
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-sacrifice of sulfide electrolytes facilitating stable solid-state sodium–sulfur batteries†
Yi
Yuan
a,
Yang
Hu
 a,
Yi
Gan
a,
Zhiliang
Dong
a,
Yijia
Wang
a,
Enzhong
Jin
a,
Mingrui
Yang
a,
Frederick Benjamin
Holness
a,
Vinicius
Martins
a,
Qingsong
Tu
*b and
Yang
Zhao
*a
a,
Yi
Gan
a,
Zhiliang
Dong
a,
Yijia
Wang
a,
Enzhong
Jin
a,
Mingrui
Yang
a,
Frederick Benjamin
Holness
a,
Vinicius
Martins
a,
Qingsong
Tu
*b and
Yang
Zhao
*a
aDepartment of Mechanical and Materials Engineering, University of Western Ontario, London, Ontario N6A 5B9, Canada. E-mail: yzhao628@uwo.ca
bDepartment of Mechanical Engineering, Rochester Institute of Technology, Rochester, NY 14623, USA. E-mail: qhteme@rit.edu
First published on 19th March 2025
Abstract
Sulfide electrolytes have emerged as the preferred choice for solid-state sodium–sulfur (Na–S) batteries due to their excellent compatibility with sulfur cathodes. Despite their advantages, such as high ionic conductivity, mechanical flexibility, and enhanced safety, challenges like narrow electrochemical stability windows and inadequate interfacial contact persist and require urgent resolution. Contrary to the conventional approach of minimizing electrolyte degradation, this study leverages the decomposition of a typically unstable sulfide electrolyte, Na3SbS4 (NAS), to enhance both cathode and anode interfaces. By elucidating the reversible self-redox mechanism of NAS, we demonstrate that a cathode composite containing NAS-S as a co-active material achieves an exceptional discharge capacity at room temperature, surpassing the theoretical specific capacity of sulfur alone. Furthermore, the strong interaction between NAS and a Na-based alloy anode leads to the in situ formation of a homogeneous interlayer. This passivation layer, acting as both an electron regulator and protective barrier, prevents further electrolyte corrosion and dendrite penetration, resulting in remarkable cycling stability. This novel approach of utilizing electrolyte decomposition offers a fresh perspective on interface engineering, advancing solid-state Na–S batteries towards practical, next-generation energy storage solutions with improved capacity output and cycle life.
Broader contextRoom-temperature sodium–sulfur batteries are regarded as ideal batteries for 100–150 years, which use low-cost and widely abundant sodium and sulfur as electrodes. Solid-state sodium–sulfur batteries with the implementation of solid-state electrolytes could further enable higher safety, good cycling stability and improved capacity. The overall electrochemical performances of solid-state sodium–sulfur batteries largely depend on the feasibility of solid electrolytes (SEs). From a conventional perspective, SE decomposition usually leads to battery performance decay and circumventing SE decomposition is an effective measure for stable cycling. Contrary to the conventional approach of minimizing electrolyte degradation, this study leverages the decomposition of a typically unstable sulfide electrolyte to enhance both cathode and anode interfaces. As a result, solid-state sodium–sulfur batteries with self-sacrificed SEs demonstrate high capacity and good stability even under low stack pressure, advancing solid-state sodium–sulfur batteries towards practical, next-generation energy storage solutions with improved capacity output and cycle life. |
Introduction
Sulfur and sulfide materials have received significant attention for their application as cathode materials for advanced sodium batteries.1–3 However, there are significant ongoing challenges that urgently need to be addressed, particularly the shuttle effects of polysulfide intermediates and dendrite penetration.4–6 By utilizing solid electrolytes (SEs) as ionic conductors and separators, solid-state Na–S batteries were relieved from the shuttle effects with enhanced safety.7 Among advanced SEs, sulfides exhibit outstanding ionic conductivity and desirable mechanical flexibility at room temperature.8 Meanwhile, the high compatibility with the sulfur-based cathode ensures the superiority of sulfides for solid-state Na–S batteries.9,10 However, the interfacial issues with both the cathode and anode hinder the practical applications of sulfide SEs. Due to the narrow electrochemical window, sulfides often undergo oxidation–reduction during electrochemical cycling, generating side products, usually with low ionic conductivity, and higher electronic conductivity compared to the original SEs.11–14 In the cathode, the decomposed byproducts block the ionic conducting pathways, severely aggravating the sluggish redox kinetics of sulfur. On the anode side, the decomposition of sulfide SEs generally forms a mixed-conducting interphase (MCI), where the electronic conductivity of the MCI highly promotes dendrite growth at the interface.15,16 Therefore, from a conventional perspective, circumventing SE decomposition is an effective measure for stable cycling. One typical approach reported in previous studies is to widen the electrochemical window by modifying the anionic dopants (e.g. oxides and halides).17,18 Another is to fabricate a sandwich-type cell with a stable second layer underneath the unstable main layer to avoid its direct contact with the Na metal/alloy anode.19–22Two typical Na-based sulfide SEs, Na3PS4 (NPS) and Na3SbS4 (NAS), have been widely used in the studies of solid-state Na–S batteries, considering their high ionic conductivity and thermal stability. NAS, as a superionic conductor, exhibits a conductivity of over 1 mS cm−1 at room temperature. Nevertheless, the presence of antimony lowers the stability, leading to a narrow electrochemical window of 1.83–1.9 V.23 In comparison, NPS exhibits a much wider electrochemical window of 1.39–2.45 V, while the ionic conductivity was reported in a lower range (0.1–0.4 mS cm−1).23–25 Both SEs are regarded as promising candidates in the applications of solid-state batteries for safety and performance reasons. In terms of long-lasting interfacial design purposes, NPS has been often used for its high stability.14,26,27 However, inspired by the deceptive Na stability of NPS observed from the experimental approach, we proposed that electrochemical stability should not be the only criterion for a reliable interface.
In this work, we demonstrated the self-sacrifice of sulfide SE through predictable decomposition to resolve intrinsic solid–solid interfacial issues, thus enabling stable solid-state Na–S batteries with high capacity (Scheme 1). Unlike the conventional perspective, NAS illustrates a reactivity-guided interface design at both the cathode and the anode. By employing S and NAS as co-active materials, an outstanding discharge capacity (over 1900 mA h g−1@0.13 mA cm−2) at room temperature was delivered, surpassing the theoretical specific capacity of sulfur (1675 mA h g−1). In further morphological and spectroscopic characterization studies, the redox mechanism of NAS was elucidated. Meanwhile, the Sb–S ionic-conducting phase was conserved, providing rapid ionic transportation during cell operation. The vigorous reaction of NAS also facilitated anode interface stabilization by initiating the in situ formation of a visible, homogeneous interlayer. Benefiting from the passivation provided by the amorphous stable phase, the parent SE layer was shielded from further corrosion and dendrite penetration. Moreover, by undergoing the simultaneous alloying process of Na–Sb and Na–Sn, the anode structure was kept intact without over-consumption of Na-ions. However, when the “stable” NPS was employed as the cathode additive, cracks were introduced due to the poor ductility of NPS during the cold pressing process, resulting in the insufficient participation of sulfur. Furthermore, with NPS as the SE layer, dendrite formation was exceedingly promoted during the initial cycles. Overall, by mediating the cathode–SE and SE–anode interfacial issues, a fresh insight into the decomposition of sulfide SE is unveiled, inspiring the practical applications of unstable sulfide SEs in long-lasting solid-state Na–S batteries.
 | ||
| Scheme 1 Schematics of interfacial features between sulfide SEs and the sulfur-based composite cathode/sodium alloy anode in solid-state Na–S batteries. | ||
Results and discussion
Mechanical and electrochemical enhancement by sulfide SEs in the cathode
Two common sulfide SEs, NAS and NPS, were used in this study, to thoroughly understand the interfacial behavior of sulfide in solid-state Na–S batteries.28–31 The composite cathode was prepared through sulfur encapsulation, forming a sulfur–carbon (S@C) composite, followed by a ball-milling process to mix with SEs. Powder X-ray diffraction (XRD) and thermogravimetric (TG) tests were performed to study the prepared S@C composite (Fig. 1a and b). From the TG result, the sulfur content in the composite was found to be approximately 60%. The XRD pattern revealed the amorphous structure of the as-prepared S@C composite. After ball-milling with NAS or NPS, the XRD results of the S@C-SE composite cathodes are shown in Fig. 1a. The features of NAS shown in the XRD pattern of the S@C-NAS cathode diminished, indicating a partial loss of structure, which was not noted for NPS in the cathode. To examine the differences between NAS and NPS in solid-state Na–S batteries, the cells were assembled, employing S@C-SE composite cathodes (SEs: NAS or NPS), NAS as the SE layer, and the Na15Sn4 (Na–Sn) alloy anode. Interestingly, a contrast in capacity utilization was observed from the galvanostatic charge–discharge (GCD) curves of S@C-NAS and S@C-NPS cathodes, which delivered initial discharge capacities of 1976 and 1246 mA h g−1, respectively (Fig. 1c). The S@C-NAS cell presented a capacity higher than the theoretical specific capacity of the 16-electron reaction from S to Na2S (1675 mA h g−1). Noticeably, from the GCD profile of S@C-NAS, a two-stage reaction was observed. Until now, the redox mechanism for solid-state Na–S remains blurry. In solid-state Li–S batteries, lack of solvation in SEs prevents the formation of long-chain polysulfides, causing the one-step solid–solid reaction between S and Li, represented in the GCD profile as a single-plateau with a mixture of Li2S2 and Li2S as the final product.7,32 The extra discharge/charge plateaus observed in this study highly suggest a side reaction other than the routine of S ↔ Na2S. XRD presented evidence pointing to the decomposition of NAS with cycling. After the initial cycle, the XRD pattern of the NAS-based cathode composite presented a hollow figure, referring to the amorphous state (Fig. S2, ESI†). The glass transition can potentially be attributed to the decomposition of SE, which will be further justified in the following section. In contrast, the features that belong to the NPS crystal structure were retained in the cathode composite after the first cycle. | ||
| Fig. 1 Fabrication of a S-based composite cathode with sulfide SEs. (a) XRD patterns of a sintered S@C composite with S reference, NAS, and NPS solid electrolytes, and the corresponding cathode composites. (b) TG curves of the S@C composite. (c) Galvanostatic charge–discharge profiles after the first cycle for NAS- and NPS-based cathodes. | ||
Scanning electron microscopy (SEM) was used to visualize the microstructure of the composite cathodes before and after the first cycle (Fig. S3, ESI†). Before cycling, a homogeneous surface was observed in the pristine S@C-NAS cathode after cold pressing, and an adequate cathode interface was maintained after the first cycle (Fig. S3a and b, ESI†). Meanwhile, cracks were randomly distributed among the surface of the S@C-NPS cathode, suggesting poor tri-phase contact before cycling and thus the low utilization of sulfur (Fig. S3c and d, ESI†). It is believed that the microstructure difference could be decided by the different ductility and brittleness of NAS and NPS.33 The criteria can be generalized into the ratio (G/B ratio) between the shear modulus (G) and the bulk modulus (B), representing whether the dominance is controlled by plasticity or fracture.34–36 The high ductility of NAS shown in SEM visualization was proved by its low intrinsic G/B ratio of 0.19, which is comparably lower than the NPS of 0.458.37 This indicates the superiority of NAS to form a uniform surface under high uniaxial pressure, and the resilience to the consistent volume change of sulfur. Therefore, the NAS with a low G/B ratio provides a solution to maintain sufficient solid–solid interfacial contact within the composite cathode, thus ensuring a well-established ionic percolation throughout the cycling.
To trace the multiple plateaus and additional capacity, differential capacity (dQ/dV) analysis was conducted for both NAS and NPS-based cathodes, emphasizing the capacity output at the platform voltages. Fig. 2a shows the dQ/dV results for the cells using S@C-NAS and NAS-C cathodes. The S@C-NAS cathode, unlike the previously described one-plateau solid conversion, featured three reduction peaks located at 1.75 V, 1.05 V (with a shoulder at 1.25 V), and 0.7 V for discharge. In the charging phase, two peaks at 1.75 and 1.05 V can each be paired up with an oxidation peak at 2.15 and 1.85 V. The reduction peak located at 0.7 V was partially terminated by the voltage cut-off, without a matching oxidation peak, likely pinpointing the reduction to Na2S and Na–Sb. Tuning the cut-off voltage to 0.7 V allowed the cessation of full conversion. Therefore, the composition of the final product can be engineered to improve the reversibility.38 When NAS was used alone as an active material to mix with carbon (NAS-C), the cell was tested, with the GCD curve and cycling performance shown in Fig. S4a and c (ESI†), respectively. The total areal loading of NAS remained the same as in the S@C-NAS cathode, tested in the identical cell configuration. An areal capacity of 0.26 mA h cm−2 was delivered by the NAS-C cathode, observed from Fig. S4a (ESI†), whereas that of the original S@C-NAS was 0.81 mA h cm−2. Although the precise percentage of NAS contribution in the composite cathode cannot be directly calculated by this comparison, its impact on overall capacity is substantial. The GCD curve of NAS-C was further converted to dQ/dV analysis. As seen from Fig. 2a, the peaks of the NAS-C cathode are aligned with S@C-NAS, with the reduction peaks at 1.6 and 1.25 V and oxidation peaks at 2.15 and 2.3 V. The shift of the first plateau to a lower voltage in the 2nd cycle again occurred, emphasizing the identical activation process. The redox peaks of the NAS-C cathode confirmed the reversible redox of NAS and located the peaks that represent this behavior. Therefore, additional peaks observed in S@C-NAS cells at 1.05 V (reduction) and 1.85 V (oxidation) correspond to the redox of sulfur, consistent with the voltage range for the conversion from Na2S2 to Na2S in the liquid state.39–41 Furthermore, cyclic voltammetry (CV) tests were conducted at room and high temperature (60 °C). At 60 °C (Fig. S5b, ESI†), the CV exhibited a three-stage discharge with a two-stage charge, matching with the dQ/dV results. The peak shifting indicates irreversibility, due to NAS decomposition facilitated by the improved thermodynamics at 60 °C. Whereas at room temperature (Fig. S5a, ESI†), blurry curves were obtained with high reversibility. Altogether, the analysis revealed a redox mechanism involving the main S to Na2S reaction and reversible NAS reactions.
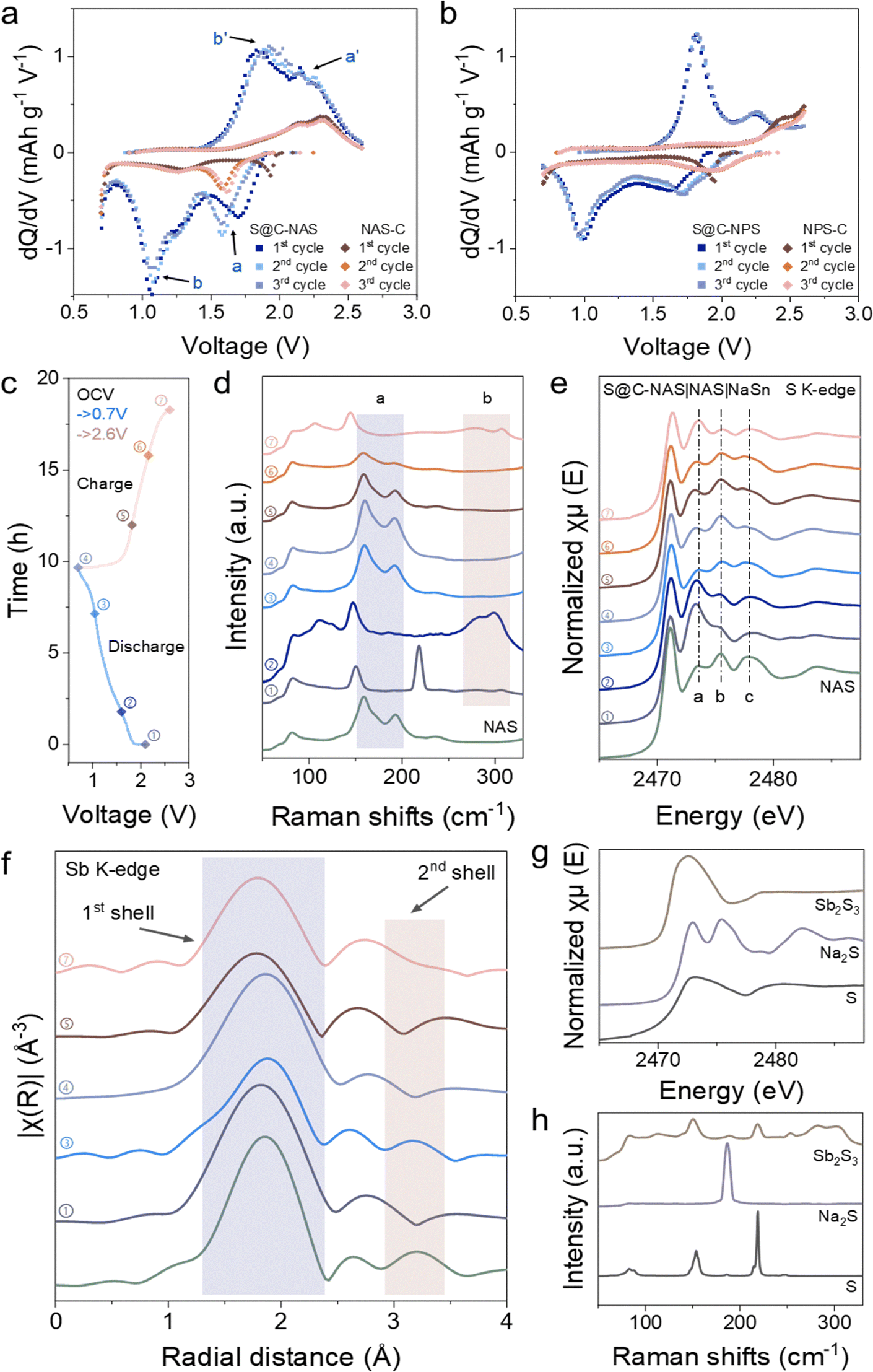 | ||
| Fig. 2 Redox pathway of the NAS-involved cathode in solid-state Na–S batteries. Differential capacity profiles of (a) NAS-based and (b) NPS-based, sulfur-contained and non-contained cathode. (c) GCD voltage profile of the S@C-NAS cathode as an indicator for DoD and SoC stages corresponding to the samples for spectroscopic characterization. (d) Ex situ Raman investigation of the S@C-NAS cathode at different stages of DoD and SoC. (e) Ex situ S K-edge XANES of the S@C-NAS cathode at the corresponding voltages. (f) Sb K-edge EXAFS in the R-space of the S@C-NAS cathode. (g) S K-edge XANES and (h) Raman spectrum of reference materials. | ||
For comparison, Fig. 2b shows the dQ/dV results for the cells with S@C-NPS and NPS-C cathodes. Similarly, two pairs of reduction–oxidation peaks (1.7–2.25 V and 1–1.8 V) were observed in the S@C-NPS cell, suggesting the inevitability of sulfide electrolyte decomposition at the cathode interface. However, by comparing the intensity, the capacity attributed to NPS decomposition was relatively low. Moreover, the NPS-C GCD profile highlighted the mismatch of NPS self-redox reactions compared to the composite cathode, suggesting the poor utilization of NPS in the overall cathode redox. The difference between the utilization of SEs (NAS and NPS) redox in cathodes can be explained by electronegativity. Compared to P (2.1)–S (2.5) bonds, electrons in Sb (1.9)–S (2.5) tend to be distributed more in the surroundings of S atoms, which makes Sb–S easier to oxidize in the charging process. Density functional theory (DFT) calculations further proved the difference between the reactivity of NAS and NPS. Tables S1 and S2 (ESI†) present the competing phases that would likely form by NAS and NPS at certain voltage ranges. Due to the much narrower voltage range, NAS was predicted to easily undergo both reductive and oxidative decomposition. Meanwhile, NAS demonstrated a multi-step reaction with several ion-conducting intermediates. In comparison, NPS presented a working voltage higher than that of the sulfur-based cathode, again proving its low contribution to the composite. This self-redox phenomenon, also observed in Li-based sulfide electrolytes like Li6PS5Cl and Li10GeP2S12, demonstrates that despite the narrow electrochemical stability window of sulfide SEs, their decomposition can be reversible and contribute to higher overall capacity.42–44
To further recognize the self-redox reactions of the NAS, various spectroscopic characterization studies were carried out. Herein, ex situ Raman spectra were recorded to trace the change of NAS-related species at different voltages (Fig. 2c). As shown in Fig. 2d and Fig. S6a (ESI†), after a ball-milling process, the S@C-NAS cathode before cycling already showed the decomposition of NAS. The dim feature peaks of SbxSy species at Raman shifts of 283 and 299 cm−1 were observed, replacing the peaks ‘a’ designated for the sodiated SbS43− tetrahedron (159.4 and 192 cm−1), the low intensity of the peaks might be due to the strong signal of pure S (150.5 and 218 cm−1). As a reference, Sb2S3 was measured (Fig. 2h), and the matching duo-peaks were observed, denoted as ‘b’. Similar Raman characteristics of SbxSy species were also described in previous studies.45 At the initiation of the discharging process, 1.6 V, representing the beginning of the first noted voltage plateau, the above-mentioned area ‘b’ for SbxSy became apparent, with the NAS features remaining unseen. In the subsequent depth of discharge (DoD) at 1 V, the Raman intensities of two characteristic NAS peaks increased. Meanwhile, the SbxSy features were not observed. This shows the sodiation of SbxSy during the first plateau as excess Na+ swarms into the cathode. In the next charging process, the Raman intensity of the sodiated SbS43− tetrahedron (‘a’) gradually disappeared, as the sign for SbxSy (‘b’) reappeared in due course at 2.6 V, the 100% state of charge (SoC). Corresponding to the group of peaks ‘a’, the main peaks of SbS43− species assigned to symmetrical/asymmetric stretches (362, 384, and 404 cm−1) shared an identical evolution trend (Fig. S6b, ESI†). Combined with the voltage profile, the Raman spectra unfolded a general mechanism for NAS involvement in the cathode reactions.
The ex situ X-ray absorption spectroscopy (XAS) study was also carried out to investigate the redox reaction of the NAS. The peak features of S K-edge X-ray absorption near edge structure (XANES) of the S@C-NAS composite cathode at various discharge/charge states confirmed the proposed catholyte redox (Fig. 2d). At first glance, the unchanged edge position during cycling suggests that the sulfur's oxidation state remained at −2 within the intact Sb–S coordination. Due to the overlapping energy ranges and complexity of the Na–S and Sb–S mixture, accurate peak identification in the near-edge region was challenging. However, by referencing NAS, S, Na2S, and Sb2S3 (Fig. 2g), the evolution trend can be elucidated. Three main peaks at post-edge were noted for the pristine NAS at 2473.5, 2475.45, and 2478 eV. Before cycling, NAS decomposed during the ball-milling process, and an intensive peak ‘a’ was detected, with a marginalized peak ‘b’ due to the partial loss of Na–S bonding. Into a deeper DoD, peak ‘a’ faded, and a feature identical to the pristine NAS was observed at 1 V after the NAS-related reaction defined in dQ/dV. At 100% DoD, Na2S formation can be recognized. During charging, peak ‘b’ gradually diminished with peak ‘a’ re-emerged, presenting Na2S consumption and sulfur formation. To isolate the electrochemical behavior of NAS, we conducted a parallel XANES analysis on the NAS-C cathode at the identical voltage regions (Fig. S7, ESI†). Once the discharge was initiated, the reacted catholyte progressively reformed to NAS, highlighted by the continuous weakening of peak ‘a’. The identical feature with the pristine NAS became visible at 1 V, in line with the evolution of the composite cathode. During charging, peak ‘a’ gradually intensified. At 100% SoC, the spectra featured with alike details (intensive peak ‘a’) to the pristine cathode sample. In the operation of both cathodes, no shifts in energy levels nor changes in specific features are observed, suggesting the capability of Sb–S coordination as an entity to accept/liberate Na-ion. Meanwhile, in the ion-conducting phase, the persistence of the SbS43− tetrahedron played a crucial role in constructing a rapid Na-ion delivery.
A similar progression trend was also recognized in Sb K-edge Extended X-ray Absorption Fine Structure (EXAFS) in R-space (Fig. 2f). FEFF modeling was conducted on the CIF data of NAS obtained from ICSD (PDF no. 01-085-8198) to ensure the correct baseline (Fig. S8, ESI†).21 The phase-uncorrected experimental data of NAS overlaps with the calculated model for both shells. The 1st shell (1.86 Å) of the Sb central atom represents the Sb–S bonding in the SbS43− tetrahedron, whereas the 2nd shell (3.2 Å) signifies Na atom on the local chemistry of Sb, interconnected by a shared S atom. Before cycling, the 2nd shell feature vanishes, indicating that Na was detached from the Sb–S coordination, testifying to the decomposition of NAS. Meanwhile, the slight leftward shift of the 1st shell implies the shrinkage of the Sb–S bonding structure, likely due to more S atoms being shared between two Sb atoms. The shorter Sb–S bonding distance suggests the formation of SbxSy species observed in the Raman spectra. Following the uptake of Na+, the 2nd shell reappeared at 1 V, and the 1st shell rebounded to its original distance, referring to the reformation of NAS after the first discharge plateau. At the deeper DoD, the cathode reduction approaches the low-voltage region, and the 2nd shell again becomes invisible. By tuning the cut-off voltage to 0.7 V, the process was terminated to avoid the destruction of the Sb–S structure. Therefore, the 1st shell representing the tetrahedron is kept intact. In the subsequent charging process, the 2nd shell is not recovered, with a noticeable shrinkage of the 1st shell appearing, suggesting the further decomposition of NAS to form a tight Sb–S structure as Na-ion departure.
Overall, the spectroscopic results together unveil the redox pathway of NAS in the S-based composite cathode. Thus, explaining the high capacity of solid-state Na–S batteries hidden behind the blurry working mechanism. Meanwhile, the reversible nature persistently preserves a sufficient ion-conducting phase to ensure the basic functionality of NAS as the electrolyte additive in the composite cathode.
Formation of a passivation layer and promotion of SE–anode stability
In the reported literature, NPS was more favorable for solid-state Na batteries, due to the wide electrochemical window (NPS: 1.06 V and NAS: 0.07 V).23 NPS has also been used as an interlayer between unstable SEs (such as NAS, NaZrYCl, NaTaCl6, etc.) and Na (metal/alloy) anodes.21,26,46 However, an opposite phenomenon was observed in this study. Different cell configurations were designed to study the anode interfaces and bulk properties of both NAS and NPS when coupling with S@C–NAS cathode and Na–Sn anode (Fig. S9, ESI†). As shown in Fig. S9a and c (ESI†), the cells heavily suffered from voltage fluctuations during charging, which eventually led to self-charging behavior when using NPS as the main layer or the interlayer. The jagged voltage profile, identified as a soft-short-circuit (SSC), was caused by the localized Na+ plating, ultimately leading to dendrite formation and irreversible cell failure.47,48 In comparison, a long-lasting cycling performance was observed when NAS was used individually (Fig. S9b, ESI†), suggesting a stable stripping/plating of Na+, antithetical to its ultra-narrow electrochemical stability window.To understand the mechanism, morphological analysis combining X-ray computed tomography (XCT), and SEM was carried out. Cells were assembled in customized Swagelok-type solid-state models, designed for XCT measurements under no stack pressure, where NAS or NPS were used as main layers, with S@C-NAS composite cathodes, and cycled 50 times. The low stack pressure was used to promote the dendrite growth for visualizing the dendrite penetration for XCT measurement.49Fig. 3a and f demonstrate the electrochemical performances of NAS and NPS-based XCT cells, respectively, in line with the results obtained in regular cell models. Fig. 3b and c show the XCT images of the NAS cell from top and side views. Interestingly, an extra layer with a uniform structure was formed in situ between the SE and the anode during the cycling. Whereas in the same configuration for the NPS cell, a nonuniform interface is observed in Fig. 3g and f. At the NPS–anode interface, various low-greyscale areas were sporadically distributed, with dendritic extension upwardly growing into the SE layer, strongly indicating the dendrite formation and the ascending penetration, in line with its electrochemical performance. A direct comparison of the surficial and cross-sectional SEM views of NAS and NPS, focusing on the interface above Na–Sn, further illustrated the previously proposed discrepancy, as shown in Fig. 3d and e. A uniform surface of the formed interlayer from the NAS cell is observed in Fig. 3d. The exceedingly compact morphology was amorphous-like, significantly different from that of the pristine crystalline NAS electrolyte (pressed pellet) (Fig. S10, ESI†). An XRD test was carried out for the self-formed interlayer, as shown in Fig. S11 (ESI†), further unveiling its amorphous nature. In contrast, hollow regions are seen at the SE–anode interface for NPS cells in Fig. 3e, potentially due to the grain boundaries in the crystal structure of NPS, thus explaining the localized growth of dendrites and the failure of NPS to inhibit further penetration.
 | ||
| Fig. 3 Visualization of the SE–anode interfacial morphology. (a) GCD profile of a NAS-based cell. (b) Planar and (c) vertical XCT images of the NAS–anode interface. Scale bar, 500 μm. Planar SEM images of the SE–anode interface of (d) NAS and (e) NPS at two scales of 3 and 50 μm. (f) GCD profile of the NPS-based cell. (g) Planar and (h) vertical XCT images of the NPS–anode interface. Scale bar, 500 μm. | ||
As extended validation, cross-sectional views of the SE–anode interface during the first cycle were evaluated by SEM (Fig. S12, ESI†). Presented in Fig. S12a (ESI†), the interlayer was accomplished even at the first discharge, and an alloy-like, porous morphology was recognized and maintained after charging. It is worth noting that the morphology of the Na–Sn alloy was preserved after cycling, indicating that the Na-ion was plated into the remaining Sn skeleton (Fig. S12b, ESI†). In contrast, massive corrosions were observed in NPS along the cracks after the first discharge, corresponding to the voltage fluctuation in the GCD profile (Fig. S12c, ESI†). In addition, a change of anode morphology was noticed, the alloy skeleton vanished at the post-charging state, signifying that the Na–Sn alloy was not formed (Fig. S12d, ESI†).
To verify the composition of the interlayer formed by NAS, an XAS study integrating Sb K-edge EXAFS in R-space with S K-edge XANES was conducted (Fig. 4a and b). In the Sb K-edge (Fig. 4a), the bonding information of Sb–S included in the 1st shell (1.86 Å) vanished, as well as the Sb–S–Na represented by the 2nd shell (3.2 Å). Instead, there was a notable appearance of the Na–Sb bonding structure at 2.69 Å before phase correction, matching the Na3Sb bonding distance. The Na3Sb FEFF model was calculated based on the CIF obtained from ICSD (PDF no. 01-074-1162).50 The formation of Na3Sb supported the speculation that Na-ion plating was promoted by undergoing co-alloying to create a homogeneous electric field. In the S K-edge of the interlayer, the Na2S formation is evident. Being an electronic insulator, a stable Na2S phase can induce passivation, avoiding further decomposition of the SE. Energy-dispersive X-ray spectroscopy (EDS) provides elemental mapping of the cross-sectional SEM image of the interlayer (Fig. S13, ESI†). In the mapping images, the uniform distribution of Sb and Sn across the interface and the anode, together with the homogeneous detection of Na, denoted the synchronous alloying of Na–Sb and Na–Sn.
 | ||
| Fig. 4 Interface evolution of NAS/NPS-Na–Sn and investigation of the NAS-formed interlayer. (a) Sb K-edge EXAFS in R-space and (b) S K-edge XANES of the in situ formed NAS–anode interlayer. (c) Galvanostatic cycling profiles of NAS and NPS at 0.51 mA cm−2–0.51 mA h cm−2, and (d) the enlargement of the interested region. Time-resolved EIS at OCV for 24 h of (e) NAS and (f) NPS symmetric cells. | ||
Na–Sn|SEs|Na–Sn symmetric cells with NAS or NPS were tested to further demonstrate the SE–anode interfacial phenomenon, contrasting SSC and SE interfacial protection (Fig. 4c and Fig. S14, ESI†). The symmetric cells were cycled at a current density of 0.51 mA cm−2 with a cut-off capacity of 0.51 mA h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2. During cycling, NPS showed steady polarization. In comparison, the cell with NAS experienced a rising overpotential but stabilized after approximately 120 hours, corresponding to our previous observation on interlayer growth. The subsequent voltage stabilization can be attributed to the passivating behavior of the stable interlayer. Although, at first glance, the Na–Sn|NPS|Na–Sn cell exhibited no augmentation of interfacial resistance, which would make NPS favorable for the alloy anode. However, the abnormal plating was noticed at the tip of each cycle in NPS in the locally magnified figure, as shown in Fig. 4d. Due to the defective Na+ deposition, the stability of NPS towards Na–Sn from the symmetric cell performance might be deceptive. This is potentially due to the MCI formed by the interfacial decomposition of NPS, which is reflected in the full cell performance as a soft breakdown.51
cm−2. During cycling, NPS showed steady polarization. In comparison, the cell with NAS experienced a rising overpotential but stabilized after approximately 120 hours, corresponding to our previous observation on interlayer growth. The subsequent voltage stabilization can be attributed to the passivating behavior of the stable interlayer. Although, at first glance, the Na–Sn|NPS|Na–Sn cell exhibited no augmentation of interfacial resistance, which would make NPS favorable for the alloy anode. However, the abnormal plating was noticed at the tip of each cycle in NPS in the locally magnified figure, as shown in Fig. 4d. Due to the defective Na+ deposition, the stability of NPS towards Na–Sn from the symmetric cell performance might be deceptive. This is potentially due to the MCI formed by the interfacial decomposition of NPS, which is reflected in the full cell performance as a soft breakdown.51
Upon the interfacial phenomenon of NAS and NPS in operating cells, we intended to investigate the formation of the interface under different voltage ranges, even at the open-circuit voltage (OCV). To probe this phenomenon, time-resolved EIS tests were conducted on the symmetric cells (Na–Sn|SEs|Na–Sn) holding at OCV for a time range of 24 hours at room temperature. After holding for an hour, a fresh semi-circle starts to emerge in the NAS-based cell, causing the internal resistance of the cell to increase, unveiling the initiation of an additional interlayer (Fig. 4e). Given another lapse of 14 hours, the complete formation of the interlayer was observed, leading to the stabilization of the overall resistance afterward. From the evolution trend of cell internal resistance, the interlayer formation was confirmed to be a one-time occurrence, proving the passivating effect previously discussed. In contrast, the NPS cell before cycling showed an unchanged Nyquist plot with stable internal resistance over 24 hours, similar to its performance in the galvanostatic electrochemical tests (Fig. 4f).
To consider all the factors that may impose influence on Na deposition in the anode, a model of the Na-ion distribution during charging was performed, in the circumstance if a small defect exists on the anode surface. Fig. S15a (ESI†) presents a homogeneous voltage distribution of NAS around the pore. Furthermore, a comparison was demonstrated between NAS and NPS of Na-ion current density distribution along the top surface of the interface, as shown in Fig. S15b (ESI†). As approaching the defect center, NPS exhibited a more inhomogeneous current distribution, potentially due to the joint effect of low bulk conductivity and the lack of surficial protection. Conventionally, a higher ionic conductivity of the SE is usually preferred, as it helps enhance Na-ion diffusion and deposition. In addition, the formation of the protective interface can further homogenize the surficial current density distribution, which is beneficial for Na deposition stability.52
Inspired by the contrasting electrochemical behaviors of NAS and NPS, which are opposite to their theoretical stability against Na/Na+, we visualized the SE–anode interfaces in solid-state Na–S batteries. Using a combination of XCT and SEM imaging, an in situ formed homogeneous interlayer was observed in NAS-based cells. In contrast, NPS cells exhibited a degraded interface with poor structural integrity, potentially explaining the promotion of dendrite penetration. In the symmetric cells, the interlayer formed by the decomposition of NAS gradually stabilized, demonstrating the passivating nature. Furthermore, owing to the sturdy mechanical properties and compact surface brought by its amorphous morphology, the self-formed interlayer highly contributes to dendrite inhibition. Benefiting from this interlayer, a long-lasting, stable solid-state Na–S battery can be achieved.
Electrochemical performance of solid-state batteries
Extended electrochemical performance tests of solid-state Na–S batteries were carried out to reveal the benefits of the NAS self-sacrifice. The cycling stability and rate performance tests were evaluated at a stack pressure of 250 MPa at room temperature, unless otherwise specified. Fig. 5a presents the long-term cycling stability of solid-state Na–S batteries with NAS and NPS-based cathodes and NAS as the SE layer at a current density of 0.127 mA cm−2. The S@C-NAS cathode delivered an ultra-high initial discharge specific capacity of 1976 mA h g−1 with an initial Coulombic efficiency (ICE) of 86.8%. After 140 cycles, the NAS cell maintained a high specific capacity of 1295 mA h g−1. In comparison, the S@C-NPS cathode displayed a much lower initial discharge specific capacity of 1246 mA h g−1. Suffering constantly from capacity fading, 728 mA h g−1 can be upheld after 140 cycles. When elevating the current density to 0.41 mA cm−2 (Fig. 5b), the NAS cell can still achieve a high specific capacity of 803 mA h g−1 after 100 cycles. Whereas only 534 mA h g−1 was maintained after 100 cycles using the NPS cathode, the fluctuating coulombic efficiency (CE) suggests an unsteady behavior (Fig. S16, ESI†). The rate performance of solid-state Na–S batteries applying NAS was further evaluated, as shown in Fig. 5c and d for the GCD curves and cycling performance, respectively. The cell exhibited a superior rate capability of 889 mA h g−1 at 0.41 mA cm−2 (0.5C) and 389 mA h g−1 at 0.82 mA cm−2 (1C) at room temperature. | ||
| Fig. 5 Electrochemical performance of NAS-aided cells with asymmetric configuration. (a) Cycling performance of NAS, and NPS-based cathode at 0.127 mA cm−2 at room temperature. (b) Cycling performance of the S@C-NAS cathode at 0.5C (0.41 mA cm−2). Rate capability shown by (c) GCD and (d) cycling performance of the S@C-NAS cathode at 0.1–1C. (e) GCD profiles of the S@C-NAS cathode operated at various stack pressures, and (f) the corresponding cycling performance at 0.127 mA cm−2. (g) GCD profiles of the S@C-NAS cathode at 0.127 mA cm−2 at 60 °C. | ||
The cathode and anode interfaces established by NAS spark the feasibility of practical solid-state Na–S batteries that operate at a low stack pressure. Fig. 5e and f exhibit the first-cycle charge–discharge profiles and cycling performances of the NAS cells at various stack pressures at 125 MPa, 62.5 MPa, and non-stack pressure (∼20 MPa). Impressively, after reducing the stack pressure to 125 MPa and 62.5 MPa, initial discharge specific capacities of 1991 and 1712 mA h g−1 were maintained. However, as expected, the ICE dropped to 79.6%/69.3%, followed by the quick capacity fading after 50 cycles. After lowering the stack pressure to 62.5 MPa, SSC was also observed during cycling, attributed to the insufficient interfacial contact of SE and anode causing the localized Na-ion deposition. Surprisingly, upon removing the stack pressure, using the upper bar of the cell model as the sole downforce, the cycling stability of the cell was remarkably enhanced. An initial discharge specific capacity of 1051 mA h g−1, and ICE of 86% is exhibited, followed by capacity retention of 88.26%/74.5% after 50/80 cycles respectively, free from the dendrite penetration. The increasing overpotential and decreasing specific capacity at non-stack pressure can be attributed to the loss of intimate interfacial contact. It is surprising that at a low stack pressure closer to atmospheric, the cell can deliver a larger number of cycles without Na reaching the cathode compared to the cells operating at 62.5 MPa and 125 MPa. Removing the external uniaxial stack allowed more space for deposited Na to elongate, alleviating the SSC phenomenon. However, this may cause poor solid–solid interfacial contact across the cell, which further explains the loss of capacity due to the joint effects of both cathode and anode sides. This is the first time to report stable and high-capacity solid-state Na–S batteries at low stack pressures, which might open a new route for future practical applications.
By elevating the temperature, redox kinetics and thermodynamics of the cathode can be promoted. To further demonstrate the existence of NAS self-redox, the cells with S@C-NAS cathode were tested at 60 °C. In the first cycle of GCD shown in Fig. 5g, there is a notable appearance of a third voltage plateau between 0.8 and 0.7 V, bringing the total initial specific capacity to an astonishing value of 2700 mA h g−1. Nevertheless, the last plateau failed to be maintained even though most of the capacity can be reversed (1917 mA h g−1@30 cycles), potentially due to the full decomposition of NAS to irreversible final products.
Conclusion
Herein, we have re-visited and re-investigated the application of NAS in solid-state Na–S batteries. Unlike the traditional understanding, we first reveal the novel mechanism of self-sacrifice and decomposition of sulfide SEs for achieving high-performance solid-state Na–S batteries. The functions of NAS can be summarized as (i) owing to high ductility, intimate interfacial contact was maintained during the volume change of sulfur to ensure the ion-conductive pathway on the cathode side. (ii) NAS can serve as a co-active material via self-redox to contribute extra reversible capacity throughout the long-term cycling. The mechanism of the SbS43− tetrahedron to undergo (de)sodiation was then unveiled by a comprehensive analysis of various characterization studies. (iii) On the anode side, an in situ formed passivation layer containing Na2S and Na3Sb was achieved between NAS and Na–Sn anodes. The self-formed interlayer with an amorphous morphology and robust mechanical properties can inhibit dendrite penetration and maintain structural integrity. Benefiting from decomposition, the NAS-aided solid-state Na–S batteries can deliver an ultra-high initial specific capacity of 1976 mA h g−1 with a capacity retention of 1295 mA h g−1 after 140 cycles, at a current density of 0.127 mA cm−2. In addition, we also demonstrated the high capacity and good stability of the solid-state Na–S batteries at low stack pressures, which is promising for future practical applications. Contrary to the traditional perspective, in this study, we utilize the intrinsic high reactivity vs. Na/Na+ of the sulfide electrolyte, NAS, to resolve critical cathode/anode interfacial issues, providing practical engineering towards a long-lasting solid-state Na–S battery.Methods
Synthesis of solid electrolytes
Two types of solid electrolytes (SEs) were prepared separately. Na3SbS4 (NAS) was prepared using the co-melting method. The starting materials, Na2S (Sigma Aldrich, 98%), Sb2S3 (Sigma Aldrich, 99.99%), and S (Sigma Aldrich, 99.99%) were well-mixed with an agate mortar, and then sealed in a quartz tube in an argon-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm). The sample was heated to 750 °C with a heating rate of 0.5 °C min−1 and rapidly quenched in liquid nitrogen. The obtained precursor was ground and pressed into a pellet under 900 MPa, followed by an annealing process at 330 °C for 2 hours also with a heating rate of 0.5 °C min−1. Na3PS4 (NPS) was synthesized by high-energy ball-milling followed by an annealing process. The starting materials, Na2S and P2S5 (Sigma Aldrich, 99%) were placed in a ZrO2 ball-milling pot (45 mL) with ZrO2 balls (50 g, 5 mm in diameter), under an argon-filled atmosphere. The process was performed using planetary ball-milling apparatus (Fritsch, Pulverisette 7) for 25 hours at 550 rpm. The obtained powder was pressed into a pellet under 900 MPa, sealed in a quartz tube, and then annealed under vacuum at 275 °C for 2 hours with a heating rate of 1 °C min−1. All the resulting products were transferred to a glovebox for further use.Preparation of the composite cathode
Sulfur-encapsulated carbon composites were first prepared through melting treatment. Starting materials, sulfur, and electro-conductive carbon Ketjen Black (KB EC-600) at a weight ratio of 60:40 were first ball-milled to achieve a well-mixed state. The obtained powders were sealed in a quartz tube, followed by an annealing process at 155 °C for 12 h under vacuum, denoted as S@C.30 The composite was then milled with the prepared NAS or NPS at a weight ratio of 2:8 for 1 hour at 300 rpm to acquire the S@C-NAS/NPS cathode composite.
Synthesis of the alloy anode
A sodium–tin (Na15Sn4) alloy was synthesized by the ball-milling method. Freshly cut Na cubes (Sigma Aldrich, 99.9%) and tin powder (Sigma Aldrich, 99.99%) were mixed with a molar ratio of 15:4 in a ZrO2 ball-milling pot with ZrO2 balls. The process was performed using a planetary ball-milling machine (Nanda QM-3SP0.4 series) at 300 rpm for 5 hours.
Electrochemical measurements
First-principles calculations
DFT calculations within the projector augmented wave (PAW) formalism were performed, as implemented in the Vienna ab initio simulation package (VASP). We employed a mixed scheme of the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional. Each calculation was performed with a k-point grid of 3 × 3 × 3 and an energy cut-off of 520 eV to maintain compatibility with the Materials Project database in constructing phase diagrams. 2 × 2 × 2 supercells of the unit cells tec-NAS were created to obtain the model cells. The cell was first relaxed and then allowed ionic movement for energy optimization under the Nose–Hoover thermostat (NVT ensemble).Thermodynamic stability analysis
For (electro)chemical stability analysis, the grand canonical phase diagram at various sodium chemical potentials was formulated. For a given sodium chemical potential μNa, we consider the grand potential ∅ of a compound following eqn (1), where c is the composition of the compound, E[c] is the enthalpy, and nNa[c] is the sodium concentration at composition c.| ∅[c,μNa] = E[c] − nNa[c]μNa | (1) |
To compute the decomposition reaction potentials, sodium chemical potentials were converted to the reaction voltage referenced by sodium metal, with eqn (2):
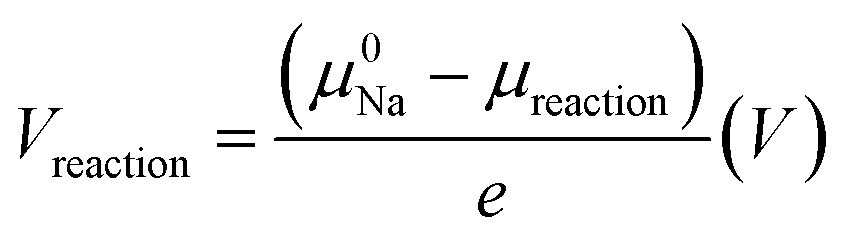 | (2) |
Modeling of Na diffusion in SE and deposition in pores
The transport of Na+ in the SE can be described by the ohmic relations:| ∇2ϕSE = 0, and i = −σNa+∇ϕSE | (3) |
The current density involved in the deposition is controlled by the surface overpotential (η) through the commonly used Butler–Volmer equation:
 | (4) |
| ict = −in = −i·nSE | (5) |
Here, ict is the current density due to charge transfer across the interface, in is the local current density normal to the interface and iexc is the reference exchange current density of the Na/Na+ reaction. αa and αc are the anodic and cathodic charge-transfer coefficients for the reaction, respectively; η is the surface overpotential: η = ϕAnode − V0, where ϕAnode is the electrode potential, which equals zero for the Na metal anode and V0 is the open circuit potential.
An in-house-developed code based on the finite element method and the MOOSE framework was implemented to solve all the PDEs numerically.52 The default values of the parameters (such as conductivities for Na-ion transport in the SE) used in this work were obtained from experimental measurements of NAS and NPS SE and are listed in Table 1.
| Name | Symbol | Value | Unit |
|---|---|---|---|
| Exchange current density at the electrode/SE interface | i k exc | 1.3 | mA cm−2 |
| Ionic conductivity of NAS | σ Na+ | 0.9 | mS cm−1 |
| Ionic conductivity of NPS | σ Na+ | 0.1 | mS cm−1 |
Characterization
TG analysis was performed using a DSC SDT Q600 thermogravimetric analyzer system (TA Instruments) to quantify the sulfur content in the S@C composite, with a heating rate of 5 °C min−1, up to 550 °C. Lab-scale XRD measurements were carried out using a Bruker AXS D8 Advance (50 kV, 6 kW mm−2) with Cu Kα radiation (λ = 1.5406 Å). Air/moisture sensitive XRD samples were covered using Kapton tape. Raman samples were sealed between two transparent microscopy slides, and spectra were recorded using a HORIBA Scientific LabRAM HR Raman spectrometer with a laser beam at 532 nm. Scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800 field-emission scanning electron microscope (acceleration voltage of 5 kV).All synchrotron-related characterization studies were carried out at the Canadian Light Source (CLS). X-ray absorption spectra (XAS) were recorded at the Soft X-ray Microcharacterization Beamline (SXRMB) for S K-edge and Hard X-ray Micro-Analysis (HXMA) for Sb K-edge. The XAS data were analyzed in the Athena program, and EXFAS data including FEFF modeling were processed in the Artemis program, both programs are from the Demeter software package. All of the sample preparations were conducted in a glovebox and transported within the aluminum-plastic bags. First, after transferring the cell model into the glovebox, tri-layer cell pellets were obtained from the ceramic die. The cathode pieces and SE–anode interfaces were detached from the pellet and stored in a 5 mL centrifuge tube for further use. For SXRMB, the samples were attached to carbon tape and measured in a vacuum chamber. For HXMA, all samples were sealed using Kapton tape and then kept in aluminum-plastic bags throughout the measurement. Synchrotron X-ray computed tomography (XCT) was carried out at the Biomedical Imaging and Therapy Facility-Wiggler Insertion Device Beamline (BMIT-ID). XCT cells were fabricated by the same cold pressing process but in a customized Swagelok-type solid-state cell with PEEK cases (Fig. S18, ESI†). The cells were cycled and the whole pellet was kept in the cell model during the XCT measurement to obtain the in situ image. To minimize damage, all cells were transported within several layers of bubble wrap. The XCT images were collected at 90 keV with an optical detector (a resolution of 3.61 μm per pixel, a magnification of 1.8×, and a field of view of 9.24 × 9.24 mm). CT reconstruction was performed using the open-source Tofu UFO-KIT software and further processed in the Fiji image processing package within ImageJ software.
Author contributions
Y. Y. conceived the idea, collected the experimental data and drafted the raw manuscript. Y. Y., Y. H., and Y. G. analyzed the data and discussed the project; E. Z., and M. Y. participated in some discussions. Y. Y., Y. G., and Y. W. collected synchrotron data; Y. Y. and Z. D. collected XRD data. Y. Y., Y. H., Y. W., Y. G., and F. B. H revised the manuscript. Q. T. performed the first principles calculations. Y. Z. supervised the entire project. All authors contributed to the discussions and revisions of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge the support from the Natural Science of Engineering Research Council of Canada (NSERC), the Canada Foundation of Innovation (CFI), and the University of Western Ontario. The synchrotron research was conducted at the SXRMB, HXAM and BMIT beamlines of the Canadian Light Source (CLS), which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. The authors also gratefully acknowledge the great help from Dr Mohsen Shakouri and Dr Qunfeng Xiao at the SXRMB beamline, Dr Ning Chen at the HXAM beamline, and Dr Arash Panahifar and Dr Sergey Gasilov at the BMIT beamline, at the Canadian Light Source. Lastly, the author greatly appreciated the contribution of Mr Han Su in designing the customized Swagelok-type solid-state cell.References
- X. Liu, Y. Li, X. Xu, L. Zhou and L. Mai, J. Energy Chem., 2021, 61, 104–134 CrossRef CAS.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS.
- J. He, A. Bhargav, J. Okasinski and A. Manthiram, Adv. Mater., 2024, 36, 2403521 CrossRef CAS.
- D. Kumar, S. K. Rajouria, S. B. Kuhar and D. K. Kanchan, Solid State Ionics, 2017, 312, 8–16 CrossRef CAS.
- L. Lin, C. Zhang, Y. Huang, Y. Zhuang, M. Fan, J. Lin, L. Wang, Q. Xie and D.-L. Peng, Small, 2022, 18, 2107368 CAS.
- S. A. Novikova, D. Y. Voropaeva and A. B. Yaroslavtsev, Inorg. Mater., 2022, 58, 333–348 CAS.
- D. Cao, X. Sun, F. Li, S.-M. Bak, T. Ji, M. Geiwitz, K. S. Burch, Y. Du, G. Yang and H. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202302363 CAS.
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Energy Storage Mater., 2018, 14, 58–74 Search PubMed.
- I.-H. Chu, C. S. Kompella, H. Nguyen, Z. Zhu, S. Hy, Z. Deng, Y. S. Meng and S. P. Ong, Sci. Rep., 2016, 6, 33733 CAS.
- D. Wang, L.-J. Jhang, R. Kou, M. Liao, S. Zheng, H. Jiang, P. Shi, G.-X. Li, K. Meng and D. Wang, Nat. Commun., 2023, 14, 1895 CAS.
- R. Xu, F. Han, X. Ji, X. Fan, J. Tu and C. Wang, Nano Energy, 2018, 53, 958–966 CAS.
- Y. Luo, X. Yang, C. Wang, A. Fraser, H. Zhang, X. Sun and X. Li, Prog. Mater. Sci., 2023, 139, 101171 CrossRef CAS.
- Z. Tong, S.-B. Wang, Y.-K. Liao, S.-F. Hu and R.-S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 47181–47196 CrossRef CAS.
- E. A. Wu, C. S. Kompella, Z. Zhu, J. Z. Lee, S. C. Lee, I.-H. Chu, H. Nguyen, S. P. Ong, A. Banerjee and Y. S. Meng, ACS Appl. Mater. Interfaces, 2018, 10, 10076–10086 CrossRef CAS.
- D. Cao, Y. Zhang, T. Ji, X. Zhao, E. Cakmak, S. Ozcan, M. Geiwitz, J. Bilheux, K. Xu, Y. Wang, K. S. Burch, Q. H. Tu and H. Zhu, Nano Lett., 2024, 24, 1544–1552 CrossRef CAS PubMed.
- F. Zhu, W. Deng, B. Zhang, H. Wang, L. Xu, H. Liu, Z. Luo, G. Zou, H. Hou and X. Ji, Nano Energy, 2023, 111, 108416 CAS.
- X. Chi, Y. Zhang, F. Hao, S. Kmiec, H. Dong, R. Xu, K. Zhao, Q. Ai, T. Terlier, L. Wang, L. Zhao, L. Guo, J. Lou, H. L. Xin, S. W. Martin and Y. Yao, Nat. Commun., 2022, 13, 2854 CrossRef CAS PubMed.
- H. Cao, M. Yu, L. Zhang, Z. Zhang, X. Yan, P. Li and C. Yu, J. Mater. Sci. Technol., 2021, 70, 168–175 CrossRef CAS.
- Y. Kim, C. Juarez-Yescas, D. W. Liao, M. K. Jangid, P. Joshi, H. Yang, B. Zahiri, P. V. Braun and N. P. Dasgupta, ACS Energy Lett., 2024, 9, 1353–1360 CrossRef CAS.
- X. Chi, Y. Zhang, F. Hao, S. Kmiec, H. Dong, R. Xu, K. Zhao, Q. Ai, T. Terlier, L. Wang, L. Zhao, L. Guo, J. Lou, H. L. Xin, S. W. Martin and Y. Yao, Nat. Commun., 2022, 13, 2854 CrossRef CAS PubMed.
- A. Banerjee, K. H. Park, J. W. Heo, Y. J. Nam, C. K. Moon, S. M. Oh, S.-T. Hong and Y. S. Jung, Angew. Chem., 2016, 128, 9786–9790 CrossRef.
- L. Ye and X. Li, Nature, 2021, 593, 218–222 CrossRef CAS.
- V. Lacivita, Y. Wang, S.-H. Bo and G. Ceder, J. Mater. Chem. A, 2019, 7, 8144–8155 RSC.
- T. Famprikis, J. A. Dawson, F. Fauth, O. Clemens, E. Suard, B. Fleutot, M. Courty, J.-N. Chotard, M. S. Islam and C. Masquelier, ACS Mater. Lett., 2019, 1, 641–646 CrossRef CAS.
- H. Nguyen, A. Banerjee, X. Wang, D. Tan, E. A. Wu, J.-M. Doux, R. Stephens, G. Verbist and Y. S. Meng, J. Power Sources, 2019, 435, 126623 CrossRef CAS.
- E. A. Wu, S. Banerjee, H. Tang, P. M. Richardson, J.-M. Doux, J. Qi, Z. Zhu, A. Grenier, Y. Li, E. Zhao, G. Deysher, E. Sebti, H. Nguyen, R. Stephens, G. Verbist, K. W. Chapman, R. J. Clément, A. Banerjee, Y. S. Meng and S. P. Ong, Nat. Commun., 2021, 12, 1256 CrossRef CAS.
- X. Fan, J. Yue, F. Han, J. Chen, T. Deng, X. Zhou, S. Hou and C. Wang, ACS Nano, 2018, 12, 3360–3368 CrossRef CAS PubMed.
- N. Tanibata, M. Deguchi, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 5232–5238 CAS.
- N. Tanibata, H. Tsukasaki, M. Deguchi, S. Mori, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2017, 311, 6–13 CAS.
- T. Ando, S. Atsushi, M. Tatsumisago and H. Akitoshi, Electrochem. Commun., 2020, 116, 5 CrossRef.
- H. Wan, W. Weng, F. Han, L. Cai, C. Wang and X. Yao, Nano Today, 2020, 33, 100860 CAS.
- S. Ohno and W. G. Zeier, Acc. Mater. Res., 2021, 2, 869–880 CAS.
- J. C. Stallard, L. Wheatcroft, S. G. Booth, R. Boston, S. A. Corr, M. F. L. De Volder, B. J. Inkson and N. A. Fleck, Joule, 2022, 6, 984–1007 CAS.
- O. N. Senkov and D. B. Miracle, Sci. Rep., 2021, 11, 4531 CAS.
- S. F. Pugh, Philos. Mag., 1954, 45, 823–843 CAS.
- R. P. Thompson and W. J. Clegg, Curr. Opin. Solid State Mater. Sci., 2018, 22, 100–108 CAS.
- M. de Jong, W. Chen, T. Angsten, A. Jain, R. Notestine, A. Gamst, M. Sluiter, C. Krishna Ande, S. van der Zwaag, J. J. Plata, C. Toher, S. Curtarolo, G. Ceder, K. A. Persson and M. Asta, Sci. Data, 2015, 2, 150009 CrossRef CAS.
- J. T. Kim, A. Rao, H.-Y. Nie, Y. Hu, W. Li, F. Zhao, S. Deng, X. Hao, J. Fu, J. Luo, H. Duan, C. Wang, C. V. Singh and X. Sun, Nat. Commun., 2023, 14, 6404 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, ChemElectroChem, 2014, 1, 1275–1280 CrossRef CAS.
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- L. Wang, T. Wang, L. Peng, Y. Wang, M. Zhang, J. Zhou, M. Chen, J. Cao, H. Fei, X. Duan, J. Zhu and X. Duan, Nat. Sci. Rev., 2022, 9, nwab050 CrossRef CAS.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS.
- J. Auvergniot, A. Cassel, D. Foix, V. Viallet, V. Seznec and R. Dedryvère, Solid State Ionics, 2017, 300, 78–85 CrossRef CAS.
- F. Han, T. Gao, Y. Zhu, K. J. Gaskell and C. Wang, Adv. Mater., 2015, 27, 3473–3483 CrossRef CAS.
- G. Xie, M. Tang, S. Xu, A. Brown and L. Sang, ACS Appl. Mater. Interfaces, 2022, 14, 48705–48714 CrossRef CAS.
- Y. Hu, J. Fu, J. Xu, J. Luo, F. Zhao, H. Su, Y. Liu, X. Lin, W. Li, J. T. Kim, X. Hao, X. Yao, Y. Sun, J. Ma, H. Ren, M. Yang, Y. Huang and X. Sun, Matter, 2024, 7, 1018–1034 CrossRef CAS.
- C. Wang, T. Deng, X. Fan, M. Zheng, R. Yu, Q. Lu, H. Duan, H. Huang, C. Wang and X. Sun, Joule, 2022, 6, 1770–1781 CrossRef CAS.
- S. Menkin, J. B. Fritzke, R. Larner, C. de Leeuw, Y. Choi, A. B. Gunnarsdóttir and C. P. Grey, Faraday Discuss., 2024, 248, 277–297 RSC.
- H. Su, Y. Zhong, C. Wang, Y. Liu, Y. Hu, J. Li, M. Wang, L. Jiao, N. Zhou, B. Xiao, X. Wang, X. Sun and J. Tu, Nat. Commun., 2024, 15, 2552 CrossRef CAS PubMed.
- G. Brauer and E. Zintl, Z. Phys. Chem., 1937, 37B, 323–352 CrossRef.
- G. Deysher, Y.-T. Chen, B. Sayahpour, S. W.-H. Lin, S.-Y. Ham, P. Ridley, A. Cronk, E. A. Wu, D. H. S. Tan, J.-M. Doux, J. A. S. Oh, J. Jang, L. H. B. Nguyen and Y. S. Meng, ACS Appl. Mater. Interfaces, 2022, 14, 47706–47715 CrossRef CAS PubMed.
- Q. Tu, L. Barroso-Luque, T. Shi and G. Ceder, Cell Rep. Phys. Sci., 2020, 1, 100106 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee06171c |
| This journal is © The Royal Society of Chemistry 2025 |