
Recent progress in the electrocatalytic applications of thiolate-protected metal nanoclusters
Yuting
Ye
and
Qing
Tang
 *
*
School of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
First published on 3rd May 2025
Abstract
Ultrasmall metal nanoclusters (NCs) with atomic precision possess a size range between individual atoms and plasmonic nanomaterials. These atomically precise materials represent an emerging class of nanocatalysts, offering unique opportunities to explore electrocatalytic properties and establish precise structure–property correlations at the atomic scale. Among the large number of metal NCs that are stabilized by various ligands, thiolate-protected metal NCs are a particularly prominent class for electrocatalytic investigations. Recent experimental and theoretical studies have demonstrated the significant potential of these materials in enhancing various electrocatalytic reactions, including hydrogen evolution, oxygen reduction and CO2 reduction reactions. However, comprehensive and in-depth discussions regarding their catalytic properties, particularly from a theoretical standpoint, are limited and require further explorations. In this review, we focus on the recent progress in thiolate-protected metal NCs in the field of electrocatalysis. The influences of structure, ligand, doping and interface control on their electrocatalytic activity/selectivity and the reaction mechanisms are discussed. Importantly, the perspectives we propose regarding future research endeavors are expected to offer valuable references for subsequent investigations in this area.
Introduction
Catalysis plays a significant role in various chemical reactions within the chemical, pharmaceutical, and agricultural industries,1 and it can be generally classified into heterogeneous catalysis, homogeneous catalysis, and biological enzyme catalysis.2 Among them, heterogeneous catalysis has garnered particular interest from researchers.3,4 However, major drawbacks of heterogeneous catalysts are their typical non-uniform morphology and distribution of active sites, which complicate the reactions and lead to product inconsistency. Therefore, the development of heterogeneous catalysts with well-defined structures and uniform active sites is crucial for advancing the understanding of their chemical processes. Heterogeneous catalysis relies exclusively on nanoparticle (NP) catalysts since they present a larger specific surface area to improve catalytic efficiency.5 Compared with conventional NPs, ligand-protected, atomically precise metal nanoclusters (NCs) have attracted significant attention recently as a novel class of nanocatalyst; their ultra-small size and well-defined atomic structures provide more advantageous platforms for investigating the actual active sites and reaction mechanisms of electrocatalysis at the atomic or molecular scale.6–12 Their ultra-small size allows them to exhibit unique electronic and optical properties, such as molecule-like energy gaps, strong photoluminescence (PL) and high catalytic properties.13–15 Moreover, the atomic-level precision of the metal clusters, owing to the ability to grow single crystals from their unique nanoparticles and to resolve their crystal structures by X-ray crystallography, could establish structure–property correlation by the close interplay between theory and experiment.8 The outermost ligands not only prevent aggregation and facilitate the separation of metal NCs but also enable the chemical functionalization of NCs that will generate interesting interface and catalytic properties.16,17Among the various stabilizing ligands, thiolates are the most widely studied ligands for NC protection, and numerous Mn(SR)m compositions, where n and m denote the numbers of metal atoms and thiolate (SR) ligands, respectively,18–20 have been successfully synthesized and crystallographically resolved using single-crystal X-ray diffraction, which holds great opportunities for fundamental catalysis research.21–28 Au, Ag, and Cu, as the three major atomically precise nanoclusters, have become research hotspots in recent years compared with Pt NCs, which must be synthesized and processed under CO or inert gas atmosphere; in addition, currently, the chemical composition of Pt NCs cannot be easily controlled with atomic precision.13,29,30 Recently, numerous comprehensive reviews have summarized the recent advances in emerging atomically precise metal NCs, thiolated gold and silver NCs in particular, in experimental syntheses and various applications in chirality, magnetism, luminescence, biological applications and catalysis.8,17,31–33 However, a systematic understanding or discussion of the correlations between the catalytic properties and electronic or interfacial structure of metal NCs, especially from the theoretical perspective, has been lacking.34–38 In this review, we focus specifically on the recent advances in the thiolate-protected atomically precise metal NCs in electrocatalysis applications. Specifically, we discuss (1) the bonding of thiolate ligands in metal NCs; (2) explore the electrocatalytic applications of metal NCs in important electrochemical processes, such as hydrogen evolution reaction (HER), oxygen reduction reaction (ORR), and carbon dioxide reduction reaction (CO2RR); and (3) conclude with some key insights and perspectives.
Bonding of thiolate ligands in metal NCs
As is well known, ligands play a critical role in stabilizing and functionalizing metal NCs.34,39 A considerable number of ligands have been reported to coordinate with metal NCs; however, there are few systematic comparisons of their binding strengths to the central atom. Tang and Jiang39 investigated the interface and adsorption strength of different types of ligands on the Au(111) surfaces using density functional theory (DFT) calculations. They found that the ligand/Au(111) binding strength follows the overall order bulky N-heterocyclic carbenes (NHCs) ≈ alkynyls > thiolates > phosphines > aryl radicals > alkylamines. The thiolates can provide a relatively stable interface bonding owing to the formation of strong covalent Au–S bonds. Note that the interface bonding of ligands depends highly on the metal type and the specific facets of the metal surfaces. Hu et al.40 theoretically examined the gold–thiolate interface and revealed that a staple motif (–RS–Au–SR–) is thermodynamically preferred on Au(111), while a bridging motif (–RS–) is favored on Au(100) and Au(110). This finding explains the experimentally observed bridging motif on the Au92(SR)44 cluster with planar (100) facets and provides insights into predicting the protective motifs of small fcc gold nanoparticles. Sun et al.41 compared the adsorption strength of the SR ligand on different coinage metals and revealed that the staple motif has higher stability on Au(111), while the bridging motif becomes more stable on Ag(111) and Cu(111).Atomically precise metal NCs exhibit distinct physicochemical properties that highly depend on their geometric structures. Therefore, controlling their structure using ligands is crucial for tuning these properties. Several types of thiolate ligands are depicted in Fig. 1. When aliphatic SRs (Fig. 1a) are used, the formation of relatively small-sized metal NCs with icosahedra is favored (e.g. [Au25(SR)18]−, [Au38(SR)24]0, and [Au144(SR)60]0).42–45 When an aromatic SR ligand is used, Au cores with FCC structures are also formed even in relatively small size ranges (as in the case for [Au23(SR)16]−, [Au28(SR)20]0, [Au36(SR)24]0, [Au42(SR)26]0, [Au44(SR)28]0, [Au52(SR)32]0 and [Au92(SR)44]0 (Fig. 1b)). The incorporation of aromatic SR ligands can also generate Au cores with decahedral (Dh), icosahedral (Ih), or hexagonal close-packed (HCP) structures in slightly larger sizes.46–51 Compared to monometallic NCs, the introduction of foreign-atom doping to form bimetallic,52–55 trimetallic,56 and even tetrametallic57 clusters is one of the most promising approaches for tailoring the electronic, optical and catalytic properties of ligand-protected metal NCs.
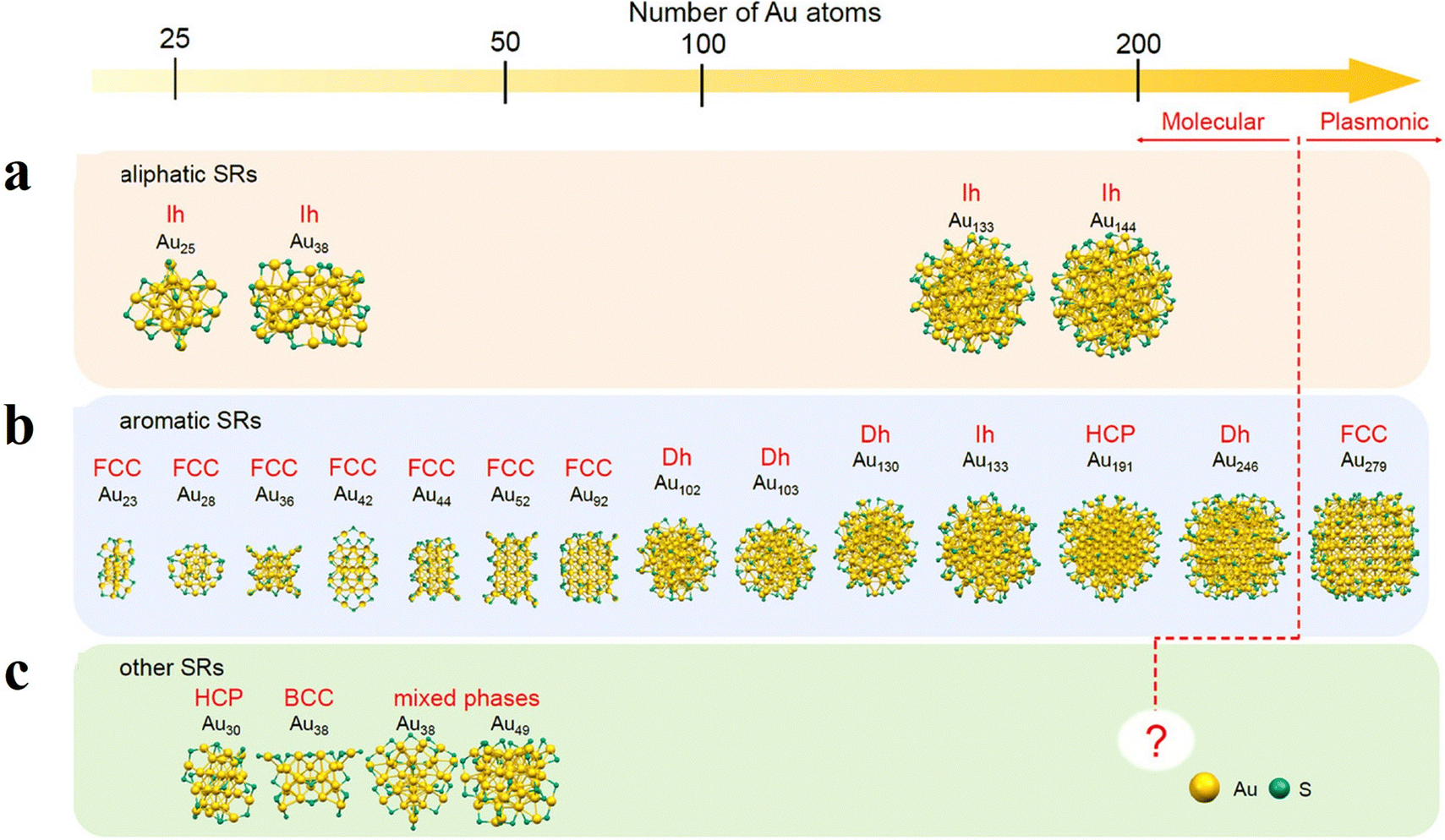 | ||
| Fig. 1 Structures of representative Aun(SR)m NCs protected by (a) aliphatic, (b) aromatic, and (c) other SRs. Adapted with permission from ref. 58 Copyright 2022, Wiley-VCH GmbH. | ||
Electrocatalytic applications of metal NCs
Electrocatalysis plays a crucial role in addressing the current environmental challenges associated with fossil fuel consumption and global warming.59 Recent studies have demonstrated that thiolate-protected metal NCs exhibit high catalytic activity in many electrocatalytic reactions.60 Herein, we mainly focus on the catalytic performance and reaction mechanism of metal NCs as electrocatalysts in the electrochemical processes of hydrogen evolution, oxygen reduction, and CO2 reduction.Hydrogen evolution reaction (HER)
Electrochemical water splitting (i.e., water electrolysis) involves two half-reactions: HER at the cathode and the oxygen evolution reaction (OER) at the anode.61 Several recent studies have reported the use of thiol-protected metal NCs as catalysts for HER and OER.62–70 HER is a process in which protons first bond to metal atoms on the NCs via the Volmer–Heyrovsky or Volmer–Tafel mechanism, followed by hydrogen formation on the metal surface and the subsequent H2 desorption. In 2017, Jin's group pioneered the study of interfacial effects in HER accelerated by Au25 NCs deposited over MoS2 nanosheets. They found that the presence of Au25 as a cocatalyst led to a lower overpotential and higher current density compared to the plain MoS2 nanosheets. They further compared the surface ligand effect (thiolate vs. selenolate) and observed that the thiol-protected Au25(SC2H4Ph)18 was more efficient in interfacial electron transfer than the selenolate-protected Au25(SePh)18 counterpart.64 Soon after, Lee and Jiang et al. reported that the electronic and electrocatalytic properties of Au25(SR)18 NC were dramatically altered by doping of a Pt atom.71 The change in the electronic configuration from superatomic 8-electron [Au25]− to 6-electron [PtAu24]0 causes the splitting of the 1P orbital, accompanying a Jahn–Teller-like distortion of the PtAu12 core. They demonstrated that the bimetallic PtAu24(SC6)18 (C6 = 1-hexanethiolate) cluster exhibits excellent catalytic activity for electrocatalytic H2 production, which is significantly higher than the pristine Au25 NC and twice as active as the benchmarking Pt nanoparticle catalyst. Their mechanistic investigations revealed that the thermo-neutral binding of hydrogen to the central Pt atom plays a key role in the superior HER activity. Later, Lee and his co-workers72 demonstrated that doping Pd into gold NCs could also considerably improve the HER activities, which follows the order of PtAu24 (Pt2Au36) > PdAu24 (Pd2Au36) > Au25 (Au38). Subsequently, Zhu et al. prepared an efficient Pt1Ag28-BTT-Mn(10) nanocatalyst that exhibited excellent performance in HER by atomic-level precision manipulation, including core alloying, ligand engineering and surface activation.73 More recently, Negishi et al.74 synthesized two new Au–Pt alloy NCs, [Au24Pt(TBBT)12(TDT)3]0 (TBBT = 4-tert-butylbenzenethiolate; TDT = thiodithiolate) and [Au24Pt(TBBT)12(PDT)3]0 (PDT = 1,3-propane-dithiolate), by the ligand-exchange reaction, which were more active in promoting HER and were, respectively, 3.5 and 4.9 times higher in the mass-based activity than the PtAu24(SC6)18. On the theoretical side, Sun et al. recently conducted first-principles calculations to reveal that the prominent HER activity of PtAu24 in acid is actually contributed not only by the central Pt atom but also by the exposed bridging Au sites.75 Their constant potential calculations with integration of ab initio molecular dynamics (AIMD) simulations revealed that H+ from the water layer preferentially attacks the S atom, weakening the Au–S bond, and eventually the thiolate ligand spontaneously desorbs from the PtAu24 surface under acidic conditions and a moderate reduction potential at URHE = −0.3 V (Fig. 2). Interestingly, the exposed dethiolated Au exhibits reaction kinetics comparable to Pt for the H2 generation via the Volmer–Heyrovsky mechanism, and the synergistic effect between them makes dethiolated PtAu24 an extraordinary HER electrocatalyst. Their predictions were further verified by electrochemical experiments and attenuated total reflection surface-enhanced infrared spectroscopy (ATR-SEIRAS) characterization. The activated PtAu24 NC afforded a lower overpotential than undoped Au25 and the commercial Pt/C, and the adsorption of Au–H* and Pt–H* bonds was evidently monitored in the HER process, confirming the Pt–Au synergistic effect. Their work indicated that the previously assumed inert Au atom capped by thiolates is greatly activated via electrochemical etching of the surface-passivated ligands, which plays an important role in the electrocatalytic process. Note that although doping Au25 with Pt or Pd tends to increase HER activity, Negishi et al.70 found that doping with Ag or Cu, instead, lowers HER activity. Honkala et al.76 theoretically explained the effects of Cu and Pd doping on the stability, hydrogen adsorption, redox potentials, and HER efficiency of Au25 NCs. Their calculations suggest that at the potential of −0.6 or −0.7 VSHE, PdAu24 mainly exists at the charge state of q = −2, while Au25 and CuAu24 exist in a different charge state of q = −1. The reaction energies and kinetic barriers for the first Volmer step in HER give the following order of activity: PdAu24 > Au25 > CuAu24, which shows a similar trend observed experimentally. They emphasized the importance of determining the charge states of a cluster catalyst under real electrochemical conditions, and the dopant can significantly affect the reaction kinetics by controlling the charge state at a given potential.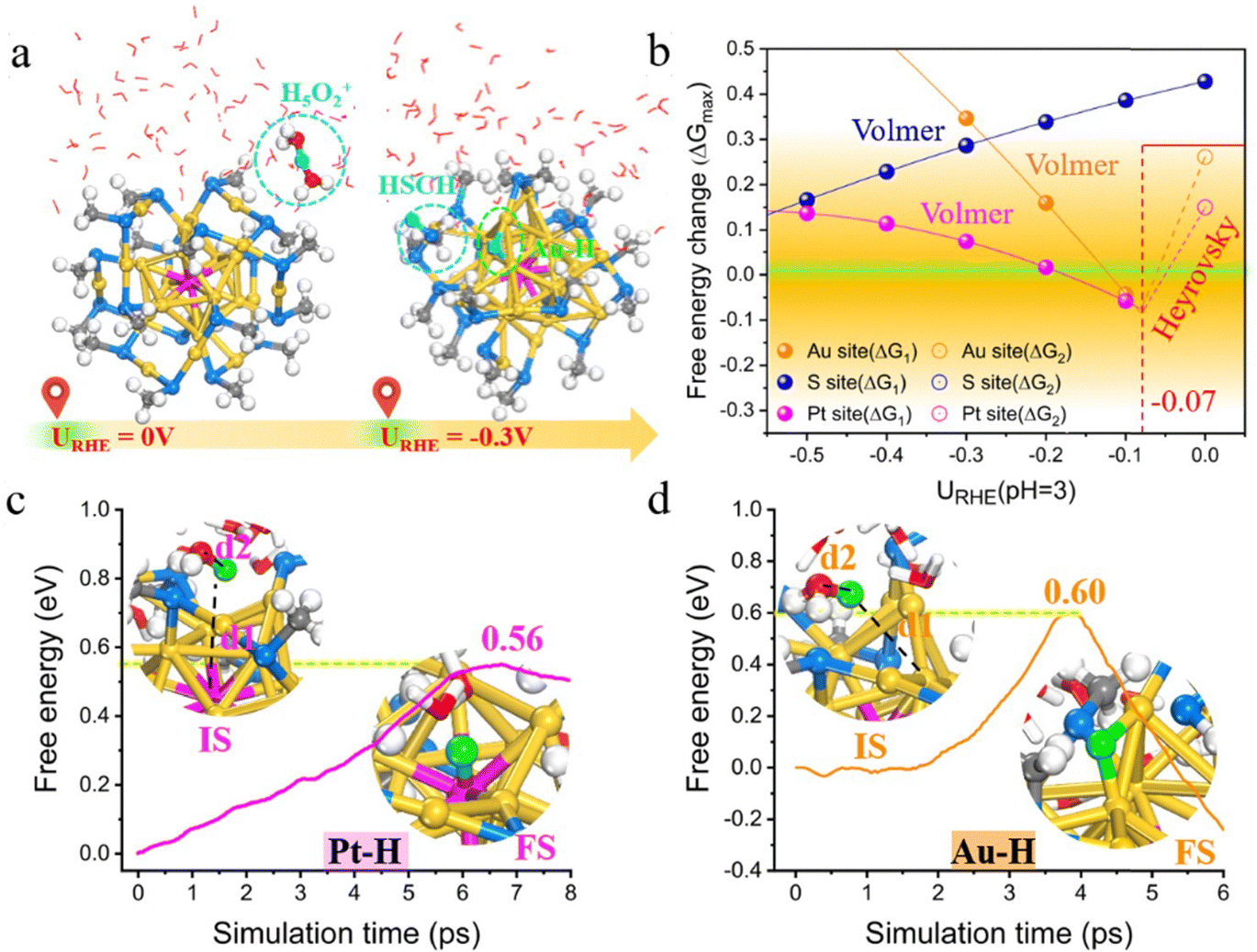 | ||
| Fig. 2 (a) Schematic of the structural evolution of PtAu24 NC with electrode potential. (b) Free energy changes in the potential-determining step (PDS) (ΔGmax) of HER as a function of the electrode potential URHE in pH = 3. (c) and (d) Free energy profiles of solvated proton (H3O+) adsorption on Pt and Au sites, respectively, in a dethiolated PtAu24(SCH3)17 during the slow-growth simulation at Ucross = −0.1 V. Adapted with permission from ref. 75 Copyright 2024, the Royal Society of Chemistry. | ||
HER performance can also be profoundly affected by the ligand effect. In this regard, Lee and Jiang et al.68 compared the electrocatalytic HER performance of Au25(SR)18 capped by three different functional groups: 1-hexanethiolate (C6S), 3-mercaptopropionic acid (MPA) and 3-mercapto-1-propanesulfonic acid (MPS). They showed that HER activity was closely related to the proton-relaying abilities of the –SR ligands, with MPS of the highest proton-releasing ability bearing higher HER activity, followed by MPA–Au25 and C6S–Au25. Moreover, Negishi et al.70 systematically investigated the impact of cluster size and ligand functional groups on the HER properties of Au NCs. Using a series of 2-phenylethanethiolate (PET)-protected Aun(SR)m NCs ([Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0) as the testing candidates, they observed that the current density and mass activities increase as the cluster size decreases (Fig. 3a and b), indicating that decreasing the cluster size induces an enhancement in the HER activity. They assumed that Au–H bonds are formed more easily on the surface of the metal core in small-sized Au NCs. Regarding the ligand effect, Negishi et al. further used the [Au25(PET)18]0, hexanethiolate (C6T)-protected [Au25(C6T)18]0, and dodecanethiolate (C12T)-protected [Au25(C12T)18]0 to evaluate the dependence of HER catalytic activity on the thiolate functional groups (Fig. 3c and d), and they observed the order of HER performance as Au25(C12T)18 < Au25(PET)18 < Au(C6T)8, indicating an improved HER activity in thinner ligand layers. Their results demonstrate that the length of the carbon tail groups can significantly affect electrocatalytic HER activity, but a detailed atomic-level understanding of the reaction kinetics and the ligand-dependent electrochemical microenvironment needs further investigations in the future. Recently, Luo et al.77 demonstrated the importance of ligand hydrophilicity and hydrophobicity in modulating the reaction interface and electrocatalytic HER performance of Au25(SR)18 NCs. Their AIMD simulations revealed that Au25 protected by hydrophilic –SCH2COOH ligands exhibits enhanced interfacial hydrogen bonding and faster kinetics in stripping the thiolate ligands than the hydrophobic –SCH2CH3 ligands, which lowers the barrier for rater-determining H2 formation by about 0.12 eV at −0.66 VSHEvia the boosted proton transfer to the active Au site. In contrast, the hydrophobic nature of the alkyl–SCH2CH3 ligands tends to reduce hydrogen bonding near the reactive Au surface and decrease the HER efficiency. Their electronic structure analysis observed a lower electron density and a higher energy of the d-band center on the active Au site in the hydrophilic Au25 cluster, which could promote the desorption of H2 product and further enhance the kinetics of hydrogen evolution. Besides HER, Xie et al.78 recently revealed that the ligand effect can induce distinct changes in the rate-determining step (RDS) of the OER process. They observed the significantly boosted OER performance of Au25(SR)18 NCs protected by para-mercaptobenzoic acid with a stronger electron-withdrawing ability by about 4–5 times higher than that of Au25 protected by 6-mercaptohexanoic acid and homocysteine ligands. They showed that the electron-withdrawing ligand establishes more positive charges on the Au(I) site in protecting the Au(I)-SR motif to assist in the adsorption of OH− species in alkaline media and to lower the barrier for OER kinetics.
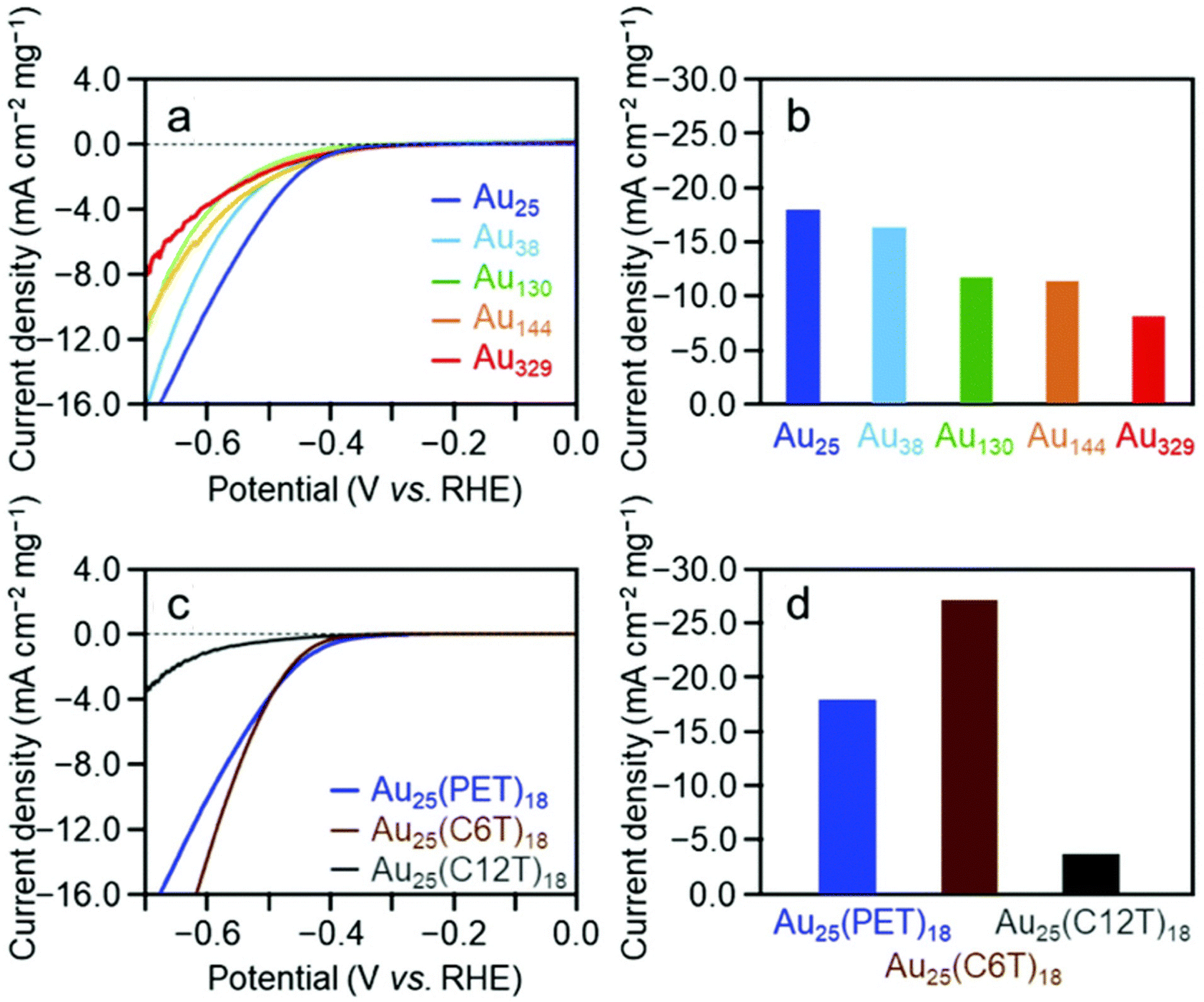 | ||
| Fig. 3 Voltammogram (a) and (c) and mass (b) and (d) HER activity of different sizes of Au NCs protected by PET ligands, and Au25 NCs capped by different ligands under N2-saturated 0.2 M HClO4 (pH = 0.7). Adapted with permission from ref. 70 Copyright 2020, the Royal Society of Chemistry. | ||
In addition to hetero-metal doping and ligand engineering, Jin et al. proposed an alternative strategy to enhance the HER activity of catalytically inert metal NCs by reducing ligand coverage and creating low-coordinated Au atoms.79 They obtained a trimeric Au36Ag2(SR)18 NC with a lower ligand-to-metal ratio and coordination number of surface Au compared with the monomeric Au25(SR)18− and dimeric Au38(SR)24 (Fig. 4a and b). As anticipated, the Au36Ag2(SR)18 exhibited remarkably high HER catalytic activity. The most feasible active sites for hydrogen adsorption were the Au atoms on the kernel's shell, i.e., Au12 shell for Au25(SR)18−, Au21 shell for Au38(SR)24, and Au27Ag2 shell for Au36Ag2(SR)18. According to their DFT calculations, the fully ligated Au36Ag2(SCH3)18 exhibited a lower hydrogen binding energy (ΔG = 0.83 eV) to form the *H intermediate than Au25(SCH3)18 (1.29 eV) and Au38(SCH3)24 (1.35 eV) (Fig. 4c and d). The enhanced HER activity of Au36Ag2(SR)18 NC was also attributed to the more negative electron affinity, which facilitated more favorable electron acceptance during the first Volmer protonation step.
 | ||
| Fig. 4 Ligand/metal ratio (m/n) versus metal number (a) and the coordination numbers of surface Au atoms in Au36Ag2(SR)18, Au25(SR)18−, and Au38(SR)24 (b). Structures of the three metal NCs upon hydrogen adsorption on the Au active site (c) and the calculated Gibbs free energy of H2 formation (d). Adapted with permission from ref. 79 Copyright 2021, ACS Publication. | ||
Oxygen reduction reaction
Atomically precise metal NCs can serve as promising catalysts for the electrochemical oxygen reduction reaction (ORR), an important yet challenging reaction in fuel cells and metal–air batteries due to their slow reaction kinetics. ORR involves either a four-electron–proton coupling to produce H2O or a two-electron–proton transfer to produce H2O2. Tang et al.80 predicted the high ORR activity of the phosphine stabilized Au22(L8)6 NC (where L8 = 1,8-bis(diphenylphosphino)), where the exposed in situ coordination unsaturated (cus) Au atoms were predicted to be the active site for oxygen activation and associative four-electron reduction to generate H2O. In their later work, Tang et al.81 theoretically predicted the doping effects on the ORR activity of thiolated Au25(SR)18 NCs. Different from the above-discussed Au22(L8)6, all surface Au atoms in Au25 are fully protected, and no cus Au atoms are present in the synthesized intact Au25 or doped MAu24 NCs. The charge state, dopant atom and ligand removal site are important tuning factors for ORR. In the case of full ligand protection, the intact Au25 and doped MAu24 NCs were predominantly selective for H2O2 production (Fig. 5a). The 2e− ORR activity decreased in the order [HgAu24]0(O) > [Au25]− > [Au25]0 > [PtAu24]0(C) > [PdAu24]0(C) > [CdAu24]0(C) > [CuAu24]−(C) > [AgAu24]−(S) > [CdAu24]−(S) > [Au25]+, where the bracketed C, S and O denote the doping site at the center of Au13 core, the surface shell of Au13 core, and the peripheral [–SR–Au–SR–Au–SR–] oligomer motif, respectively. The [HgAu24(SCH3)18]0(O) candidate exhibited particularly the highest activity for H2O2 production with an ultralow overpotential of 0.08 V. When the protective thiolates were partially removed, either by removing an –SR ligand to expose the metal site or by cleaving the organic –R moiety to expose the S site, the O2 adsorption and binding of ORR intermediates (*OOH, *O, and *OH) were largely strengthened. This, in turn, led to a shift in ORR selectivity from a 2e− reduction to generate H2O2 in fully ligand-protected NCs to a 4e− reduction to form H2O in dethiolated NCs (Fig. 5b). In particular, dethiolated [HgAu24(SCH3)17]0 after removing one exterior −SCH3 ligand displayed the highest 4e− ORR activity with a small overpotential of 0.43 V. Their electronic structural analysis revealed that the distinguished ORR activity in Hg-doped Au25 was primarily attributed to the 6s state of the staple-doped Hg atom near the Fermi level. In fully protected [HgAu24(SCH3)18]0, the staple Hg served as the active site, contributing its electrons to strengthen the interaction with the *OOH intermediate and improving the 2e− ORR performance for H2O2 formation (Fig. 5c). In the dethiolated [HgAu24(SCH3)17]0 system, the Hg 6s electronic state shifted to a lower energy position (Fig. 5d), which enhanced the adsorption of O2 and ORR intermediates without overbinding the key *OH intermediate, thereby leading to optimal adsorption and superior 4e− catalytic behavior over the other transition metal dopants. In the same year, Zhu's team introduced a 1,1-bis-(diphenylphosphino)ferrocene (DPPF) ligand into M1Ag21 (M = Au/Ag), forming two novel ligands. The catalytic activity of M1Ag21 was confirmed to be derived from the M13 core and the DPPF ligand by DFT calculations, and M1Ag21 can be used as an efficient oxygen reduction reaction catalyst in alkaline solution.82 Furthermore, Tong et al. reported for the first time on oxygen reduction electrocatalysis in nanoclusters with more than 100 Ag atoms, Ag213(Adm-S)44Cl33 (Ag213), coprotected by bulky thiolates and chlorides. They found that halides play a key role here.83 In addition, Pei et al.84 theoretically investigated the effect of grafted single-atom catalysts on tailoring the electrocatalytic performance of Au25 NCs. They predicted that the Au25(SR)18 NCs with grafted Ni2+ single atom via l-cysteine ligand as the bridging ligand showed excellent bi-functional activities with a low overpotential of 0.48 for OER and 0.27 V for ORR.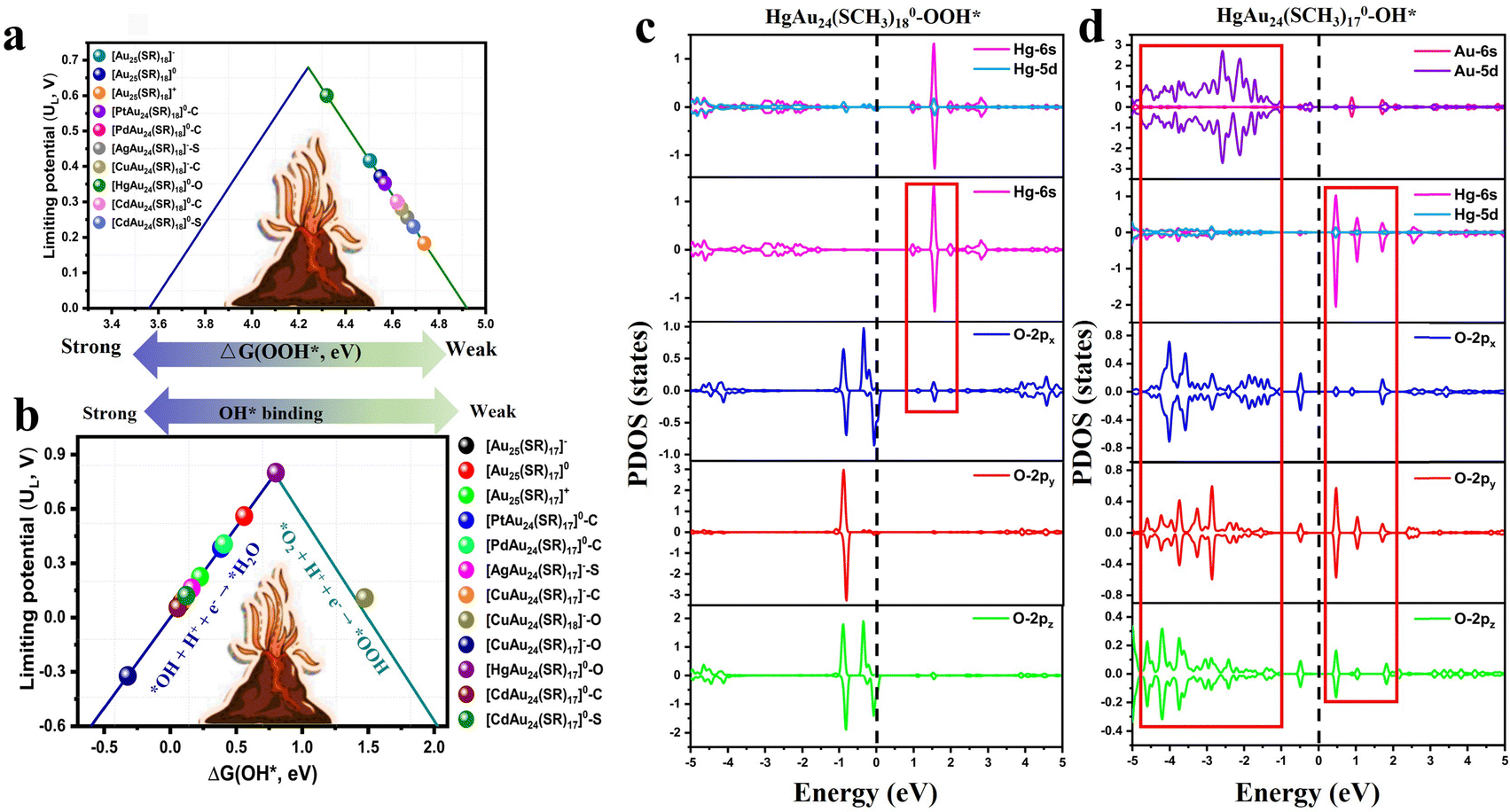 | ||
| Fig. 5 Volcano relation between the limiting potential (UL) and ΔG*OOH/ΔG*OH for fully protected (a) and dethiolated (b) NCs. The inset in (b) shows the structure of a dethiolated [HgAu24(SCH3)17]0. Projected density of states for *OOH/*OH adsorbed on [HgAu24(SCH3)18]0–O (c) and [HgAu24(SCH3)17]0–O (d). Adapted with permission from ref. 81 Copyright 2021, ACS Publication. | ||
Experimentally, Jin et al.85 studied the ORR performance of heterometal-doped Ag25(SR)18 NCs, including Ag25(SR)18, Au1Ag24(SR)18, Pd1Ag24(SR)18, and Pt1Ag24(SR)18. The Ag25 NC is structurally parallel to Au25, with Au, Pd and Pt dopant atoms exclusively located at the center of the Ag13 icosahedral core. The electrochemical ORR experiments indicated a two-electron reduction mechanism in all these NCs, leading to the formation of H2O2 product, with the rate-determining step likely being the first proton–electron transfer to oxygen (*OOH formation). They found that the Pd1Ag24 and Pt1Ag24 NCs exhibited better ORR performance, with about ∼100 mV lower onset potential compared with Au1Ag24 and Ag25. Their difference in ORR activity was further explained via DFT calculations. They proposed that the exposure of sulfur active sites via partial –CH3 removal was responsible for catalytic ORR activity. In another experimental report, Xie et al.86 demonstrated the composition-dependent catalytic performance of AuxAg25−x(MHA)18 (x = 0–25 and MHA = 6-mercaptohexanoic acid) NCs for ORR. They showed that the dominant ORR product changes from H2O to H2O2 with the increasing number of Au in AuxAg25−x NCs. As shown in Fig. 6, as the doping content of Au atoms in AuxAg25−x NCs increases, both the onset potential (Fig. 6a) and the half-wave potential (Fig. 6b) of ORR continue to decrease. Moreover, the number of electrons transferred gradually decreases from 3.9 in Ag25 to 2.1 in Au25 (Fig. 6c), and the corresponding H2O2 selectivity increases from 8% in Ag25 to 97% in Au25 (Fig. 6d), leading to the alteration of the main reaction product from water to hydrogen peroxide. They calculated the Gibbs free energy of *OOH adsorption (ΔGOOH*) and found that Au25 is more favorable for H2O2 formation owing to its optimal ΔGOOH* close to the ideal value of 4.22 eV, while the Ag alloying makes the ΔGOOH* deviate from the optimal value and leads to the production of water in Ag-rich alloy NCs. In line with this finding, Zhu et al.87 reported that the heavy doping of Ag atoms in icosahedral Au13 NCs (Au7Ag6) also resulted in good ORR activity via a four-electron reduction process. Moreover, Teranishi et al.88 recently explored the reactivity of thiolate-protected Au25 NCs in an air-oxidation reaction. Their quantitative kinetic analysis of the reversible oxidation between anionic Au25− and neutral Au025 revealed that the affinity for O2 is regulated by the electronic and/or steric effects of the R moieties of the thiolate ligands. The Au25 NCs protected by rigid aromatic thiolate ligands exhibited higher O2 binding constants than those capped by flexible aliphatic thiolate ligands. In another recent study, Ma et al.89 employed a single-nanoparticle collision electrochemistry method to directly monitor the dynamic structural evolution of Au25(PPh3)10(SC2H4Ph)5Cl22+ NCs during the ORR process. Their electrochemical experiments observed a characteristic current signal with continuous “ON–OFF” switches and fluctuations in the “ON” state during oxygen reduction. They correlated these signals with the dynamic and reversible dissociation and reforming of Au–ligand bonds, which was confirmed by the AIMD simulations of Au252+ NC in aqueous water. The desorption of surface Cl, thiolate or phosphine ligands in the Au252+ NC at an ORR potential of −500 mV vs. Ag/Ag2O was proposed as the possible origin of the active catalytic state.
 | ||
| Fig. 6 (a) ORR polarization curves of each AuxAg25−x(MHA)18 NC sample in O2-saturated 1.0 M KOH at a scan rate of 5 mV s−1 with a rotating rate of 1600 rpm. (b) Comparison of the half-wave potential of each NC catalyst. Calculated electron transfer number (c) and H2O2 selectivity (d) during the potential sweep of each NC sample. Adapted with permission from ref. 86 Copyright 2024, Tsinghua University Press. | ||
Although thiolate-protected group XI elements have been successfully reported in the last few decades, few studies have been conducted on Pd analogs. In 2022, Wu's team obtained the first atomic-level monodisperse thiolated Pd nanoclusters.90 In contrast to newly synthesized Pd2 complexes, ≈1.4 nm Pd NPs, and even commercial Pt/C catalysts for ORR, this Pd nanocluster contains a fully +6 charged ditetrahedral metal core and a +3 charged interstitial B atom (forming a Pd6B triangular prism). A comparison of Pd8, Pd2, and Pd NPs reveals that Pd8 nanoclusters have lower electron transfer resistance at the Pd site and have the best electrocatalytic activity for ORR. This work heralds a new field in the synthesis of thiolated palladium NCs.
CO2 reduction reaction (CO2RR)
Electrocatalytic CO2 conversion offers a promising solution to mitigate ecological changes caused by global warming and provides great sustainability in producing useful fuels and chemicals. Gold-based NCs have garnered particular attention for CO2RR owing to their high activity and selectivity toward CO, which is intrinsically attributed to their weak binding with the critical *CO intermediate.91–100 Recent studies have reported that various factors, such as the core size, surface structure, heterometal doping, and ligand type, could modulate the CO2RR performance.101Thiolated gold NCs are the most widely explored catalysts for CO2RR, both experimentally and theoretically, but the current understanding of the real active site and electrocatalytic mechanism remains ambiguous. Taking the recently reported studies as examples, Jin et al. demonstrated that single-atom Pd substitution can substantially improve the CO2RR selectivity of Au25(SR)18 over a wide potential range, while the reactivity of undoped Au25 declined at higher potentials with considerable H2 evolution.93 The DFT-calculated reaction thermodynamics indicated that the exposed surface S atoms of the thiolate ligand (–CH3 removal) served as the active sites for selective CO2 reduction, whereas the exposed undercoordinated Au active sites (–SCH3 removal) favored the H2 evolution. They further obtained Cd-modified Au19Cd2(SR)16 by surface doping of Cd into the Au23(SR)16, which exhibited about twice the CO selectivity and activity compared to the undoped Au23.96 They hypothesized that the thiolated Au NCs should contain S active sites, which are exposed by the desorption of the –R group at moderate overpotentials, while the complete desorption of thiolate ligands exposes the Au active site at sufficiently high overpotentials. Jin et al. also investigated the size and isomer effects of gold NCs protected by the same thiolate ligands in CO2RR.102 They investigated two groups of size series: Au38(SR)24, Au144(SR)60, Au333(SR)79; and Au28(SR)20, Au36(SR)24, Au279(SR)84. The electrochemical tests indicated that the CO2RR selectivity was not directly dependent on the cluster size, while the CO2RR activity increased for smaller NCs, suggesting that the surface-to-volume ratio and the population of exposed S active sites were the dominant factors for the catalytic activity. They further studied two Au38(SR)24 isomers with the same size and composition but different core and surface staple motifs.102 The Au38Q showed higher activity and selectivity towards CO than Au38T. Using the *COOH formation energy as an energy descriptor, their DFT results revealed that the formation of the key *COOH intermediate at the exposed S active site was more favorable on Au38Q compared to Au38T. Note that the postulation about the active site solely relies on thermodynamic calculations based on the computational hydrogen electrode (CHE) model, which predicted that the ligand removal mode leading to the formation of dealkylated S is thermodynamically more plausible than the one leading to the exposure of a metal atom, while kinetic investigations under realistic electrochemical conditions are lacking.96,103
However, recent experimental evidence has challenged the above argument that dealkylated S is the active site responsible for the observed CO2RR performance.98,104,105 Lee et al.98 monitored the CO2RR activity and chemical composition of the Au25(SR)18− NCs using electrochemical impedance spectroscopy (EIS) and X-ray photoelectron spectroscopy (XPS). They found a dramatic increase in the initial CO current density (jCO) after electrochemical activation (Fig. 7a) and observed a gradual reduction in the S/Au ratio as the activation time increased (Fig. 7b), indicating instead that the dethiolated Au serves as the active site. The chemical composition was determined to be Au25(SR)12 after 60 min of activation at −0.46 V, implying that approximately six thiolate ligands were removed from each Au25 catalyst. Considering the high structural symmetry of Au25, they inferred that one SR ligand could be detached from each Au2(SR)3 staple motif in this cluster. A similar trend was also found in other thiolate-protected Au NCs, such as Au38(SR)24 and Au144(SR)60, where the number of detached SR ligands increased to 9 and 27, respectively. They further revealed that the CO2RR activity was mainly affected by the total number of dethiolated sites and that Au144 exhibited the highest activity among the three Au NCs. In their subsequent work, Lee's group105 performed the EXAFS (extended X-ray absorption fine structure) spectroscopic analysis of a smaller thiolate-protected Au4Ni2(SR)8 NC and observed a decrease in the coordination number of Au–S and Ni–S bonds after electrochemical activation, suggesting that the thiolate ligands can be readily removed.
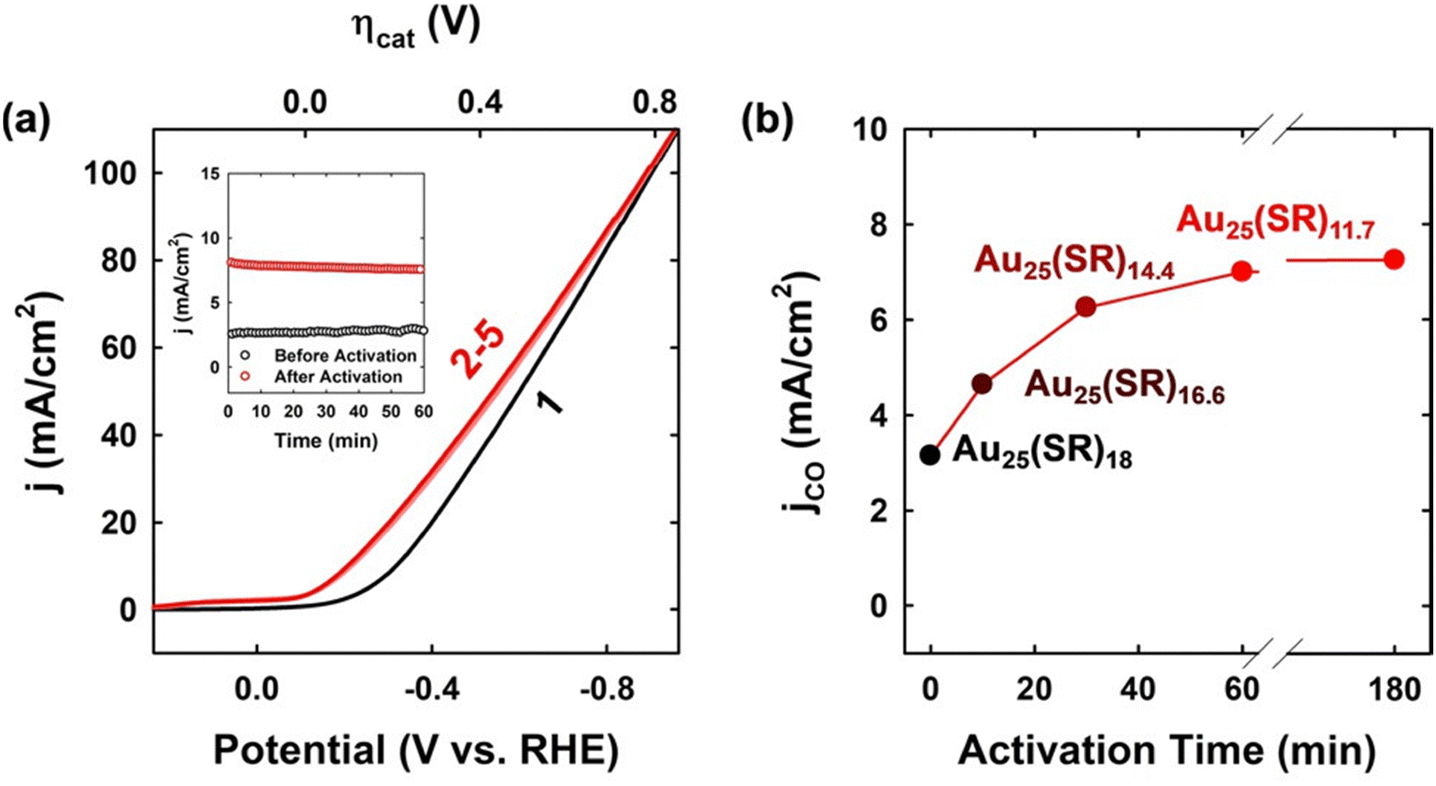 | ||
| Fig. 7 (a) LSV curves recorded during the first (black) and second to fifth (red) sweeps on Au25/GDE in a 1.0 M KOH electrolyte solution at 50 mV s−1. The inset shows the jCO values determined at −0.16 V vs. RHE on Au25 in the same solution before (black) and after (red) electrochemical activation. (b) jCO values measured on Au25/GDE at −0.16 V vs. RHE in the 1.0 M KOH electrolyte solution after electrochemical activation at −0.46 V vs. RHE for 0, 10, 30, and 60 min. The NC compositions obtained using XPS are shown in the graph. Adapted with permission from ref. 98 Copyright 2021, Wiley-VCH GmbH. | ||
In response to the controversy between the prior thermodynamic viewpoint (which favors S as the active center) and electrochemical experiments (which favors Au as the active center), Sun et al.106 recently investigated the mechanism of ligand etching in Au25(SR)18. Their study found that the simplified charge-neutral method based on the traditional CHE model yields a strong thermodynamic preference for S–C breaking (–R removal). In contrast, the constant potential calculations coupled with an explicit solvent model of the electrochemical interface predict, instead, the preference for Au–S breaking (–SR removal). Their AIMD simulations (Fig. 8) of the Au25/water interface at 300 K observed spontaneous barrier-less –SR detaching at the reduction potential (URHE) in the range of −0.3 to −1.0 V, which was initiated by the selective attack of protons from H3O+ (acidic condition) or water dissociation (neutral or basic condition) onto the S atom from the staple motif, followed by the breaking of the surface and staple Au–S bond; finally, the formed free thiol molecule dissolved in solution. Their dynamic simulations verified the facile dethiolation of the SR ligands in the experiment, and the exposed surface Au could be catalytically active sites. Recently, Alfonso group107 also confirmed this view by the Au25 NC in the −1 valence state, where the exposed Au site after the dethiolation is both active and selective for CO2 reduction. These studies clearly account for the importance of accurate simulation of electrochemical interfaces and the consideration of reaction kinetics to rationalize experimental observations. Besides electrochemical biasing, Zhu et al.108 recently showed that thiolate ligands can be partially detached by applying the acid etching method. Under the acid solution, they observed interesting charge-state-mediated etching dynamics. The anionic and neutral Au25(SR)q18 (q = −1, 0) NCs favorably react with the acid and partially remove the thiolate ligands, while the cationic Au25(SR)18+ is acid-resistant. They further predicted that when the Au25 NCs are supported on the graphene substrate, the reactive vacancy defect in graphene induces the breaking of local Au–S motifs from the cluster surface and exposes the undercoordinated Au site to promote CO2RR.109
 | ||
| Fig. 8 (a) Two electrochemical ligand removal modes on Au25(SCH3)18: removal of an entire –SCH3 ligand (dethiolation) or cleavage of the –CH3 moiety (dealkylation) to produce an exposed Au or S atom active site. AIMD snapshots of ligand removal under an acid system (b) and an alkaline system (c). Adapted with permission from ref. 106 Copyright 2023, the Royal Society of Chemistry. | ||
The electrochemical CO2RR activity of Au25 NCs can be delicately modulated by interface and ligand engineering. The presence of alkali cations in the electrolyte has been well recognized to play a crucial role in affecting the reaction dynamics of CO2RR, and much research attention has been paid to increasing the local cation concentration to boost electrocatalytic performance.110 Recently, Lee group reported a strategy to engineer the local environment of Au25 NCs by introducing a cation-relaying ligand, 6-mercaptohexanoic acid (MHA), bearing carboxylate groups to enhance the local alkaline cation concentrations in the proximity of the MHA-Au25 electrocatalyst.111 As shown in Fig. 9a, the dethiolated Au active sites in the MHA-Au25 NC is sufficiently open to allow small reactants such as CO2 and H2O to freely access the catalytic site, and more importantly, the presence of the anionic terminal (MHA) ligands attracts the electrolyte cations and relays them to the catalytic site, leading to an increased cation concentration around the reaction interface. As a comparison, they also synthesized the Au25 NCs protected with neutral hydrophobic 1-hexanethiolat (HT) ligands. The elemental mapping analysis revealed that the local cation concentrations in MHA-Au25 were significantly higher than those in HT-Au25. By comparing the partial current density for CO (jCO) of the two Au25 NCs in different alkali metal electrolytes, they found that the jCO value for MHA-Au25 was significantly higher than that for HT-Au25 (Fig. 9b and c) when the catholyte pH exceeds the pKa of MHA, where the CO selectivity remained >80% in the potential ranging from −1.0 to −1.4 V in different catholytes containing 1.0 M MCl (M = Li+, Na+, K+, or Cs+), demonstrating the significant cation-relaying effect of the anionic terminal groups. Their mechanistic investigations of CO2-to-CO conversion on the Au25 NCs revealed that the RDS process is governed by the cation-coupled second electron transfer step (*COOH + e− → *COOH−), and the CO2RR activities increased in the order Li+ < Na+ < K+ < Cs+. Obviously, their results showed that the larger cations, such as K+ and Cs+, are more significant in boosting the CO2RR performances, which could be attributed to their high binding ability to the reaction intermediates.
 | ||
| Fig. 9 (a) Schematic of electrocatalytic CO2 reduction reaction occurring on an Au25 NC protected by cation-relaying ligands (only one ligand is shown for clarity). jCO values and the corresponding CO selectivity measured for (b) HT-Au25/GDE and (c) MHA-Au25/GDE in different catholytes containing 1.0 M MCl (M = Li+, Na+, K+, or Cs+). Adapted with permission from ref. 111 Copyright 2024, Wiley-VCH GmbH. | ||
However, recent advancements have also revealed that protecting SR ligands can be tailored to enhance the activity and selectivity in CO2RR, which is achieved by creating a hydrophobic electrocatalytic surface or increasing the local CO2 concentration to facilitate the mass transfer of CO2 molecules. For example, the theoretical simulations by Luo et al. showed that Au25 protected by hydrophobic ligands exhibited enhanced CO2RR activity by minimizing water interference to stabilize the key *COOH intermediate and lower the barrier for the rate-determining step of CO formation. They tested the electrocatalytic activity using synthesized [Au25(MPA)18]− (MPA = mercaptopropionic acid), [Au25(MHA)18]− (MHA = 6-mercaptohexanoic acid) and [Au25(SC6H13)18]− as model catalysts and well validated the higher faradaic efficiency and current density for CO production in the hydrophobic ligand-protected Au25 NCs. Moreover, the reaction temperature is another critical parameter in modulating the local microenvironment. Although electrochemical experiments are typically conducted at room temperature, in practice, thermal losses and/or hot feedstocks always lead to different degrees of heating in electrolytic cells. Experiments by Koper et al.112 revealed that there exists a compromise between the increase in reaction kinetics and the decrease in CO2 solubility with increasing temperature, and overheating decreases the CO2RR activity despite the more efficient mass transport properties. Sun et al.113 combined simulations and experiments to probe the temperature-induced interface effect on the CO2RR performance of hydrophobic Au25(SR)18 NCs. They explored three different temperatures (300 K, 330 K, and 350 K) and found that the temperature induces great disturbance of the hydrogen bonding at the Au25/water interface and affects the proton transfer kinetics for the formation of *COOH and *CO species. At a moderate temperature of 330 K, the energy barrier for the CO production is greatly reduced, and the hydrophobic Au25 NCs exhibit the best performance with a high CO faradaic efficiency of ∼93% and a CO partial current density that is about 2 times higher than that at room temperature. If the temperature further increases (350 K), it damages the hydrogen bond network to decrease the catalytic performance or even cause deactivation. Therefore, establishing a suitable reaction temperature is essential for achieving optimal catalytic activity and selectivity in CO2-to-CO conversion. Furthermore, Liu et al.114 developed an alternative strategy for the interface regulation of the reaction microenvironment by surface ligand engineering (Fig. 10). In their method, they precisely incorporated the pyrimidine-containing 2-thiouracil-5-carboxylic acid (TCA) ligand into the protecting layer of Au25 NCs. The strengthened affinity between the electronegative Nδ− of the pyrimidine ring and the electropositive Cδ+ of CO2 leads to a faster increase in the local concentration of CO2 reactants at the cluster surface (Fig. 10a), which consequently accelerates the mass transfer of CO2 and promotes the subsequent kinetics of CO2 reduction to deliver a close-to-unit FECO of 98.6% at −0.9 V (Fig. 10b). As another promising strategy of surface-ligand engineering, Zhao et al. utilized a ligand exchange method to anchor the thiol-functionalized terpyridine metal complexes into the ligand shell of Au25(PET)18 NCs (PET = Phenethyl mercaptan), resulting in a series of new hybrid catalysts, denoted as Au25–M (M = Fe, Ni, Ru, and Co).115 This approach enhances the photocoupled electrocatalytic CO2 reduction by providing additional active sites and facilitating the electron transfer for CO2RR. These findings demonstrate the effectiveness of surface engineering in electrocatalytic reactions.
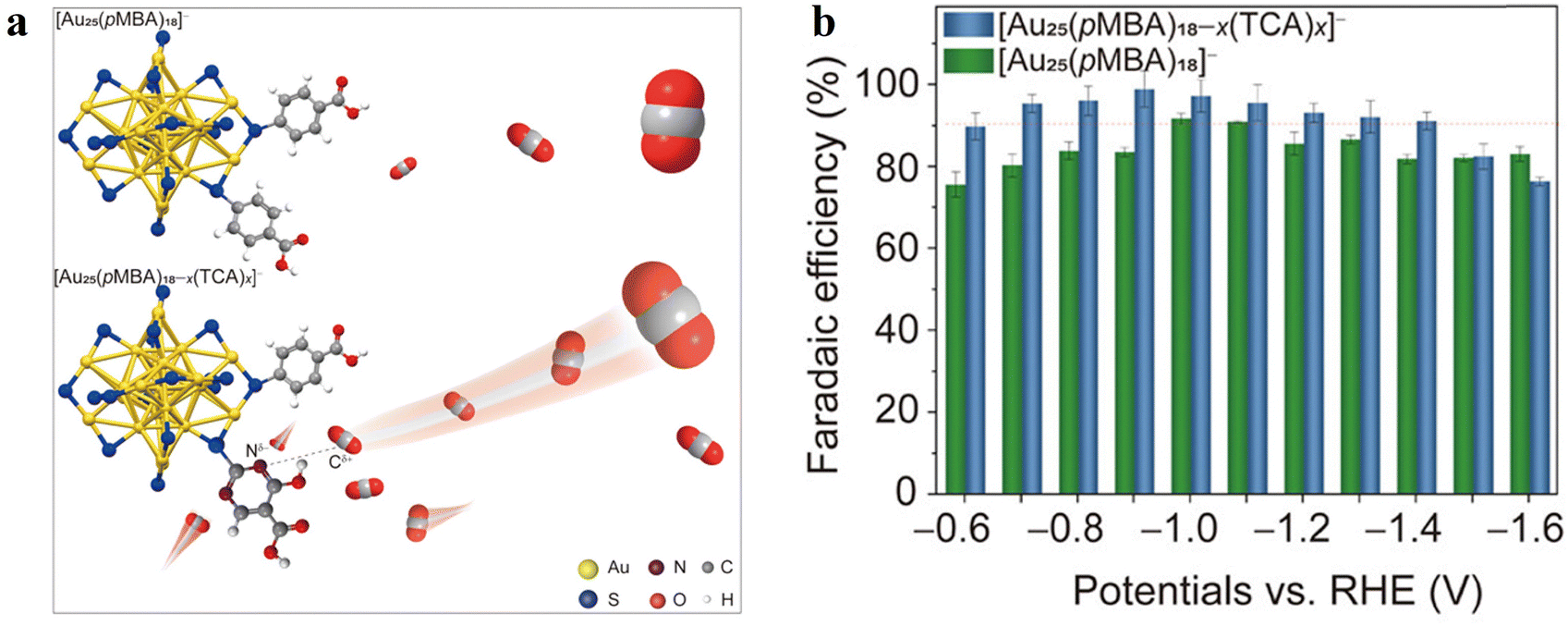 | ||
| Fig. 10 (a) Schematic of the affinity between Au25 NCs and CO2 molecules. (b) Faradaic efficiency (FE) of CO during the CO2RR. Adapted with permission from ref. 114 Copyright 2024, ACS Publication. | ||
Compared to the widely studied gold NCs, thiolate-protected Ag NCs are much less explored in the electrocatalytic CO2RR process.33,110,116–119 One particular representative composition is Ag25(SR)18, which is structurally identical to that of Au25(SR)18, thus providing an ideal model for investigating metal-dependent electrocatalytic performances. Lee group compared the CO2RR activities of Au25(SEtPh)18− (SEtPh = 2-phenylethanethiolate) and Ag25(SPhMe2)18− (SPhMe2 = 2,4-dimethylbenzenethiolate) NCs in an alkaline flow electrolyzer.120 The two NCs produced CO as the main reduction product, but the CO2RR activity of Au25 was significantly higher than that of Ag25. They then replaced the protecting Ag12(SR)18 shell of the Ag25 NC with the more active Au12(SR)18 shell, and the generated core–shell AuAg12Au12 NC exhibited dramatically enhanced CO2RR activity compared to the pristine Ag25. Huang group reported an Au-doped Ag NC with an open-shell structure, [AuAg26(S-Adm)18S]− (denoted as AuAg26; S-Adm = 1-adamantanethiolate), which exhibited superior CO2RR performance than Ag25 and Au21 NCs owing to the exposed facets without being protected by thiolate ligands.121 Bootharaju and co-workers designed two atomically precise Ag25(SR)18 NCs with identical sizes but different surface microenvironments.122 They reported that the locally induced hydrophobicity by bulky alkyl 2-isopropylbenzenethiolate (IPBT) ligands in Ag25 NC promotes the surface interactions with water and dramatically enhances the CO2RR activity with over 90% faradaic efficiency for CO (FECO) compared to the 66% FECO in Ag25 capped by captopril thiolates with confined hydrophilicity.
Theoretically, Ye et al.123 compared the interfacial stability and electrocatalytic activity in CO2RR between Ag25 and Au25, which were identical structurally and ligand-wise, differing only in the core metal. Their simulations revealed that Ag25(SR)18 exhibits distinct leaching behavior and electrocatalytic mechanisms in CO2 electroreduction compared to its golden analogue Au25(SR)18. Under the applied reduction potential, the Ag–S interface becomes destabilized, and the water molecule preferentially attacks the S atom from the exterior thiolate ligand initially coordinated to two staple Ag atoms, leading to the detachment of the exterior SR ligand (Fig. 11a). In contrast, in Au25, the water is more inclined to attack the interior thiolate ligand (Fig. 11b). The difference in the attacking sites is correlated to the electronic state of the different S sites. In Ag25, the exterior S site carries a more negative charge compared to the interior S site, while in Au25, the interior S site is more negatively charged. The S site with a more negative charge dictates higher reactivity. They revealed that the stripping of the SR ligand from the Ag–S interface is kinetically more difficult than that from the Au–S interface, which is possibly related to the stronger and more ionic Ag–S bond. After the removal of one SR ligand, the dethiolated metal atoms serve as catalytically active sites. The position of the removed ligand varies, which leads to a different linkage for the subsequent steps in CO2RR (Fig. 11c–f). The dethiolated Ag sites effectively promote CO2 electroreduction to CO while suppressing competitive H2 formation, but the potential-determining step of CO2 reduction differs significantly between Ag25 and Au25. In Ag25, the highest potential barrier corresponds to the *COOH formation, while in Au25, the most difficult step is the *CO formation. This set of results suggests that the rate-determining steps and the electrocatalytic activity differ for NCs with different cores, which can be utilized to regulate electrocatalytic behaviors via metal core or active site engineering.
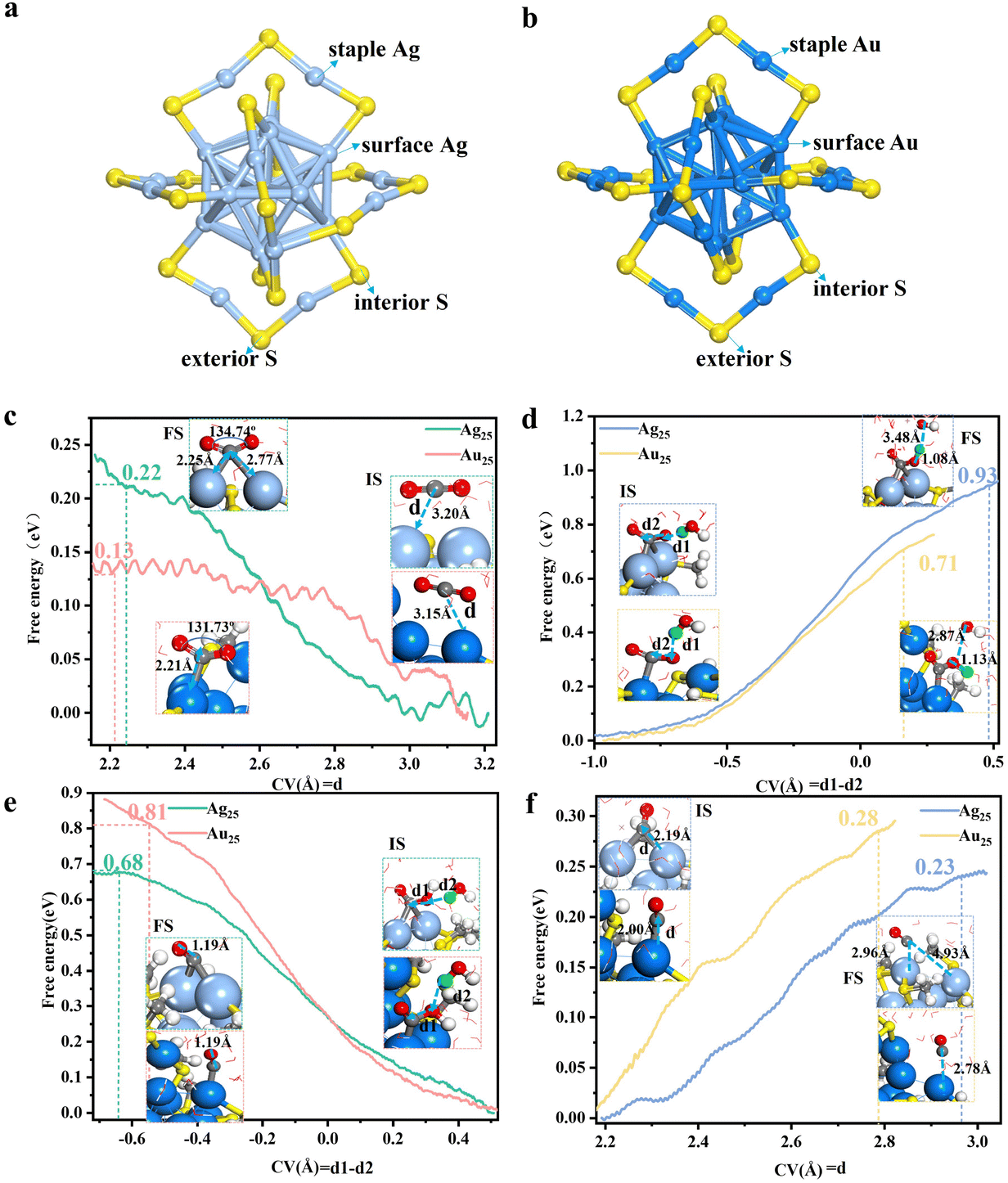 | ||
| Fig. 11 Schematic of the Ag–S (Au–S) framework structure of (a) Ag25(SR)18 and (b) Au25(SR)18 NCs. Comparison of the integral free energy curves between Ag25 and Au25 in CO2 activation (c), *COOH formation (d), *CO formation (e) and CO desorption (f) through constrained AIMD simulations. The local structures of the initial state (IS), final state (FS) and the defined collective variable (CV, d1–d2 or d) are shown as insets. Adapted with permission from ref. 123 Copyright 2025, the Royal Society of Chemistry. | ||
The thiolate-protected Cu NCs with accurate compositions and atomically precise structures have attracted increasing attention in various electrocatalytic reactions.124–130 Protected by the organic ligands, the Cu atoms in Cu NCs typically exhibit a stable +1 valence, and their electronic structure can be easily tuned through metal doping or ligand engineering.131–134 In 1989, Hori et al. found that when used as cathodes, copper electrodes exhibited catalytic activity for the conversion of CO2 to alkanes, olefins, and alcohols.135 This discovery prompted an increasing number of researchers to focus on the copper-based materials for CO2RR.129,132,136–141 Understanding the catalytic mechanism of Cu-catalyzed CO2RR helps design catalysts that reduce onset overpotential and enhance the product selectivity. Robinson et al. reported an ultrasmall Cu13(MBO)12H NC consisting of 13 Cu atoms and 12 2-mercaptobenzoxazole ligands, which exhibits oxygen reduction activity and produces H2 and CO as the major products in CO2RR.142 Huang et al.143 utilized the S- and N-containing ligands to synthesize a small Cu6 NC with symmetry-broken Cu–S2N1 active sites (named Cu6-(MBD)6, MBD = 2-mercaptobenzimidazole) to shift the product of CO2RR from HCOOH/CO to higher-value hydrocarbons, such as CH4 and C2H4. The symmetry-broken Cu–S2N1 active sites facilitate specific coordination between the C atom of CO2 and the Cu–S2N1 site, significantly reducing the formation barrier of *COOH and favoring *CO formation to form deep reduced hydrocarbons while hindering the HCOOH pathway. Moreover, Tan et al. demonstrated that decorating an odd number of Cu atoms onto the icosahedral Au13 surface could lead to asymmetric surfaces to enhance the CO2RR efficiency.134 They synthesized a series of Au13Cux (x = 1–4) NCs to investigate the relationship between the degree of symmetry breaking and electrocatalytic activity. Their results showed that Au13Cu3 with the highest degree of symmetry breaking and an unsaturated coordinated Cu1S2 site benefits the CO2 adsorption and shows superior CO2RR activity for CO formation.
Recently, Zang et al. revealed the synergistic effect of Cu+ and neighboring S sites at the Cu–S interface of Cu4(MMI)4 and Cu8(MMI)4(tBuS)4 NCs (MMI = 2-mercapto-1-methylimidazole) in reducing CO2 to deep-reduced products (Fig. 12a).144 Owing to its ultra-small size, it exposes unique metal sites in low coordination environments, which are ideal for enhancing the adsorption of reaction intermediates.143 Furthermore, the S sites can serve as the potential active centers for water activation.145,146 As shown in Fig. 12b, the experimentally measured Faraday efficiency (FE) for CH4 reaches a maximum of ∼53.7% at −1.2 VRHE for Cu4, which indicates that CH4 is the main product of the Cu4 electrocatalyst. Comparatively, Cu8 exhibits a remarkable preference for C2+ products, achieving a maximum FE of 58.5% at −1.3 VRHE. Their DFT calculations showed that the CO2 molecule spontaneously adsorbs onto the Cu sites of Cu4, with an adsorption free energy of −0.46 eV (Fig. 12c). The formation of *CHO is commonly considered the rate-determining step in CH4 production, which displays a reaction barrier notably lower than that for *COH formation, CH3OH formation or CO desorption, indicating the low CO selectivity and more favorable CH4 formation. They also found that the formation of the *CHOCO dimer via C–C coupling is energetically less favorable, consistent with the experimentally observed suppressed production of C2+ products on Cu4. Based on their in situ ATR-SEIRAS characterizations and theoretical calculations, they proposed a possible mechanism for CH4 formation on Cu4: CO2 → *COOH → *CO → *CHO → *CH2O → *OCH3 → CH4 (Fig. 12d). They stated that the S sites facilitate H2O dissociation to generate *H species, which directly participate in the protonation process on adjacent Cu sites for the protonation of *CO to *CHO. Their study highlights the important role of Cu–S dual sites in promoting CO2 electroreduction toward high-value hydrocarbon products.
 | ||
| Fig. 12 (a) Schematic structure of Cu4 and Cu8. (b) Faraday efficiency (FE) values of CO2RR products on the Cu4 in the flow cell at various applied potentials. (c) Calculated Gibbs free energy diagram of CO2RR for Cu4. The * denotes the adsorbed intermediate on the active sites. (d) Proposed mechanism of Cu4 for the formation of CH4, along with the optimized structures of the reaction intermediates. Adapted with permission from ref. 144 Copyright 2024, Wiley-VCH GmbH. | ||
Negishi et al. used a one-pot reduction strategy to synthesize three different Cu NCs successfully:147 [Cu11(PTT)9(PPh3)6]2+ (Cu11PTT) (PTT: p-toluenethiolate); [Cu11(ABT)9(PPh3)6]2+ (Cu11ABT) (ABT: m-aminobenzethiolate) NCs, which share a similar structural architecture but differ in their thiolate ligand systems; and [Cu18H2(PTT)15(PPh3)6] (Cu18PTT) NC, which features the same PTT ligand as Cu11PTT but adopts a distinct structural architecture. Interestingly, the Cu11 NC with the p-toluenethiol ligand exhibits selectivity toward HCOOH production, while substituting p-toluenethiol with m-aminobenzethiol shifted the selectivity to the competitive side reaction, which demonstrates that the interplay between the core structure confinement and surface ligand environment governs their catalytic behavior. Therefore, the specific electrocatalytic applications of Cu NCs are regulated by precise control of their structure and surface chemistry. Notably, the current experimental and theoretical investigations on the electrocatalytic applications of thiolated Ag- and Cu-based NCs are still in the infant stage. In this regard, unlocking the correlation between the interface structure, catalytic activity, and the atomic level reaction mechanism of metal NCs represents a key challenge to be accomplished for future studies.
Note that the support plays a critical role in modulating the interfacial and catalytic properties of metal nanoclusters.148 Taking the extensively studied Au25(SR)18 as an example, Tsukuda et al.149 chose layered double hydroxide (LDH) as the support, focusing on the interaction of Au25 with the support. Following appropriate thermal aging, some of the pMBA (p-mercaptobenzoic acid) ligands remain owing to the electrostatic interaction between the cationic LDH and the deprotonated pMBA. This cluster–support interface improves the robustness during the catalytic reaction for 5-hydroxymethylfurfural oxidation. Tsukuda and his colleagues150 also selectively removed thiolate ligands exposed to vacuum from Au25(SR)18 NCs by calcination, where the partially thiolated Au25 NCs were successfully supported on porous carbon via van der Waals (vdW) interactions, which inhibited the diffusion and aggregation of Au25 NCs and exhibited higher durability in the catalytic aerobic oxidation of alcohols. Moreover, Yang et al.151 used sulfur-doped graphene (S-G) as a support for Au25(PET)18 (PET = phenylethanethiol) and found that the sulfur dopant on S-G partially replaces the thiol ligand to bind with the staple Au atom. This interfacial behavior achieves the anchoring of Au25(PET)18 on S-G and exhibits high stability in the electrocatalytic nitrogen reduction reaction (NRR). Particularly, they demonstrated that the anchored Au25 clusters preserve ∼80% NRR activity after four days of continuous operation, which is a significant improvement compared to the unloaded clusters tested under the same NRR conditions. In most cases, the support materials regulate the catalytic properties by modulating the electronic properties, the support-induced strain and the formation of interface active sites.
Summary and outlook
Atomically precise metal NCs exhibit unique geometrical, electronic and catalytic properties that differ from those of discrete atoms and plasmonic nanoparticles. Several strategies, including ligand removal, ligand engineering, atom-precise alloying/doping, and interface engineering, have been introduced to modulate the electronic and catalytic performance of metal NCs. This work provides a brief review of the latest developments in thiolate-protected metal NC systems for electrocatalytic applications. The results show that ligand effects, doping, size effects, and synergistic interactions play key roles in determining catalytic activity and selectivity, contributing significantly to the atomic-level understanding of the reaction mechanism.However, several challenges remain, and a few perspectives are discussed herein. First, atomic-level doping is one of the most popular strategies for manipulating novel alloy NCs. Although most dopants are concentrated in elements such as Pd, Pt, Cu, Ag/Au, Ir, Cd, and Hg, introducing other cheaper elements such as Fe, Mn, Ni, Co, Ga, In, and Tl into novel alloy NCs continues to pose significant challenges. Second, owing to the unique and tunable physicochemical properties of atomically precise metal NCs, they have significant potential to expand the current electrocatalytic applications to other complex multi-electron reduction processes, such as the nitrogen (nitrite) reduction reaction or C–N coupling reactions, to achieve more highly valued chemicals. Third, in addition to the structure of the cluster catalysts, the electrolyte environment is an important factor in mediating ion transport and diffusion. It greatly affects electrocatalytic activity and selectivity by tuning the local microenvironment at the electric double layer. Nevertheless, the influence of electrolyte effects, such as the cation or anion type, concentration and charge state as well as the underlying regulation mechanism, has been less explored. Finally, when elucidating the electrocatalytic mechanism, most DFT studies rely on the reaction thermodynamics calculated based on the charge-neutral model, which neglects the reaction kinetics and does not consider the influence of potential and explicit solvation. A typical example is the controversial debate on the active site of thiolated gold NCs in electrochemical CO2RR. The implementation of electrochemical potential and kinetic information plays an essential role. Thus, to resolve the site-specific active site and reaction mechanism, it is necessary to apply computational methods with higher accuracy to simulate the realistic electrochemical interface of metal NCs under electrolytes and probe the structure–property relation. Moreover, advanced in situ electrochemical characterization techniques are important in monitoring the evolution of the cluster structure and identifying the key reaction intermediates/products to determine the reaction process. To date, hundreds of atomically precise metal NCs have been structurally resolved, and new NC structures continue to emerge. Establishing a clear correlation between structure and catalytic properties is crucial for discovering novel heterogeneous catalysts with unique catalytic functions. Machine learning is considered one of the most promising approaches to accelerate the screening process. Therefore, developing efficient algorithms and descriptors to speed up the identification of potential candidates for experimentation is critical. However, this requires the development of more efficient methods and new strategies, including hybrid approaches that address simulation challenges across different lengths and timescales152
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22473017) and Chongqing Science and Technology Commission (CSTB2024NSCQ-MSX0250).References
- S. Wacławek, V. V. Padil and M. Černík, Ecol. Chem. Eng. S, 2018, 25, 9–34 Search PubMed.
- F. Zaera, Chem. Rev., 2022, 122, 8594–8757 CrossRef CAS PubMed.
- P. Harris, Interdiscip. Sci. Rev., 1998, 23, 184 Search PubMed.
- B. Cornils and W. A. Hermann, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, VCH, Weinheim, 1996, vol. 2, p. 1048 Search PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- X. Yuan and M. Zhu, Inorg. Chem. Front., 2023, 10, 3995–4007 RSC.
- A. Baghdasaryan and T. Bürgi, Nanoscale, 2021, 13, 6283–6340 RSC.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- X. Kang and M. Zhu, Chem. Mater., 2020, 33, 39–62 CrossRef.
- M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. D. M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews, S. Barcikowski, M. M. Kappes, W. J. Parak, C. M. Aikens and I. Chakraborty, J. Am. Chem. Soc., 2021, 143, 9405–9414 CrossRef CAS PubMed.
- X. Zou, X. Kang and M. Zhu, Chem. Soc. Rev., 2023, 52, 5892–5967 RSC.
- D. Bain, S. Maity and A. Patra, Phys. Chem. Chem. Phys., 2019, 21, 5863–5881 RSC.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- Y.-P. Xie, Y.-L. Shen, G.-X. Duan, J. Han, L.-P. Zhang and X. Lu, Mater. Chem. Front., 2020, 4, 2205–2222 RSC.
- R. Kobayashi, Y. Nonoguchi, A. Sasaki and H. Yao, J. Phys. Chem. C, 2014, 118, 15506–15515 CrossRef CAS.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- M. Hesari and Z. Ding, Acc. Chem. Res., 2017, 50, 218–230 CrossRef CAS PubMed.
- N. Hirata, M. Sato, E. Tsunemi, Y. Watanabe, H. Tsunoyama, M. Nakaya, T. Eguchi, Y. Negishi and A. Nakajima, J. Phys. Chem. C, 2017, 121, 10638–10644 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed.
- E. Porret, L. Sancey, A. Martín-Serrano, M. I. Montañez, R. Seeman, A. Yahia-Ammar, H. Okuno, F. Gomez, A. Ariza, N. Hildebrandt, O.-B. Fleury, J.-L. Coll and X. L. Guével, Chem. Mater., 2017, 29, 7497–7506 CrossRef CAS.
- X. Zhu, L. Chen, Y. Liu and Z. Tang, Polyoxometalates, 2023, 2, 9140031 CrossRef.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- Z. Wu and R. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS PubMed.
- N. De Silva and L. F. Dahl, Inorg. Chem., 2005, 44, 9604–9606 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni, T. Lovato and S. Zacchini, Inorg. Chem., 2013, 52, 4384–4395 CrossRef CAS PubMed.
- T. Chen, H. Lin, Y. Cao, Q. Yao and J. Xie, Adv. Mater., 2022, 34, 2103918 CrossRef CAS PubMed.
- J. Yang, F. Yang, C. Zhang, X. He and R. Jin, ACS Mater. Lett., 2022, 4, 1279–1296 CrossRef CAS.
- X. Liu, T. Ki, G. Deng, S. Yoo, K. Lee, B.-H. Lee, T. Hyeon and M. S. Bootharaju, Nanoscale, 2024, 16, 12329–12344 RSC.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- Z. Lei, X.-K. Wan, S.-F. Yuan, J.-Q. Wang and Q.-M. Wang, Dalton Trans., 2017, 46, 3427–3434 Search PubMed.
- L. Qin, G. Ma, L. Wang and Z. Tang, J. Energy Chem., 2021, 57, 359–370 CrossRef CAS.
- Q. Tang and D.-E. Jiang, Chem. Mater., 2017, 29, 6908–6915 CrossRef CAS.
- G. Hu, R. Jin and D.-E. Jiang, Nanoscale, 2016, 8, 20103–20110 RSC.
- F. Sun and Q. Tang, Nanoscale, 2021, 13, 819–831 RSC.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 8710–8713 CrossRef CAS PubMed.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- S. Zhuang, L. Liao, J. Yuan, N. Xia, Y. Zhao, C. Wang, Z. Gan, N. Yan, L. He, J. Li, H. Deng, Z. Guan, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 4510–4514 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320, 238–250 CrossRef.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- S. Sharma, W. Kurashige, K. Nobusada and Y. Negishi, Nanoscale, 2015, 7, 10606–10612 RSC.
- S. Sharma, S. Yamazoe, T. Ono, W. Kurashige, Y. Niihori, K. Nobusada, T. Tsukuda and Y. Negishi, Dalton Trans., 2016, 45, 18064–18068 RSC.
- S. Hossain, D. Hirayama, A. Ikeda, M. Ishimi, S. Funaki, A. Samanta, T. Kawawaki and Y. Negishi, Aggregate, 2023, 4, e255 Search PubMed.
- Q. Zhu, X. Huang, Y. Zeng, K. Sun, L. Zhou, Y. Liu, L. Luo, S. Tian and X. Sun, Nanoscale Adv., 2021, 3, 6330–6341 RSC.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 Search PubMed.
- M. H. Naveen, R. Khan and J. H. Bang, Chem. Mater., 2021, 33, 7595–7612 Search PubMed.
- G. Ma, F. Wang, R. Jin, B. Guo, H. Huo, Y. Dai, Z. Liu, J. Liu and S. Li, Int. J. Mol. Sci., 2025, 26, 1582 Search PubMed.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 Search PubMed.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 Search PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-E. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- D. Eguchi, M. Sakamoto and T. Teranishi, Chem. Sci., 2018, 9, 261–265 RSC.
- T. C. Jones, L. Sumner, G. Ramakrishna, M. B. Hatshan, A. Abuhagr, S. Chakraborty and A. Dass, J. Phys. Chem. C, 2018, 122, 17726–17737 CrossRef CAS.
- K. Kwak, W. Choi, Q. Tang, D.-E. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- L. Sumner, N. A. Sakthivel, H. Schrock, K. Artyushkova, A. Dass and S. Chakraborty, J. Phys. Chem. C, 2018, 122, 24809–24817 CrossRef CAS.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-E. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- H. Shen, Q. Zhu, J. Xu, K. Ni, X. Wei, Y. Du, S. Gao, X. Kang and M. Zhu, Nanoscale, 2023, 15, 14941–14948 RSC.
- M. Sera, S. Hossain, S. Yoshikawa, K. Takemae, A. Ikeda, T. Tanaka, T. Kosaka, Y. Niihori, T. Kawawaki and Y. Negishi, J. Am. Chem. Soc., 2024, 146, 29684–29693 CrossRef CAS PubMed.
- F. Sun, L. Qin, Z. Tang and Q. Tang, Chem. Sci., 2024, 15, 16142–16155 RSC.
- O. López-Estrada, N. Mammen, L. Laverdure, M. M. Melander, H. Häkkinen and K. Honkala, ACS Catal., 2023, 13, 8997–9006 CrossRef.
- L. Luo, X. Zhou, Y. Chen, F. Sun, L. Wang and Q. Tang, Chem. Sci., 2025, 16, 3598–3610 RSC.
- Z. Liu, H. Tan, B. Li, Z. Hu, D.-E. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- C. Deng, F. Li and Q. Tang, J. Phys. Chem. C, 2019, 123, 27116–27123 Search PubMed.
- F. Sun, C. Deng, S. Tian and Q. Tang, ACS Catal., 2021, 11, 7957–7969 CrossRef CAS.
- X. Zou, S. He, X. Kang, S. Chen, H. Yu, S. Jin, D. Astruc and M. Zhu, Chem. Sci., 2021, 12, 3660–3667 RSC.
- C.-G. Shi, J.-H. Jia, Y. Jia, G. Li and M.-L. Tong, CCS Chem., 2023, 5, 1154–1162 CrossRef CAS.
- B. Huang, X. Zhao and Y. Pei, Mol. Catal., 2023, 540, 113030 CrossRef CAS.
- S. Li, A. V. Nagarajan, S. Zhao, G. Mpourmpakis and R. Jin, J. Phys. Chem. C, 2021, 125, 24831–24836 CrossRef CAS.
- C. Mu, B. Wang, Q. Yao, Q. He and J. Xie, Nano Res., 2024, 17, 9490–9497 CrossRef CAS.
- M. Zhou, K. Li, Y. Pei, S. Jin and M. Zhu, J. Phys. Chem. Lett., 2023, 14, 11715–11724 CrossRef CAS PubMed.
- W. Suzuki, R. Takahata, Y. Mizuhata, N. Tokitoh, S. Xue and T. Teranishi, Chem. Sci., 2024, 15, 18896–18902 RSC.
- Z. Sun, J. Wang, L. Su, Z. Gu, X.-P. Wu, W. Chen and W. Ma, J. Am. Chem. Soc., 2024, 146, 20059–20068 CrossRef CAS PubMed.
- S. Zhuang, D. Chen, Q. You, W. Fan, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2022, 134, e202208751 CrossRef.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- B. Kim, H. Seong, J. T. Song, K. Kwak, H. Song, Y. C. Tan, G. Park, D. Lee and J. Oh, ACS Energy Lett., 2019, 5, 749–757 Search PubMed.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 Search PubMed.
- A. V. Nagarajan, R. Juarez-Mosqueda, M. J. Cowan, R. Jin, D. R. Kauffman and G. Mpourmpakis, SN Appl. Sci., 2020, 2, 1–12 Search PubMed.
- W. Choi, H. Seong, V. Efremov, Y. Lee, S. Im, D.-H. Lim, J. S. Yoo and D. Lee, J. Chem. Phys., 2021, 155, 014305 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 Search PubMed.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Angew. Chem., Int. Ed., 2021, 133, 14684–14691 Search PubMed.
- Y. Sun, X. Liu, K. Xiao, Y. Zhu and M. Chen, ACS Catal., 2021, 11, 11551–11560 CrossRef CAS.
- M. Rybacki, A. V. Nagarajan and G. Mpourmpakis, Environ. Sci.: Nano, 2022, 9, 2032–2040 RSC.
- F. Sun and Q. Tang, Nanotechnology, 2021, 32, 352001 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, 134, e202211771 CrossRef.
- J. McKay, M. J. Cowan, C. A. Morales-Rivera and G. Mpourmpakis, Nanoscale, 2021, 13, 2034–2043 Search PubMed.
- S. Chen, M. Li, S. Yu, S. Louisia, W. Chuang, M. Gao, C. Chen, J. Jin, M. B. Salmeron and P. Yang, J. Chem. Phys., 2021, 155, 051101 CrossRef CAS PubMed.
- H. Seong, Y. Jo, V. Efremov, Y. Kim, S. Park, S. M. Han, K. Chang, J. Park, W. Choi, W. Kim, H. C. Chang, J. S. Yoo and D. Lee, J. Am. Chem. Soc., 2023, 145, 2152–2160 CrossRef CAS PubMed.
- F. Sun, L. Qin, Z. Tang, G. Deng, M. S. Bootharaju, Z. Wei, Q. Tang and T. Hyeon, Chem. Sci., 2023, 14, 10532–10546 Search PubMed.
- D. Alfonso, Chem. Phys. Lett., 2024, 850, 141462 Search PubMed.
- P. Zhu, X. Zhu, X. Zhou, F. Sun, Y. Chen, L. Wang, Z. Tang and Q. Tang, Small, 2025, 21, 2411226 Search PubMed.
- P. Zhu, Y. Chen and Q. Tang, Nanoscale, 2025, 17, 9490–9501 RSC.
- Y. Chen, X. Zhou, X. Liu, Z. Tang, L. Wang and Q. Tang, J. Am. Chem. Soc., 2025, 147, 2699–2713 Search PubMed.
- S. M. Han, M. Park, J. Kim and D. Lee, Angew. Chem., Int. Ed., 2024, e202404387 Search PubMed.
- R. E. Vos and M. T. Koper, ChemElectroChem, 2022, 9, e202200239 Search PubMed.
- F. Sun, X. Zhou, L. Qin, Z. Tang, L. Wang and Q. Tang, ACS Catal., 2025, 15, 4605–4617 CrossRef CAS.
- Z. Liu, J. Chen, B. Li, D.-E. Jiang, L. Wang, Q. Yao and J. Xie, J. Am. Chem. Soc., 2024, 146, 11773–11781 CrossRef CAS PubMed.
- J. Zhao, A. Ziarati, A. Rosspeintner and T. Bürgi, Angew. Chem., Int. Ed., 2024, 136, e202316649 Search PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 128, 9900–9904 CrossRef.
- W. Rong, H. Zou, W. Zang, S. Xi, S. Wei, B. Long, J. Hu, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 466–472 CrossRef CAS PubMed.
- L. Chen, F. Sun, Q. Shen, L. Qin, Y. Liu, L. Qiao, Q. Tang, L. Wang and Z. Tang, Nano Res., 2022, 15, 8908–8913 CrossRef CAS.
- D. Yang, J. Wang, Q. Wang, Z. Yuan, Y. Dai, C. Zhou, X. Wan, Q. Zhang and Y. Yang, ACS Nano, 2022, 16, 15681–15704 CrossRef CAS PubMed.
- H. Seong, M. Choi, S. Park, H.-W. Kim, J. Kim, W. Kim, J. S. Yoo and D. Lee, ACS Energy Lett., 2022, 7, 4177–4184 CrossRef CAS.
- X. Lin, W. Ma, K. Sun, B. Sun, X. Fu, X. Ren, C. Liu and J. Huang, J. Phys. Chem. Lett., 2020, 12, 552–557 CrossRef PubMed.
- S. Yoo, S. Yoo, G. Deng, F. Sun, K. Lee, H. Jang, C. W. Lee, X. Liu, J. Jang, Q. Tang, Y. J. Hwang, T. Hyeon and M. S. Bootharaju, Adv. Mater., 2024, 36, 2313032 CrossRef CAS PubMed.
- Y. Ye, X. Zhou, Y. Chen, L. Wang and Q. Tang, Chem. Commun., 2025, 61, 2790–2793 RSC.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. Liu, D. Lee and D.-E. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 Search PubMed.
- Y. E. Shi, J. Ma, A. Feng, Z. Wang and A. L. Rogach, Aggregate, 2021, 2, e112 CrossRef CAS.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- C. Dong, R. W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 Search PubMed.
- G. Li, J. Hou, X. Lei, D. Li, E. Yu, W. Hu, X. Cai, X. Liu, M. Chen and Y. Zhu, Angew. Chem., Int. Ed., 2023, 135, e202216735 CrossRef.
- S. Li, X. Yan, J. Tang, D. Cao, X. Sun, G. Tian, X. Tang, H. Guo, Q. Wu, J. Sun, J. He and H. Shen, Chem. Mater., 2023, 35, 6123–6132 CrossRef CAS.
- J. Sun, X. Yan, L. Wang, Z. Xie, G. Tian, L. Wang, A. He, S. Li, Q. Guo, L. Chao, J. He and H. Shen, Inorg. Chem., 2023, 62, 9005–9013 CrossRef CAS PubMed.
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2022, 145, 3401–3407 CrossRef PubMed.
- M. Ding, L. Tang, X. Ma, C. Song and S. Wang, Commun. Chem., 2022, 5, 172 CrossRef CAS PubMed.
- X. Ma, F. Sun, L. Qin, Y. Liu, X. Kang, L. Wang, D.-E. Jiang, Q. Tang and Z. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC.
- Y. Tan, G. Sun, T. Jiang, D. Liu, Q. Li, S. Yang, J. Chai, S. Gao, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317471 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- T.-D. Nguyen-Phan, C. Wang, C. M. Marin, Y. Zhou, E. Stavitski, E. J. Popczun, Y. Yu, W. Xu, B. H. Howard, M. Y. Stuckman, I. Waluyo, P. R. Ohodnicki Jr and D. R. Kauffman, J. Mater. Chem. A, 2019, 7, 27576–27584 RSC.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- L. J. Liu, Z. Y. Wang, Z. Y. Wang, R. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 134, e202205626 Search PubMed.
- Y. Wang, Y. Zhu, X. Zhu, J. Shi, X. Ren, L. Zhang and S. Li, ACS Catal., 2022, 13, 714–724 CrossRef.
- S. Ajmal, G. Yasin, A. Kumar, M. Tabish, S. Ibraheem, K. A. Sammed, M. A. Mushtaq, A. Saad, Z. Mo and W. Zhao, Coord. Chem. Rev., 2023, 485, 215099 Search PubMed.
- J. Du, B. Cheng, H. Yuan, Y. Tao, Y. Chen, M. Ming, Z. Han and R. Eisenberg, Angew. Chem., Int. Ed., 2023, 62, e202211804 CrossRef CAS PubMed.
- H. Han, Y. Yao, A. Bhargava, Z. Wei, Z. Tang, J. Suntivich, O. Voznyy and R. D. Robinson, J. Am. Chem. Soc., 2020, 142, 14495–14503 CrossRef CAS PubMed.
- Q. J. Wu, D. H. Si, P. P. Sun, Y. L. Dong, S. Zheng, Q. Chen, S. H. Ye, D. Sun, R. Cao and Y. B. Huang, Angew. Chem., Int. Ed., 2023, 135, e202306822 CrossRef.
- J. K. Li, J. P. Dong, S. S. Liu, Y. Hua, X. L. Zhao, Z. Li, S. N. Zhao, S. Q. Zang and R. Wang, Angew. Chem., Int. Ed., 2024, 63, e202412144 CrossRef CAS PubMed.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- M. Wang, S. Liu, B. Chen, M. Huang and C. Peng, Chem. Eng. J., 2023, 451, 139056 CrossRef CAS.
- S. Biswas, Y. Shingyouchi, M. Kamiyama, M. K. Jena, M. Ogami, T. Kawawaki, B. Pathak and Y. Negishi, Small, 2025, 2500302 CrossRef PubMed.
- J. Hu, Y. Liu, A. Al-Salihy, Y. Zhou, W. Fan, P. Tao, H. Huang, H. Li, M. Chen and S. Li, ChemCatChem, 2025, 17, e202401904 CrossRef CAS.
- S. Masuda and T. Tsukuda, ACS Catal., 2023, 13, 16179–16187 CrossRef CAS.
- K. Sakamoto, S. Masuda, S. Takano and T. Tsukuda, ACS Catal., 2023, 13, 3263–3271 CrossRef CAS.
- M. Li, B. Zhang, T. Cheng, S. Yu, S. Louisia, C. Chen, S. Chen, S. Cestellos-Blanco, W. A. Goddard III and P. Yang, Nano Res., 2021, 14, 3509–3513 CrossRef CAS.
- S. Malola and H. Häkkinen, Nat. Commun., 2021, 12, 2197 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |