
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatial tuning of adsorption enthalpies by exploiting spectator group effects in organosilica carbon capture materials†
Mario
Evers
a,
Karin
Hauser
 b,
Wolfgang G.
Hinze
b,
Nele
Klinkenberg
b,
Yasar
Krysiak
a,
Daniel
Mombers‡
a and
Sebastian
Polarz‡
*a
b,
Wolfgang G.
Hinze
b,
Nele
Klinkenberg
b,
Yasar
Krysiak
a,
Daniel
Mombers‡
a and
Sebastian
Polarz‡
*a
aInstitute of Inorganic Chemistry, Leibniz-University of Hannover, Callinstrasse 9, 30167 Hannover, Germany. E-mail: sebastian.polarz@aca.uni-hannover.de
bDepartment of Chemistry, University of Konstanz, Universitaetsstrasse 10, 78457 Konstanz, Germany
First published on 4th April 2024
Abstract
Functional gradient materials can process more complex tasks than a mixture of their homogeneous analogs. Generating such materials is difficult as it necessitates spatial control over chemical and/or structural properties. A gradient is a unique degree of freedom in hierarchical material architectures, and as such, biology has managed exploiting the full potential of graded structures. For instance, despite being present at a comparably low concentration (approaching 420 ppm in 2023), plants are capable of capturing carbon dioxide from the air. Binding occurs in the carboxysome, a complex entity characterized by pores with engineered surfaces composed of shell proteins that create a concentration gradient of CO2 towards an enzyme responsible for the first conversion step. The current paper hypothesizes that porous organosilica materials can mimic some of the features of the mentioned biological paragon. Primary amines as sites for interacting with CO2 are surrounded by spectator groups on bifunctional surfaces. It is found that the proper choice of the spectator group almost doubles the adsorption enthalpy. Above a critical density, the hydrophobic moieties create a quasi-solvent layer on the surfaces in which CO2 molecules dissolve. When the density of the spectator groups gradually changes inside a graded organosilica monolith, one obtains zones varying systematically in adsorption enthalpy. Directionality in affinity towards CO2 and controlled transport properties are realized.
Introduction
Scientific investigations in different fields clearly show that the atmosphere's composition, the abundance of so-called greenhouse gases such as carbon dioxide (CO2), correlates with the medium temperature at the earth's surface. With growing populations and economies, one cannot foresee when there will be a turning point in the increase in the CO2 concentration which has already reached 419 ppm in the meantime. Given that industrial plants, transportation, and animal husbandry will continue to emit greenhouse gases for the next few decades, it is obvious that trapping those gases before they reach the atmosphere represents an indispensable step in all future climate management scenarios. An even larger challenge is to reduce the atmospheric concentration by adsorption of CO2 despite it being only a minor constituent of air. This is where carbon capture materials (CCMs) become important.1,2 The optimization of the uptake capacity for CO2 is one important task in the development of CCMs, and impressive progress has been made and is reported in the literature.3 As expected, the specific surface area combined with the density of CO2-binding functionalities determines the uptake capacity. For instance, microporous materials such as metal–organic frameworks (MOFs) have proven highly valuable CCMs.4 However, considering the huge amount of CO2, it is questionable that the long-term storage of solid, gas-loaded adsorbents is the best way. This poses the question of whether uptake capacity is a CCM's most important aspect. For sure, it is not the only aspect.To learn about what other features beyond capacity are important for future technical solutions to the problem, it is worth considering how nature deals with atmospheric CO2. Plants can bind CO2 from the air under ambient conditions. Fixation occurs in the carboxysome and is succeeded by the redox-activated conversion of CO2.5,6 Overall, the carboxysome acts as a specialized compartment that creates a concentration gradient of CO2 around the enzyme responsible for the conversion reaction.7 Carbon dioxide from the surrounding environment diffuses into the carboxysome through small pores with engineered surfaces composed of shell proteins. Once inside the carboxysome, carbon dioxide reacts with water to form bicarbonate ions. This reaction is facilitated by the enzyme carbonic anhydrase. The bicarbonate ions are channeled towards succeeding enzymes. An alternative enzyme is crotonyl-CoA carboxylase/reductase (CCR).8 The newest results have shown that the enzyme contains a hydrophobic CO2-binding pocket in the active site.9 The surface of the cavity is characterized by four amino acids, most importantly phenylalanine which efficiently excludes water from the active site and therefore limits unwanted irreversible side reactions.
Multiple conclusions can be drawn from those biological examples for effectively removing CO2 from a gas phase, which are also relevant for developing and improving technical systems. The biological approach indicates that capacity can be a minor aspect. In essence, the chemical structures of the surfaces of the adsorbent material have to be engineered much more precisely. For capturing CO2 close to ambient conditions, preferably even from normal air, and for transportation of the loaded CCM, the adsorption enthalpy ΔHads must not be too low. However, even for a CCM with high uptake capacity, at a certain point, its regeneration is vital and must be possible without significant energy consumption. Thus, strongly bound carbon dioxide is not always preferred. The range in which the CO2 medium is strongly attached to the surface is between physi- and chemisorption, corresponding to −100 kJ mol−1 > ΔHads < −50 kJ mol−1. The precise adjustment of ΔHads(CO2) cannot be managed by pore-size engineering alone, but rather control over active surface functionalities attached to the pore-walls is necessary. The chemical structure of the surfaces in a CCM has many more implications, most importantly regarding selectivity. In a mixture, different compounds compete for adsorption sites, and it is particularly difficult to separate weakly interacting and low concentrated species. CO2 can interact with Lewis-basic centers, such as primary amine groups, as do water molecules (H2O) via hydrogen binding.10
Special adsorbent materials with bifunctional surfaces have already been discussed in the literature.11 In the so-called synergistic approach, bifunctional surfaces can exhibit special effects, where different functional groups work cooperatively to enhance CO2 adsorption. For example, combining amine groups (such as primary, secondary, or tertiary amines) with other functional groups such as carboxyl (–COOH), hydroxyl (–OH), or imidazole (–NH) on a surface can create more favorable interactions with CO2. The amine groups can form chemical bonds (amine–CO2 adducts), while the other functional groups provide additional sites for physical interactions (such as hydrogen bonding) with CO2 molecules. The presence of a secondary group can also affect the adsorption capacity. Bifunctional surfaces can serve as both adsorbents and catalysts for CO2 conversion. The functional groups on the surface can activate CO2 molecules, making them more reactive for subsequent chemical transformations. This allows the possibility of utilizing the adsorbent–catalyst synergy to capture and convert CO2 simultaneously.
For the current paper, the most important aspect is that bifunctional surfaces can also enhance the recyclability of the adsorbent, i.e., the release of adsorbed CO2, which can be facilitated by controlling the desorption temperature or exploiting the functional groups' reversible nature. It is well known in the literature that modifying surfaces with non-polar groups such as hydrocarbons results in hydrophobicity.12 However, how these groups affect ΔHads(CO2) has not been investigated in-depth. The latter aspect represents the focus of the paper presented herein. Because the mentioned hydrophobic groups cannot interact with CO2via Lewis acid-base pair formation or hydrogen bonding, we call them spectator groups. How do spectator groups change ΔHads(CO2)?
Nanoporous CCMs with bifunctional surfaces are required to investigate the formulated question. Although porous organosilica materials may not have specific surface areas as high as those for microporous materials such as zeolites, MOFs, or charcoal and consequently fall behind regarding uptake capacity, it has been proven that an extremely rich surface chemistry is possible.13–16 A special approach involves the use of sol–gel precursors containing a bridging organic (functional) unit RF: (RO)3Si–RF–Si(OR)3.15 The undiluted precursor is converted to organosilica materials RFSi2O3 with a very high content of organic functionality. The sol–gel process can be performed in the presence of a template which generates porosity after removal. Bi- and multifunctional materials can be prepared by mixing different precursors containing RF1,2, in the sol. This approach is known in the literature as the co-condensation route. Several years ago, we were able to show that the distribution of two different functionalities RF1 = –NH2 and RF2 = –COOH is statistical.17 The composition of the material (RF1)x(RF2)1−x Si2O3 can be varied freely from x = 0 to x = 1, directly affecting the distance between RF1 and RF2 and their intermolecular interaction. Another approach to introduce bifunctionality into a porous organosilica material is the so-called post-modification. If RF1 is reactive and can be converted to RF2, one can also obtain (RF1)x(RF2)1−xSi2O3. The composition depends on the degree of conversion and, thus, on parameters such as the concentration of reactants, temperature, etc. In our research, we could establish RF1 (=–N3, –CH = CH2, –C≡CH, –SH, etc.) representing a group capable of undergoing click-chemistry, which was used for the preparation of a multitude of mono- and bifunctional organosilica materials.18–23
Although many papers exist in the literature on bifunctional organosilica materials,24–27 only a few papers deal with the importance of the secondary group in adsorption processes and CCM applications in particular. In a recent paper, we investigated the role of RF1 = –COOH next to combined surface-bound primary amines RF2 = –NH2 for the properties of porous organosilica materials in CO2 adsorption.28 It was found that there is a cooperative binding of CO2 if RF1 and RF2 are next to each other on the surface. RF1 = –COOH is obviously not a spectator group as it interacts directly with surface-bound CO2 molecules. In the latter paper, we have already recognized that passive neighboring groups also seem to influence ΔHads(CO2), but the effect was not studied in detail.
Here, we propose a porous organosilica material with regions differing in the presence of the spectator group. These regions would then be connected to different ΔHads(CO2) values. So-called functional gradient materials (FGMs) are expected to fulfill more complex tasks compared to homogeneous materials as discussed in the nice review article by Pragya et al.29 FGMs have also been proposed for their large potential in carbon capture applications,3,30–33 but there are only a few experimental realizations yet. We could already show that organosilica materials containing chemical and/or structural gradients can be prepared using click-chemistry.18,22,28 For instance, ref. 18 described the attachment of dye molecules to the surfaces of azide-functionalized organosilica molecules using the copper-catalyzed Huisgen cycloaddition reaction. The conversion of the azide groups was managed to depend on the position of the materials, and this allowed the introduction of the chemical gradient.
The current paper is organized as follows. In the first section, we present the preparation and characterization of amine-functionalized organosilica materials with aerogel structures containing a broad selection of spectator groups (see Chart 1). The adsorption properties of the materials are investigated with particular emphasis on identifying connections between the nature of the spectator groups and ΔHads(CO2) will be identified. In the final section of the paper, we approach a gradient material and want to prove that there is a spatial dependency of ΔHads(CO2).
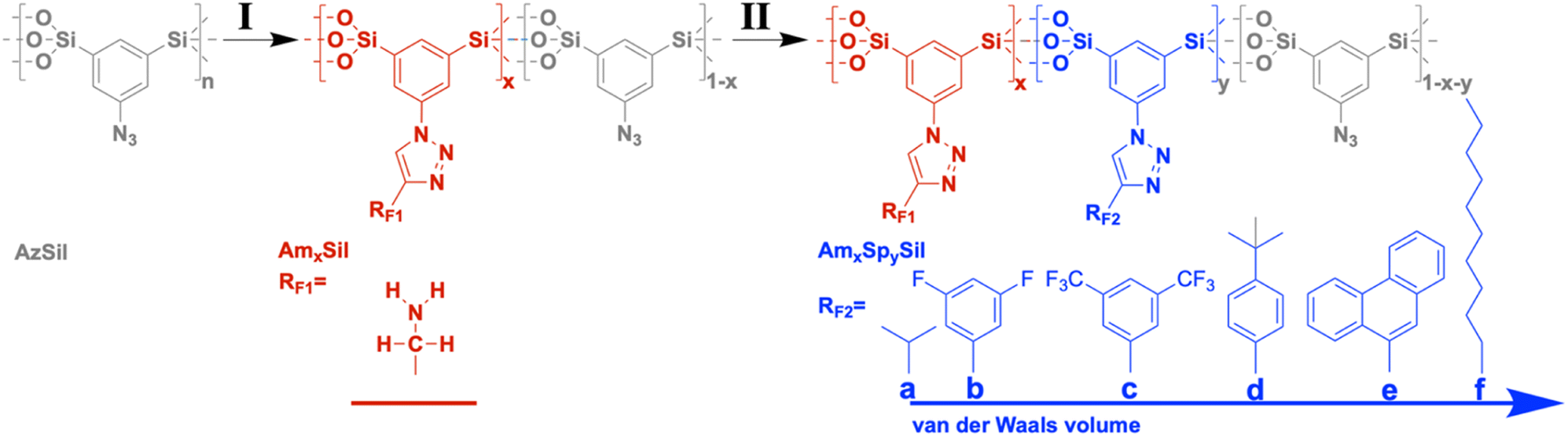 | ||
| Chart 1 Two-step synthesis of amine-modified organosilica materials containing hydrophobic moieties (RF2) as spectator groups. | ||
Results and discussion
Preparation of bifunctional materials
Starting from the azide-modified organosilica aerogel (=AzSil; for characterization, see ESI Fig. S1†)18,28,34 the target materials were prepared in a two-step process shown in Chart 1; see also the experimental part. The first step involves the attachment of primary amine groups from the copper-catalyzed click reaction of AzSil with a tert-butyloxycarbonyl (boc) protected propargylamine. After deprotection, one obtains AmxSil with the subscript defining the degree of conversion of the azide groups. The analytical data of this reference material are given in ESI Fig. S2.† The residual azide groups in AmxSil were then reacted in a second step with terminal alkynes to introduce RF2. At this point, all accessible azide groups will be consumed. However, a small portion might still be present, for instance, inside the pore walls. Depending on the spectator group (=RF2), those materials are denoted as AmxSp(a–f)ySil.Data for the materials AmxSp(a–g)ySil are summarized in the ESI Fig. SI-3–8.† The analysis of the chemical composition of AmxSp(d)ySil is discussed as a representative case. The total degree of azide functionalization (x + y) can be determined from Fourier-transform infrared (FT-IR) spectroscopy and thermogravimetric analysis (TGA). Fig. 1a shows the FT-IR fingerprint region of AmxSp(d)ySil in comparison to AzSil and Am0.57Sil as references. The spectra were referenced to the intensity of the Si–O–Si band. Because the siloxane network is not affected by the chemical reactions (Chart 1), the band is suitable for comparing the samples among each other. The assignment of the bands is shown in more detail in ESI Fig. S6† as AmxSp(d)ySil contains all relevant groups, residual azide, the triazole ring, the amine group and tert-butylphenyl as a SG. The signal at 2115 cm−1 is characteristic of the azide group. It can be seen that the band is very weak for AmxSp(d)ySil. The decrease in the intensity (peak area) indicates that 80% (= x + y) of the azide groups have been converted. Thermogravimetric analysis (TGA) can be used as an independent method to determine the sum x + y. Fig. 1b shows the TGA analysis of AmxSp(d)ySil performed in an atmosphere containing 50% O2 and 50% N2. After loss of residual water in the temperature range below T = 120 °C, one sees two main steps with a maximum in the first derivative of the TGA data at T1 = 416 °C and T2 = 546 °C. The first main decomposition step is at the same temperature compared to AmxSil materials. The missing decomposition step at T = 295 °C agrees with the almost quantitative conversion of the azide functionalities, which has been deduced from IR spectroscopy. We conclude that T2 corresponds to the presence of SP(d) and a ratio of Am to Sp(d) equal to ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The conversion of AmxSp(d)ySil to SiO2 corresponds to a mass-loss of 60.8% and thus x + y = 0.77. Because the separate values of x and y cannot be determined from TGA data with absolute precision, we adopted a method developed by us in the past to determine the composition by liquid nuclear magnetic resonance (NMR) spectroscopy.35 Organosilica materials (AmxSPyAz1−x−ySi2O3) can be dissolved quantitatively at elevated pH values (>10) resulting in deprotonated silicic acid species AmSi2(OH)n(O−)m, SPSi2(OH)n(O−)m, AzSi2(OH)n(O−)m (Fig. 1c). These species are soluble in deuterated water, D2O, and the quantitative evaluation of the 1H-NMR spectra allows us to deduce the values for x and y. For instance, the signals at δ = 8.04 can be assigned to the RF1 = Am group, at 7.38 to RF1 = SP(d) and at 7.83 to residual Az. For the concrete assignment of the signals in the aromatic region, refer to ESI Fig. S6.† The described analysis results in a composition Am0.48Sp(d)0.42Sil and agrees with the values obtained from the TGA measurement. The results for the analysis of the composition for the other samples AmxSP(a,b,c,e,f) are shown in Fig. 1d and the original data are shown in ESI Fig. SI-3–8.†
:1. The conversion of AmxSp(d)ySil to SiO2 corresponds to a mass-loss of 60.8% and thus x + y = 0.77. Because the separate values of x and y cannot be determined from TGA data with absolute precision, we adopted a method developed by us in the past to determine the composition by liquid nuclear magnetic resonance (NMR) spectroscopy.35 Organosilica materials (AmxSPyAz1−x−ySi2O3) can be dissolved quantitatively at elevated pH values (>10) resulting in deprotonated silicic acid species AmSi2(OH)n(O−)m, SPSi2(OH)n(O−)m, AzSi2(OH)n(O−)m (Fig. 1c). These species are soluble in deuterated water, D2O, and the quantitative evaluation of the 1H-NMR spectra allows us to deduce the values for x and y. For instance, the signals at δ = 8.04 can be assigned to the RF1 = Am group, at 7.38 to RF1 = SP(d) and at 7.83 to residual Az. For the concrete assignment of the signals in the aromatic region, refer to ESI Fig. S6.† The described analysis results in a composition Am0.48Sp(d)0.42Sil and agrees with the values obtained from the TGA measurement. The results for the analysis of the composition for the other samples AmxSP(a,b,c,e,f) are shown in Fig. 1d and the original data are shown in ESI Fig. SI-3–8.†
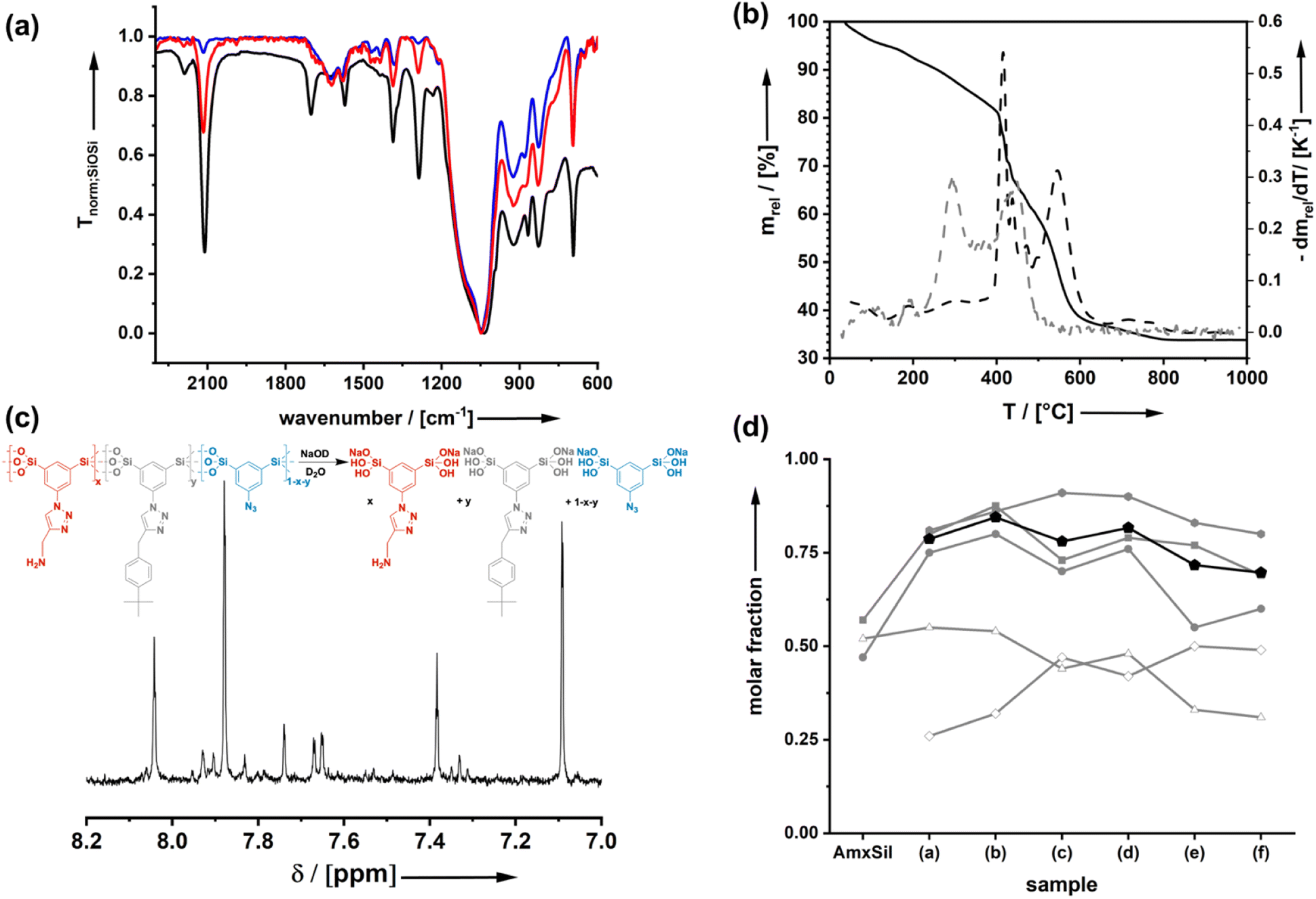 | ||
| Fig. 1 (a) FT-IR spectra of AzSil (black), AmSil (red) and AmxSpy(d)Sil (blue; RF2 = tert-butyl phenyl). (b) TGA trace (solid line) for the oxidative decomposition of AmSp(d)Sil (black) and the first derivative (dashed line) in comparison to the decomposition steps of AmSil (grey). (c) 1H-NMR spectrum (aromatic region) of the dissolved AmxSpy(d)Sil sample. (d) Results for the composition analysis of the different samples; squares (x + y determined from IR), circles (x + y determined from TGA), hashes (x determined from NMR), triangles (y determined from NMR), hexagons (x + y determined from NMR), and pentagons (x + y; averaged value). | ||
The total conversion (x + y) is quite constant for all bifunctional samples. Because the first click-reaction was performed under the same conditions, the number of amine groups also varies only a little. Because of the different kinetics with the second click-step for the attachment of the different RF2 groups, it is very hard to avoid that there is a certain variance. In particular, sample AmxSP(c) is distinct from the others.
The structure of the materials and effects of the functionalization process were investigated by scanning electron microscopy (SEM) as shown in Fig. 2a–c and ESI Fig. S3–8.†
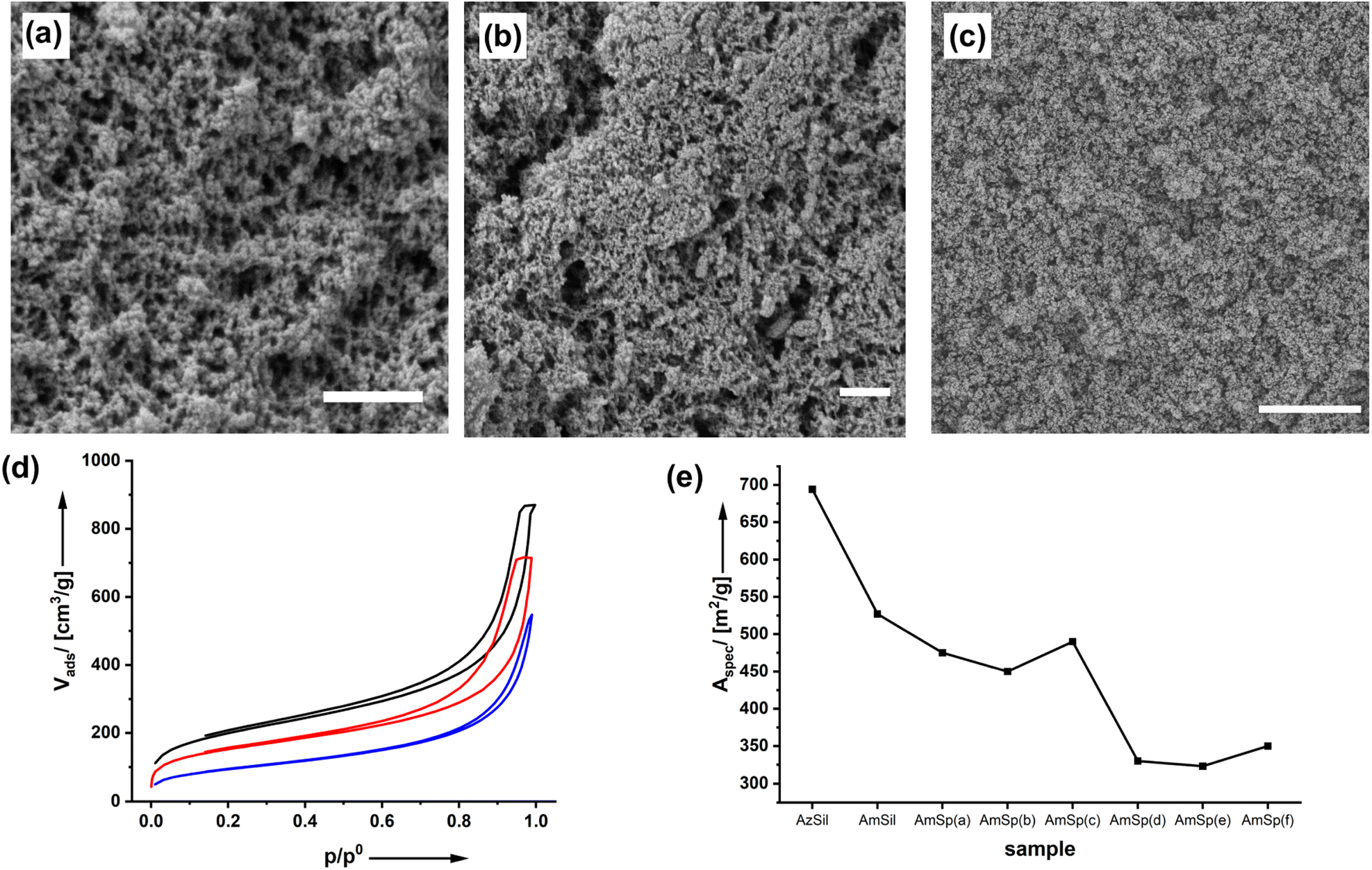 | ||
| Fig. 2 SEM micrograph of AzSil (a; scalebar = 1 μm), AmSil (b; scalebar = 1 μm) and Am0.48Sp(d)0.42Sil (c; scalebar = 5 μm). (d) Nitrogen physisorption isotherms (adsorption and desorption) of AzSil (black), AmSil (red) and Am0.48Sp(d)0.42Sil (blue). (e) Specific surface area of the different organosilica samples as determined by N2 physisorption measurements. | ||
The starting material, the monolithic AzSil body (ESI Fig. S1†), possesses an aerogel structure after supercritical drying. The first postmodification step to AmSil has no marked effects on the pore structure, as can be seen from Fig. 2b, and the monolithic shape is retained (ESI Fig. S2†). However, introducing the hydrophobic tert-butyl phenyl group in AmSp(d)Sil during the second modification step (Chart 1) leads to partial densification. The material is still highly porous, but in particular, fewer pores with diameters >100 nm seem to be present.
The material's porosity can be characterized by nitrogen physisorption measurements shown in Fig. 2d. All materials feature a type II isotherm. In interpreting those isotherms, one has to consider that the attachment of organic groups to the existing pore walls increases the mass of the aerogels. Because the density of the material enters the calculations of values from isotherm plots, for parameters such as the specific surface area, for instance, it is expected that the curves for AmSil and AmSp(d)Sil are shifted to lower values. In agreement with this, lower values for the specific surface area Aspec are also expected (Fig. 2e). However, the drop in Aspec from 527 m2 g−1 for AmSil to 330 m2 g−1 for AmSp(d) as an example cannot be explained by the gain of mass alone. As we have already assumed based on the SEM measurements, the post-functionalization process seems to cause a certain degree of compaction of the aerogel. Another consequence of the described compaction is that the micropores in the system almost vanish. The values for Vads close to p/p0 = 0 are indicative of the presence of micropores. One can see in Fig. 2d that there are notable micropores in AzSil. In comparison, the amount of micropores is much lower in AmSil and AmSp(d)Sil. One can also see from Fig. 1e that sample AmxSP(c) shows a little bit different behavior compared to the others.
Hydrophobicity of materials containing spectator groups
All spectator groups shown in Chart 1 are hydrophobic, and the uptake of water (H2O) indicates the prepared materials' hydrophobicity. Therefore, we have measured H2O vapor adsorption curves (Fig. 3; ESI Fig. S9†). One can see that the introduction of amino groups does not change the hydrophobicity/hydrophilicity of the material. The H2O uptake isotherms for AzSil and AmSil are almost identical. Because all spectator groups (a–f) are non-polar, we expect that the water uptake will be reduced because of enhanced hydrophobicity. From chemical intuition, one would sort the samples a → f → d → e → b → c in an order of increasing hydrophobicity. Neither the latter nor the order of the van der Waals volumes reflect the empirical order of the water uptake capacities (Fig. 3b). The attachment of the tert-butyl phenyl groups leads to an overall lowest water uptake at all pressures. If the adsorption of water is an indication of the hydrophobicity of the material, one can conclude that AmSp(d)Sil is the most hydrophobic material within the series presented here.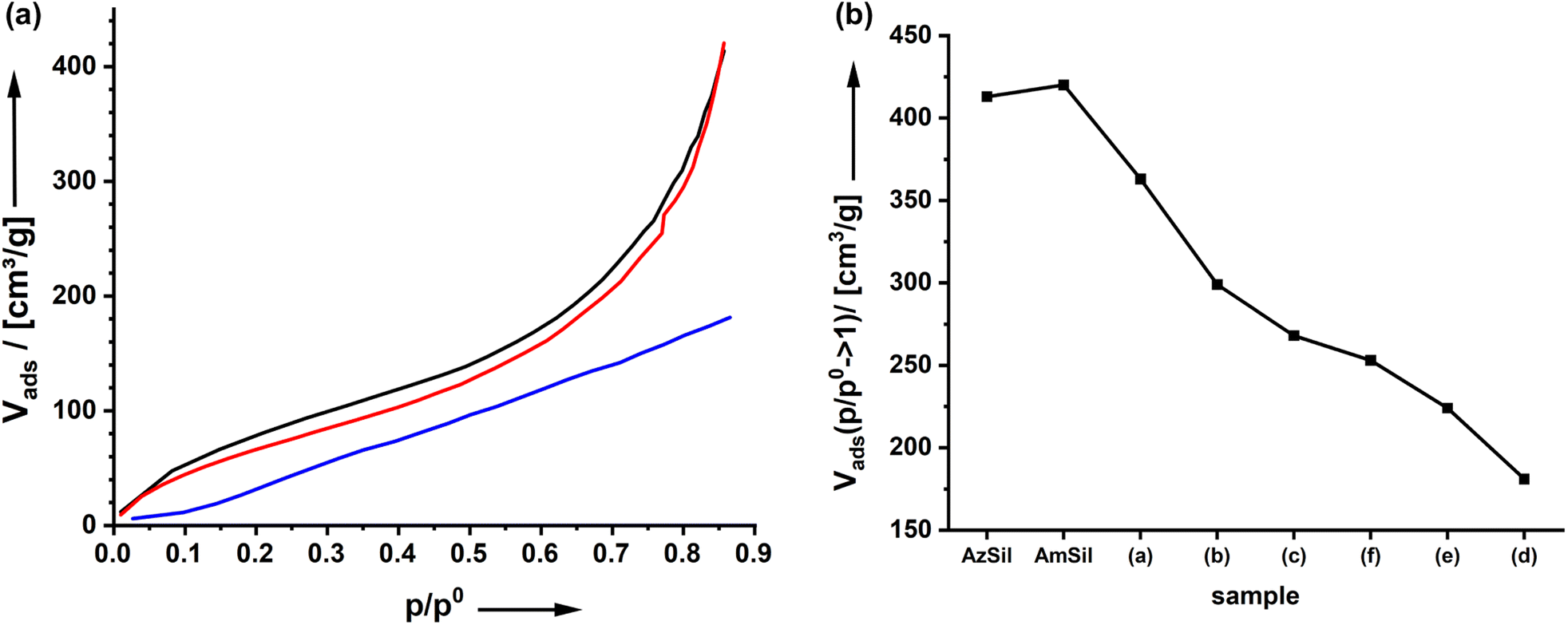 | ||
| Fig. 3 (a) H2O vapor adsorption of AzSil (black), AmSil (red) and Am0.48Sp(d)0.42Sil (blue). (b) Water uptake at p/p0 = 0.86 for different organosilica materials. | ||
Carbon capture properties of amine-modified organosilica materials containing spectator groups
It is expected that the presence of a hydrophobic group on the surfaces of amine-modified organosilica materials will have consequences for water adsorption. However, none of the spectator groups can interact specifically with carbon dioxide. Thus, the current paragraph addresses how the presence of spectator groups influences the adsorption of CO2.Fig. 4a shows the adsorption isotherm of CO2 on AmSp(d)Sil. The data for the other samples are given in ESI Fig. S10.† The isotherms display a Henry-type behavior. The adsorbed volume at p = 1 atm = 760 mm Hg at T = 0 °C was used to compare the uptake capacity. With values of cap = 1.0–1.75 mmol CO2 per gram of material (Fig. 4b), the organosilica aerogels' storage capacity is only average. Materials known in the literature with much higher specific surface area, such as MOFs, possess much better values up to 5 mmol g−1 under analogous conditions.36 As stated in the paper's introductory section, the uptake capacity is not the focus of the current work.
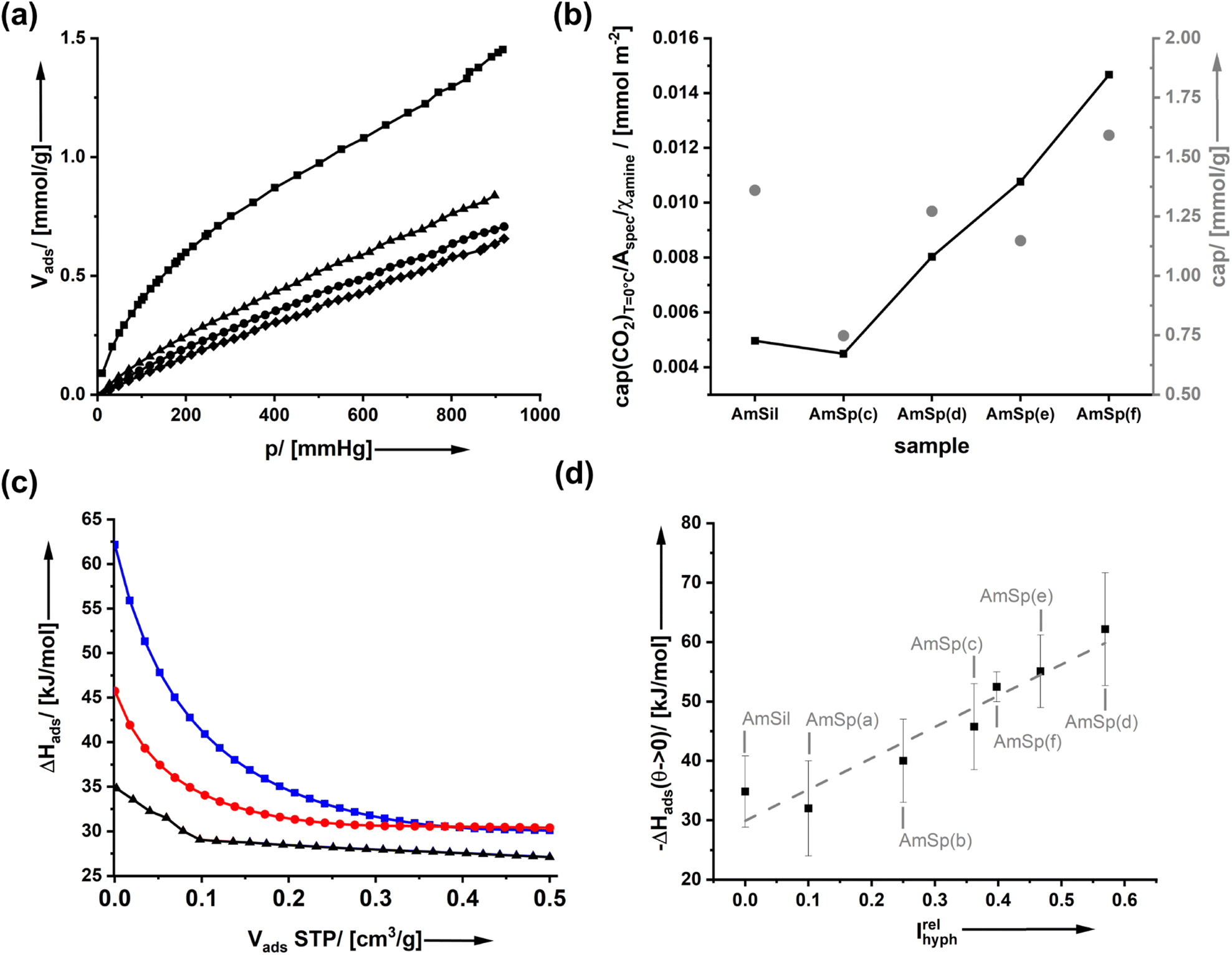 | ||
| Fig. 4 (a) CO2 adsorption isotherms of AmSp(d)Sil measured at T = 0 °C (squares), 30 °C (triangles), 40 °C (circles) and 50 °C (hashes). (b) Comparison of the CO2 uptake capacity between different organosilica materials; grey circles = capacity in mmol CO2 per gram material; black line = data referenced to Aspec and χamine of the respective sample. (c) isosteric heat of CO2 adsorption determined from T-dependent adsorption measurements for three exemplary materials; black triangles = AmSil; red circles AmSp(c); blue squares AmSp(d). (d) Correlation between the isosteric adsorption heat of CO2 on different organosilica aerogels and the correlation with the relative hydrophobicity index Irelhyph. | ||
The ideal model system would display a constant RF1:RF2 ratio whilst also the internal surface area Aspec is the same for all materials. The differences in kinetics when using different click reagents lead to a variation in both parameters as documented in Fig. 1d and 2e. It is thus difficult to compare the results directly. Therefore, in Fig. 4b, a second graph shows where cap was divided by Aspec and χamine. Compared to AmSil, the spectator groups have a positive effect on the uptake capacity of the material.
To determine the effectiveness of a CO2 molecule binding to an amine when the spectators are next to it, we have used T-dependent adsorption measurements (ESI Fig. S10†) to determine the isosteric adsorption enthalpy ΔHads(CO2). Fig. 4c shows the results of three materials as an example (AmSil; AmSp(c); AmSp(d)). One sees that there is a pronounced effect of the spectator groups on the adsorption enthalpy. The highest values for ΔHads are always found at low surface coverage, corresponding to low Vads values at STP (standard temperature and pressure). This behavior is nothing unusual because, for most adsorbents, the most reactive sites are typically occupied first. However, one can see that the presence of RF2 = tert-butyl phenyl in AmSP(d)Sil almost doubles ΔHads(CO2) compared to AmSil.
Even for higher surface coverages, there is a notable difference in ΔHads(CO2) between AmSil and AmSP(d)Sil (Fig. 4c). We tried to identify a systematic correlation between the nature of the spectator group and ΔHads(CO2). One idea is that the hydrophobicity next to the amine site can be a deciding factor. The adsorption of water (Fig. 3) is a good way to quantify the hydrophobicity. The material with the highest tendency to adsorb water is the least hydrophobic in the series (AmSil), and its adsorption value Vads,H2O(max) at p/p0 = 0.86 is used as a reference point for the calculation of the relative hydrophobicity index Irelhyph = 1 − Vads,H2O/Vads,H2O(max). The values for ΔHads(CO2)Vads→0 is then plotted as a function of Irelhyph in Fig. 4d. Interestingly, there is a linear correlation between the hydrophobicity and ΔHads(CO2). Again, one has to consider that the samples are not absolutely constant in RF1:RF2, and sample AmxSP(c) deviates more than the others. However, taking into account the large error bars, the correlation is still reliable. What are the possible reasons for the unusual behavior? we propose two alternative hypotheses.
(a) The initial step in the chemisorption of the CO2 molecules is the Lewis-acid-base reaction with lone pairs at the amine groups, which attack the carbon atom as an electrophilic center. If the nucleophilicity of the amine group is influenced in some way by the spectator groups, one could explain differences in ΔHads(CO2). More specifically, if the amine becomes more nucleophilic, the bond between C–N becomes stronger. However, one would then expect that the stretching frequency in the IR spectra of the resulting carbamic acid species should differ from material to material and should be shifted to higher wavenumbers the higher the ΔHads(CO2) value is. However, after close inspection of the IR data, we cannot see any frequency shift of the carbamate signals. An alternative explanation for the observed phenomena has to be found.
(b) CO2 is an unpolar molecule; thus, its solubility is higher in less polar media. We hypothesize that the hydrophobic groups on the surface of the organosilica material act as a quasi-solvent, which may even increase the local concentration of CO2 and allow the molecule to bind more tightly to the amine functionalities. Similar effects are known in the literature, for instance, for films formed by ionic liquids on the surfaces of porous hosts.37 In our case, the additional van der Waals (vdW) interactions with the quasi-solvent layer contribute to the adsorption enthalpy.
Molecular modeling calculations (Fig. 5b) support the view that the amine functionality is integrated inside a quite dense phase of the spectator groups forming the outer surface of the pores. Because a carbon dioxide molecule approaching the surface goes through different stages in the adsorption process, we expect that effects can be seen spectroscopically.
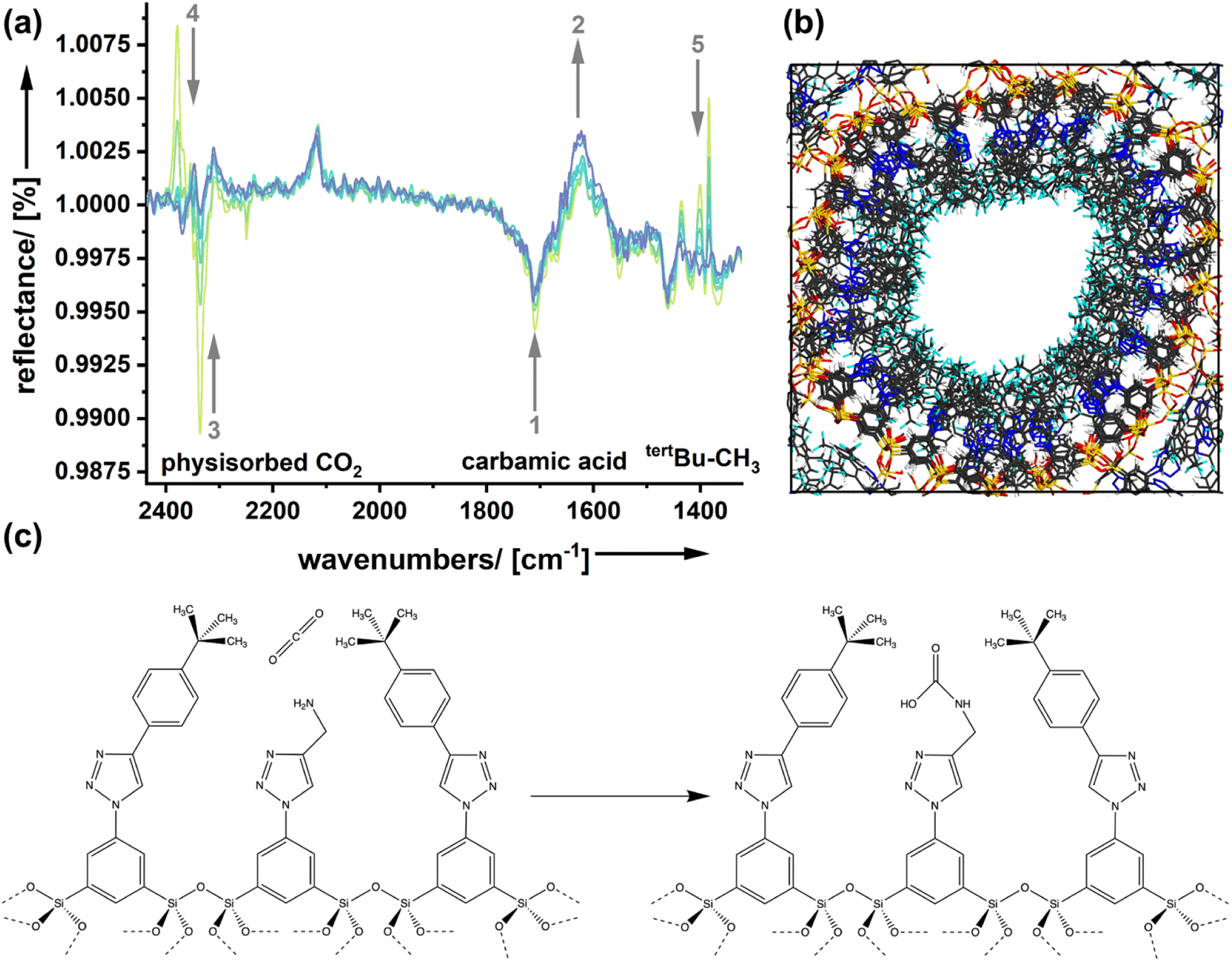 | ||
| Fig. 5 (a) DRIFTS measurements performed for AmSp(d)Sil; t = 1 min (yellow) to 15 min (blue). (b) Molecular modelling of the surfaces of one pore in an organosilica material. The spectator group extends the primary amine function, and the tert-butyl phenyl groups in AmSp(d) Sil form a disordered layer that is liquid-like. (c) Schematic representation of the CO2 uptake proposed for amine-modified organosilica surfaces containing spectator groups (RF2 = tert-BuPh; AmSp(d)Sil). The first step is the quasi-solvation of CO2 in the surface's hydrophobic domains, which is associated with ΔHqsolv. The following Lewis-acid reaction between the amino group and carbon dioxide followed by carbamic acid formation is also known from the literature and contributes to ΔHchemisorb. Both enthalpies contribute to ΔHadsorb. However, only ΔHqsolv depends on the nature of the spectator group, respectively, and its hydrophobicity. | ||
Fig. 5a shows DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy) spectra of the sample AmSp(d)Sil, which has shown the strongest spectator group effect in the determination of the adsorption enthalpies (Fig. 4d). The sample was treated with carbon dioxide (p = 1 bar) first. Because the CO2 in the chamber's atmosphere is irrelevant and interferes with the signals for bound CO2, the system was flushed with dry nitrogen. Spectra were recorded at different times and are shown in Fig. 5a. The decrease in the intensity of a reflection signal, indicated by the grey, downward arrows, means that there is more absorption and the corresponding species is forming. Vice versa, signals with increasing reflection intensity (upward arrows) stand for species that disappear. Signal assignment was performed according to the extensive literature on amine-modified materials used for carbon dioxide adsorption.38–40 In good agreement with the literature is the existence and formation of carbamic acid species (see also Fig. 5b), or carbamate–ammonium pairs seen in the region 1750–1550 (signals ‘1’ and ‘2’). Because those species are more tightly bound, there is only a small change in the corresponding DRFITS signals with time. The region of 2340 cm−1 is characteristic of physically adsorbed species ‘3’. The reflectance increases, which is not a surprise as physisorbed CO2 is only weakly bound in such large pores as those present in the organosilica aerogels presented here. However, we see one more signal in the same spectral region but shifted to slightly higher wavenumbers (‘4’; 2377 cm−1). It seems that this species forms with time as its reflectance decreases. We assume that the vdW interactions with the spectator groups change the vibrations of CO2 slightly, which would result in a small signal shift to higher wavenumbers. This carbon dioxide species could be the one that resides in a quasi-solvated state in the hydrophobic domain close to the silica surface (Fig. 5b, left image). The latter interpretation is consistent with the intensity changes seen in the fingerprint region (‘5’). The signals in this region are present in the pure, carbon-dioxide free AmSp(d)Sil material and correspond to the tertbutyl-CH3 groups. A change in this signal is not expected unless the presence of carbon dioxide has an influence.
There is now sufficient evidence that the quasi-solvation of CO2 in the hydrophobic domain of the organosilica surfaces (Fig. 5b) is associated with an enthalpy ΔHqsolv. ΔHqsolv depends on the hydrophobicity of the spectator group and contributes to the adsorption enthalpy.
Materials consisting of zones of different adsorption enthalpies
The van der Waals interactions associated with ΔHqsolv can be strong, but due to the distance (d) dependency (∝ 1/d6) only when the surface-bound CO2 and the spectator groups are close to each other. From our previous work on bifunctional organosilica materials, which were investigated by electron spin resonance (ESR) spectroscopy,17 we know that the distribution of the functionalities on the pore-surfaces is statistical. This means that the average distance between equal groups e.g. RF1, is a function of composition x,y. The modification of organosilica materials via click-chemistry allows the preparation of monolithic materials with controlled chemical gradients as we showed in several past papers.18,22,28 We adopted the method here and have prepared a material containing a gradient in the tert-butyl phenyl spectator group. Fig. 6a–d demonstrate this for an exemplary, fixed amine density in Amx=0.5SP(d)ySil materials. One can see that the probability is very high that each amine group has at least one further amine group as a next neighbor. However, for a low density of the spectator groups (x ≪ 0.5) one can see that only a few amine groups have a spectator as a neighbour. For moderate densities (x < 0.5), the probability is high that most amines have at least one neighbouring spectator. For x = 0.5, most amines are surrounded by two spectator groups. Vice versa, lowering x and increasing y further would reduce the amine–amine contacts and increase the number of spectator groups in the direct vicinity.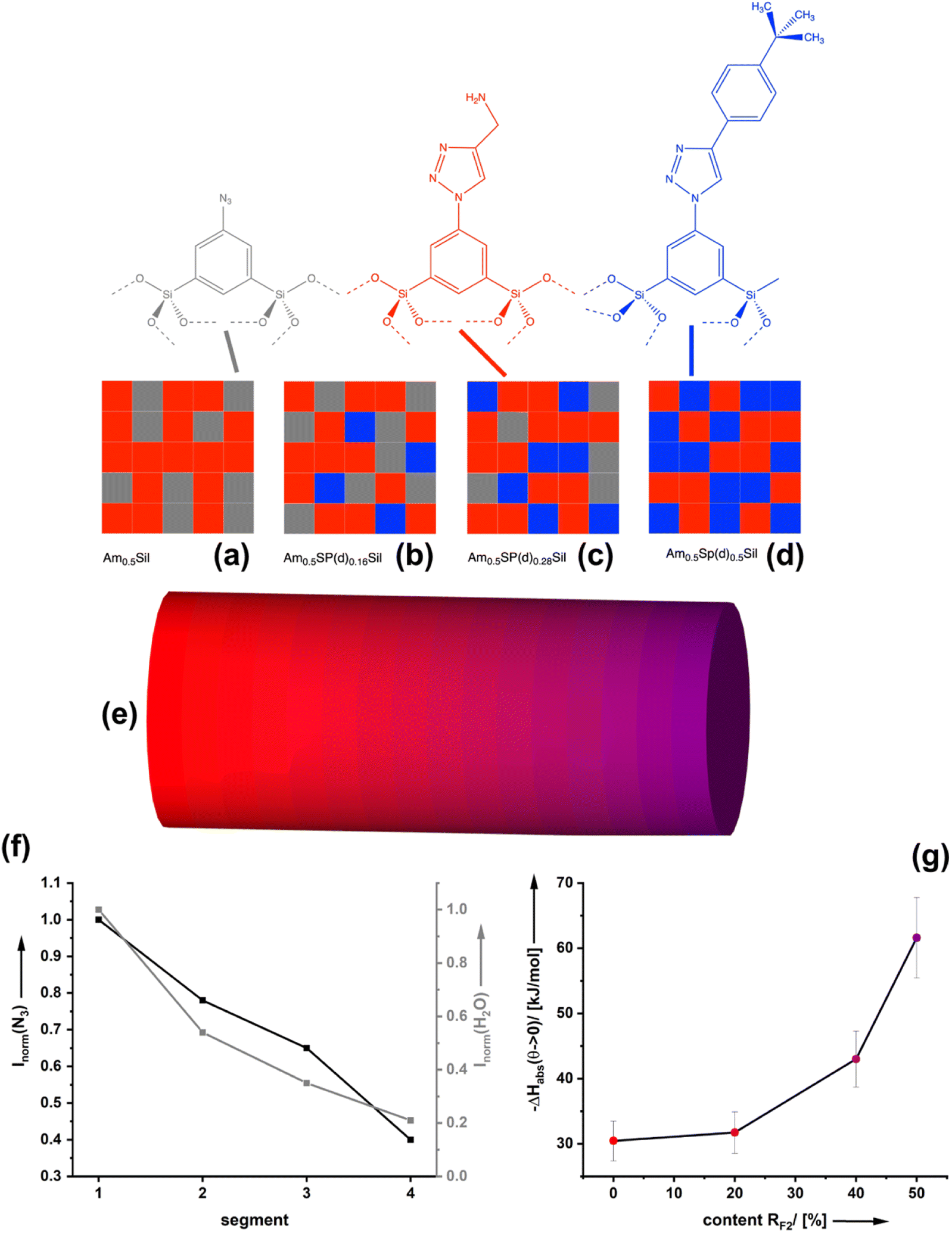 | ||
| Fig. 6 (a–d) Schematic representation of the chemical nature of the pore-surfaces of Amx=0.5Sp(d)ySil materials which are constant in the amine density (red squares) and vary systematically in the spectator group (blue). For high y-values every amine has a spectator group as a neighbor and many have even two neighbors. (e) Schematic representation of a monolithic organosilica material containing a gradient in the spectator group RF2 = tert-butyl phenyl. (f) Evaluation of FT-IR spectra of different segments associated with an increasing ratio of RF2. (g) CO2 adsorption enthalpies of materials associated with an increasing ratio of RF2. | ||
For characterization, different areas of the resulting monolith (Fig. 6b) were characterized by IR spectroscopy (see Fig. 6c and ESI S11†). The decreasing intensity of the azide vibration demonstrates the successive presence of the spectator group at the surfaces. The more tert-butyl phenyl groups are present, the weaker the vibration of bands (3382 cm−1) associated with the presence of water. The latter result proves that the zones containing the spectator groups are more and more hydrophobic. Next, we have investigated the CO2 adsorption isotherms for materials differing in the ratio of amine functionalities to tert-butyl phenyl groups RF1:RF2 (ESI Fig. S11† and 6d). According to our current knowledge, each composition of AmxSp(d)ySil should lead to a different CO2 adsorption enthalpy. Starting from the value of a material containing no spectator groups Am0.48Sp(d)0Sil (ΔHads ≈ 30 kJ mol−1), we see that a low density of the spectator group (Fig. 6b) has only a minor effect. The tert-butyl phenyl sites are more or less isolated on the surface, which is insufficient to induce the quasi-solvent effect. A higher density of spectator groups seems necessary (Fig. 6c and d), and then the quasi-solvent effect manifests by increasing values for ΔHads.
Experimental
Chemical synthesis
All starting chemicals and solvents have been purchased from Sigma Aldrich or TCI and were used after purification and drying.AzSil material. 117.2 μL (0.22 mmol, 116 mg) 1,3-bis-tri-isopropoxysilylphenyl-5-azide are dissolved in 1 mL ethanol. 50 μL aqueous HCl is added and left stirring for 1 h. Then, 50 μL aqueous NH3 (25 wt%) is added under vigorous stirring. The solution is transferred into a sealed 3 mL vial. Gelation takes place within 15 minutes. The gel is aged for 20 h at room temperature, after which the monolith is removed from the vial. Solvent exchange is performed by placing the monolith in acetone for 3 days. Fresh solvent is used multiple times a day. Finally, an aerogel is prepared by supercritical extraction with liquid CO2.
Supercritical drying. A monolith is placed into a sample holder filled with acetone. The holder was transferred into a dryer chamber and closed. The chamber was filled with liquid CO2 (p ∼60 bar). The acetone was exchanged with liquid CO2 by removing the acetone through the release valve. After complete removal of acetone from the chamber, it was heated to 42 °C (p ∼90 bar). The CO2, which is supercritical under these conditions, was released throughout 4–5 h. After completely releasing CO2, the dried monoliths were removed from the dryer chamber.
AmSil material. Depending on the desired degree of azide functionalization and the mass of the dry monolith, one can use different amounts of the catalyst and the alkyne (0.5–2 eq.). For instance, (0.2–0.4 mmol, = 1–2 eq.) copper(I)-tetrakis(acetonitrile) hexafluorophosphate is dissolved in 10 mL acetone. N-Boc-propargylamine (0.11–0.44 mmol, = 0.5–2 eq) is added to the solution. The AzSil monolith (0.2 mmol, = 1 eq.) is then placed in the reaction solution. While stirring for 48 hours, one has to be careful that there is no mechanical damage from the stirring bar. For removal of the Boc protection group, the monolith was added to a 4 M HCl in acetone solution for two days. This solution was exchanged once after 24 h. After another washing step with acetone the monolith was placed in a solution of 500 μL NH3 (25%) in 10 mL acetone for 24 h. This step was repeated twice. An aerogel is prepared by supercritical extraction with liquid CO2.
AmSp(x) bifunctional materials. The general procedure is similar to the one described before. However, the AmSil monolith still containing the Boc-group is used for the second post-functionalization step. The corresponding alkyne (1–3 eq.) was added together with the new catalyst dissolved in 30 mL acetone, and the mixture was reacted for 90 hours at room temperature. Afterward, deprotection and supercritical drying are performed as described before.
Gradient materials. A boc-protected AmSil material is the starting point. Ethanol is added to obtain a wet monolith. The monolith is placed inside a shrinking tube to avoid accessing the reaction solution from the side.28 One side of the tube is sealed and the other side is placed inside the reaction solution containing the catalyst and the spectator group alkyne. After the completion of the reaction the monolith was removed from the shrinking tube by dipping it into dichloromethane for 15 min. Thereby, the shrinking tube was swollen, and the monolith was removed. The solvent was exchanged for acetone. Afterward, deprotection and supercritical drying are performed as described before.
Analytical methods
ATR-IR spectroscopy was performed on a Bruker Tensor with an ATR unit. All spectra were background corrected and normalized to the Si–O–Si vibration at 1044 cm−1. Thermogravimetric analysis measurements were performed using a Netzsch STA 409 PC LUXX with a heating rate of 5 K min−1 until 300 °C and 10 K min−1 for 300–1000 °C. NMR measurements (1H, 13C) were performed on a Bruker Ascend 400 MHz spectrometer. Scanning electron microscopy was performed on a Hitachi Regulus SU8200. Before conducting any gas adsorption measurement, the samples were degassed at 85 °C with the build-in degas system of an ASAP 2020 for 6 h. For temperature-dependent measurements the samples were degassed in between each measurement. The N2 measurements were performed at 77 K using liquid nitrogen and the CO2 measurements were performed at 0–50 °C using a Micromeritics Iso Controller Sub-Ambient to maintain a constant temperature. The equilibration interval was 10 s. The measurements were blank corrected by subtracting the blank measurement of an empty sample tube measured under identical conditions. The heat of adsorption was determined with the plugin of the MicroActive (4.06) software using the Clausius–Clapeyron-equation and isotherms at 0–50 °C.For the DRIFT measurements, a Vertex 80v FTIR spectrometer from Bruker was used, equipped with a reaction chamber (Praying Mantis, DRP-M-16) and a gas flow cell (CHC-CHA-4), both from Harrick. To produce the samples, the silica material was ground with a pestle and mortar. The resulting powder was mixed with KBr to yield a mixture with 1% weight of the material. The mixture was again ground to minimize the size of the silica and the KBr grains. This is necessary to prevent artifact formation in the DRIFT spectra. KBr for infrared spectroscopy from Roth was used. A stock of the sample was produced, but each experiment was conducted with a new sample from the stock. The stocks were kept at 80 °C in a drying oven over night and desiccated for at least 15 min prior to the measurements, to remove crystal water from KBr. For the measurement, a set of three grids was placed in a CHC gas cell to prevent the powder from leaking out of the sample holder when the gas flow was applied. The pore size of the smallest grid (from Plano) was approximately 4 μm. To stabilize this fragile grid, it was placed between the two others with bigger pore sizes (from Harrick). To apply the gas flow, two mass flow controllers (Bronkhorst, El Flow Select F-201CV) were used, one for N2 and one for CO2. First, the sample was purged with nitrogen for 15–30 min and afterward the background signal was recorded. Gas mixtures of N2 and CO2 with different partial pressures were then applied for five minutes each. Each of the CO2-adsorption steps was followed by a purging step, conducted with pure nitrogen for 15 min. The total flow rate was always 0.5 L min−1. For the gas mixtures, the rates of the two gases were adjusted to result in partial pressures of 0.2, 0.4, 0.6, 0.8 and 1 bar of CO2. IR measurements were performed with a spectral resolution of 4 cm−1 and baseline correction.
Conclusions
Porous materials containing primary amine groups are well known in the literature for their capability to adsorb and reversibly bind carbon dioxide. For any adsorbent, co-adsorption of competing molecules is a problem, and in the case of carbon-capture materials, the presence of water is an issue. One expects that the more hydrophobic a surface is, the less the co-adsorption of water contributes. We have shown here that the relevance of hydrophobic groups goes much further.Organosilica materials characterized by bifunctional materials have been prepared using click-chemistry. The active group, a primary amine responsible for the interaction with CO2, is next to the hydrophobic group. We found that the density of this so-called spectator group, as well as its nature, is used to determine the adsorption enthalpy ΔHads(CO2). The presented data indicate that it is not the aliphatic or aromatic character of the hydrophobic entities that is the most important factor, nor is it a steric influence. The capability of a spectator group to make the surface more hydrophobic is essential. Above a certain density of the spectator groups on the surface, they act like a quasi-solvent. The solvation enthalpy ΔHsolv(CO2) contributes and is higher the more hydrophobic the spectator group is. The effect allows for almost doubling the adsorption enthalpy. The latter observation represents the first main result of our study: one can now precisely adjust the adsorption enthalpy of CO2 on amine-modified materials. The described findings agree with research by others on the solubility of CO2 in ionic liquids (ILs), where hydrophobicity is also an important factor and influences enthalpic factors directly.40–43 Considering the latter work and the fact that liquid CO2 shows preferable interactions with perfluorinated groups, it is promising for the future to prepare a material containing more extended fluorinated alkyls compared to AmxSP(b,c).
Our definition of a spectator group is that it does not directly interact with CO2. As a consequence, one would expect that the effects on the adsorption enthalpy are only weak. However, the current results show that the seemingly “spectator” groups are equally important compared to the active CO2 binding site (the amine groups). Unlike chemisorption, where one can also increase the adsorption enthalpy, the described quasi-solvent effects are fully reversible.
We developed an additional approach based on our experience with gradient materials. An organosilica monolith comprising a fixed amine “background” and a gradient in the hydrophobic spectator group was prepared. It can be shown that the ΔHsolv(CO2) becomes a function of the location. The different zones of the material correlate with different adsorption enthalpies. We expect a material associated with a high value for ΔHads(CO2) to have a high affinity and can exhibit selectivity. In contrast, low values for ΔHads(CO2) mean the opposite. Thus, we hope to be able to show in the future that the gradient material is capable of separating gas mixtures and transporting the different species in opposite directions.
Author contributions
ME: investigation (materials); KH: funding acquisition, supervision (DRIFTS); WGH: investigation (DRIFTS), formal analysis (DRIFTS); YK: project administration, supervision (materials); DM: investigation (materials), formal analysis; NK: methodology (materials); SP: conceptualization, data curation, funding acquisition, methodology, project administration, supervision (materials), visualization, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by a grant from the Deutsche Forschungsgemeinschaft (DFG): Grant numbers PO 780/23-1 and HA 5142/6-1. We thank Marvin Träger, Katharina Rüther and Andreas Schneider for the molecular modelling calculations.Notes and references
- D. D'Alessandro, B. Smit and J. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- R. Ben-Mansour, M. Habib, O. Bamidele, M. Basha, N. Qasem, A. Peedikakkal, T. Laoui and M. Ali, Appl. Energy, 2016, 161, 225–255 CrossRef CAS.
- Z. Zhang, Z. Cano, D. Luo, H. Dou, A. Yu and Z. Chen, J. Mater. Chem. A, 2019, 7, 20985–21003 RSC.
- B. Chen, S. Xiang and G. Qian, Accounts Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- J. Borden and D. Savage, Curr. Opin. Microbiol., 2021, 61, 58–66 CrossRef CAS PubMed.
- S. Tanaka, C. Kerfeld, M. Sawaya, F. Cai, S. Heinhorst, G. Cannon and T. Yeates, Science, 2008, 319, 1083–1086 CrossRef CAS PubMed.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed.
- T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina and T. J. Erb, Science, 2016, 354, 900–904 CrossRef CAS PubMed.
- G. M. M. Stoffel, D. A. Saez, H. DeMirci, B. Vögeli, Y. Rao, J. Zarzycki, Y. Yoshikuni, S. Wakatsuki, E. Vöhringer-Martinez and T. J. Erb, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13964–13969 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- X. Zhang, F. Shi, J. Niu, Y. Jiang and Z. Wang, J. Mater. Chem., 2008, 18, 621–633 RSC.
- F. Hoffmann and M. Froeba, Chem. Soc. Rev., 2011, 40, 608–620 RSC.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634–2642 CrossRef CAS PubMed.
- P. Van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- Y. Chen and J. Shi, Adv. Mater., 2016, 28, 3235–3272 CrossRef CAS PubMed.
- A. Kuschel, M. Drescher, T. Kuschel and S. Polarz, Chem. Mater., 2010, 22, 1472–1482 CrossRef CAS.
- A. Schachtschneider, M. Wessig, M. Spitzbarth, A. Donner, C. Fischer, M. Drescher and S. Polarz, Angew. Chem., Int. Ed., 2015, 54, 10465–10469 CrossRef CAS PubMed.
- J. Gehring, D. Schleheck, M. Luka and S. Polarz, Adv. Funct. Mater., 2014, 24, 1140–1150 CrossRef CAS.
- J. Gehring, D. Schleheck, B. Trepka and S. Polarz, ACS Appl. Mater. Interfaces, 2015, 7, 1021–1029 CrossRef CAS PubMed.
- J. Gehring, B. Trepka, N. Klinkenberg, H. Bronner, D. Schleheck and S. Polarz, J. Am. Chem. Soc., 2016, 138, 3076–3084 CrossRef CAS PubMed.
- H. Bronner, A.-K. Holzer, A. Finke, M. Kunkel, A. Marx, M. Leist and S. Polarz, RSC Adv., 2020, 10, 17327–17335 RSC.
- D. Kollofrath, M. Geppert and S. Polarz, Chemsuschem, 2020, 13, 5100–5111 CrossRef CAS PubMed.
- N. Pal, J. Lee and E. Cho, Nanomaterials, 2020, 10, 2122 CrossRef CAS PubMed.
- E. Cho, D. Kim and M. Jaroniec, J. Phys. Chem. C, 2008, 112, 4897–4902 CrossRef CAS.
- P. Bhanja, A. Modak, S. Chatterjee and A. Bhaumik, ACS Sustainable Chem. Eng., 2017, 5, 2763–2773 CrossRef CAS.
- T. Asefa, M. Kruk, M. MacLachlan, N. Coombs, H. Grondey, M. Jaroniec and G. Ozin, J. Am. Chem. Soc., 2001, 123, 8520–8530 CrossRef CAS.
- N. Klinkenberg, A. Klaiber, M. Müller and S. Polarz, Microporous Mesoporous Mater., 2020, 294, 109879 CrossRef CAS.
- A. Pragya and T. K. Ghosh, Adv. Mater., 2023, 2300912 CrossRef CAS PubMed.
- P. Kortunov, L. Heinke, S. Vasenkov, C. Chmelik, D. Shah, J. Karger, R. Rakoczy, Y. Traa and J. Weitkamp, J. Phys. Chem. B, 2006, 110, 23821–23828 CrossRef CAS PubMed.
- Z. Liu, M. Meyers, Z. Zhang and R. Ritchie, Prog. Mater. Sci., 2017, 88, 467–498 CrossRef CAS.
- C. Zhang, Y. Xie, C. Xie, H. Dong, L. Zhang and J. Lin, MRS Bull., 2022, 47, 432–439 CrossRef.
- E. Lopez-Maya, C. Montoro, V. Colombo, E. Barea and J. Navarro, Adv. Funct. Mater., 2014, 24, 6130–6135 CrossRef CAS.
- N. Klinkenberg, S. Kraft and S. Polarz, Adv. Mater., 2021, 33, 2007734 CrossRef CAS PubMed.
- M. Luka and S. Polarz, Microporous Mesoporous Mater., 2013, 171, 35–43 CrossRef CAS.
- S. Mahajan and M. Lahtinen, J. Environ. Chem. Eng., 2022, 10, 108930 CrossRef CAS.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martín-Matute and N. Hedin, Langmuir, 2011, 27, 11118–11128 CrossRef CAS PubMed.
- N. Hedin and Z. Bacsik, Curr. Opin. Green Sustainable Chem., 2019, 16, 13–19 CrossRef.
- Z. Bacsik and N. Hedin, Vib. Spectrosc., 2016, 87, 215–221 CrossRef CAS.
- A. Yokozeki, M. B. Shiflett, C. P. Junk, L. M. Grieco and T. Foo, J. Phys. Chem. B, 2008, 112, 16654–16663 CrossRef CAS PubMed.
- M. J. Muldoon, S. N. V. K. Aki, J. L. Anderson, J. K. Dixon and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 9001–9009 CrossRef CAS.
- L. A. Blanchard and J. F. Brennecke, Ind. Eng. Chem. Res., 2001, 40, 287–292 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01381f |
| ‡ SP and DM contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |