availability
This review paper does not contain any unpublished data. All figures included were reproduced from previously published sources, with appropriate citations and permissions secured. No new data were generated or analyzed for this work.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
B. A. acknowledges the support of the Australian Research Council (ARC) through the DECRA program (DE210100662). The authors acknowledge the Sydney Nano Institute and the Hunter Medical Research Institute through the Precision Medicine Research Program. E. A. acknowledges the support of the Faculty of Engineering Research Scholarship from The University of Sydney.References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- X. Zhang, F. Centurion, A. Misra, S. Patel and Z. Gu, Molecularly targeted nanomedicine enabled by inorganic nanoparticles for atherosclerosis diagnosis and treatment, Adv. Drug Delivery Rev., 2022, 194, 11470 Search PubMed.
- C. Domingues, A. Santos, C. Alvarez-Lorenzo, A. Concheiro, I. Jarak, F. Veiga, I. Barbosa, M. Dourado and A. Figueiras, Where Is Nano Today and Where Is It Headed? A Review of Nanomedicine and the Dilemma of Nanotoxicology, ACS Nano, 2022, 16(7), 9994–10041 CrossRef CAS PubMed.
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Polymeric Nanoparticles for Drug Delivery, Chem. Rev., 2024, 124(9), 5505–5616 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- L. Hong, W. Li, Y. Li and S. Yin, Nanoparticle-based drug delivery systems targeting cancer cell surfaces, RSC Adv., 2023, 13(31), 21365–21382 RSC.
- S. A. Dilliard and D. J. Siegwart, Passive, active and endogenous organ-targeted lipid and polymer nanoparticles for delivery of genetic drugs, Nat. Rev. Mater., 2023, 8(4), 282–300 CrossRef CAS PubMed.
- W. Zhang, A. Mehta, Z. Tong, L. Esser and N. H. Voelcker, Development of Polymeric Nanoparticles for Blood–Brain Barrier Transfer—Strategies and Challenges, Adv. Sci., 2021, 8(10), 2003937 CrossRef CAS PubMed.
- S. Y. Ong, D. C. Zhang, D. X. Dong and P. S. Q. Yao, Recent Advances in Polymeric Nanoparticles for Enhanced Fluorescence and Photoacoustic Imaging, Angew. Chem., Int. Ed., 2021, 60(33), 17797–17809 CrossRef CAS PubMed.
- S. Suárez-García, R. Solórzano, F. Novio, R. Alibés, F. Busqué and D. Ruiz-Molina, Coordination polymers nanoparticles for bioimaging, Coord. Chem. Rev., 2021, 432, 213716 CrossRef.
- X. Chen, S. Hussain, A. Abbas, Y. Hao, A. H. Malik, X. Tian, H. Song and R. Gao, Conjugated polymer nanoparticles and their nanohybrids as smart photoluminescent and photoresponsive material for biosensing, imaging, and theranostics, Microchim. Acta, 2022, 189(3), 83 CrossRef CAS PubMed.
- T. Chen, Y. Peng, M. Qiu, C. Yi and Z. Xu, Heterogenization of homogeneous catalysts in polymer nanoparticles: From easier recovery and reuse to more efficient catalysis, Coord. Chem. Rev., 2023, 489, 215195 CrossRef CAS.
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Role of Polymer Structures in Catalysis by Transition Metal and Metal Oxide Nanoparticle Composites, Chem. Rev., 2019, 120(2), 1350–1396 CrossRef PubMed.
- J. Zhu, X. Zhao, N. Wu, D. Wu, X. Huang, Z. Nie and D. Chen, Constructing a Thin Layer of the Concentrated Polyelectrolyte Solution on the Hydrogel Surface That Can Considerably Improve the Solar Evaporation Efficiency, Macromolecules, 2024, 57(20), 9811–9822 CrossRef CAS.
- G. Zhang, Q. Li, E. Allahyarov, Y. Li and L. Zhu, Challenges and Opportunities of Polymer Nanodielectrics for Capacitive Energy Storage, ACS Appl. Mater. Interfaces, 2021, 13(32), 37939–37960 CrossRef CAS PubMed.
- M. Yang, M. Guo, E. Xu, W. Ren, D. Wang, S. Li, S. Zhang, C.-W. Nan and Y. Shen, Polymer nanocomposite dielectrics for capacitive energy storage, Nat. Nanotechnol., 2024, 19, 588–603 CrossRef CAS PubMed.
- J. Zhu, Z. Xiao, F. Song, X. Huang, D. Chen and Z. Nie, Amphiphilic Janus patch-grafted hydrogels for salt-rejecting solar water desalination, J. Mater. Chem. A, 2024, 12(28), 17142–17150 RSC.
- J. Zhu, D. Wu, X. Huang, D. Chen and Z. Nie, Hydrogen-bond disruption in molecularly engineered Janus evaporators for enhanced solar desalination, Soft Matter, 2025 Search PubMed.
- D. Wu, J. Zhu, J. Xu, X. Zhao, O. Jiang, X. Huang, Z. Nie and D. Chen, Recyclable Amphiphilic Magnetic-responsive Mixed-Shell Nanoparticles With High Interfacial Activity Comparable to Janus Particles for Oily Water Purification, Macromol. Rapid Commun., 2024, 2400734 CrossRef PubMed.
- S. Kang, T. W. Yoon, G.-Y. Kim and B. Kang, Review of Conjugated Polymer Nanoparticles: From Formulation to Applications, ACS Appl. Nano Mater., 2022, 5(12), 17436–17460 CrossRef CAS.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Functional nanoparticles through π-conjugated polymer self-assembly, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- J. W. Hickey, J. L. Santos, J.-M. Williford and H.-Q. Mao, Control of polymeric nanoparticle size to improve therapeutic delivery, J. Controlled Release, 2015, 219, 536–547 CrossRef CAS PubMed.
- L. Lv, Z. Zhang, H. Li, X. Huang and D. Chen, Endowing Polymeric Assemblies with Unique Properties and Behaviors by Incorporating Versatile Nanogels in the Shell, ACS Macro Lett., 2019, 8(10), 1222–1226 CrossRef CAS PubMed.
- J.-M. Williford, J. L. Santos, R. Shyam and H.-Q. Mao, Shape control in engineering of polymeric nanoparticles for therapeutic delivery, Biomater. Sci., 2015, 3(7), 894–907 RSC.
- L. L. Haidar, M. Bilek and B. Akhavan, Surface Bio-engineered Polymeric Nanoparticles, Small, 2024, 20(21), 2310876 CrossRef CAS PubMed.
- E. Austria, M. Bilek, P. Varamini and B. Akhavan, Breaking biological barriers: Engineering polymeric nanoparticles for cancer therapy, Nano Today, 2025, 60, 102552 CrossRef CAS.
- S. Chu, X. Shi, Y. Tian and F. Gao, pH-Responsive Polymer Nanomaterials for Tumor Therapy, Front. Oncol., 2022, 12, 855019 CrossRef CAS PubMed.
- A. Wang, H. Gao, Y. Sun, Y.-l. Sun, Y.-W. Yang, G. Wu, Y. Wang, Y. Fan and J. Ma, Temperature- and pH-responsive nanoparticles of biocompatible polyurethanes for doxorubicin delivery, Int. J. Pharm., 2013, 441(1–2), 30–39 CrossRef CAS PubMed.
- M. Li, G. Zhao, G. Zhao, W.-K. Su, W.-K. Su and Q. Shuai, Enzyme-Responsive Nanoparticles for Anti-tumor Drug Delivery, Front. Chem., 2022, 8, 647 CrossRef PubMed.
- Y. Liu, G. Yang, T. Baby, Tengjisi, D. Chen, D. A. Weitz and C.-X. Zhao, Stable Polymer Nanoparticles with Exceptionally High Drug Loading by Sequential Nanoprecipitation, Angew. Chem., 2020, 132(12), 4750–4758 CrossRef.
- K. C. d. Castro, J. M. Costa and M. G. N. Campos, Drug-loaded polymeric nanoparticles: a review, Int. J. Polym. Mater. Polym. Biomater., 2020, 71(1), 1–13 Search PubMed.
- K. Wróbel, A. Deręgowska, G. Betlej, M. Walczak, M. Wnuk, A. Lewińska and S. Wołowiec, Cytarabine and dexamethasone-PAMAM dendrimer di-conjugate sensitizes human acute myeloid leukemia cells to apoptotic cell death, J. Drug Delivery Sci. Technol., 2023, 81, 104242 CrossRef.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Intramolecular Cross-Linking Methodologies for the Synthesis of Polymer Nanoparticles, Chem. Rev., 2015, 114(3), 878–961 CrossRef PubMed.
- U. Rehman, N. Parveen, A. Sheikh, M. A. S. Abourehab, A. Sahebkar and P. Kesharwani, Polymeric nanoparticles-siRNA as an emerging nano-polyplexes against ovarian cancer, Colloids Surf., B, 2022, 218, 112766 CrossRef CAS PubMed.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Polymeric Nanostructures for Imaging and Therapy, Chem. Rev., 2015, 115(19), 10967–11011 CrossRef CAS PubMed.
- M. Machtakova, H. Therien-Aubin and K. Landfester, Polymer nano-systems for the encapsulation and delivery of active biomacromolecular therapeutic agents, Chem. Soc. Rev., 2022, 51, 128 RSC.
- X. Huang, J. Li, G. Li, B. Ni, Z. Liang, H. Chen, C. Xu, J. Zhou, J. Huang and S. Deng, Cation-free siRNA-cored nanocapsules for tumor-targeted RNAi therapy, Acta Biomater., 2023, 161, 226–237 CrossRef CAS PubMed.
- Lakshmi, S. Singh, K. Shahc and H. K. Dewangan, Dual Vinorelbine bitartrate and Resveratrol loaded polymeric aqueous core nanocapsules for synergistic efficacy in breast cancer, J. Microencapsulation, 2022, 39(4), 299–313 CrossRef CAS PubMed.
- C. L. Ventola, Progress in Nanomedicine: Approved and Investigational Nanodrugs, Pharm. Ther., 2017, 42(12), 742 Search PubMed.
- S. Alven and B. A. Aderibigbe, The Therapeutic Efficacy of Dendrimer and Micelle Formulations for Breast Cancer Treatment, Pharmaceutics, 2020, 12(12), 1212 CrossRef CAS PubMed.
- Y. Fujiwara, H. Mukai, T. Saeki, J. Ro, Y.-C. Lin, S. E. Nagai, K. S. Lee, J. Watanabe, S. Ohtani, S. B. Kim, K. Kuroi, K. Tsugawa, Y. Tokuda, H. Iwata, Y. H. Park, Y. Yang and Y. Nambu, A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients, Br. J. Cancer, 2019, 120, 475–480 CrossRef CAS PubMed.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel, Br. J. Cancer, 2005, 92, 1240–1246 CrossRef CAS PubMed.
- K. Kato, K. Chin, T. Yoshikawa, K. Yamaguchi, Y. Tsuji, T. Esaki, K. Sakai, M. Kimura, T. Hamaguchi, Y. Shimada, Y. Matsumura and R. Ikeda, Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer, Invest. New Drugs, 2012, 30, 1621–1627 CrossRef CAS PubMed.
- S. Mignani, X. Shi, J. Rodrigues, H. Tomas, A. Karpus and J.-P. Majoral, First-in-class and best-in-class dendrimer nanoplatforms from concept to clinic: Lessons learned moving forward, Eur. J. Med. Chem., 2021, 219, 113456 CrossRef CAS PubMed.
- C. M. Patterson, S. B. Balachander, I. Grant, P. Pop-Damkov, B. Kelly, W. McCoull, J. Parker, M. Giannis, K. J. Hill, F. D. Gibbons, E. J. Hennessy, P. Kemmitt, A. R. Harmer, S. Gales, S. Purbrick, S. Redmond, M. Skinner, L. Graham, J. P. Secrist, A. G. Schuller, S. Wen, A. Adam, C. Reimer, J. Cidado, M. Wild, E. Gangl, S. E. Fawell, J. Saeh, B. R. Davies, D. J. Owen and M. B. Ashford, Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy, Commun. Biol., 2021, 4(1), 112 CrossRef CAS PubMed.
- M. Konopleva, N. Jain, C. L. Andersen, N. C. Francisco, N. Elgeioushi, R. Hobson, M. Scott, J. Stone, S. Sharma, P. M. Gutierrez, R. Tibes, B. Davies, T. Winkler, G. Fabbri, F. Z. Cader and N. McNeer, NIMBLE: A Phase I/II Study of AZD0466 Monotherapy or in Combination in Patients with Advanced Hematological Malignancies, Blood, 2021, 138, 2353 CrossRef.
- Y. Wei, X. Gu, Y. Sun, F. Meng, G. Storm and Z. Zhong, Transferrin-binding peptide functionalized polymersomes mediate targeted doxorubicin delivery to colorectal cancer in vivo, J. Controlled Release, 2020, 319, 407–415 CrossRef CAS PubMed.
- T. S. Zavvar, M. Babaei, K. Abnous, S. M. Taghdisi, S. Nekooei, M. Ramezani and M. Alibolandi, Synthesis of multimodal polymersomes for targeted drug delivery and MR/fluorescence imaging in metastatic breast cancer model, Int. J. Pharm., 2020, 578, 119091 CrossRef CAS PubMed.
- Y. Deng, C. Tan, S. Huang, H. Sun, Z. Li, J. Li, Z. Zhou and M. Sun, Site-Specific Polyplex on CCR7 Down-Regulation and T Cell Elevation for Lymphatic Metastasis Blocking on Breast Cancer, Adv. Healthcare Mater., 2022, 11(22), 2201166 CrossRef CAS PubMed.
- T. Jeon, R. Goswami, H. Nagaraj, Y. A. Cicek, V. Lehot, J. Welton, C. J. Bell, J. Park, D. C. Luther, J. Im, C. M. Rotello, J. Mager and V. M. Rotello, Engineered Zwitterionic Diblock Copolymer-siRNA Polyplexes Provide Highly Effective Treatment of Triple-Negative Breast Cancer in a 4T1 Murine Model, Adv. Funct. Mater., 2024, 34(42), 2406763 CrossRef CAS PubMed.
- D. D. V. Hoff, M. M. Mita, R. K. Ramanathan, G. J. Weiss, A. C. Mita, P. M. LoRusso, H. A. Burris, III, L. L. Hart, S. C. Low, D. M. Parsons, S. E. Zale, J. M. Summa, H. Youssoufian and J. C. Sachdev, Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors, Clin. Cancer Res., 2016, 22(13), 3157–3163 CrossRef PubMed.
- L. L. Haidar, M. Baldry, S. T. Fraser, B. B. Boumelhem, A. D. Gilmour, Z. Liu, Z. Zheng, M. M. M. Bilek and B. Akhavan, Surface-Active Plasma-Polymerized Nanoparticles for Multifunctional Diagnostic, Targeting, and Therapeutic Probes, ACS Appl. Nano Mater., 2022, 5(12), 17576–17591 CrossRef CAS.
- L. Haidar, Y. Wang, A. Gilmour, E. Austria, B. B. Boumelhem, N. A. Khan, A. Fadzil, S. T. Fraser, M. Bilek and B. Akhavan, Direct covalent attachment of fluorescent molecules on plasma polymerized nanoparticles: A simplified approach for biomedical applications, J. Mater. Chem. B, 2025, 13(5), 1666–1680 RSC.
- M. Santos, P. L. Michael, E. C. Filipe, A. H. P. Chan, J. Hung, R. P. Tan, B. S. L. Lee, M. Huynh, C. Hawkins, A. Waterhouse, M. M. M. Bilek and S. G. Wise, Plasma Synthesis of Carbon-Based Nanocarriers for Linker-Free Immobilization of Bioactive Cargo, ACS Appl. Nano Mater., 2018, 1(2), 580–594 CrossRef CAS.
- P. L. Michael, Y. T. Lam, J. Hung, R. P. Tan, M. Santos and S. G. Wise, Comprehensive Evaluation of the Toxicity and Biosafety of Plasma Polymerized Nanoparticles, Nanomaterials, 2021, 11(5), 1176 CrossRef CAS PubMed.
- Z. E. Clayton, M. Santos, H. Shah, J. Lu, S. Chen, H. Shi, S. Kanagalingam, P. L. Michael, S. G. Wise and J. J. H. Chong, Plasma polymerized nanoparticles are a safe platform for direct delivery of growth factor therapy to the injured heart, Front. Bioeng. Biotechnol., 2023, 11, 1127996 CrossRef PubMed.
- M. Santos, P. L. Michael, T. C. Mitchell, Y. T. Lam, T. M. Robinson, M. J. Moore, R. P. Tan, J. Rnjak-Kovacina, K. S. Lim and S. G. Wise, On-Demand Bioactivation of Inert Materials With Plasma-Polymerized Nanoparticles, Adv. Mater., 2024, 2311313 CrossRef CAS PubMed.
- D. Sivadasan, M. H. Sultan, O. Madkhali, Y. Almoshari and N. Thangavel, Polymeric Lipid Hybrid Nanoparticles (PLNs) as Emerging Drug Delivery Platform—A Comprehensive Review of Their Properties, Preparation Methods, and Therapeutic Applications, Pharmaceutics, 2021, 13(8), 1291 CrossRef CAS PubMed.
- A. Musyanovych and K. Landfester, Polymer Micro- and Nanocapsules as Biological Carriers with Multifunctional Properties, Macromol. Biosci., 2014, 14(4), 458–477 CrossRef CAS PubMed.
- O. S. A. Abed, C. S. Chaw, L. Williams and A. A. Elkordy, PEGylated polymeric nanocapsules for oral delivery of trypsin targeted to the small intestines, Int. J. Pharm., 2021, 592, 120094 CrossRef PubMed.
- R. H. Utama, Y. Jiang, P. B. Zetterlund and M. H. Stenzel, Biocompatible Glycopolymer Nanocapsules via Inverse Miniemulsion Periphery RAFT Polymerization for the Delivery of Gemcitabine, Biomacromolecules, 2015, 16(7), 2144–2156 CrossRef CAS PubMed.
- A. Fahmi, M. Abdur-Rahman, O. Mahareek and M. A. Shemis, Synthesis, characterization, and cytotoxicity of doxorubicin-loaded polycaprolactone nanocapsules as controlled anti-hepatocellular carcinoma drug release system, BMC Chem., 2022, 16(1), 95 CrossRef CAS PubMed.
- A. C. S. Oliveira, P. M. Oliveira, M. Cunha-Filho, T. Gratieri and G. M. Gelfuso, Latanoprost Loaded in Polymeric Nanocapsules for Effective Topical Treatment of Alopecia, AAPS PharmSciTech, 2020, 21, 1–7 CrossRef PubMed.
- H. Zakaria, R. E. Kurdi and D. Patra, Curcumin-PLGA based nanocapsule for the fluorescence spectroscopic detection of dopamine, RSC Adv., 2022, 12(43), 28245–28253 RSC.
- S. B. Alam, G. Soligno, J. Yang, K. C. Bustillo, P. Ercius, H. Zheng, S. Whitelam and E. M. Chan, Dynamics of Polymer Nanocapsule Buckling and Collapse Revealed by In Situ Liquid-Phase TEM, Langmuir, 2022, 38(23), 7168–7178 CrossRef CAS PubMed.
- S. Nicolas, M.-A. Bolzinger, L. P. Jordheim, Y. Chevalier, H. Fessi and E. Almouazen, Polymeric nanocapsules as drug carriers for sustained anticancer activity of calcitriol in breast cancer cells, Int. J. Pharm., 2018, 550(1–2), 170–179 CrossRef CAS PubMed.
- F. S. Poletto, E. Jäger, L. Cruz, A. R. Pohlmann and S. S. Guterres, The effect of polymeric wall on the permeability of drug-loaded nanocapsules, Mater. Sci. Eng., C, 2008, 28(4), 472–478 CrossRef CAS.
- M. F. Kabil, S. A. A. Gaber, M. A. Hamzawy, I. M. El-Sherbiny and M. Nasr, Folic/lactobionic acid dual-targeted polymeric nanocapsules for potential treatment of hepatocellular carcinoma, Drug Delivery Transl. Res., 2024, 14(5), 1338–1351 CrossRef CAS PubMed.
- S. Jiang, W. Li, J. Yang, T. Zhang, Y. Zhang, L. Xu, B. Hu, Z. Li, H. Gao, Y. Huang and S. Ruan, Cathepsin B-Responsive Programmed Brain Targeted Delivery System for Chemo-Immunotherapy Combination Therapy of Glioblastoma, ACS Nano, 2024, 18(8), 6445–6462 CrossRef CAS PubMed.
- S. K. Hari, A. Gauba, N. Shrivastava, R. M. Tripathi, S. K. Jain and A. K. Pandey, Polymeric micelles and cancer therapy: an ingenious multimodal tumor-targeted drug delivery system, Drug Delivery Transl. Res., 2023, 13(1), 135–163 CrossRef PubMed.
- R. F. Riedel, V. S. Chua, T. Kim, J. Dang, K. Zheng, A. Moradkhani, A. Osada and S. P. Chawla, Results of NC-6300 (nanoparticle epirubicin) in an expansion cohort of patients with angiosarcoma, Oncologist, 2022, 27(10), 809-e765 CrossRef PubMed.
- T. S. v. Solinge, L. Nieland, E. A. Chiocca and M. L. D. Broekman, Advances in local therapy for glioblastoma—taking the fight to the tumour, Nat. Rev. Neurol., 2022, 18, 221–236 CrossRef PubMed.
- D.-W. Kim, S.-Y. Kim, H.-K. Kim, S.-W. Kim, S. W. Shin, J. S. Kim, K. Park, M. Y. Lee and D. S. Heo, Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer, Ann. Oncol., 2007, 18(12), 2009–2014 CrossRef PubMed.
- T. Negishi, F. Koizumi, H. Uchino, J. Kuroda, T. Kawaguchi, S. Naito and Y. Matsumura, NK105, a paclitaxel-incorporating micellar nanoparticle, is a more potent radiosensitising agent compared to free paclitaxel, Br. J. Cancer, 2006, 95(5), 601–606 CrossRef CAS PubMed.
- Y. Jiang, Z. Jiang, M. Wang and L. Ma, Current understandings and clinical translation of nanomedicines for breast cancer therapy, Adv. Drug Delivery Rev., 2022, 180, 114034 CrossRef CAS PubMed.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. D. Favero, L. Cantù and S. Nicoli, Polymeric micelles in drug delivery: An insight of the techniques for their characterization and assessment in biorelevant conditions, J. Controlled Release, 2021, 332, 312–336 CrossRef CAS PubMed.
- G. Miao, Y. He, K. Lai, Y. Zhao, P. He, G. Tan and X. Wang, Accelerated blood clearance of PEGylated nanoparticles induced by PEG-based pharmaceutical excipients, J. Controlled Release, 2023, 363, 12–26 CrossRef CAS PubMed.
- Z. Li, M. Liu, L. Ke, L.-J. Wang, C. Wu, C. Li, Z. Li and Y.-L. Wu, Flexible polymeric nanosized micelles for ophthalmic drug delivery: research progress in the last three years, Nanoscale Adv., 2021, 3(18), 5240–5254 RSC.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Glutathione responsive polymers and their application in drug delivery systems, Polym. Chem., 2017, 8(1), 97–126 RSC.
- P. Chen, Z. Wang, X. Wang, J. Gong, J. Sheng, Y. Pan, D. Zhu and X. Liu, ROS–Responsive Ferrocenyl Amphiphilic PAMAM Dendrimers for On–Demand Delivery of siRNA Therapeutics to Cancer Cells, Pharmaceutics, 2024, 16(7), 936 CrossRef CAS PubMed.
- S. Zhang, J. Lv, P. Gao, Q. Feng, H. Wang and Y. Cheng, A pH-Responsive Phase-Transition Polymer with High Serum Stability in Cytosolic Protein Delivery, Nano Lett., 2021, 21(18), 7855–7861 CrossRef CAS PubMed.
- A. Bohr, N. Tsapis, C. Foged, I. Andreana, M. Yang and E. Fattal, Treatment of acute lung inflammation by pulmonary delivery of anti-TNF-α siRNA with PAMAM dendrimers in a murine model, Eur. J. Pharm. Biopharm., 2020, 156, 114–120 CrossRef CAS PubMed.
- S. K. Wong, I. Zainol, M. P. Ng, C. H. Ng and I. H. Ooi, Dendrimer-like AB2-type star polymers as nanocarriers for doxorubicin delivery to breast cancer cells: synthesis, characterization, in vitro release and cytotoxicity studies, J. Polym. Res., 2020, 27, 1–19 CrossRef.
- M. Fatima, A. Sheikh, N. Hasan, A. Sahebkar, Y. Riadi and P. Kesharwani, Folic acid conjugated poly(amidoamine) dendrimer as a smart nanocarriers for tracing, imaging, and treating cancers over-expressing folate receptors, Eur. Polym. J., 2022, 170, 111156 CrossRef CAS.
- A. P. Dias, S. d. S. Santos, J. V. d. Silva, R. Parise-Filho, E. I. Ferreira, O. E. Seoud and J. Giarolla, Dendrimers in the context of nanomedicine, Int. J. Pharm., 2020, 573, 118814 CrossRef CAS PubMed.
- B. D. Kelly, V. McLeod, R. Walker, J. Schreuders, S. Jackson, M. Giannis, C. Dietinger, P. Reitano, R. Pathak, S. Xia, A. Cargill, A. Seta, R. Hufton, G. Heery, C. Cullinane and D. J. Owen, Anticancer activity of the taxane nanoparticles, DEP® docetaxel and DEP® cabazitaxel, Cancer Res., 2020, 80, 1716 CrossRef.
- Starpharma, DEP® cabazitaxel Phase 2 clinical trial completes enrolment and treatment (ASX Announcement), 2023. https://www.starpharma.com/news/story/dep-cabazitaxel-phase-2-clinical-trial-completes-enrolment-and-treatment-asx-announcement. (2023).
- B. Surekha, N. S. Kommana, S. K. Dubey, A. V. P. Kumar, R. Shukla and P. Kesharwani, PAMAM dendrimer as a talented multifunctional biomimetic nanocarrier for cancer diagnosis and therapy, Colloids Surf., B, 2021, 204, 111837 CrossRef CAS PubMed.
- H. Yang, Targeted nanosystems: Advances in targeted dendrimers for cancer therapy, Nanomedicine, 2016, 12(2), 309–316 CrossRef CAS PubMed.
- C. L. Waite, S. M. Sparks, K. E. Uhrich and C. M. Roth, Acetylation of PAMAM dendrimers for cellular delivery of siRNA, BMC Biotechnol., 2009, 9, 1–10 CrossRef PubMed.
- J. Wang, B. Li, L. Qiu, X. Qiao and H. Yang, Dendrimer-based drug delivery systems: history, challenges, and latest developments, J. Biol. Eng., 2022, 16(1), 18 CrossRef PubMed.
- E. Scarpa, J. L. Bailey, A. A. Janeczek, P. S. Stumpf, A. H. Johnston, R. O. C. Oreffo, Y. L. Woo, Y. C. Cheong, N. D. Evans and T. A. Newman, Quantification of intracellular payload release from polymersome nanoparticles, Sci. Rep., 2016, 6(1), 1–13 CrossRef PubMed.
- C. Katterman, C. Pierce and J. Larsen, Combining Nanoparticle Shape Modulation and Polymersome Technology in Drug Delivery, ACS Appl. Bio Mater., 2021, 4(4), 2853–2862 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, Polymersomes for drug delivery: Design, formation and characterization, J. Controlled Release, 2012, 161(2), 473–483 CrossRef CAS PubMed.
- F. Porta, D. Ehrsam, C. Lengerke and H. E. M. z. Schwabedissen, Synthesis and Characterization of PDMS–PMOXA-Based Polymersomes Sensitive to MMP-9 for Application in Breast Cancer, Mol. Pharm., 2018, 15(11), 4884–4897 CrossRef CAS PubMed.
- Y. Zou, Y. Sun, B. Guo, Y. Wei, Y. Xia, Z. Huangfu, F. Meng, J. C. M. v. Hest, J. Yuan and Z. Zhong, α3β1 Integrin-Targeting Polymersomal Docetaxel as an Advanced Nanotherapeutic for Nonsmall Cell Lung Cancer Treatment, ACS Appl. Mater. Interfaces, 2020, 12(13), 14905–14913 CrossRef CAS PubMed.
- L. Simón-Gracia, H. Hunt, P. D. Scodeller, J. Gaitzsch, G. B. Braun, A.-M. A. Willmore, E. Ruoslahti, G. Battaglia and T. Teesalu, Paclitaxel-Loaded Polymersomes for Enhanced Intraperitoneal Chemotherapy, Mol. Cancer Ther., 2016, 15(4), 670–679 CrossRef PubMed.
- H. E. Colley, V. Hearnden, M. Avila-Olias, D. Cecchin, I. Canton, J. Madsen, S. MacNeil, N. Warren, K. Hu, J. A. McKeating, S. P. Armes, C. Murdoch, M. H. Thornhill and G. Battagli, Polymersome-Mediated Delivery of Combination Anticancer Therapy to Head and Neck Cancer Cells: 2D and 3D in Vitro Evaluation, Mol. Pharm., 2014, 11(4), 1176–1188 CrossRef CAS PubMed.
- E. Rideau, F. R. Wurm and K. Landfester, Giant polymersomes from non-assisted film hydration of phosphate-based block copolymers, Polym. Chem., 2018, 9, 5385–5394 RSC.
- S. Matoori and J.-C. Leroux, Twenty-five years of polymersomes: lost in translation?, Mater. Horiz., 2020, 7, 1297–1309 RSC.
- M. Fonseca, I. Jarak, F. Victor, C. Domingues, F. Veiga and A. Figueiras, Polymersomes as the Next Attractive Generation of Drug Delivery Systems: Definition, Synthesis and Applications, Materials, 2024, 17(2), 319 CrossRef CAS PubMed.
- Y. Zhu, B. Yang, S. Chen and J. Du, Polymer vesicles: Mechanism, preparation, application, and responsive behavior, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, A holistic approach to targeting disease with polymeric nanoparticles, Nat. Rev. Drug Discovery, 2015, 14(4), 239–247 CrossRef CAS PubMed.
- W. R. Gombotz and D. K. Pettit, Biodegradable Polymers for Protein and Peptide Drug Delivery, Bioconjugate Chem., 1995, 6, 332–351 CrossRef CAS PubMed.
- D. Demir-Dora and F. Öner, Development and evaluation of polyethylenimine polyplexes as non-viral vectors for delivery of plasmid DNA encoding shRNA against STAT3 activity into triple negative breast cancer cells, J. Drug Delivery Sci. Technol., 2023, 82, 104113 CrossRef CAS.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Challenges in DNA Delivery and Recent Advances in Multifunctional Polymeric DNA Delivery Systems, Biomacromolecules, 2017, 18(8), 2231–2246 CrossRef CAS PubMed.
- S. B. Ebrahimi and D. Samanta, Engineering protein-based therapeutics through structural and chemical design, Nat. Commun., 2023, 14(1), 2411 CrossRef CAS PubMed.
- A. Zielinska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology, Molecules, 2020, 25(16), 3731 CrossRef CAS PubMed.
- Y. Bobde, S. Biswas and B. Ghosh, PEGylated N-(2 hydroxypropyl) methacrylamide-doxorubicin conjugate as pH-responsive polymeric nanoparticles for cancer therapy, React. Funct. Polym., 2020, 151, 104561 CrossRef CAS.
- X. Wang, Y. Chen, Y. Xiong, L. Zhang, B. Wang, Y. Liu and M. Cui, Design and Characterization of Squaramic Acid-Based Prostate-Specific Membrane Antigen Inhibitors for Prostate Cancer, J. Med. Chem., 2023, 66(10), 6889–6904 CrossRef CAS PubMed.
- Y. J. Ryu, S. Y. Lim, Y. M. Na, M. H. Park, S. Y. Kwon and J. S. Lee, Prostate-specific membrane antigen expression predicts recurrence of papillary thyroid carcinoma after total thyroidectomy, BMC Cancer, 2022, 22(1), 1278 CrossRef CAS PubMed.
- R. A. v. Eerden, R. H. Mathijssen and S. L. Koolen, Recent Clinical Developments of Nanomediated Drug Delivery Systems of Taxanes for the Treatment of Cancer, Int. J. Nanomed., 2020, 15, 8151–8166 CrossRef PubMed.
- H. He, L. Liu, E. E. Morin, M. Liu and A. Schwendeman, Survey of Clinical Translation of Cancer Nanomedicines - Lessons Learned from Successes and Failures, Acc. Chem. Res., 2019, 52(9), 2445–2461 CrossRef CAS PubMed.
- J. V. Nauman, P. G. Campbell, F. Lanni and J. L. Anderson, Diffusion of Insulin-Like Growth Factor-I and Ribonuclease through Fibrin Gels, Biophys. J., 2007, 92(12), 4444–4450 CrossRef CAS PubMed.
- C. I. C. Crucho and M. T. Barros, Polymeric nanoparticles: A study on the preparation variables and characterization methods, Mater. Sci. Eng., C, 2017, 80, 771–784 CrossRef CAS PubMed.
- U. C. Palmiero, M. Sponchioni, N. Manfredini, M. Maraldi and D. Moscatelli, Strategies to combine ROP with ATRP or RAFT polymerization for the synthesis of biodegradable polymeric nanoparticles for biomedical applications, Polym. Chem., 2018, 9(30), 4084–4099 RSC.
- S. Kumari, S. Raturi, S. Kulshrestha, K. Chauhan, S. Dhingra, K. András, K. Thu, R. Khargotra and T. Singh, A comprehensive review on various techniques used for synthesizing nanoparticles, J. Mater. Res. Technol., 2023, 27, 1739–1763 CrossRef CAS.
- P. Rafiei and A. Haddadi, A robust systematic design: Optimization and preparation of polymeric nanoparticles of PLGA for docetaxel intravenous delivery, Mater. Sci. Eng., C, 2019, 104, 109950 CrossRef CAS PubMed.
- M. Gao, Y. Yang, A. Bergfel, L. Huang, L. Zheng and T. M. Bowden, Self-assembly of cholesterol end-capped polymer micelles for controlled drug delivery, J. Nanobiotechnol., 2020, 18(1), 1–10 CrossRef PubMed.
- R. Maji, C. A. Omolo, N. Agrawal, K. Maduray, D. Hassan, C. Mokhtar, I. Mackhraj and T. Govender, pH-Responsive Lipid−Dendrimer Hybrid Nanoparticles: An Approach To Target and Eliminate Intracellular Pathogens, Mol. Pharm., 2019, 16(11), 4594–4609 CrossRef CAS PubMed.
- R. Martı-Centelles, J. Rubio-Magnieto and B. Escuder, A minimalistic catalytically-active cell mimetic made of a supra-molecular hydrogel encapsulated into a polymersome, Chem. Commun., 2020, 56(92), 14487–14490 RSC.
- A. Mohameda, N. K. Kundab, K. Rossa, G. A. Hutcheona and I. Y. Saleem, Polymeric nanoparticles for the delivery of miRNA to treat Chronic Obstructive Pulmonary Disease (COPD), Eur. J. Pharm. Biopharm., 2019, 136, 1–8 CrossRef PubMed.
- A. M. Pineda-Reyes, M. H. Delgado, M. d. l. L. Zambrano-Zaragoza, G. Leyva-Gomez, N. Mendoza-Munoz and D. Quintanar-Guerrero, Implementation of the emulsification-diffusion method by solvent displacement for polystyrene nanoparticles prepared from recycled material, RSC Adv., 2021, 11(4), 2226–2234 RSC.
- L. Miao, J. Su, X. Zhuo, L. Luo, Y. Kong, J. Gou, T. Yin, Y. Zhang, H. He and X. Tang, mPEG5k-b-PLGA2k/PCL3.4k/MCT Mixed Micelles as Carriers of Disulfiram for Improving Plasma Stability and Antitumor Effect in Vivo, Mol. Pharm., 2018, 15(4), 1556–1564 CrossRef CAS PubMed.
- M. B. David-Naim, E. Grad, G. Aizik, M. M. Nordling-David, O. Moshel, Z. Granot and G. Golomb, Polymeric nanoparticles of siRNA prepared by a double-emulsion solvent-diffusion technique: Physicochemical properties, toxicity, biodistribution and efficacy in a mammary carcinoma mice model, Biomaterials, 2017, 145, 154–167 CrossRef PubMed.
- K. Letchford and H. Burt, A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed.
- W. Ye, F. Zhu, Y. Cai, L. Wang, G. Zhang, G. Zhao, X. Chu, Q. Shuai and Y. Yan, Improved paclitaxel delivery with PEG-b-PLA/zein nanoparticles prepared via flash nanoprecipitation, Int. J. Biol. Macromol., 2022, 221, 486–495 CrossRef CAS PubMed.
- N. Zhang, Y. Fan, H. Chen, S. Trépout, A. Brûletd and M.-H. Li, Polymersomes with a smectic liquid crystal structure and AIE fluorescence, Polym. Chem., 2022, 13(8), 1107–1115 RSC.
- G. Bovone, E. A. Guzzi and M. W. Tibbitt, Flow-based reactor design for the continuous production of polymeric nanoparticles, AIChE J., 2019, 65(12), e16840 CrossRef CAS.
- S. Shingdilwar, S. Dolui and S. Banerjee, Facile Fabrication of Functional Mesoporous Polymer Nanospheres for CO2 Capture, Ind. Eng. Chem. Res., 2022, 61(2), 1140–1147 CrossRef CAS.
- Y.-L. Lo, X.-S. Huang, H.-Y. Chen, Y.-C. Huang, Z.-X. Liao and L.-F. Wang, ROP and ATRP fabricated redox sensitive micelles based on PCL-SS-PMAA diblock copolymers to co-deliver PTX and CDDP for lung cancer therapy, Colloids Surf., B, 2021, 198, 111443 CrossRef CAS PubMed.
- A. Kumar, S. V. Lale, S. Mahajan, V. Choudhary and V. Koul, ROP and ATRP Fabricated Dual Targeted Redox Sensitive Polymersomes Based on pPEGMA-PCL-ss-PCL-pPEGMA Triblock Copolymers for Breast Cancer Therapeutics, ACS Appl. Mater. Interfaces, 2015, 7(17), 9211–9227 CrossRef CAS PubMed.
- C. Nehate, A. A. M. Raynold and V. Koul, ATRP Fabricated and Short Chain Polyethylenimine Grafted Redox Sensitive Polymeric Nanoparticles for Codelivery of Anticancer Drug and siRNA in Cancer Therapy, ACS Appl. Mater. Interfaces, 2017, 9(45), 39672–39687 CrossRef CAS PubMed.
- P. Sun, Y. Wang, S. Yang, X. Sun, B. Peng, L. Pan, Y. Jia, X. Zhang and C. Nie, Molecularly Imprinted Polymer Nanospheres with Hydrophilic Shells for Efficient Molecular Recognition of Heterocyclic Aromatic Amines in Aqueous Solution, Molecules, 2023, 28(5), 2052 CrossRef CAS PubMed.
- S. E. Ahmed, N. L. Fletcher, A. R. Prior, P. Huda, C. A. Bell and K. J. Thurecht, Development of targeted micelles and polymersomes prepared from degradable RAFT-based diblock copolymers and their potential role as nanocarriers for chemotherapeutics, Polym. Chem., 2022, 13, 4004 RSC.
- M. Banaei and M. Salami-Kalajahi, Synthesis of poly(2-hydroxyethyl methacrylate)-grafted poly(aminoamide) dendrimers as polymeric nanostructures, Colloid Polym. Sci., 2015, 293, 1553–1559 CrossRef CAS.
- A. B. Cook, R. Peltier, M. Hartlieb, R. Whitfield, G. Moriceau, J. A. Burns, D. M. Haddleton and S. Perrier, Cationic and hydrolysable branched polymers by RAFT for complexation and controlled release of dsRNA, Polym. Chem., 2018, 9, 4025 RSC.
- P. Michael, Y. T. Lam, E. C. Filipe, R. P. Tan, A. H. P. Chan, B. S. L. Lee, N. Feng, J. Hung, T. R. Cox, M. Santos and S. G. Wise, Plasma polymerized nanoparticles effectively deliver dual siRNA and drug therapy in vivo, Sci. Rep., 2020, 10(1), 1–14 CrossRef PubMed.
- C. N. Cheaburu-Yilmaz, H. Y. Karasulu and O. Yilmaz, Nanoscaled dispersed systems used in drug-delivery applications, Polym. Nanomater. Nanother., 2019, 437–468 CAS.
- N. Mendoza-Muñoz, S. Alcalá-Alcalá and D. Quintanar-Guerrero, Preparation of Polymer Nanoparticles by the Emulsification-Solvent Evaporation Method: From Vanderhoff's Pioneer Approach to Recent Adaptations, Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development, Springer, Cham, 2016, pp. 87–121 Search PubMed.
- D. Quintanar-Guerrero, E. Allemann, E. Doelker and H. Fessi, A mechanistic study of the formation of polymer nanoparticles by the emulsification-diffusion technique, Colloid Polym. Sci., 1997, 275(7), 640–647 CrossRef CAS.
- Y. N. Konan, R. Gurny and E. Allemann, Preparation and characterization of sterile and freeze-dried sub-200 nm nanoparticles, Int. J. Pharm., 2002, 233(1–2), 239–252 CrossRef CAS PubMed.
- S. Galindo-Rodriguez, E. Allémann, H. Fessi and E. Doelker, Physicochemical Parameters Associated with Nanoparticle Formation in the Salting-Out, Emulsification-Diffusion, and Nanoprecipitation Methods, Pharm. Res., 2004, 21, 1428–1439 CrossRef CAS PubMed.
- B. Kupikowska-Stobba and M. Kasprzak, Fabrication of nanoparticles for bone regeneration: new insight into applications of nanoemulsion technology, J. Mater. Chem. B, 2021, 9(26), 5221–5244 RSC.
- X. Yan, J. Bernard and F. Ganachaud, Nanoprecipitation as a simple and straightforward process to create complex polymeric colloidal morphologies, Adv. Colloid Interface Sci., 2021, 294, 102474 CrossRef CAS PubMed.
- U. Bilati, E. Allémann and E. Doelker, Nanoprecipitation versus emulsion-based techniques for the encapsulation of proteins into biodegradable nanoparticles and process-related stability issues, AAPS PharmSciTech, 2005, 6(4), E594–E604 CrossRef PubMed.
- A. Alshamsan, Nanoprecipitation is more efficient than emulsion solvent evaporation method to encapsulate cucurbitacin I in PLGA nanoparticles, Saudi Pharm. J., 2014, 22(3), 219–222 CrossRef PubMed.
- L. Silverman, G. Bhatti, J. E. Wulff and M. G. Moffitt, Improvements in Drug-Delivery Properties by Co-Encapsulating Curcumin in SN-38-Loaded Anticancer Polymeric Nanoparticles, Mol. Pharm., 2022, 19(6), 1866–1881 CrossRef CAS PubMed.
- K. M. Pustulka, A. R. Wohl, H. S. Lee, A. R. Michel, J. Han, T. R. Hoye, A. V. McCormick, J. Panyam and C. W. Macosko, Flash Nanoprecipitation: Particle Structure and Stability, Mol. Pharm., 2013, 10(11), 4367–4377 CrossRef CAS PubMed.
- K. Matyjaszewski, Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives, Macromolecules, 2012, 45(10), 4015–4039 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, A comparison of RAFT and ATRP methods for controlled radical polymerization, Nat. Rev. Chem., 2021, 5(12), 859–869 CrossRef CAS PubMed.
- J. Ran, L. Wu, Z. Zhang and T. Xu, Atom transfer radical polymerization (ATRP): A versatile and forceful tool for functional membranes, Prog. Polym. Sci., 2014, 39(1), 124–144 CrossRef CAS.
- M. Mirzaeinejad, Y. Mansoori and B. Koohi-Zargar, New acrylamide-based monomer containing metal chelating units: Homopolymer grafted magnetite nanoparticles via ATRP for the magnetic removal of Co(II) ions, Polym. Adv. Technol., 2017, 29(4), 1206–1218 CrossRef.
- G. B. Desmet, N. D. Rybel, P. H. M. V. Steenberge, D. R. D′hooge, M.-F. Reyniers and G. B. Marin, Ab initio-Based Kinetic Modeling to Understand RAFT Exchange: The Case of 2-Cyano-2-Propyl Dodecyl Trithiocarbonate and Styrene, Macromol. Rapid Commun., 2017, 39(2), 1700403 CrossRef PubMed.
- V. K. Patel, N. K. Vishwakarma, A. K. Mishra, C. S. Biswas and B. Ray, (S)-2-(ethyl propionate)-(O-ethyl xanthate)- and (S)-2-(Ethyl isobutyrate)-(O-ethyl xanthate)-mediated RAFT polymerization of vinyl acetate, J. Appl. Polym. Sci., 2012, 125(4), 2946–2955 CrossRef CAS.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Progress and Perspectives Beyond Traditional RAFT Polymerization, Adv. Sci., 2020, 7(20), 2001656 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, RAFT Polymerization and Some of its Applications, Chem. – Asian J., 2013, 8(8), 1634–1644 CrossRef CAS PubMed.
- V. Hasirci, P. Y. Huri, T. E. Tanir, G. Eke and N. Hasirci, Polymer Fundamentals: Polymer Synthesis, in Comprehensive Biomaterials II, ed. P. Ducheyne, Elsevier, 2017, pp. 478–506 Search PubMed.
- F. Bensebaa, Wet production methods, Interface Sci. Technol., 2013, 19, 85–146 Search PubMed.
- K. Kollbek, P. Jabłoński, M. Perzanowski, D. Święch, M. Sikora, G. Słowik, M. Marzec, M. Gajewska, C. Paluszkiewiczc and M. Przybylski, Inert gas condensation made bimetallic FeCu nanoparticles – plasmonic response and magnetic ordering, J. Mater. Chem. C, 2024, 12(7), 2593–2605 RSC.
- M. Fernández-Arias, M. Boutinguiza, J. d. Val, A. Riveiro, D. Rodríguez, F. Arias-González, J. Gil and J. Pou, Fabrication and Deposition of Copper and Copper Oxide Nanoparticles by Laser Ablation in Open Air, Nanomaterials, 2020, 10(8), 300 CrossRef PubMed.
- L. Cui, X. Ren, J. Wang and M. Sun, Synthesis of homogeneous carbon quantum dots by ultrafast dual-beam pulsed laser ablation for bioimaging, Mater. Today Nano, 2020, 12, 100091 CrossRef.
- R. Kumar, R. Pandey, E. Joanni and R. Savu, Laser-induced and catalyst-free formation of graphene materials for energy storage and sensing applications, Chem. Eng. J., 2024, 497, 154968 CrossRef CAS.
- N. Patelli, F. Cugini, D. Wang, S. Sanna, M. Solzi, H. Hahn and L. Pasquini, Structure and magnetic properties of Fe-Co alloy nanoparticles synthesized by pulsed-laser inert gas condensation, J. Alloys Compd., 2022, 890, 161863 CrossRef CAS.
- V. Piotto, L. Litti and M. Meneghetti, Synthesis and Shape Manipulation of Anisotropic Gold Nanoparticles by Laser Ablation in Solution, J. Phys. Chem. C, 2020, 124(8), 4820–4826 CrossRef CAS.
- H.-Y. Park, I. Jang, N. Jung, Y.-H. Chung, J. Ryu, I. Y. Cha, H.-J. Kim, J. H. Jang and S. J. Yoo, Green synthesis of carbon-supported nanoparticle catalysts by physical vapor deposition on soluble powder substrates, Sci. Rep., 2015, 5(1), 14245 CrossRef PubMed.
- M. Malekzadeh, P. Rohani, Y. Liu, A. Raszewski, F. Ghanei and M. T. Swihart, Laser pyrolysis synthesis of zinc-containing nanomaterials using low-cost ultrasonic spray delivery of precursors, Powder Technol., 2020, 376, 104–112 CrossRef CAS.
- A. Hassani, M. Karaca, S. Karaca, A. Khataee, Ö. Açışlı and B. Yılmaz, Preparation of magnetite nanoparticles by high-energy planetary ball mill and its application for ciprofloxacin degradation through heterogeneous Fenton process, J. Environ. Manage., 2018, 211, 53–62 CrossRef CAS PubMed.
- M. Zhianmanesh, A. Gilmour, M. M. M. Bilek and B. Akhavan, Plasma surface functionalization: A comprehensive review of advances in the quest for bioinstructive materials and interfaces, Appl. Phys. Rev., 2023, 10(2), 021301 CAS.
- A. H. Shaker, K. A. Aadim and M. H. Nida, Spectroscopic analysis of zinc plasma produced by alternating and direct current jet, J. Opt., 2024, 53, 1273–1281 CrossRef.
- B. Akhavan, K. Jarvis and P. Majewski, Development of Oxidized Sulfur Polymer Films through a Combination of Plasma Polymerization and Oxidative Plasma Treatment, Langmuir, 2014, 30(5), 1444–1454 CrossRef CAS PubMed.
- B. Akhavan, S. G. Wise and M. M. M. Bilek, Substrate-Regulated Growth of Plasma-Polymerized Films on Carbide-Forming Metals, Langmuir, 2016, 32(42), 10835–10843 CrossRef CAS PubMed.
- B. Akhavan, M. Croes, S. G. Wise, C. Zhai, J. Hung, C. Stewart, M. Ionescu, H. Weinans, Y. Gan, S. A. Yavari and M. M. M. Bilek, Radical-functionalized plasma polymers: Stable biomimetic interfaces for bone implant applications, Appl. Mater. Today, 2019, 16, 456–473 CrossRef.
- C. A. C. Stewart, B. Akhavan, M. Santos, J. Hung, C. L. Hawkins, S. Bao, S. G. Wise and M. M. M. Bilek, Cellular responses to radical propagation from ion-implanted plasma polymer surfaces, Appl. Surf. Sci., 2018, 456, 701–710 CrossRef CAS.
- O. Sharifahmadian, C. Zhai, J. Hung, G. Shineh, C. A. C. Stewart, A. A. Fadzil, M. Ionescu, Y. Gan, S. G. Wise and B. Akhavan, Mechanically robust nitrogen-rich plasma polymers: Biofunctional interfaces for surface engineering of biomedical implants, Mater. Today Adv., 2021, 12, 100188 CrossRef CAS.
- P. Pleskunov, D. Nikitin, R. Tafiichuk, A. Shelemin, J. Hanuš, I. Khalakhan and A. Choukourov, Carboxyl-Functionalized Nanoparticles Produced by Pulsed Plasma Polymerization of Acrylic Acid, J. Phys. Chem. B, 2018, 122(14), 4187–4194 CrossRef CAS PubMed.
- P. Pleskunov, D. Nikitin, R. Tafiichuk, A. Shelemin, J. Hanus, J. Kousal, Z. Krtous, I. Khalakhan, P. Kus, T. Nasu, T. Nagahama, C. Funaki, H. Sato, M. Gawek, A. Schoenhals and A. Choukourov, Plasma Polymerization of Acrylic Acid for the Tunable Synthesis of Glassy and Carboxylated Nanoparticles, J. Phys. Chem. B, 2020, 124, 668–678 CrossRef CAS PubMed.
- Y. T. Lam, B. S. L. Lee, J. Hung, P. Michael, M. Santos, R. P. Tan, R. Liu and S. G. Wise, Delivery of Therapeutic miRNA via Plasma-Polymerised Nanoparticles Rescues Diabetes-Impaired Endothelial Function, Nanomaterials, 2023, 13(16), 2360 CrossRef CAS PubMed.
- H. Yasuda, Plasma polymerization, Academic Press, 1985 Search PubMed.
- H. Kobayashi, A. T. Bell and M. Shen, Formation of an Amorphous Powder During the Polymerization of Ethylene in a Radio-Frequency Discharge, J. Appl. Polym. Sci., 1973, 17, 885–892 CrossRef CAS.
- H. Kobayashi, A. T. Bell and M. Shen, Plasma Polymerization of Saturated and Unsaturated Hydrocarbons, Macromolecules, 1974, 7(3), 277–283 CrossRef CAS.
- R. Rudda, A. Obrusníkb, P. Zikánb, C. Halla, P. Murphya, D. Evansa and E. Charrault, Plasma gas aggregation cluster source: Influence of gas inlet configuration and total surface area on the heterogeneous aggregation of silicon clusters, Surf. Coat. Technol., 2019, 364, 1–6 CrossRef.
- A. Choukourov, P. Pleskunov, D. Nikitin, V. Titov, A. Shelemin, M. Vaidulych, A. Kuzminova, P. Solař, J. Hanuš, J. Kousal, O. Kylián, D. Slavínská and H. Biederman, Advances and challenges in the field of plasma polymer nanoparticles, Beilstein J. Nanotechnol., 2017, 8(1), 2002–2014 Search PubMed.
- O. Kylián, A. Shelemin, P. Solar, P. Pleskunov, D. Nikitin, A. Kuzminova, R. Štefaníková, P. Kúš, M. Cieslar, J. Hanuš, A. Choukourov and H. Biederman, Magnetron Sputtering of Polymeric Targets: From Thin Films to Heterogeneous Metal/Plasma Polymer Nanoparticles, Materials, 2019, 12(15), 2366 CrossRef PubMed.
- M. Vasquez-Ortega, M. Ortega, J. Morales, M. G. Olayo, G. J. Cruz and R. Olayo, Core–shell polypyrrole nanoparticles obtained by atmospheric pressure plasma polymerization, Polym. Int., 2014, 63(12), 2023–2029 CrossRef CAS.
- B. Hu, X.-L. Shi, T. Cao, M. Li, W. Chen, W.-D. Liu, W. Lyu, T. Tesfamichael and Z.-G. Chen, Advances in flexible thermoelectric materials and devices fabricated by magnetron sputtering, Small Sci., 2023, 2300061 Search PubMed.
- Q. Ren, L. Qin, F. Jing, D. Cheng, Y. Wang, M. Yang, D. Xie, Y. Leng, B. Akhavan and N. Huang, Reactive magnetron co-sputtering of Ti-xCuO coatings: Multifunctional interfaces for blood-contacting devices, Mater. Sci. Eng., C, 2020, 116, 111198 Search PubMed.
- R. Ganesan, B. Akhavan, M. A. Hiob, D. R. McKenzie, A. S. Weiss and M. M. M. Bilek, HiPIMS carbon coatings show covalent protein binding that imparts enhanced hemocompatibility, Carbon, 2018, 139, 118–128 CrossRef CAS.
- Z. Ye, Y. Bao, Z. Chen, H. Ye, Z. Feng, Y. Li, Y. Zeng, Z. Pan, D. Ouyang, K. Zhang, X. Liu and Y. He, Recent advances in the metal/organic hybrid nanomaterials for cancer theranostics, Coord. Chem. Rev., 2024, 504, 215654 CrossRef CAS.
- E. S. Austria, R. B. Lamorena and S. D. Arco, Synthesis and functionalization of magnetic nanoparticles for glycoprotein and glycopeptide enrichment: A review, Microchem. J., 2024, 201, 110663 Search PubMed.
- D. Tao, Y. Tang, B. Zou and Y. Wang, Mesoporous Magnetic/Polymer Hybrid Nanoabsorbent for Rapid and Efficient Removal of Heavy Metal Ions from Wastewater, Langmuir, 2024, 40(5), 2773–2780 CrossRef CAS PubMed.
- M. Santos, B. Reeves, P. Michael, R. Tan, S. G. Wise and M. M. M. Bilek, Substrate geometry modulates self-assembly and collection of plasma polymerized nanoparticles, Commun. Phys., 2019, 2(1), 1–11 CrossRef.
- E. Delikonstantis, F. Cameli, M. Scapinello, V. Rosa, K. M. V. Geem and G. D. Stefanidis, Low-carbon footprint chemical manufacturing using plasma technology, Curr. Opin. Chem. Eng., 2022, 38, 100857 CrossRef.
- I. B. Denysenko, E. v. Wahl, M. Mikikian, J. Berndt, S. Ivko, H. Kersten, E. Kovacevic and N. A. Azarenkov, Plasma properties as function of time in Ar/C2H2 dust-forming plasma, J. Phys. D: Appl. Phys., 2020, 53(13), 135203 CrossRef CAS.
- B. Nisol, S. Watson, S. Lerouge and M. R. Wertheimer, Energetics of Reactions in a Dielectric Barrier Discharge with Argon Carrier Gas: III Esters, Plasma Processes Polym., 2016, 13(9), 900–907 CrossRef CAS.
- J. Weerasinghe, W. Li, R. Zhou, R. Zhou, A. Gissibl, P. Sonar, R. Speight, K. Vasilev and K. K. Ostrikov, Bactericidal Silver Nanoparticles by Atmospheric Pressure Solution Plasma Processing, Nanomaterials, 2020, 10(5), 874 CrossRef CAS PubMed.
- S. A. Al-Bataineh, A. A. Cavallaro, A. Michelmore, M. N. Macgregor, J. D. Whittle and K. Vasilev, Deposition of 2-oxazoline-based plasma polymer coatings using atmospheric pressure helium plasma jet, Plasma Processes Polym., 2019, 16(10), 1900104 CrossRef.
- Z. Chen, G. Chen, R. Obenchain, R. Zhang, F. Bai, T. Fang, H. Wang, Y. Lu, R. E. Wirz and Z. Gu, Cold atmospheric plasma delivery for biomedical applications, Mater. Today, 2022, 54, 153–188 CrossRef CAS.
- R. Brandenburg, Dielectric barrier discharges: progress on plasma sources and on the understanding of regimes and single filaments, Plasma Sources Sci. Technol., 2017, 26(5), 053001 CrossRef.
- G. J. Cruz, M. G. Olayo, O. G. López, L. M. Gómez, J. Morales and R. Olayo, Nanospherical particles of polypyrrole synthesized and doped by plasma, Polymer, 2010, 51(19), 4314–4318 CrossRef CAS.
- C. T. H. Tran, A. D. Gilmour, B. B. Boumelhem, S. T. Fraser and M. M. M. Bilek, Plasma-Activated Coated Glass: A Novel Platform for Optimal Optical Performance and Cell Culture Substrate Customization, Small Sci., 2024, 4(4), 2300228 CrossRef CAS PubMed.
- D. Hegemann, B. Nisol, S. Gaiser, S. Watson and M. R. Wertheimer, Energy Conversion Efficiency in Low- and Atmospheric-Pressure Plasma Polymerization Processes with Hydrocarbons, Phys. Chem. Chem. Phys., 2019, 21(17), 8698–8708 RSC.
- M. Asadian, K. V. Chan, T. Egghe, Y. Onyshchenko, S. Grande, H. Declercq, P. Cools, R. Morent and N. D. Geyter, Acrylic acid plasma polymerization and post-plasma ethylene diamine grafting for enhanced bone marrow mesenchymal stem cell behaviour on polycaprolactone nanofibers, Appl. Surf. Sci., 2021, 563, 150363 CrossRef CAS.
- B. Akhavan, B. Menges and R. Förch, Inhomogeneous Growth of Micrometer Thick Plasma Polymerized Films, Langmuir, 2016, 32(19), 4792–4799 CrossRef CAS PubMed.
- S. M. Palardonio, J. Magdaleno and R. Vasquez, Deposition and morphology of direct current plasma-polymerized aniline, J. Vac. Sci. Technol., B:Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 2023, 41(3), 034201 CAS.
- D. Hegemann, E. Bülbül, B. Hanselmann, U. Schütz, M. Amberg and S. Gaiser, Plasma polymerization of hexamethyldisiloxane: Revisited, Plasma Processes Polym., 2020, 18(2), 2000176 CrossRef.
- B. Akhavan, K. Jarvis and P. Majewski, Evolution of Hydrophobicity in Plasma Polymerised 1,7-Octadiene Films, Plasma Processes Polym., 2013, 10(11), 1018–1029 CrossRef CAS.
- B. Akhavan, K. Jarvis and P. Majewski, Hydrophobic Plasma Polymer Coated Silica Particles for Petroleum Hydrocarbon Removal, ACS Appl. Mater. Interfaces, 2013, 5(17), 8563–8571 CrossRef CAS PubMed.
- B. Akhavan, K. Jarvis and P. Majewski, Tuning the hydrophobicity of plasma polymer coated silica particles, Powder Technol., 2013, 249, 403–411 CrossRef CAS.
- B. Akhavan, K. Jarvis and P. Majewski, Plasma polymerization of sulfur-rich and water-stable coatings on silica particles, Surf. Coat. Technol., 2015, 264, 72–79 CrossRef CAS.
- B. Akhavan, K. Jarvis and P. Majewski, Development of negatively charged particulate surfaces through a dry plasma-assisted approach, RSC Adv., 2015, 5(17), 12910–12921 RSC.
- P. A. F. Herbert, L. O'Neill, J. Jaroszyńska-Wolińska, C. P. Stallard, A. Ramamoorthy and D. P. Dowling, A Comparison between Gas and Atomized Liquid Precursor States in the Deposition of Functional Coatings by Pin Corona Plasma, Plasma Processes Polym., 2011, 8(3), 230–238 CrossRef CAS.
- E. Sainz-García, F. Alba-Elías, R. Múgica-Vidal and M. Pantoja-Ruiz, Promotion of tribological and hydrophobic properties of a coating on TPE substrates by atmospheric plasma-polymerization, Appl. Surf. Sci., 2016, 371, 50–60 CrossRef.
- C. W. Karl, W. Rahimi, S. Kubowicz, A. Lang, H. Geisler and U. Giese, Surface Modification of Ethylene Propylene Diene Terpolymer Rubber by Plasma Polymerization Using Organosilicon Precursors, ACS Appl. Polym. Mater., 2020, 2(9), 3789–3796 CrossRef CAS.
- C. Pattyn, E. Kovacevic, S. Hussain, A. Dias, T. Lecas and J. Bernd, Nanoparticle formation in a low pressure argon/aniline RF plasma, Appl. Phys. Lett., 2018, 112(1), 013102 CrossRef.
- J. Friedrich, Mechanisms of Plasma Polymerization – Reviewed from a Chemical Point of View, Plasma Processes Polym., 2011, 8(9), 783–802 CrossRef CAS.
- H. Yasuda and Y. Matsuzawa, Economical Advantages of Low-Pressure Plasma Polymerization Coating, Plasma Processes Polym., 2005, 2(6), 507–512 CrossRef CAS.
- L. Boufendi, M. C. Jouanny, E. Kovacevic, J. Berndt and M. Mikikian, Dusty plasma for nanotechnology, J. Phys. D: Appl. Phys., 2011, 44(17), 174035 CrossRef.
- B. Chutia, T. Deka, Y. Bailung, S. K. Sharma and H. Bailung, A nanodusty plasma experiment to create extended dust clouds using reactive argon acetylene plasmas, Phys. Plasmas, 2021, 28(6), 063703 CrossRef CAS.
- C. Hollenstein, C. Deschenaux, D. Magni, F. Grangeon, A. Affolter, A. A. Howling and P. Fayet, On the powder formation in industrial reactive RF plasmas, Front. Dusty Plasmas, 2000, 169–176 CAS.
- A. Bouchoule, Dusty plasmas, Phys. World, 1993, 6(8), 47 CrossRef CAS.
- S. V. Vladimirov and K. Ostrikov, Self-Organization and Dynamics of Nanoparticles in Chemically Active Plasmas for Low-Temperature Deposition of Silicon and Carbon-Based Nanostructured Films, Plasmas Polym., 2003, 8, 135–152 CrossRef CAS.
- Y. Chen, Y. Huang, Q. Li, Z. Luo, Z. Zhang, H. Huang, J. Sun, L. Zhang, R. Sun, D. J. Bain, J. F. Conway, B. Lu and S. Li, Targeting Xkr8 via nanoparticle-mediated in situ co-delivery of siRNA and chemotherapy drugs for cancer immunochemotherapy, Nat. Nanotechnol., 2023, 18, 193–204 CrossRef CAS PubMed.
- M. Zhang, X. Zhang, S. Huang, Y. Cao, Y. Guo and L. Xu, Programmed nanocarrier loaded with paclitaxel and dual-siRNA to reverse chemoresistance by synergistic therapy, Int. J. Biol. Macromol., 2024, 261, 129726 CrossRef CAS PubMed.
- X. Wang, J. Shi, M. Huang, J. Chen, J. Dan, Y. Tang, Z. Guo, X. He and Q. Zhao, TUBB2B facilitates progression of hepatocellular carcinoma by regulating cholesterol metabolism through targeting HNF4A/CYP27A1, Cell Death Dis., 2023, 14(3), 179 CrossRef CAS PubMed.
- D. Shin, J. Park, D. Han, J. H. Moon, H. S. Ryu and Y. Kim, Identification of TUBB2A by quantitative proteomic analysis as a novel biomarker for the prediction of distant metastatic breast cancer, Clin. Proteomics, 2020, 17, 1–19 CrossRef PubMed.
- Y. Zhou, X. He, W. Zhang, W. Zhang, H. Zhao, X. Zhou, Q. Gu, H. Shen, H. Yang, X. Liu, L. Huang and Q. Shi, Cell-recruited microspheres for OA treatment by dual-modulating inflammatory and chondrocyte metabolism, Mater. Today Bio, 2024, 27, 101127 CrossRef CAS PubMed.
- L. Khalef, R. Lydia, K. Filicia and B. Moussa, Cell viability and cytotoxicity assays: Biochemical elements and cellular compartments, Cell Biochem. Funct., 2024, 42(3), e4007 CrossRef CAS PubMed.
- F. Bonnier, M. E. Keating, T. P. Wróbel, K. Majzner, M. Baranska, A. Garcia-Munoz, A. Blanco and H. J. Byrne, Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models, Toxicol. in Vitro, 2015, 29(1), 124–131 CrossRef CAS PubMed.
- M. Sajid, S. Mehmood, Y. Yuan, T. Yue, M. Z. Khalid, A. Mujtaba, S. A. Alharbi, M. J. Ansari, A. Zinedine and J. M. Rocha, Biosafety measures for Alicyclobacillus spp. strains across various levels of biohazard, Food Chem. Toxicol., 2024, 191, 114840 CrossRef CAS PubMed.
- X. Li, Y. Zhao, J. Yin and W. Lin, Organic fluorescent probes for detecting mitochondrial membrane potential, Coord. Chem. Rev., 2020, 420, 213419 CrossRef CAS.
- S. K. Filippov, R. Khusnutdinov, A. Murmiliuk, W. Inam, L. Y. Zakharova, H. Zhang and V. V. Khutoryanskiy, Dynamic light scattering and transmission electron microscopy in drug delivery: a roadmap for correct characterization of nanoparticles and interpretation of results, Mater. Horiz., 2023, 10(12), 5354–5370 RSC.
- S. Zhang and C. Wang, Effect of stirring speed on particle dispersion in silica synthesis, Nano-Struct. Nano-Objects, 2023, 35, 100994 CrossRef CAS.
- S. B. Oudih, D. Tahtat, A. N. Khodja, M. Mahlous, Y. Hammache, A.-E. Guittoum and S. K. Gana, Chitosan nanoparticles with controlled size and zeta potential, Polym. Eng. Sci., 2023, 63(3), 1011–1021 CrossRef.
- Z. Németh, I. Csóka, R. S. Jazani, B. Sipos, H. Haspel, G. Kozma, Z. Kónya and D. G. Dobó, Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives, Pharmaceutics, 2022, 14(9), 1798 CrossRef PubMed.
- A. Carone, S. Emilsson, P. Mariani, A. Désert and S. Parola, Gold nanoparticle shape dependence of colloidal stability domains, Nanoscale Adv., 2023, 5(7), 2017–2026 RSC.
- T. Zuo, Y. Liu, M. Duan, X. Pu, M. Huang, D. Zhang and J. Xie, Platelet-derived growth factor PDGF-AA upregulates connexin 43 expression and promotes gap junction formations in osteoblast cells through p-Akt signaling, Biochem. Biophys. Rep., 2023, 34, 101462 CAS.
- S. Thavapalachandran, S. M. Grieve, R. D. Hume, T. Y. L. Le, K. Raguram, J. E. Hudson, J. Pouliopoulos, G. A. Figtree, R. P. Dye, A. M. Barry, P. Brown, J. Lu, S. Coffey, S. H. Kesteven, R. J. Mills, F. N. Rashid, E. Taran, P. Kovoor, L. Thomas, A. R. Denniss, E. Kizana, N. S. Asli, M. Xaymardan, M. P. Feneley, R. M. Graham, R. P. Harvey and J. J. H. Chong, Platelet-derived growth factor-AB improves scar mechanics and vascularity after myocardial infarction, Sci. Transl. Med., 2020, 12(524), eaay2140 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |