
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly acidic N-triflylphosphoramides as chiral Brønsted acid catalysts: the effect of weak hydrogen bonds and multiple acceptors on complex structures and aggregation†
Markus
Hecht
a,
Philipp
Dullinger
 b,
Wagner
Silva
a,
Dominik
Horinek
b and
Ruth M.
Gschwind
*a
b,
Wagner
Silva
a,
Dominik
Horinek
b and
Ruth M.
Gschwind
*a
aInstitut für Organische Chemie, Universität Regensburg, D-93053, Regensburg, Germany. E-mail: ruth.gschwind@chemie.uni-regensburg.de
bInstitute of Physical and Theoretical Chemistry, University of Regensburg, D-93053, Germany
First published on 29th April 2024
Abstract
N-Triflylphosphoramides (NTPAs) represent an important catalyst class in asymmetric catalysis due to their multiple hydrogen bond acceptor sites and acidity, which is increased by several orders of magnitude compared to conventional chiral phosphoric acids (CPAs). Thus, NTPAs allow for several challenging transformations, which are not accessible with CPAs. However, detailed evidence on their hydrogen bonding situation, complex structures and aggregation is still lacking. Therefore, this study covers the hydrogen bonding behavior and structural features of binary NTPA/imine complexes compared to their CPA counterparts. Deviating from the single-well potential hydrogen bonds commonly observed in CPA/imine complexes, the NTPA/imine complexes exhibit a tautomeric equilibrium between two proton positions. Low-temperature NMR at 180 K supported by computer simulations indicates a OHN hydrogen bond between the phosphoramide oxygen and the imine, instead of the mostly proposed NHN H-bond. Furthermore, this study finds no evidence for the existence of dimeric NTPA/NTPA/imine complexes as previously suggested for CPA systems, both synthetically and through NMR studies.
Introduction
Building molecules with high levels of stereoselectivity and under mild reaction conditions is a major challenge in organic chemistry. Among the metal-free classes of catalysts, chiral Brønsted acids have found widespread applications.1–3 In particular, 1,1′-Bi-2-naphthol (BINOL)-derived chiral phosphoric acids (CPAs) constitute a class of highly enantioselective catalysts available for asymmetric transformations, including Mannich-reactions, transfer hydrogenations, Strecker reactions, and many others.1,4–7 Within the vast variety of reactions, nucleophilic additions to imines developed into the central mechanistic model system for CPAs as Brønsted acid catalysts.8,9 Extensive NMR studies in binary CPA and N-arylimine complexes have been conducted previously, which proved the formation of hydrogen bond assisted ion pairs in these CPA/substrate complexes (Fig. 1A), supported by a network of CH–π and π–π interactions.10–12 For the CPA/imine complexes, four core structures with strong POHN hydrogen bonds were observed experimentally.11 Besides spectroscopic and experimental investigations, machine learning approaches have been key to a deeper understanding of reactivity and stereoselectivity in CPA-catalyzed reactions.13,14 Goodman et al. linked the stereoselectivity to the steric properties of the 3,3′-substituents and developed a model for predicting suitable CPAs based on reactant structures.15,16 Furthermore, Sigman et al. developed a data-driven prediction model to clarify the impact of various parameters on the stereoselectivity.17–19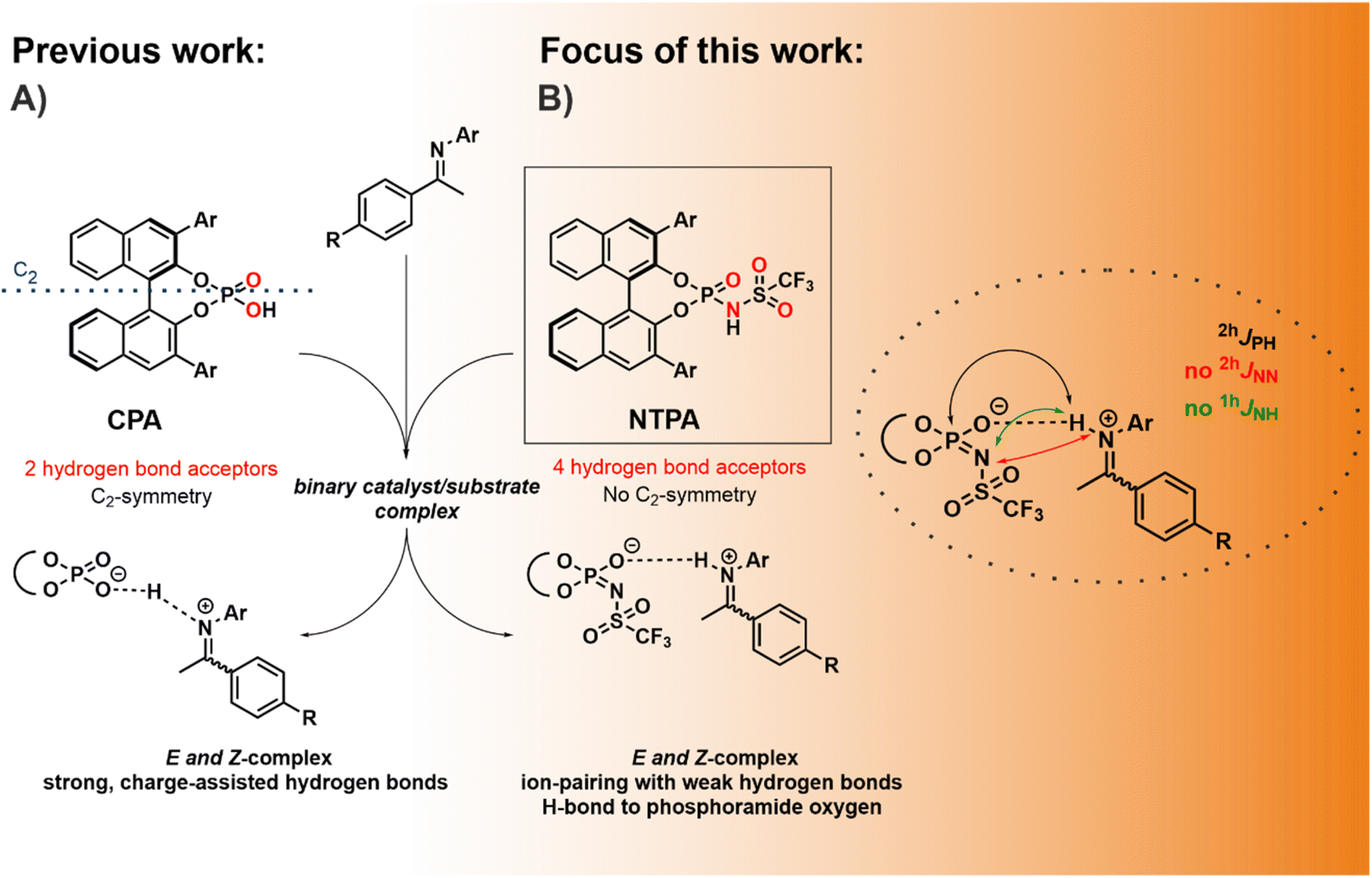 | ||
| Fig. 1 (A) Previously binary complexes between CPAs and imines have been studied proving the formation of strong, charge-assisted hydrogen bonds. For the CPA/imine complexes, two different orientations of each imine isomer (E and Z) inside the binary complex were detected. (B) The focus of this work was a hydrogen bond and a structural analysis of NTPA/imine complexes addressing the influence of increased acidity of the catalyst, multiple hydrogen bond acceptors and the missing C2 symmetry. | ||
However, the investigated classical chiral phosphoric acid catalysts are still limited to reactive substrates.2 For this purpose, the groups of List and Yamamoto reported the synthesis of other highly acidic catalysts such as disulfonimides (DSIs),20 imidodiphosphorimidates (IDPis)21 or N-triflylphosphoramides (NTPAs).22 These catalysts vary significantly from CPAs due to their increased acidity, chiral microenvironment, and the presence of multiple hydrogen bond acceptor sites, potentially enabling both OHN and NHN hydrogen bonds.23 For DSIs detailed investigations were conducted by our group revealing the existence of binary DSI/imine complex structures with hydrogen bonds to both acceptors, the nitrogen and the oxygen of the DSI.24,25 Furthermore, IDPi-catalyzed transformations were closely investigated by the group of List in an attempt to locate and characterize ionic or covalent species.26,27 However, the structure and hydrogen bond situation of NTPAs has to the best of our knowledge not been intensively studied yet. Several challenging transformations such as cycloadditions or addition reactions to imines which are not suitable with CPAs could be realized with NTPAs.23,28,29 Exemplarily, the group of Yamamoto reported an asymmetric Mukaiyama–Mannich reaction, in which less reactive aldimines, without an N-(2-hydroxyphenyl) moiety, are activated by NTPA.30 Subsequently, the question arises how the use of a stronger Brønsted acid such as NTPA influences the structure of the reaction intermediates and the hydrogen bond strength.31 Furthermore, the incorporation of a N-trifluoromethanesulfonyl group into the CPA system breaks the C2-symmetry of the catalyst and introduces multiple hydrogen bond acceptors (Fig. 1B). Hence, there is the question which of the several hydrogen bond acceptors is included in the resulting hydrogen bond. So far, Yamamoto et al. and other groups suggested that the negative charge is located at the nitrogen and consequently a hydrogen bond exists between the nitrogen of the NTPA catalyst and the substrate.30,32–34 However, Yoon et al. isolated a substrate-acid complex indicating a hydrogen bond between a protonated imidazolium and the phosphoramide oxygen of the NTPA catalyst via X-ray crystallography.35 Nevertheless, crystal structures do not always align with intermediates observed in solution.36 Moreover, it is also possible that the catalyst forms two hydrogen bond with one substrate, leading to a bifunctional activation mode.23 Therefore, the question remains what kind of H-bonding is active in NTPAs in solution. Low-temperature NMR spectroscopy at 180 K was proven to be an excellent tool to investigate the occurring intermediates as well as potential hydrogen bonds as sensitive experimental indicator for the binding situation within the binary complexes.10,12
Therefore, binary complexes of a NTPA catalyst with 11 different N-arylimines were investigated by low-temperature NMR. First, an in-depth analysis of the hydrogen bond situation in these binary NTPA/imine complexes was performed. The hydrogen bond was characterized by analyzing 1H and 15N chemical shifts in a Steiner–Limbach correlation and compared with the corresponding CPA/imine complexes.37–39 Second, various two-dimensional NMR-techniques were applied at 180 K for a chemical shift assignment of the NTPA/imine systems. Based on the assignment, characteristic NOE cross signals between the catalyst and substrate were analyzed. Structures for the binary complexes based on NOE cross signals were supported by molecular dynamics (MD)-simulations. In particular, the effect of the increased number of hydrogen bond acceptors of the NTPA compared to the CPA is addressed. Finally, the NTPA catalyst was examined for its potential involvement in a dimeric reaction pathway in the Mukaiyama–Mannich reaction.
Results and discussion
Model system
In order to compare the hydrogen bonding in binary NTPA/imine complexes with the previously investigated CPA/imine complexes,10–12 a comprehensive H-bond analysis was conducted. As catalyst we selected the commercially available NTPA 1 (Fig. 2a), which provides the opportunity for 19F NMR investigations, and eleven imines 2–12 (Fig. 2b) with different electronic and steric properties. 15N labeling of the imines allowed the access to the 15N chemical shifts which were necessary for the determination of the hydrogen bond strength. Whereas imines 2–12 were used to cover a broad range of basicities for a H-bond analysis using the Steiner–Limbach curve, the methoxy-substituted N-arylimine 2 was chosen for structural investigations due to the reduced chemical shift overlap in the binary complex. Furthermore, 15N labeling of NTPA 1 enabled deeper insights into a catalytic system with multiple hydrogen bond acceptors and clarification which of the hydrogen bond acceptors is included in the resulting hydrogen bond. Both, intense NOE cross peaks between imine 2 and the BINOL backbone of the NTPA as well as computer simulations were used for the structure elucidation. Finally, the NTPA 1 was examined in an enantioselective Mukaiyama–Mannich reaction to address possible reaction intermediates.30 For NMR measurements CD2Cl2 was chosen as solvent, since it provides the best chemical shift dispersion and smallest line widths. All NMR spectra were recorded at 180 K to reach the slow exchange regime of the hydrogen-bonded protons (see ESI Fig. S1–S11 and S12–S14†).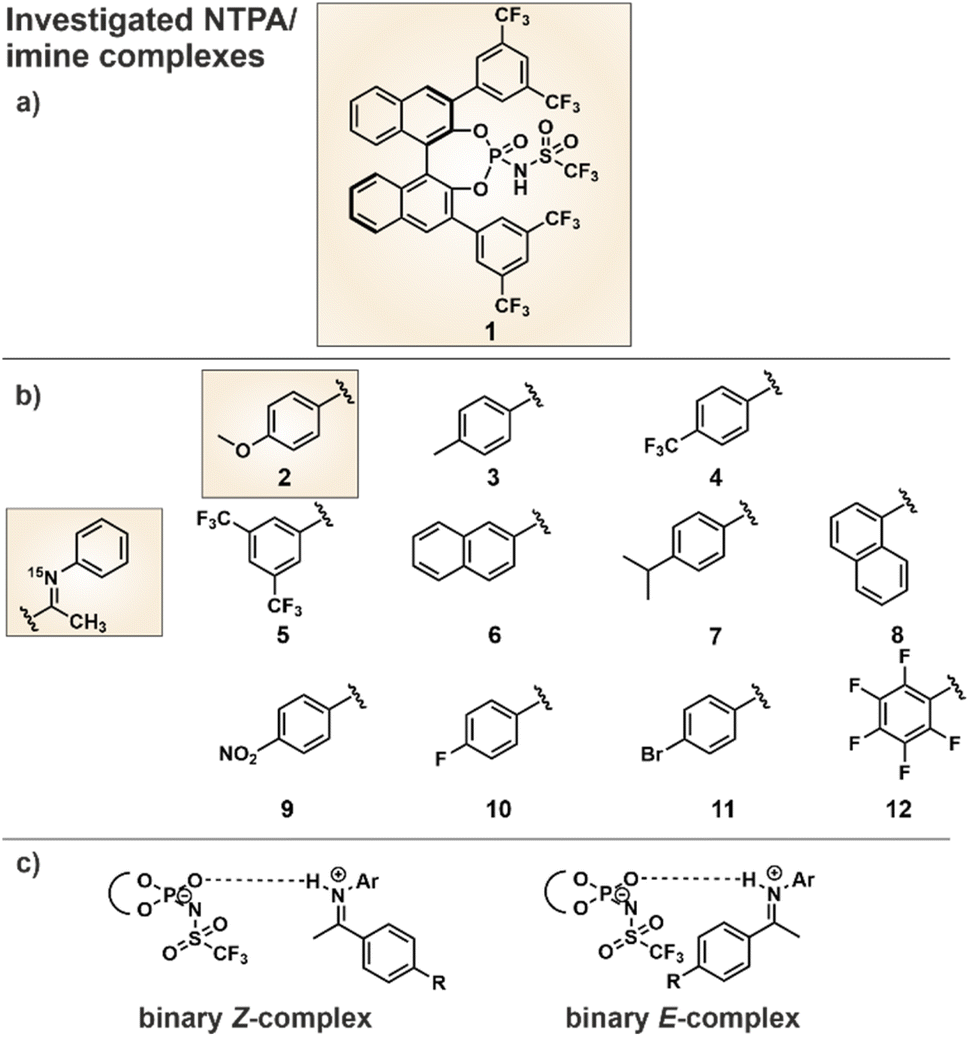 | ||
| Fig. 2 (a) NTPA catalyst 1 was selected due to its 3,3′-substituents, which provide the opportunity for 19F NMR measurements. (b) Structures of the imines 2–12 for the hydrogen bond analysis to cover a broad basicity range. Imine 2 was investigated in detail with NTPA 1 to gain structural insights into the binary NTPA/imine complexes. (c) The focus of this work was the NMR-spectroscopic investigation of the binary NTPA/imine complexes to clarify which of the several hydrogen bond acceptors is included in the weak hydrogen bond. The imine exists either as E- or Z-isomer. | ||
Hydrogen bond analysis
The use of a Brønsted acid with a stronger external acidity such as NTPA 1 raised the question of whether binary complexes show similar hydrogen bonds to those present in the CPA/imine complexes or whether pure ion pairs without hydrogen bond contributions are formed. In general, the position of the proton within a hydrogen bond hinges on both the acidity of the hydrogen bond donor and the basicity of the hydrogen bond acceptor. The strongest hydrogen bonds are formed with the proton shifted towards the center of the hydrogen bond. With increasing acidity of the H-bond donor a hydrogen bond assisted ion-pair is formed. Finally, also a pure ion pair without hydrogen bonding can be generated by shifting the proton completely to the acceptor. The amount of proton transfer between CPA and imine was previously interpreted by the analysis of different binary CPA/imine complexes regarding 1H and 15N chemical shifts as well as 1hJNH coupling constants in a Steiner–Limbach correlation.10,12,38 The correlation was developed by Limbach and Denisov in their studies of OHN hydrogen bonds in pyridine acid complexes.37–39 Notably, our recent studies have demonstrated that this correlation is also applicable to the POHN hydrogen bonds in CPA/imine complexes.10,12 For CPA/imine complexes, the data points derived from the hydrogen bond proton and the 15N chemical shift of the hydrogen bond acceptor revealed a parabolic dependency between these two chemical shifts (Fig. 3 orange points). This suggests the existence of a single-well hydrogen bond, where the maximum of the parabolic correlation curve represents the formation of the strongest hydrogen bond.38 In general, the calculated parabolic correlation curve is valid only if the H-bond proton always moves in a single-well potential. When an equilibrium between two proton positions exists, indicating a double-well potential, the correlation between the two chemical shifts results in a straight line.38 In Fig. 3, the Steiner Limbach plot of NTPA 1 is compared with that of the CPA TRIP, which is known to provide extremely strong hydrogen bonds. To cover a broad basicity range, binary complexes of NTPA 1 and TRIP with the imines 2–12 from Fig. 2 were employed.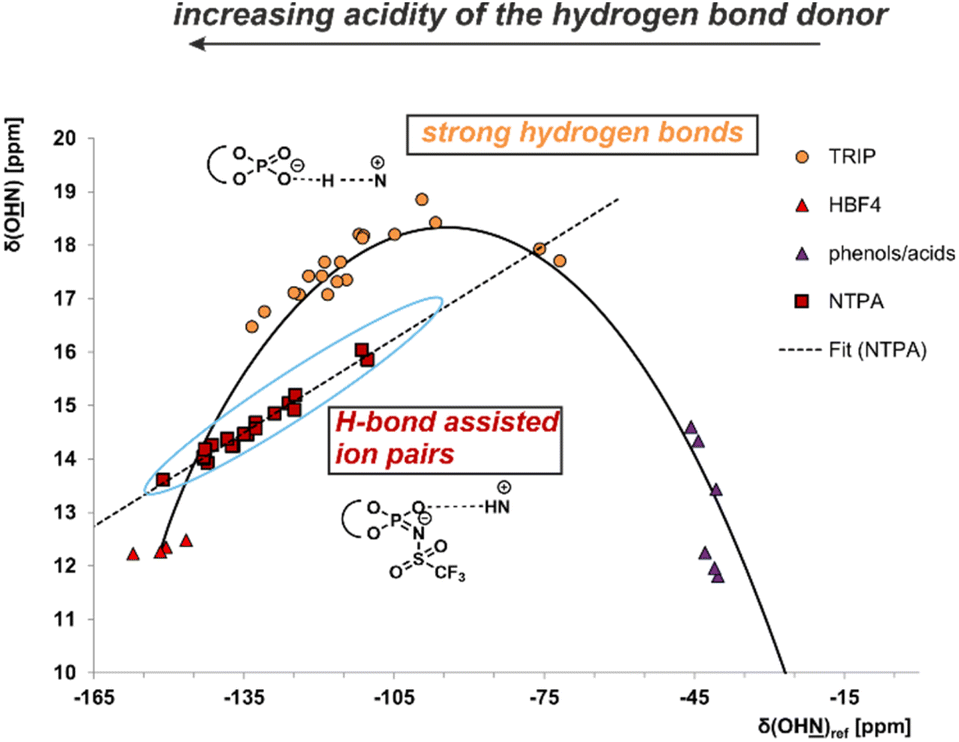 | ||
Fig. 3 Plot of δ(OH![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) )ref against δ(O )ref against δ(O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) N) for various binary NTPA 1/imine and CPA/imine complexes. The 15N chemical shifts are referenced by δ(OH)ref = δ(OH)obs – 340.8 ppm. 340.8 ppm is the chemical shift of the free imine showing the strongest hydrogen bond in the previous work. Data points of phenols/carboxylic acids, tetrafluoroboric acid and the CPA TRIP are from previous studies (see ESI†). Binary E- and Z-complexes of NTPA 1 and imines 2–12 are depicted in red rectangles and show a deviation from the correlation curve. This is an indication for a double-well potential, which means the proton is either located at the proton acceptor or donor. N) for various binary NTPA 1/imine and CPA/imine complexes. The 15N chemical shifts are referenced by δ(OH)ref = δ(OH)obs – 340.8 ppm. 340.8 ppm is the chemical shift of the free imine showing the strongest hydrogen bond in the previous work. Data points of phenols/carboxylic acids, tetrafluoroboric acid and the CPA TRIP are from previous studies (see ESI†). Binary E- and Z-complexes of NTPA 1 and imines 2–12 are depicted in red rectangles and show a deviation from the correlation curve. This is an indication for a double-well potential, which means the proton is either located at the proton acceptor or donor. | ||
Whereas the CPA/imine systems show chemical shifts for the hydrogen bond proton above 16 ppm and follow a parabolic curve revealing very strong hydrogen bonds (Fig. 3), the NTPA/imine E- and Z-complexes exhibit high field shifts for both 1H and 15N (for 1H and 15N spectra see ESI Fig. S1–S11†). As a result, these complexes were positioned far down on the left side of the Steiner–Limbach curve close to the almost pure ion pair with HBF4. The position of the NTPA complexes showed that the proton within the hydrogen bond is significantly shifted toward the imine nitrogen suggesting weak or no hydrogen bonds. However, upon closer examination of the Steiner–Limbach curve, there's another significant difference between the CPA/imine complexes and the current NTPA/imine complexes. In contrast to the CPA systems, which aligned perfectly with the parabolic Steiner–Limbach correlation curve, the data points of the NTPA systems formed a straight line, deviating from the parabolic dependency.
This was the first time we observed such a trend within our catalyst systems, and it can be interpreted as explained above: the parabolic correlation curve is valid only for strong H-bond and not for a proton-transfer equilibrium between two tautomeric forms.37,40 Therefore, the deviation of NTPA 1/imine complexes from the parabolic 1H/15N chemical shift correlation curve is an indication for a double-well potential in these complexes.37,40 This means, the proton is either located at the nitrogen of the imine or at the nitrogen/oxygen of the NTPA catalyst and upon variation of the imine basicity only the relative populations of the two positions are changed and not the hydrogen bond itself, which explains the linear correlation using the different imines. Indeed, upon examination of the 1hJNH coupling constants of the binary NTPA 1/imine complexes, coupling constants of 1hJNH ∼ 87–91 Hz are detected, which are larger than for all CPA/imine complexes (1hJNH between 82 and 86 Hz).121hJNH coupling constants are a fundamental factor for the binding strengths between proton and nitrogen of the imine. The observed 1hJNH coupling constants for the NTPA 1/imine complexes differ only about 2–5 Hz from the completely protonated HBF4/imine complexes. Again, this suggests very weak hydrogen bonds and a tautomeric equilibrium between two proton positions.
Structure elucidation and hydrogen bond position
To corroborate the double-well potential and to distinguish between hydrogen-bond assisted ion pairs and pure ion pairs, the scalar couplings within and through the hydrogen bonds were investigated beyond the dominating 1hJNH to the imine nitrogen. Additionally, analyzing these hydrogen bond scalar couplings could provide insights into the question which of the multiple hydrogen bond acceptors is involved in the potential hydrogen bond. However, compared to strong single-well potential hydrogen bonds a magnetization transfer through weak hydrogen bonds is typically quite challenging. Nonetheless, in order to clarify whether a scalar coupling can be detected in the hydrogen bond, we conducted 2D 1H,31P HMBC experiments as well as modified 1D 1H,31P HMBC experiments as developed by Löhr et al. (for details see ESI Fig. S14†).41,42 Originally these pulse sequences were applied for the detection of scalar couplings across NH⋯OP and OH⋯OP hydrogen bonds in flavoproteins.41 Our group also employed the modified 1H,31P HMBC experiments for the detection of 2hJPH scalar coupling in CPA/imine complexes.11As the binary complex between NTPA 1 and imine 2 produced spectra with good signal dispersion in the hydrogen bond region, this system was chosen for the investigations (Fig. 4a).
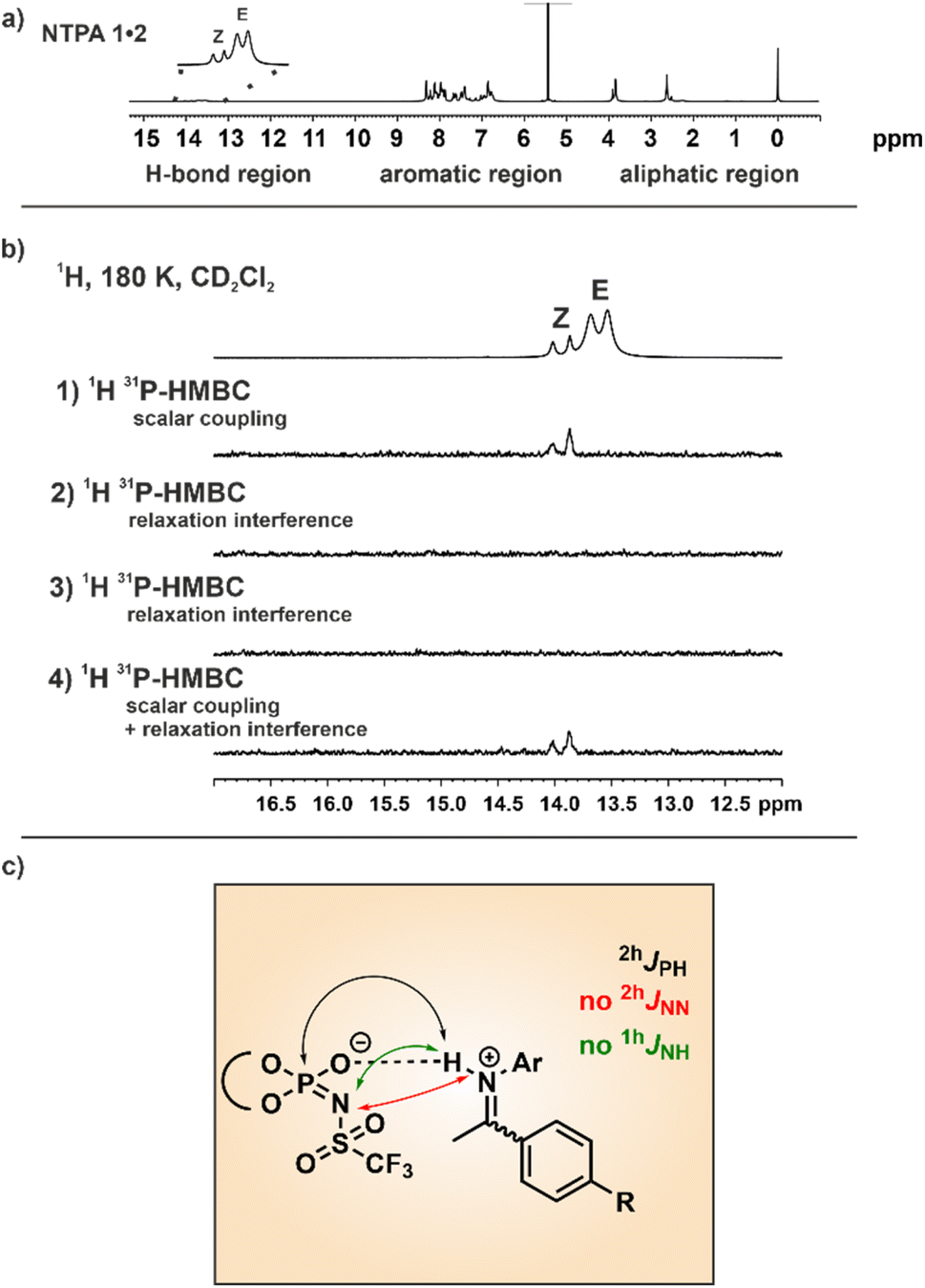 | ||
| Fig. 4 (a) 1H NMR spectra of the binary complexes between NTPA 1 and imine 2 at 180 K (600 MHz). Generally, the 1H NMR spectra can be divided into an H-bond region, an aromatic region, and an aliphatic region. (b) 1H,31P HMBC H-bond section of the NTPA 1/imine 2 complex at 180 K and 600 MHz in CD2Cl2. (1) Polarization transfer is mediated only by scalar coupling (2) and (3) polarization transfer is mediated by 1H-CSA and 1H,31P-DD cross relaxation. (4) Polarization transfer is mediated by the sum of scalar coupling and the 1H-CSA and 1H,31P-DD cross relaxation. (c) Binary complex of NTPA 1 and imine 2. 2hJPH coupling was detected in a 1H, 31P HMBC experiment, whereas neither the 2hJNN coupling nor the 1hJNH coupling could be detected in a 1D 15N or 2D 1H,15N-HSQC spectrum. | ||
The results from the modified 1H,31P-HMBC experiments demonstrated that scalar coupling is a decisive factor for the magnetization transfer in the NTPA 1/Z-imine complex, opposed to chemical shift anisotropy (CSA) or dipole interactions (Fig. 4b). The doublet corresponding to the NTPA 1/Z-imine hydrogen bond was readily apparent in the modified 1H,31P-HMBC sequence only allowing magnetization transfer through scalar coupling. No signals with the modified 1H,31P-HMBC sequence only allowing magnetization transfer through 1H chemical shift anisotropy (1H-CSA) and 1H,31P dipolar interactions (DD) cross relaxation were detected. This means, that 1H-CSA and DD cross relaxation is not the main origin of the magnetization transfer but scalar coupling. Detecting these cross peaks was an unexpected achievement. From the NMR spectra we cannot differentiate, whether the origin is a real 2hJPH transfer with the proton attached to the imine nitrogen or whether it comes from the low populated PXH position of the double well potential. The detection of cross peaks exclusively in the binary Z-complex can be attributed to two phenomena. First, the E-complex is involved in an exchange with the free imine (not active for Z) resulting in partial decoupling and therefore missing cross peaks. This is obvious from EXSY spectra (see ESI Fig. S15 and S16†) and results in an exchange broadened line width of the hydrogen-bonded proton in the 1H spectrum (half line widths E-complex ∼ 50.1 Hz, Z-complex ∼ 24.7 Hz). Secondly, the observed 1hJNH coupling constants for the NTPA 1/imine Z-complexes are slightly smaller than for the binary E-complexes, indicating a stronger hydrogen bond and thus supporting potentially 2hJPH magnetization transfer.
To conclude, our findings thus far demonstrate the presence of a weak hydrogen bond, characterized by a tautomeric equilibrium between two proton positions, in NTPA/imine complexes with a strong preference for the protonated imine. This phenomenon differs from what was observed in previously studied CPA systems.10,12 Nevertheless, despite demonstrating the presence of a weak hydrogen bond with the 1H,31P HMBC experiments, there was still no evidence regarding which hydrogen bond acceptor is involved in the hydrogen bond. So far, mostly the negative charge was predicted to be at the nitrogen and consequently the hydrogen bond was proposed between the nitrogen of the NTPA catalyst and the substrate.30,32–34 However, in principle, there are four potential hydrogen bond acceptors including the phosphoramide oxygen or nitrogen, as well as the triflyl oxygens. Based on the finding of the 1H,31P HMBC experiments, we can ignore the triflyl oxygens and suggest either the phosphoramide nitrogen or oxygen as the most likely hydrogen bond acceptor. To gain further insights, 15N labeling of NTPA 1 was necessary. This way, potential trans-hydrogen bond scalar couplings (1hJNH and 2hJNN) could be detected and therefore prove a hydrogen bond between the imine and nitrogen of the NTPA, as it was proposed previously.30,32–34 Hence, the BINOL-based 15N-labeled NTPA 1 was synthesized according to the procedure of Yamamoto et al.22 Since first trans-hydrogen bond scalar couplings were measured between nitrogen atoms of Watson–Crick base pairs in 15N-labeled RNA with large coupling scalar couplings of 2hJNN ≈ 7 Hz, also in the NTPA complexes strong trans-hydrogen bond 2hJNN scalar couplings were expected.43
Moreover, concerning the NTPA complexes, these scalar couplings through hydrogen bonds should serve as the most effective indicator for detecting the presence of weak hydrogen bonds to the NTPA nitrogen, considering the presence of multiple other hydrogen bond acceptors. Specifically, in case of a hydrogen bond to the NTPA nitrogen, these 1hJNH scalar coupling are expected to be larger than the 2hJPH scalar couplings already detected in the modified 1H,31P-HMBC experiments. However, even with 15N-labeled NTPA 1 neither the 2hJNN coupling nor the 1hJNH coupling between the acidic proton and NTPA nitrogen were detectable in a 1D 15N or 2D 1H,15N-HSQC spectrum (see ESI Fig. S12†) Two reasons account for this phenomenon. First, the 15N signal of the free labeled NTPA is sharp (half line widths ∼ 3.4 Hz), while the 15N signal of the NTPA in the binary complex is broadened significantly (half line widths ∼ 53.7 Hz, for spectra see ESI Fig. S13†). This line broadening suggests again exchange processes and therefore missing cross peaks via partial decoupling. These exchange processes may derive from either an exchange of the hydrogen bond proton acceptor (nitrogen and oxygen) or conformational exchange.
Secondly, nitrogen might not be the main contributor in hydrogen bonding. The expected scalar coupling for a NHN hydrogen bond is around one order of magnitude larger than for a PXH situation. Furthermore, the 15N line widths of both E- and Z-complexes is only double of that of 31P in the binary Z-complex. Thus, the modified 1H,31P-HMBC experiments indicate that, at least for the binary Z-complex, cross peaks should be observable in the 2D 1H,15N-HSQC spectrum as well if a hydrogen bond exists between the imine and nitrogen of the NTPA. Combining then the results of the 1H,31P-HMBC and the 2D 1H,15N-HSQC spectrum, the phosphoramide oxygen must be regarded as the main hydrogen bond acceptor. This contradicts previous suggestions, which proposed a hydrogen bond between the imine and nitrogen of the NTPA.30,32–34
To corroborate the position of the hydrogen bond, further structural investigations of the NTPA/imine complexes via low-temperature NMR spectroscopy and molecular dynamics (MD)-simulations were applied. In previous studies of CPA/imine complexes, we identified the presence of two different orientations for each isomer within the binary complexes, independent of the CPA and the substitution of the N-arylimine.11,44 Even at 180 K, the different orientations exhibit fast exchange on the NMR time scale.11,44 However, due to significantly weaker hydrogen bonds in the NTPA/imine complexes and the multiple hydrogen bond acceptors, additional structures compared to CPAs were expected. Still, to achieve a full structure elucidation, 1H,1H–COSY, 1H,1H-TOCSY, 1H,1H-NOESY, 1H,19F-HOESY, 1H,13C-HSQC, 1H,13C-HMBC, 1H,15N-HMBC and 1H,31P-HMBC were performed with 15N-labeled imine 2 and NTPA 1 at 180 K.
Based on intense NOEs between imine 2 and the BINOL backbone of the NTPA 1 (for spectra see ESI Fig. S17–S21†), computer simulations suggest a structure motif for the binary E-complex as shown in Fig. 5 top (for details see ESI†). Various structures with a hydrogen bond to the phosphoramide oxygen were obtained from classical molecular dynamics simulations: one of the main structures resembles the type I E (here type I E0), another one the type II E (here type II E0) of the CPA complexes. The individual species were compared to the NOE data, which allowed for the identification of type I E0 shown in the Fig. 5 top as the most likely candidate for the experimental data. Again, the hydrogen bond was located between the phosphoramide oxygen and the nitrogen of the imine. The type I E0 species was additionally confirmed to correspond to a density functional minimum structure (B3LYP def2-TZVP with implicit CPCM solvent – for details see ESI†). Furthermore, displacing the H-bond towards N− revealed again the type I E0 structure as the primary minimum (details see ESI†). This suggests an H-bond as shown in Fig. 5. These findings for the binary NTPA 1/E-imine complement the hydrogen bond situation identified in the NTPA 1/Z-imine complex, as confirmed by scalar coupling in a 1H,31P-HMBC experiment.
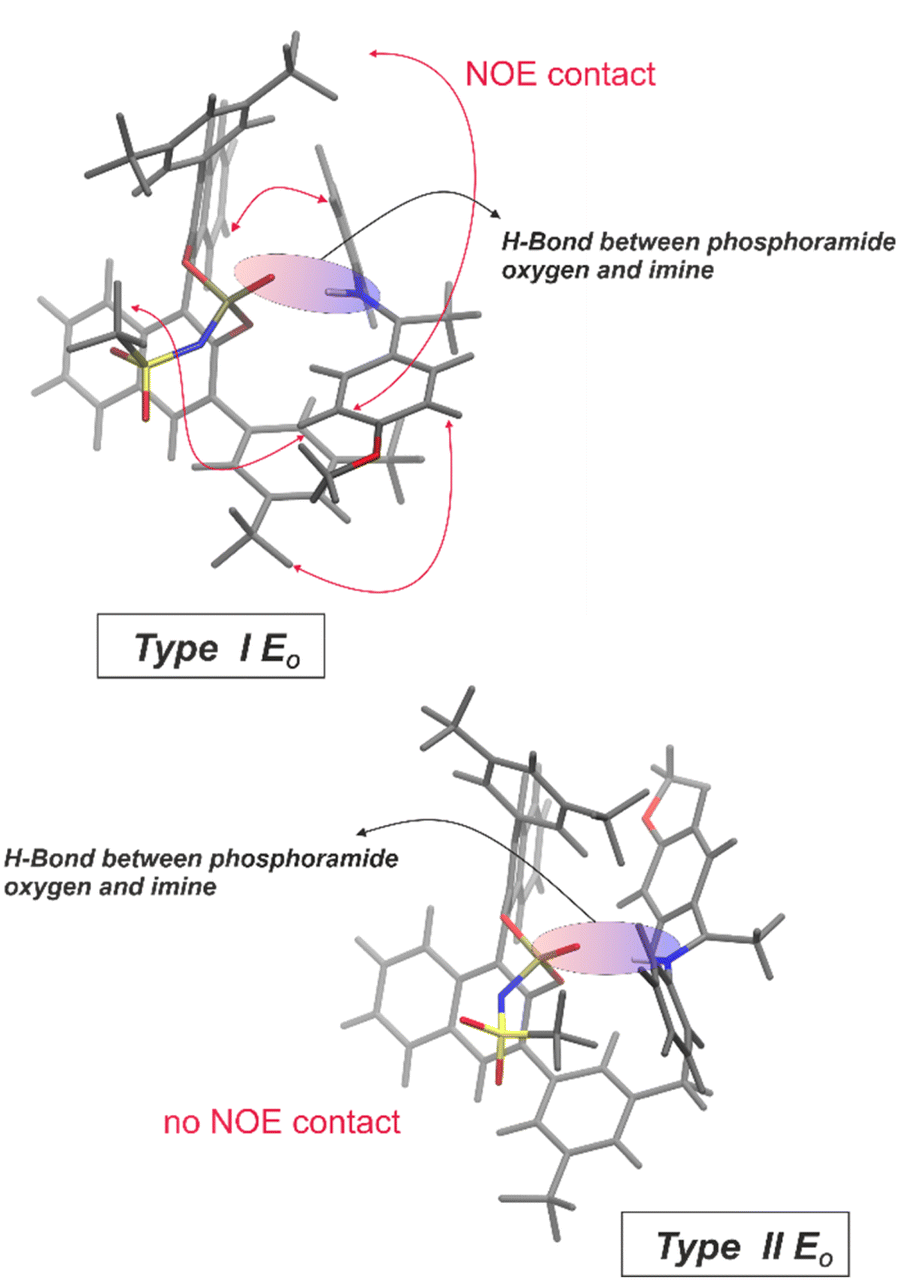 | ||
| Fig. 5 Structures for the oxygen-based H-binding motif of the binary E-complex: inter-atomic distances obtained from molecular dynamics simulations are consistent with the intense NOE contacts for the type I E0 structure (for details and assignment of the substructures to the NOE signals see ESI†). No NOE contacts were found for the type II E0 structure, obtained by rotating the imine leading to the exclusion of structure motif II. | ||
For the first time, there is now evidence that in NTPA catalyzed reactions, intermediates have a strong ion-pair character supported by an extremely weak hydrogen bond between the phosphoramide oxygen and the nitrogen of the imine. This finding opposes the previously proposed activation mode for NTPA-catalyzed reactions suggesting that the negative charge and consequently the hydrogen bond exists between the nitrogen of the NTPA catalyst and the substrate.30,32–34 Our detailed hydrogen bond analysis, as well as structural investigations via low-temperature NMR at 180 K supported by computer simulations suggest an ion-pair character in these binary complexes supported by a weak hydrogen bond between the phosphoramide oxygen and the nitrogen of the imine. Whereas Yoon et al. also isolated a substrate-acid complex indicating a hydrogen bond between a protonated imidazolium and the phosphoramide oxygen of the NTPA catalyst via X-ray crystallography,35 our findings confirm this binding situation in solution. We expect that these findings aid in understanding reaction intermediates, potentially shedding light on the factors contributing to the enantioselectivity of reactions, which is essential for further catalyst development.
Screening for dimers in NTPA catalyzed reactions
In recent research, the transfer hydrogenation of imines with Hantzsch Ester (HE) under CPA catalysis was adopted as a model system for in-depth mechanistic studies. In our NMR studies, we observed the presence of intermediates not only involving a single catalyst molecule but also those involving two catalyst molecules.45,46 Furthermore, Niemeyer et al. revealed that supramolecular linked CPAs show cooperativity effects in the transformation of quinolines.45 This discovery raised the question, whether competing monomeric and dimeric reaction pathways may be a general feature in Brønsted acid catalysis. Lately, Franta et al. demonstrated that a dimeric pathway is active for a broad variety of CPAs typically used in the synthesis of chiral amines leading to opposite enantioslectivities.47 Additonally, dimeric formation could be observed by low-temperature NMR at 180 K.This raised the question of whether the observed phenomenon is also applicable to NTPAs, addressing the presence of a sterically demanding N-trifluoromethanesulfonyl group and the absence of a C2-symmetry. Therefore, we examined NTPA 1 in an enantioselective Mukaiyama–Mannich reaction developed by the group of Yamamoto to address possible reaction intermediates (Table 1).30 Significantly, the challenge of achieving enantioselective Mukaiyama–Mannich reactions catalyzed by chiral phosphoric acids, which previously needed a 2-hydroxyphenyl moiety on the aldimine, was solved by taking advantage of a NTPA catalyst.30 Previously, a NTPA catalyst bearing 2,4,6-trimethyl-3,5-dinitrophenyl substituents at the 3,3′-positions enabled the reactions with excellent enantioselectivities up to 95% ee.30
|
|
|||
|---|---|---|---|
| Entry | Temp. [°C] | Cat. loading [%] | ee [%] |
| 1 | r.t. | 0.1 | No conversion |
| 2 | r.t. | 10 | 72 |
| 3 | r.t. | 25 | 66 |
| 4 | −10 | 0.1 | No conversion |
| 5 | −10 | 10 | 76 |
| 6 | −10 | 25 | 80 |
| 7 | 80 | 1 | 56 |
| 8 | 80 | 10 | 56 |
| 9 | −80 | 25 | 86 |
To check, whether intermediates involving two catalyst molecules are present in the NTPA/imine complexes too, we conducted both a synthetic study and an NMR study. In this study, we once more employed NTPA 1 featuring 3,5-bis trifluoromethyl phenyl substituents at the 3,3′-positions, as in previously investigated CPA systems these substituents showed a general preference for dimerization.47 In the synthetic part we aimed to achieve an inversion of enantioselectivity by varying temperature and catalyst loading in the Mukaiyama–Mannich reaction, as it was previously seen in the transfer hydrogenation of imines under CPA catalysis.47 During the reaction, we applied temperatures ranging from −80 °C to +80 °C. Contrary to our expectations of observing an inversion, we merely observed a decrease in enantioselectivity from 86% ee to 56% ee (Table 1). When the catalyst loadings were varied from 0.1% to 25%, only minor differences in enantioselectivity were detected. Consequently, the synthetic approach did not reveal any indication of a dimeric pathway.
Subsequently, the selection of low-temperature NMR was chosen to examine the existence of dimers. Best conditions for the study were determined to be a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of catalyst to either quinoline or imine, as established in previous research.45,47,48 However, no 2:1 NTPA/NTPA/imine dimer formation with ketimines or aldimines was observed in the 1H-NMR spectrum (Fig. 6). Previously, a characteristic pattern for dimers could be identified in the hydrogen bond region of the 1H-spectrum using CPAs (Fig. 6). We assume that the difference in hydrogen bond strength and steric demands of the N-trifluoromethanesulfonyl group is responsible for this phenomenon. Our findings suggest only the presence of the monomeric pathway for NTPA based Brønsted acid catalysis reactions.
:1 ratio of catalyst to either quinoline or imine, as established in previous research.45,47,48 However, no 2:1 NTPA/NTPA/imine dimer formation with ketimines or aldimines was observed in the 1H-NMR spectrum (Fig. 6). Previously, a characteristic pattern for dimers could be identified in the hydrogen bond region of the 1H-spectrum using CPAs (Fig. 6). We assume that the difference in hydrogen bond strength and steric demands of the N-trifluoromethanesulfonyl group is responsible for this phenomenon. Our findings suggest only the presence of the monomeric pathway for NTPA based Brønsted acid catalysis reactions.
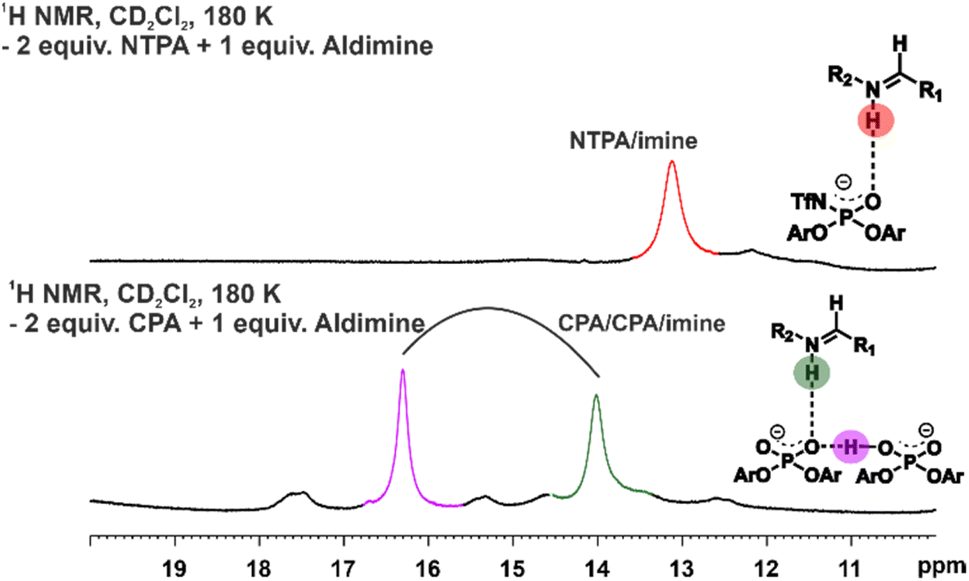 | ||
| Fig. 6 Low-temperature NMR at 180 K was used to investigate the presence of dimers in a 2:1 ratio of catalyst to imine. In the samples with a 2:1 ratio of NTPA to imine, no dimeric formation was identified. However, when employing a 2:1 ratio with CPA to imine, dimer formation became evident in the 1H NMR spectrum. | ||
Conclusions
The investigation into the application of NTPAs as catalysts in asymmetric catalysis has provided valuable insights into their structural behavior and reactivity. By conducting an in-depth analysis of the strength and positioning of hydrogen bonds within binary NTPA/imine complexes, distinct characteristics of NTPAs have emerged in comparison to CPAs. Notably, our study has unveiled the presence of a weak hydrogen bond between the phosphoramide oxygen and the imine, characterized by a proton-transfer equilibrium between two tautomeric forms, challenging previous assumptions regarding the activation mechanism of NTPAs. Also computer simulations support an ion-pair character in these binary complexes and predict a weak hydrogen bond between the phosphoramide oxygen and the nitrogen of the imine. Additionally, our investigation of the NTPA-catalyzed Mukaiyama–Mannich reaction revealed that the coexistence of monomeric and dimeric reaction pathways, previously observed in CPA-catalyzed reactions, does not apply to NTPAs. This difference is attributed to the unique properties of the sterically demanding N-trifluoromethanesulfonyl group resulting in higher acidity of the catalyst and therefore weaker hydrogen bonds. Ongoing investigations aim to further elucidate the distinctive behavior of NTPAs, facilitating their application in asymmetric catalysis.Data availability
Necessary data is given in the ESI.† For further information ask the corresponding author.Author contributions
M. H. and R. G. conceived and conceptualized the project. M. H. planned and performed all experiments. Analysis of all experiments was done by M. H., W. S. and R. G. All calculations were performed by P. D. and analyzed by P. D. and D. H. Visualization was done by M. H. and P. D., interpretation of results, writing and revision of the manuscript was done by all authors. R. G. and D. H. provided funding and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financed by the German Science Foundation (DFG; RTG 2620) project number 426795949. P. D. thanks the German Academic Scholarship Foundation for funding.References
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed.
- M. Yamanaka, J. Itoh, K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2007, 129, 6756 CrossRef CAS PubMed.
- S. J. Connon, Angew. Chem., Int. Ed., 2006, 45, 3909 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS PubMed.
- N. Sorgenfrei, J. Hioe, J. Greindl, K. Rothermel, F. Morana, N. Lokesh and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 16345 CrossRef CAS PubMed.
- J. Greindl, J. Hioe, N. Sorgenfrei, F. Morana and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 15965 CrossRef CAS PubMed.
- K. Rothermel, M. Melikian, J. Hioe, J. Greindl, J. Gramüller, M. Žabka, N. Sorgenfrei, T. Hausler, F. Morana and R. M. Gschwind, Chem. Sci., 2019, 10, 10025 RSC.
- L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741 CrossRef PubMed.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910 CrossRef CAS PubMed.
- J. P. Reid and J. M. Goodman, Chem.–Eur. J., 2017, 23, 14248 CrossRef CAS PubMed.
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343 CrossRef CAS PubMed.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803 CrossRef CAS PubMed.
- A. J. Neel, A. Milo, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 3863 CrossRef CAS PubMed.
- P. García-García, F. Lay, P. García-García, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2009, 48, 4363 CrossRef PubMed.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200 CrossRef CAS PubMed.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626 CrossRef CAS PubMed.
- G. Caballero-García and J. M. Goodman, Org. Biomol. Chem., 2021, 19, 9565 RSC.
- K. Rothermel, M. Žabka, J. Hioe and R. M. Gschwind, J. Org. Chem., 2019, 84, 13221 CrossRef CAS PubMed.
- M. Žabka and R. M. Gschwind, Chem. Sci., 2021, 12, 15263 RSC.
- H. Zhou, Y. Zhou, H. Y. Bae, M. Leutzsch, Y. Li, C. K. De, G.-J. Cheng and B. List, Nature, 2022, 605, 84 CrossRef CAS PubMed.
- R. Properzi, P. S. J. Kaib, M. Leutzsch, G. Pupo, R. Mitra, C. K. De, L. Song, P. R. Schreiner and B. List, Nat. Chem., 2020, 12, 1174 CrossRef CAS PubMed.
- A. Borovika and P. Nagorny, Tetrahedron, 2013, 69, 5719 CrossRef CAS.
- T. Hashimoto, H. Nakatsu, K. Yamamoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 9730 CrossRef CAS PubMed.
- F. Zhou and H. Yamamoto, Angew. Chem., Int. Ed., 2016, 55, 8970 CrossRef CAS PubMed.
- K. Kaupmees, N. Tolstoluzhsky, S. Raja, M. Rueping and I. Leito, Angew. Chem., Int. Ed., 2013, 52, 11569 CrossRef CAS PubMed.
- S. P. Bew, J. Liddle, D. L. Hughes, P. Pesce and S. M. Thurston, Angew. Chem., Int. Ed., 2017, 56, 5322 CrossRef CAS PubMed.
- P. C. Knipe and M. D. Smith, Org. Biomol. Chem., 2014, 12, 5094 RSC.
- H.-H. Liao, A. Chatupheeraphat, C.-C. Hsiao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 15540 CrossRef CAS PubMed.
- E. M. Sherbrook, M. J. Genzink, B. Park, I. A. Guzei, M.-H. Baik and T. P. Yoon, Nat. Commun., 2021, 12, 5735 CrossRef CAS PubMed.
- S. O. Garbuzynskiy, B. S. Melnik, M. Y. Lobanov, A. V. Finkelstein and O. V. Galzitskaya, Proteins, 2005, 60, 139 CrossRef CAS PubMed.
- S. Sharif, G. S. Denisov, M. D. Toney and H.-H. Limbach, J. Am. Chem. Soc., 2007, 129, 6313 CrossRef CAS PubMed.
- H.-H. Limbach, M. Pietrzak, S. Sharif, P. M. Tolstoy, I. G. Shenderovich, S. N. Smirnov, N. S. Golubev and G. S. Denisov, Chem.–Eur. J., 2004, 10, 5195 CrossRef CAS PubMed.
- H. Benedict, I. G. Shenderovich, O. L. Malkina, V. G. Malkin, G. S. Denisov, N. S. Golubev and H.-H. Limbach, J. Am. Chem. Soc., 2000, 122, 1979 CrossRef CAS.
- S. Sharif, G. S. Denisov, M. D. Toney and H.-H. Limbach, J. Am. Chem. Soc., 2006, 128, 3375 CrossRef CAS PubMed.
- F. Löhr, S. G. Mayhew and H. Rüterjans, J. Am. Chem. Soc., 2000, 122, 9289 CrossRef.
- G. Federwisch, R. Kleinmaier, D. Drettwan and R. M. Gschwind, J. Am. Chem. Soc., 2008, 130, 16846 CrossRef CAS PubMed.
- A. J. Dingley and S. Grzesiek, J. Am. Chem. Soc., 1998, 120, 8293 CrossRef CAS.
- M. Melikian, J. Gramüller, J. Hioe, J. Greindl and R. M. Gschwind, Chem. Sci., 2019, 10, 5226 RSC.
- D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme, H. Zhu, R. M. Gschwind and J. Niemeyer, Chem. Sci., 2020, 11, 4381 RSC.
- J. Gramüller, M. Franta and R. M. Gschwind, J. Am. Chem. Soc., 2022, 144, 19861 CrossRef PubMed.
- M. Franta, J. Gramüller, P. Dullinger, S. Kaltenberger, D. Horinek and R. M. Gschwind, Angew. Chem., Int. Ed., 2023, 62, e202301183 CrossRef CAS PubMed.
- R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 11456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01939c |
| This journal is © The Royal Society of Chemistry 2024 |