
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(N-acyliminophosphoranes): main chain functionalized poly(ylides) with pH-dependent degradation†
Kevin
Neumann
 *,
Dulce M.
Sánchez-Cerrillo
and
Daria R.
Galimberti
*,
Dulce M.
Sánchez-Cerrillo
and
Daria R.
Galimberti
Institute for Molecules and Materials, Radboud University, The Netherlands. E-mail: kevin.neumann@ru.nl
First published on 15th May 2025
Abstract
Zwitterionic polymers have gained attention for various applications but are typically based on non-degradable polyolefin backbones. Here, we introduce an Fe(II)-catalyzed step-growth polymerization that enables N-acyliminophosphorane formation, incorporating ylides directly into the polymer main chain. This design imparts pH-responsive degradability in aqueous environments, and opens opportunities for stimuli-responsive polymer systems.
Hydrophilic yet charge-neutral polymers have gained significant interest across the chemical sciences due to their strong hydration, which often imparts antifouling properties.1 Among them, zwitterionic polymers—particularly polybetaines with their equimolar balance of positive and negative charges—have found numerous applications in nanomedicine.2,3 As a closely related species to polybetaines, our group introduced poly(ylides), a polymer class which also displays an overall charge neutral nature while bearing highly polar residues in the form of ylides with zwitterionic character.4,5 We have shown that stabilized sulfur and phosphorus-derived poly(ylides) display bactericidal properties when applied as coatings while not affecting mammalian cells.6,7 A common drawback of the vast majority of zwitterionic polymers is their polyolefin-based backbone, most commonly being derived from polyacrylamides and polyacrylates with zwitterionic side chains.8 The inherent lack of degradability associated with polyolefin backbones limits the use of polybetaines and poly(ylides) for medicinal applications, for example as components for biomaterials, nanocarriers, protein-polymer or drug-polymer conjugates. Not only does the absence of degradability impact possible pathways for drug release, such as controlled release, but it may also lead to long-term tissue accumulation. Despite the need for main chain-functionalized polymers, only a handful of examples exists of main chain-functionalized zwitterionic polymers, including phospholipids, polysquaraines, and, more recently diisothiocyanate-derived polymers.9–12 Even less prevalent in literature are examples in which such functionalized backbone offers sufficient degradation kinetics under physiological conditions. In 2018, the group J. Johnson reported readily accessible polymeric NHCs as a form of polybetaines applicable to depolymerisation at elevated temperatures.13 While these main chain zwitterionic supramolecular polymers are promising scaffolds for applications in nanotechnology, applications in (nano)medicine require sensitivity to physiological triggers such as pH and redox potential.
We envisioned incorporating ylide functionalities via a step-growth polymerization mechanism would enable the development of polymers with multiple ylide residues integrated into the main chain. Inspired by the recent report of conjugated poly(iminophosphoranes),14 we turned our attention to N-acyliminophosphoranes, which have been extensively studied as small molecules but remained so far unexplored in polymer chemistry.15 We further hypothesized that the molecular structure of N-acyliminophosphoranes could be tailored to generate pH-sensitive ylide linkages, ultimately allowing access to pH-degradable polymers for potential applications in (nano)medicine (Fig. 1).
 | ||
| Fig. 1 (A) In contrast to previously employed ylide residues for poly(ylide), imino phosphoranes offer the possibility for main chain incorporation. (B) Stability depends strongly on the residues displayed on the phosphorus. (C) Hydration behaviour of imino phosphorane obtained by MD simulations (D) Designed synthesis route towards poly(iminophosphoranes). | ||
At the onset of our research, we explored the chemical space of N-acyliminophosphoranes and their potential as pH-responsive ylide residues. While acyliminophosphoranes have gained significant attention in recent years as promising components in drug design, there is limited information on their structure–property relationship, particularly in the context of stability in aqueous environment. Most attention is given to complexes of iminophosphoranes with metals due to their potential anticancer properties.16 Since the vast majority of research has focused on triaryl phosphine-derived iminophosphoranes, such as N-acylimino(triphenylphosphoranes), we were motivated to revisit the possibility of tailoring stability in aqueous environment by modifying the substituents on the phosphorus residue. N-Acyliminophosphoranes 1 and 2 were readily accessible displaying alkyl and aryl substituents at the phosphorus, respectively. In addition, ylide 1 was derived from an aromatic carboxylic acid while ylide 2 derived from an aliphatic acid, which—alongside the altered substituents at the phosphorus—provided two examples with opposite chemical stabilisation. Interestingly, we observed that ylide 1 slowly degraded even in protic organic solvents, whereas the triphenyl-substituted ylide remained stable in a buffered aqueous environment (TRIS buffer, pH 7.4) for at least seven days (Fig. 1B). This strong contrast in stability was attributed to the electron-deficient nature of the aryl ester for labile ylide 1, as well as the electron-deficient nature of the phosphorus substituents in the case of ylide 2, making an overall neutral resonance structure preferable.
To gain further insights into the molecular structure and aqueous interaction of iminophosphoranes, we conducted molecular dynamics simulations. Recently, we reported that ylides exhibit an environmentally sensitive dipole.17 Similarly, here, we observed that iminophosphoranes exhibit comparable behaviour, with dipoles similar to those of recently reported sulfur ylides both in the hydrophobic environment and in an aqueous solution (Fig. 1C, ESI†).17 These findings suggest that the degree of charge separation depends on the solvent and reflects the balance between ylide and ylene structures. To illustrate this, we chose to depict the ylide form rather than the ylene form.
Next, we tuned our attention to identifying suitable polymerisation conditions. We were intrigued by a recent report that access small molecule N-acyliminophosphoranes from commercially available phosphines and acyloxyamide in presence of transition metal catalysts.18 According to previous literature, the metal catalyst enables formation of an iron–nitrenoid species with subsequent nucleophilic addition of phosphine. We environed that such imidization reaction would be suitable for a step-growth polymerisation mechanism (Fig. 1D). For this purpose, we accessed difunctional acyloxyamide monomer 1 (M1) from pimelic acid by establishing a one-pot protocol. Conveniently, a variety of difunctional phosphines are commercially available. Given our findings that stability is strongly impacted by substituents on the phosphorus residue, we chose 1,2-bis(diphenylphosphino)ethane as the second monomer (M2) which displays two aryl and one alkyl residue. Our first trials of step-growth polymerisation included the use of equimolar amount of M1 and M2 in acetonitrile with varying amounts of FeCl2 as catalysts (20 and 40 mol%, Table 1) under nitrogen atmosphere. Since no indication of conversion could be observed by neither GPC or 31P-NMR, polymerisation in 1,4-dioxane and DCE as alternative solvents were examined. We speculated that in particular the difunctional phosphine M2 requires a more apolar solvent, while the activation of M1 by Fe(II) requires non-nucleophilic solvents. Indeed, reactions carried out in either solvent provided first evidence of polymerisations albeit with low conversion of 80% and 82% for 1,4-dioxane and DCE, respectively (entry 3 and 4).
| Solvent | Conc. of cat. [mol%] | Time [h] | Cat. | Additive/comment | Conv. [%] | DP | Mn [kg mol−1] | |
|---|---|---|---|---|---|---|---|---|
| 1 | CH3CN | 20 | 8 | FeCl2 | — | None | n/a | n/a |
| 2 | CH3CN | 40 | 8 | FeCl2 | — | None | n/a | n/a |
| 3 | 1,4-Dioxane | 20 | 8 | FeCl2 | — | 80 | 5.2 | 2.7 |
| 4 | DCE | 20 | 8 | FeCl2 | — | 82 | 5.6 | 3.1 |
| 5 | DCE | 20 | 20 | FeCl2 | — | 91 | 6.5 | 3.6 |
| 6 | DCE | 20 | 20 | FeCl2 | Fe(C5H5)2 | 86 | 4.7 | 2.6 |
| 7 | 1,4-Dioxane | 20 | 20 | Fe(OAc)2 | — | None | n/a | n/a |
| 8 | 1,4-Dioxane | 20 | 20 | FeCl2 | N2 flow | >95 | 9.8 | 5.4 |
The conversion was determined by comparing CH2 signals adjacent to the phosphorus of M2 with the signals formed in the polymer (Fig. 2b).19 Alternatively, comparing the signals of P(III) and P(V) in 31P-NMR spectra afforded very similar results (Fig. 2c). In both cases, around 20% of remaining phosphine monomer was detected, thus another panel of polymerisations were left for reaction for 20 hours. After 20 hours, significantly higher conversion (>90%) towards N-acyliminophosphoranes was observed, resulting in a calculated degree of polymerisation of 6.5 and a calculated Mn of 3565 Da. The degree of polymerization was estimated by determining the relative amounts of the two possible end groups, namely phosphine oxide and primary amide, in relation to the polymer main chain (ESI). Analysis with 31P-NMR revealed that only negligible amounts of starting material were left after 20 hours, but notable amounts of phosphine oxide were observed. The formation of phosphine oxide as an end-group functionality during work-up was anticipated. However, if generated during polymerization, its presence would significantly hinder the progress of step-growth polymerization. Electron-rich phosphines are highly susceptible to oxidation in the presence of trace oxygen. To rule out oxygen as the cause of oxidation during polymerization, we conducted the reaction in the presence of ferrocene, which has recently been reported as an effective oxygen scavenger that prevents phosphine oxidation.20 No differences were observed; in fact, the conversion was lower compared to the polymerization without ferrocene. Additionally, the amount of phosphine oxide detected remained unchanged between samples taken after 8 and 20 hours, making us consider the possibility that acid may trigger a Wittig-like reaction, resulting in a phosphine oxide and a nitrile (ESI). During the catalytic activation of acyloxyamide, two equivalents of HCl are released from FeCl2. To assess whether a weaker acid could mitigate acid-triggered degradation and yield a higher degree of polymerization, we conducted the reaction using Fe(OAc)2. However, no polymerization was observed, indicating that FeCl2 is essential for efficient conversion with the developed protocol. As an alternative strategy to prevent acid-triggered degradation, the polymerization was performed under a nitrogen stream to ensure rapid removal of the generated HCl. This approach led to full conversion, as confirmed by 31P NMR, with significantly reduced phosphine oxide formation (Fig. 2c).
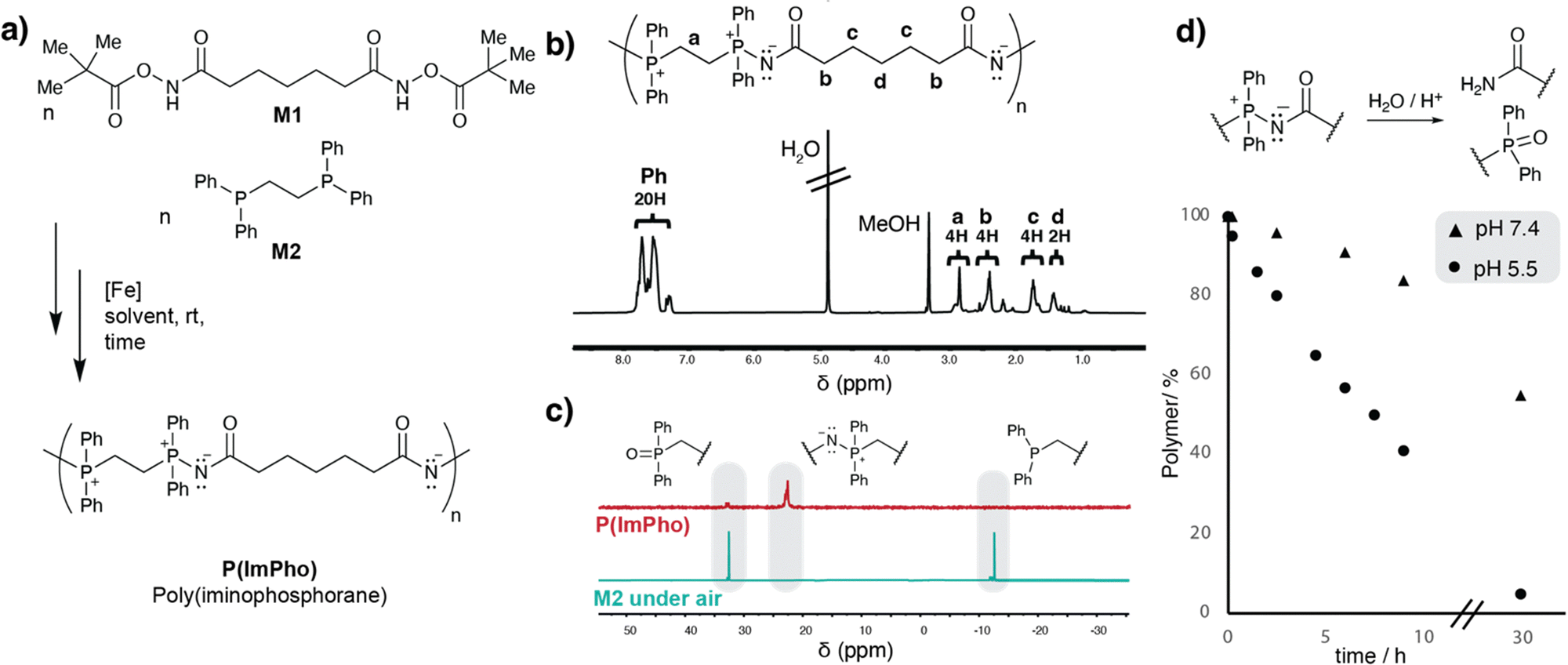 | ||
| Fig. 2 (a) Polymerisation of P(ImPho) with screened variables (b) representative 1H-NMR of P(ImPho) (c) representative 31P-NMR of P(ImPho) in comparison with M2 exposed to air (d) hydrolysis monitored over time at different pH values. | ||
End-group analysis via1H-NMR indicated a molecular weight of Mn = 5.4 kDa, while GPC confirmed the presence of higher-molecular-weight polymers with fewer oligomeric species.
Next, we examined the degradation behavior of poly(iminophosphoranes) under aqueous conditions. Since these polymers feature ylide residues along the main chain, hydrolysis induces depolymerization, which could enable applications in nanomedicine and biomaterials science. To assess stability, we incubated poly(iminophosphoranes) in aqueous environments at pH 7.4 and pH 5.5, using DMSO as a co-solvent. Monitoring via31P-NMR revealed a strongly pH-dependent degradation profile, with kobs = 0.0003 min−1 at pH 7.4 and kobs = 0.0017 min−1 at pH 5.5. This corresponds to a 5.5-fold increase in half-life from 6.8 hours at pH 5.5 to 38.5 hours at pH 7.4. Notably, this pronounced pH sensitivity in the range between ∼5 and 7.4 is particularly relevant for biomedical applications, where controlled degradation in response to pH shifts—such as those occurring in the endosomal pathway or the tumor microenvironment—can be leveraged for targeted release.
Hydrophilic yet charge-neutral polymers, such as zwitterionic polymers and polymeric ylides, hold great promise for diverse applications but are often non-depolymerizable due to polyolefin backbones. Here, we introduce a Fe(II)-catalyzed step-growth polymerization that, for the first time, incorporates ylides into the polymer's main chain. Notably, we identified that iminophosphorane ylide residues with alkyl substituents undergo controlled, pH-dependent degradation in aqueous environment. While currently only low molecular weight polymers are accessible, we envision that this strategy significantly expands the scope of degradable zwitterionic materials, offering new opportunities for stimuli-responsive and environmentally compatible polymer systems.
We would like to thank the NMR and mass spectrometry facility at Radboud University for their support. This work was funded by Radboud University (start-up-package).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- N. Bayliss and B. V. K. J. Schmidt, Prog. Polym. Sci., 2023, 147, 101753 Search PubMed.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 Search PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59–63 Search PubMed.
- K. Neumann, Biomater. Sci., 2024, 12, 5481–5490 Search PubMed.
- G. Poulladofonou and K. Neumann, Polym. Chem., 2022, 13, 4416–4420 Search PubMed.
- B. B. Berking, G. Poulladofonou, D. Karagrigoriou, D. A. Wilson and K. Neumann, Angew. Chem., Int. Ed., 2023, 62, e202308971 Search PubMed.
- D. Karagrigoriou, B. B. Berking, Q. Wang, D. M. Sánchez-Cerrillo, D. R. Galimberti, D. A. Wilson and K. Neumann, ACS Macro Lett., 2023, 12, 1608–1613 Search PubMed.
- Q. Li, C. Wen, J. Yang, X. Zhou, Y. Zhu, J. Zheng, G. Cheng, J. Bai, T. Xu, J. Ji, S. Jiang, L. Zhang and P. Zhang, Chem. Rev., 2022, 122, 17073–17154 Search PubMed.
- T. Nakaya and Y. J. Li, Prog. Polym. Sci., 1999, 24(1), 143–181 Search PubMed.
- T. Nakaya, M. Yasuzawa and M. Imoto, Macromolecules, 1989, 22, 3180–3181 Search PubMed.
- J. Eldo and A. Ajayaghosh, Chem. Mater., 2002, 14, 410–418 Search PubMed.
- A. Yamauchi, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2145–2148 Search PubMed.
- N. M. Gallagher, A. V. Zhukhovitskiy, H. V.-T. Nguyen and J. A. Johnson, Macromolecules, 2018, 51(8), 3006–3015 Search PubMed.
- T. Kotnik, A. Debuigne, J. De Winter, M. Huš, A. Pintar and S. Kovačič, Commun. Chem., 2025, 8, 15 Search PubMed.
- R. Bielsa, R. Navarro, T. Soler and E. P. Urriolabeitia, Dalton Trans., 2008, 1787–1794 Search PubMed.
- M. Carreira, R. Calvo-Sanjuan, M. Sanau, I. Marzo and M. Contel, Organometallics, 2012, 31, 5772–5781 Search PubMed.
- B. Berking, D. Karagrigoriou, D. R. Galimberti, B. H. E. Zhang, D. A. Wilson and K. Neumann, ACS Langmuir, 2025, 41(13), 8627–8636 Search PubMed.
- S. Lin, B. Lin, Z. Zhang, J. Chen, Y. Luo and Y. Xia, Org. Lett., 2022, 24(17), 3302–3306 Search PubMed.
- K. Ratzenböck, D. Pahovnik and C. Slugovc, Polym. Chem., 2020, 11, 7476 Search PubMed.
- F. Horký, R. Franz, C. Bruhn and R. Pietschnig, Chem. – Eur. J., 2023, 29(68), e202302518 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01640a |
| This journal is © The Royal Society of Chemistry 2025 |