
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzole reagents enabled ligation of peptide acyl pyrazoles for chemical protein synthesis†
Peisi
Liao
and
Chunmao
He
 *
*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: hecm@scut.edu.cn
First published on 25th April 2024
Abstract
Native chemical ligation (NCL) has been playing an increasingly important role in chemical protein synthesis (CPS). More efficient ligation methods that circumvent the requirement of a peptidyl thioester and thiol additive—which allow the following desulfurization or refolding in one pot—are urgently needed for the synthesis of more complex protein targets and in large quantities. Herein, we discover that the weak acyl donor peptidyl N-acyl pyrazole can be activated by azole reagents like 3-methylpyrazole or imidazole to facilitate its ligation directly with an N-terminal cysteine peptide. As it requires no thioester or thiol additive, this ligation strategy can be conveniently combined with metal-free desulfurization (MFD) or oxidative protein folding to allow various one-pot protocols. The utility and generality of the strategy are showcased by the total synthesis of ubiquitin via an N-to-C sequential ligation–MFD strategy, the semi-synthesis of the copper protein azurin, and the efficient assembly of a sulfated hirudin variant and the cyclotide kalata B1, all in a one-pot fashion.
Introduction
Chemical protein synthesis (CPS), enabled by chemo-selective peptide ligation reactions like native chemical ligation (NCL), has contributed enormously to the generation of proteins with specific structural modifications and facilitated the selective introduction of unnatural amino acids, biophysical probes, and post-translational modifications.1 Since its inception by Kent and co-workers,2 and with great contributions from many research groups, NCL has become the most powerful and dominant method for CPS.3 With the increasing demand for large and complex proteins, the reactivity of peptidyl thioesters (e.g., alkyl thioesters)4 is usually too low to enable NCL under challenging conditions, and thiol additives—with 4-mercaptophenylacetic acid (MPAA) being the most widely employed and gold standard catalyst—have been added to generate more reactive acyl donors to facilitate the ligation.3b,5 A significant advancement in CPS is the development of metal-free desulfurization (MFD) chemistry, which broadened the scope of NCL by allowing ligation sites beyond Cys.6 However, the use of MPAA—due to its inherent free radical quenching properties7—is problematic when NCL needs to be combined with MFD in one pot, which is a highly desirable approach to achieve high synthetic efficiency. In this context, a number of strategies have been developed to enable the one-pot NCL–MFD operation, including removal of MPAA through extensive extraction/dialysis and solid-phase techniques.8 Recently, several thiol reagents (poor radical scavengers) have been used to replace MPAA in facilitating the one-pot NCL–MFD protocol, including 2-mercaptoethanesulfonate sodium salt (MESNa), trifluoroethanethiol (TFET), methyl thioglycolate (MTG), 2-sulfanylmethyl-4-dimethylaminopyridine (SMDMAP) and thiocholine (Fig. 1A-i) (see Table S1† for detailed information).9 It is worth noting that sequential C-to-N or N-to-C ligation of multiple segments (≥3) is essential in the efficient synthesis of complex proteins; these thiol additives, however, may complicate the kinetically controlled NCL reactions where two or more peptide thioester segments with different reactivities are involved.3b,9b,10 Alternatively, non-thiol reagents such as imidazole11 and 2-methyl imidazole12 have also been shown to serve the purpose (Fig. 1A-ii). These aforementioned additive strategies, however, require the pre-formed peptidyl thioesters or their surrogates, where, although many excellent synthetic methods are available,3b,4c,13 operational issues and limitations are still present, such as relatively complicated post-SPPS manipulations, insolubility of protected peptides, use of large amounts of oxidants and cryogenic conditions, instability of the peptide thioester during storage, etc. Thus, ligation methods that do not require thioesters and thiol additives would be highly efficient in streamlining one-pot NCL–MFD and thus the protein synthesis. To this end, another azole additive (i.e., 1,2,4-triazole) has been employed to assist the one-pot NCL–MFD of peptidyl N-acyl-N′-methyl-benzimidazolinone (MeNbz) (Fig. 1B-i).14 It is worth mentioning that strained peptidyl β-thiolactones are versatile acyl donors allowing ligation–MFD at Val/Pro-Xaa sites, where no additive is needed for the ligation.6g,15 Unfortunately, these peptide thioester surrogates can only be obtained via Fmoc-SPPS but not recombinantly, which may limit their application in the synthesis of complex and large proteins.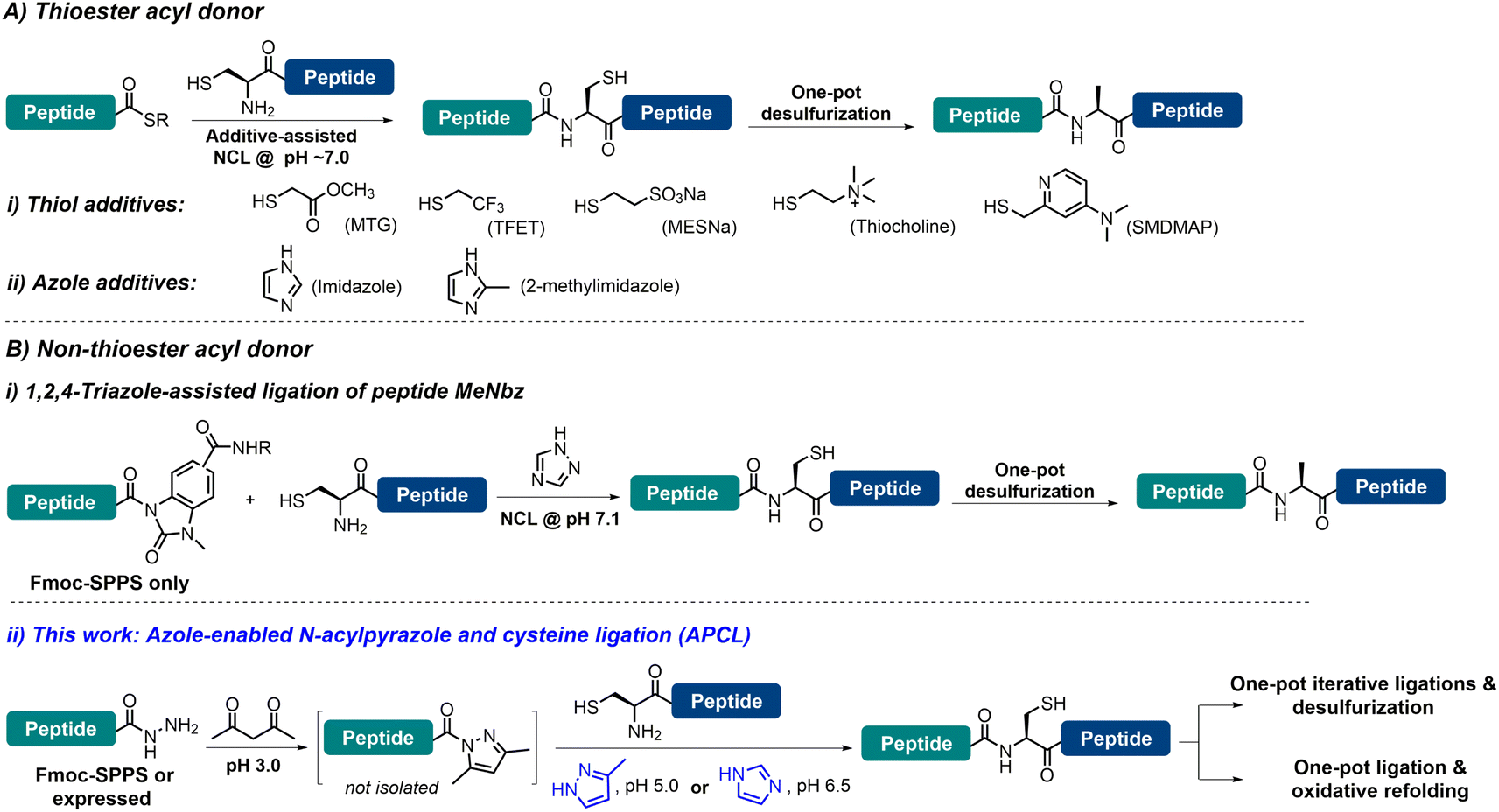 | ||
| Fig. 1 (A) Selected examples of the one-pot NCL–MFD strategy using a C-terminal α-thioester as the acyl donor. (B) (i) One-pot NCL–MFD strategy using C-terminal MeNbz as the acyl donor; (ii) the principles of the azole reagents enabled the APCL strategy and its following one-pot operations described in this work. | ||
Meanwhile, peptide hydrazides have been developed as extremely powerful peptidyl thioester surrogates,13j,16 due to their facile access through Fmoc-SPPS or recombinant expression, stability, and convenient thioester transformation.17 Recently, Dawson et al.18 developed a mild and efficient condensation activation method for peptide hydrazides, where the weakly activated peptide N-acyl pyrazoles—generated with a stoichiometric amount of acetylacetone (acac)—can be readily exchanged to peptide thioesters in situ, preferably with MPAA, for NCL. Importantly, as peptide thioesters are generated with the addition of stoichiometric quantities of acac, multiple ligations can be allowed sequentially in one pot through this mild activation strategy. We thus envision that if MPAA can be replaced by thiol-free additives compatible with MFD, it would enable (sequential) ligations at other sites than Cys in one pot, and together with the absence of requirement of a thioester, it should help to greatly simplify the purification procedure and increase synthetic efficiency. Moreover, refolding of synthetic proteins is a key step enabling them, a large portion of which contain one or more disulfides, to regain natural conformation and biological activity.19 Unfortunately, most current synthetic methods—either involving large equivalents of thiol additives or reducing reagents like TCEP (tris(2-carboxyethyl)phosphine hydrochloride)—require an extra HPLC purification or dialysis step, and are not compatible with direct dilution for protein refolding in one pot. Thus, a ligation strategy that completely eliminates the presence of any thiol agent and avoids the use of TCEP would be highly valuable in facilitating one-pot ligation and protein refolding.
Herein, we report that azole reagents (i.e., imidazole (pKa 6.99) and 3-methylpyrazole (pKa 3.54)) are efficient non-thiol additives facilitating the ligation between peptide N-acyl pyrazoles and another peptide bearing N-terminal Cys, and we term this strategy N-acyl pyrazole and cysteine ligation (APCL) (Fig. 1B-ii). While peptide N-acyl pyrazoles can be readily obtained through addition of acac, no further exchange into a thioester is needed; rather it can react directly with N-terminal Cys peptides at low pH (5.0–6.5) with the help of 3-methylpyrazole or imidazole, forming native peptide bonds. As such, APCL is compatible with subsequent one-pot MFD or protein refolding (in cases where TCEP is not used) operations. The utility and generality of the strategy are demonstrated through the total synthesis of ubiquitin via an N-to-C sequential ligation–MFD strategy, the semi-synthesis of the copper protein azurin, via a ligation–folding strategy, and the efficient synthesis of a sulfated hirudin (sY63-HV1) protein and the cyclotide kalata B1 (kB1), all in a one-pot fashion.
Results and discussion
Conditions screening of the APCL strategy
The weakly activated peptide N-acyl pyrazole can be readily transformed into a peptide thioester/selenoester for ligation,18,20 and initially we were interested to see whether its ligation to another Cys-peptide could occur without any additive. Our study began with the ligation between model peptides H-CTFPGASALMKGTLTLK-NH2 (1) and Fmoc-RPGKVTEQA-NHNH2 (2). The ligation involves two steps: peptide hydrazide 2 was firstly converted to the corresponding N-acyl pyrazole 2a at pH 3.0, and Cys-peptide 1 was then added and the ligation was carried out at various pHs ranging between 3.0 and 6.5 for 3 h, before the yield was determined by HPLC. It was observed that the ligation yield was pH-dependent and surprisingly high at pH 6.5 (41%) (Table S2,† entries 1–4), which encouraged us to further improve the reaction. To this end, N-acyl heterocycles such as N-acyl imidazoles are well known strong acylating agents;21 it was thus hoped that azole reagents would replace the pyrazole moiety to activate the peptidyl acyl group22 and prompt its ligation with Cys-peptide directly.A series of azole reagents including imidazole, 1,2,4-triazole, pyrazole, 3-methylpyrazole and 3,5-dimethylpyrazole were screened for their ability to facilitate the ligation at pH 5.0 and 6.5, which are relatively low compared to traditional NCL in order to minimize possible hydrolysis of 2 while maintaining the nucleophilicity of N-terminal Cys of peptide 1 at a reasonably high level. As shown in Fig. 2 and Table S2,† we were delighted to observe that all of the ligations performed in the presence of 2.5 M 3-methylpyrazole at pH 5.0 and 2.5 M imidazole at pH 6.5, with or without the reductant TCEP, gave very high HPLC yields after 3 h, rivalling the gold standard catalyst MPAA (compare Table S2,† entry 13 vs. 5; entry 16 vs. 6). This suggests that pairing the pKa of the azole reagents and the working pH is the key to balancing the acylating power of the corresponding peptide acyl intermediates and N-terminal Cys's nucleophilicity, which ensures a high ligation/hydrolysis ratio. Further, the effect of the additive concentration was also probed and both additives worked best at 2.5 M (Fig. S28†), agreeing well with the literature.5b,11a,12,14 It is worth mentioning that both additives are inexpensive and highly water-soluble, which elute early and will not affect the HPLC trace. Importantly, the epimerization issue was studied for both additives, and no epimerization at the ligation site was observed during the process (Fig. S31†). Further control ligations were carried out between peptide 2 and peptide 1a with Acm-protected N-terminal Cys or peptide 1b with N-terminal Ser, and only complete hydrolysis of peptide 2 could be observed in both cases (Table S3 and Fig. S32–S35†). This suggests that, instead of direct aminolysis, the azole enabled APCL proceeds through a thiol capture and S → N acyl shift reaction pathway similar to NCL, which ensures the chemo- and stereo-selectivity of the ligation.
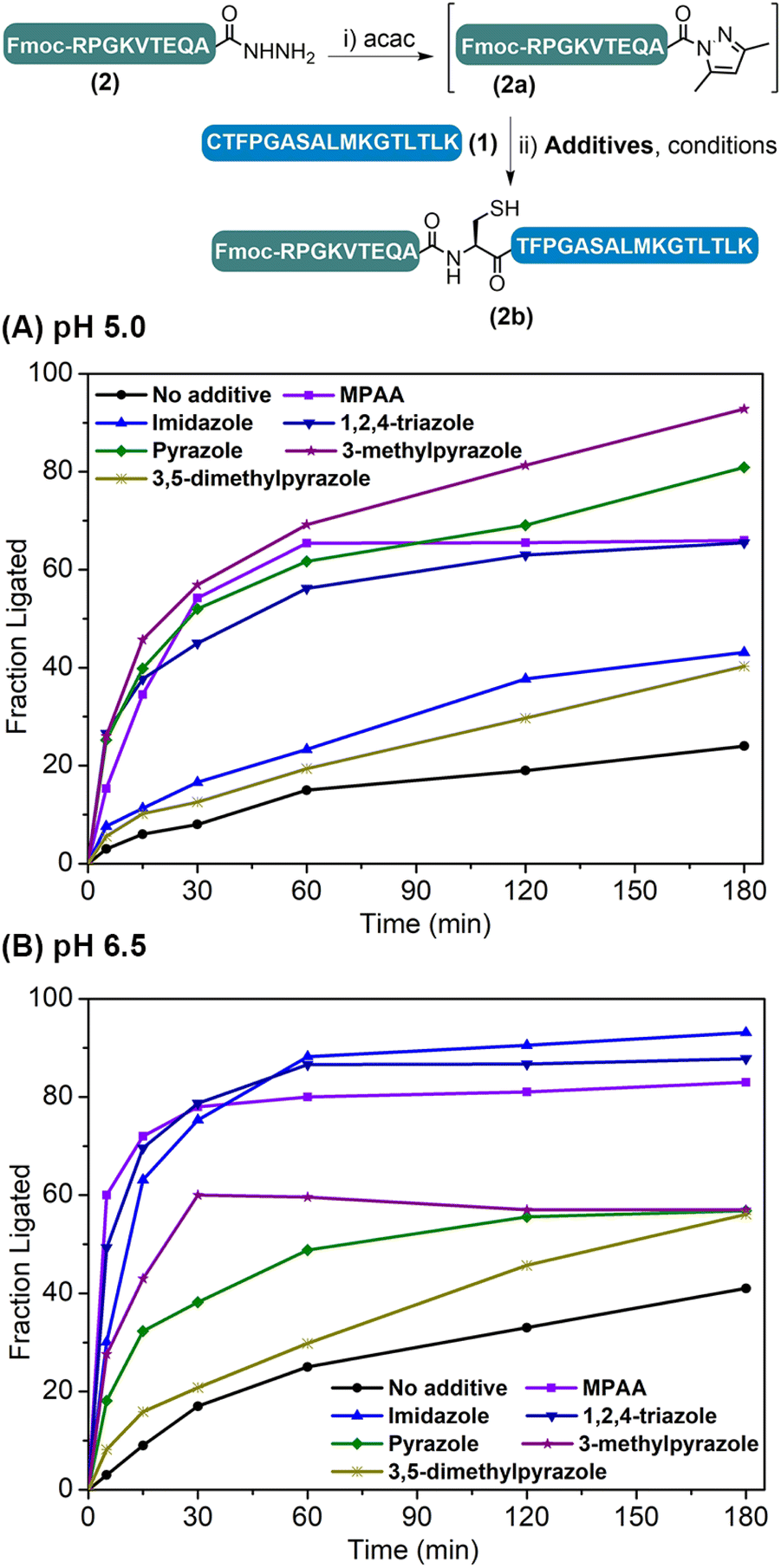 | ||
| Fig. 2 Time-course analysis of APCL with different additives at pH 5.0 (A) and 6.5 (B). Conditions: (i) 6 M Gdn·HCl, 0.2 M Na2HPO4, pH 3.0, 2.5 eq. of acac, 37 °C, 40 min; (ii) 6 M Gdn·HCl, 0.2 M Na2HPO4, 20 mM TCEP, 2.5 M additives (or 100 mM MPAA), 37 °C, pH 5.0 or 6.5, 3 h. | ||
The scope of the APCL strategy
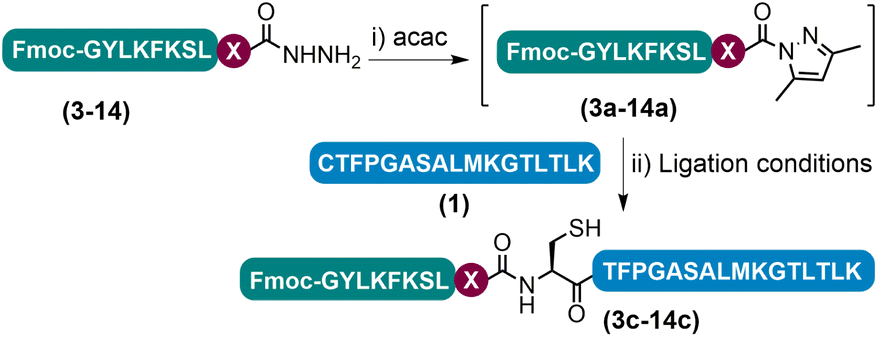
Having determined that the APCL strategy could be used as a viable method for peptide ligation, we proceeded to investigate the scope of the strategy by varying the C-terminal residue of the hydrazide segments (Tables 1 and S4†). The initial acac activation was carried out for 15–150 minutes at pH 3.0, and the following ligation at most C-terminal residues tested could be completed within 3 h with decent isolated yields (47–69%), except for the sterically demanding residues like Val and Pro. Ligation at Glu was complicated by side chain cyclization and isomerization and other known problematic sites like Asp and Asn for obtaining the corresponding peptide hydrazides were not included in the test.13j,18 Notably, in the case of C-terminal Lys peptide 9, although an excellent isolated yield of 67% was obtained when using 3-methylpyrazole as the additive, a noticeable amount of side-chain lactamization side product (9′′ in Fig. S42†) could be observed,23 which may be sequence specific as this side-reaction was not as obvious in other peptides bearing C-terminal Lys residues (vide infra). In addition, a similar level of lactamization could be observed when conducting the ligation with the conventional NaNO2-MPAA method (data not shown). For the C-terminal His peptide 14, a certain amount of side-chain cyclized intermediate resulting from the nucleophilic substitution of N-acyl pyrazoles by the imidazole side-chain was observed during the acac activation (14′′ in Fig. S47†); subsequent ligation, however, proceeded smoothly affording a high isolated yield (47%). Lastly, sensitive residues like C-terminal Cys and Trp and internal Cys are well tolerated. Collectively, these results demonstrate the practicability of the azole reagent enabled APCL strategy for peptide ligation.|
|
|||
|---|---|---|---|
| Entry | Ligation site (X) | Ligation time (h) | Isolated yieldd (%) |
| a Conditions: (i) 6 M Gdn·HCl, 0.2 M Na2HPO4, 1.5–2.5 eq. of acac, 37 °C, pH 3.0; (ii) 6 M Gdn·HCl, 0.2 M Na2HPO4, 20 mM TCEP, 2.5 M azoles, pH 5 or 6.5, 37 °C. b 3-Methylpyrazole additive, pH 5.0. c Imidazole additive, pH 6.5. d The weighed isolated yield of the ligation product. e Sequence of the model peptide hydrazide 3*: Fmoc-GCGYLKFKSLA-NHNH2. | |||
| 1 | Ala (3) | 3 | 48b |
| 2 | Leu (4) | 3 | 47b |
| 3 | Gly (5) | 3 | 57b |
| 4 | Trp (6) | 4 | 69b |
| 5 | Cys (7) | 3 | 48b |
| 6 | Phe (8) | 3 | 61b |
| 7 | Lys (9) | 2 | 67b |
| 8 | Pro (10) | 15 | 14c |
| 9 | Val (11) | 6 | 26b |
| 10 | Glu (12) | 2 | 5b |
| 11 | Gln (13) | 2 | 54c |
| 12 | His (14) | 0.5 | 47c |
| 13 | — (3*)e | 3 | 65b |
One-pot ligation–MFD chemistry
With the knowledge that non-thiol azole additives may not interfere with any subsequent MFD, we next carried out model peptide tests to investigate their suitability for one-pot ligation–MFD chemistry. In the initial screening, unwanted degradation (data not shown) of the peptides occurred at above pH 6.5 (required for MFD) in the presence of the 3-methylpyrazole but not imidazole; hence it was decided that imidazole could be a more suitable catalyst compatible with the one-pot ligation–MFD protocol. In this case, the ligated mixture of 2b (containing 2.5 M imidazole) was subjected directly to MFD (see the ESI† for experimental details), where additional TCEP (final conc. 200 mM), the radical initiator VA-044 (2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride) (25 eq.) and the hydrogen source t-BuSH (tert-butylthiol) (5%, v/v) were added and incubated at 37 °C for 8 h to ensure complete desulfurization. Gratifyingly, the one-pot ligation–MFD proceeded smoothly in the presence of imidazole, giving the desired product 2c in 85% HPLC yield (Fig. S50†).Total synthesis of ubiquitin through the one-pot sequential ligation and MFD strategy
As previously reported peptide hydrazides could be converted into acyl pyrazoles efficiently with a stoichiometric amount of activating agent,18 which suggested a potential to use this chemistry, powered by imidazole catalysis, to perform multiple sequential ligations and MFD without intermediate purification steps. We attempted to apply this strategy to the synthesis of ubiquitin, which is an important and commonly used model protein consisting of 76 amino acid residues. A number of successful synthetic strategies have been reported, including three-segment ligation,9d,24 two-segment ligation25 and direct Fmoc-SPPS.26 Herein, we document its synthesis by an assembly of three peptide segments in the N → C direction, followed by a one-pot MFD protocol. Specifically, the full-length ubiquitin was disconnected into three segments at Lys27–Ala28 and Phe45–Ala46. Notably, Ala28 and Ala46 were incorporated as Cys during Fmoc-SPPS and could be desulfurized after ligation to restore the Ala side-chains (Fig. 3). The required fragments were prepared by Fmoc-SPPS, including ubiquitin(1–27)-NHNH2 (16) (Fig. S51†), ubiquitin(28–45) (17) containing N-terminal Cys and C-terminal hydrazide moieties (Fig. S52†), and ubiquitin(46–76) (18) containing N-terminal Cys (Fig. S53†). Peptide segment 16 (1.0 eq.) was firstly converted into the corresponding acyl pyrazole ligated with peptide 17 (0.7 eq.) in the presence of imidazole. Despite the desired ligation product 19 being the major product, C-terminal lactamization of 16 (∼10%) could be observed.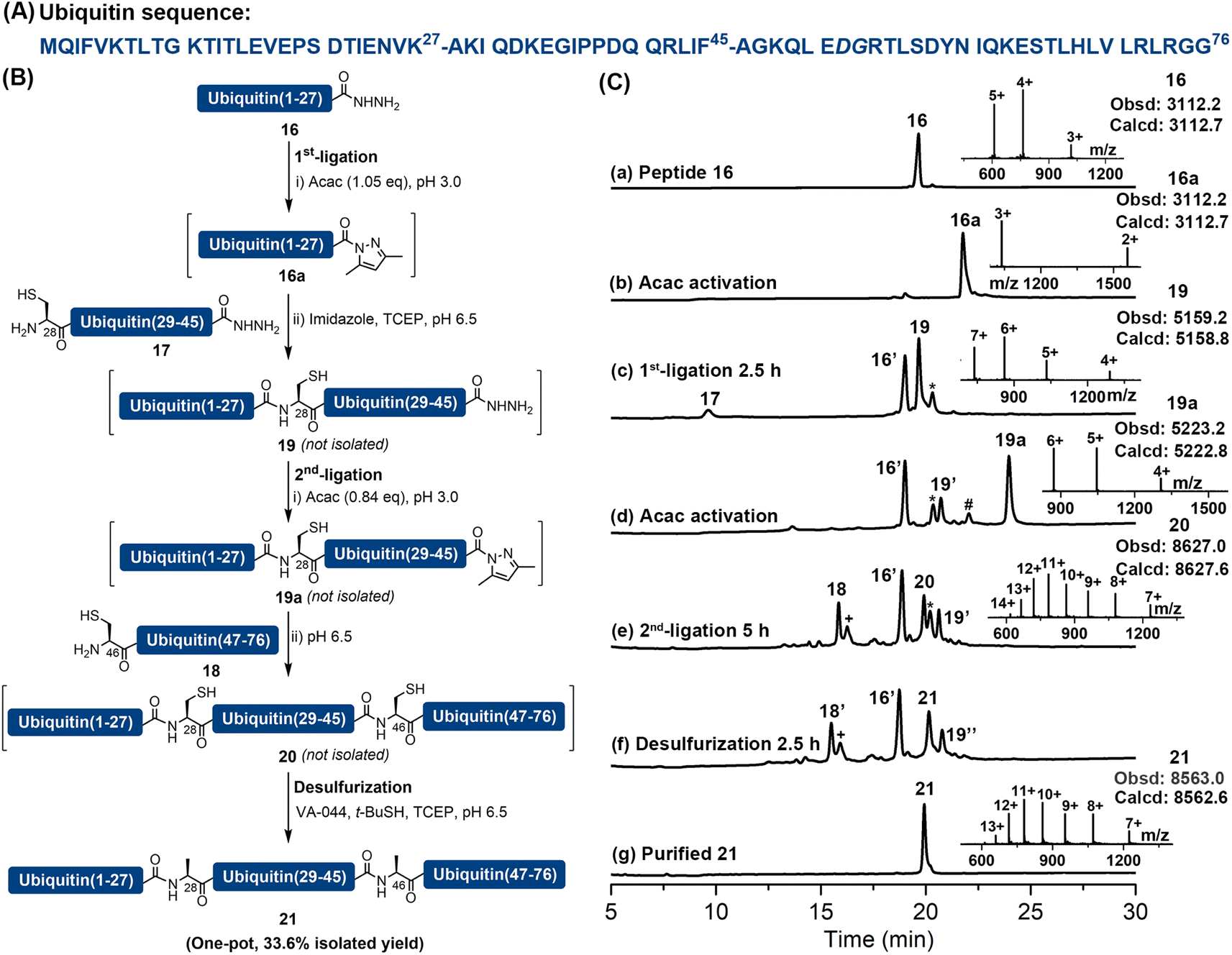 | ||
| Fig. 3 (A) The sequence of ubiquitin (21), the ligation sites are indicated with dashes, any dipeptide used in the SPPS is in italic. (B) Scheme for the synthesis of ubiquitin 21via a one-pot sequential ligation–MFD in the N-to-C direction. (C) HPLC traces (214 nm) and ESI-MS characterization of the major reaction steps. (a–c) 1st ligation, 16a is the peptide acyl pyrazole derivative of 16, 19 is the ligation product, 16′ is the hydrolysis by-product and the peak * denotes C-terminal side-chain lactamization of Lys27. (d) Acac activation, 19a denotes the acyl pyrazole product of 19, 19′ is the hydrolysis by-product, and the peak # denotes a hydrazone intermediate. (e) 2nd ligation at 5 h, 20 is the product, the peak + corresponds to a +82 Da adduct to the remaining peptide 18. (f) Desulfurization at 2.5 h, 21 is the desired product, 18′ and 19′′ are desulfurized versions from the remaining 18 and 19′, respectively. (g) Analytical HPLC trace and ESI-MS of the purified full-length protein 21. | ||
After completion of the first ligation (confirmed by HPLC), the resulting mixture was treated with additional acac at pH 3.0 to obtain intermediate 19a with a C-terminal acyl pyrazole moiety. Segment 18 (0.63 eq.) was then added and its pH was adjusted to 6.5. Upon completion, the ligation mixture was incubated with additional TCEP, t-BuSH and VA-044 for 2.5 h to achieve the desulfurization. Gratifyingly, after only one HPLC purification step, full-length ubiquitin (21) was obtained with an isolated yield of 33.6% (one pot over five steps, calculated based on the limiting Cys-peptide 18) (Fig. 3 and S54–S56†). In contrast to other additive-assisted one-pot ligation–desulfurization methods, as no thiol reagents were added or generated in the reaction system our strategy did not require additional treatments such as increased temperature (40–65 °C)9d,11b,27 or prolonged reaction time (12–48 h)9d,e,11 to obtain satisfactory desulfurization results. This case illustrates the convenience and utility of the imidazole-aided APCL strategy in facilitating sequential ligations and MFD in one pot, which does not require intermediate purification steps or other operations like extraction and heating.
Semi-synthesis of azurin through a one-pot expressed protein ligation and refolding
The scope and utility of the APCL strategy was then extended to protein semi-synthesis, where a more challenging protein, the Pseudomonas aeruginosa azurin, was targeted. It is a bule copper protein, consisting of 128 amino acids, involved in electron transfer.28 Here, based on the available ligation site we decided that the full-length azurin could be disconnected into two segments at Phe111–Cys112. The N-terminal segment azurin(1–111)-NHNH222 (Fig. S57 and S58†) was generated through hydrazine cleavage of the corresponding E. coli expressed azurin(1–111)–intein fusion protein as reported in the literature,13j,29 while azurin(112–128) segment 23 containing an N-terminal Cys residue was synthesized by Fmoc-SPPS (Fig. S59†). To increase the overall synthetic yield, we aimed to carry out protein ligation and refolding in one pot. The 3-methylpyrazole enabled APCL strategy can be achieved at low pH (5.0) where Cys-containing peptide segments are more resistant to oxidation than at neutral pH, thus it offers an opportunity to perform the ligation without any reducing agent, i.e., TCEP, after which the following refolding would be allowed in one pot. To this end, the protein hydrazide segment 22 was firstly treated with acac to transform it to its corresponding N-acyl pyrazole (22a) within ∼1.5 h, as judged by HPLC. Without any reducing agents like TCEP, the azurin(112–128) segment 23 and 3-methylpyrazole were added, and the pH was adjusted to 5.0 to initiate the ligation. Interestingly, the exchanged peptide N-acyl 3-methylpyrazole intermediate 22b can be clearly observed with HPLC-ESI-MS, highlighting a plausible azole-exchanging and acyl-activating mechanism. As shown in Fig. 4C-d, the ligation went to completion within 2 h, and product 24 was co-eluted with hydrolysis and truncation (at Met13) by-products. Nevertheless, without any purification, dialysis, or ultracentrifugation treatment, a rapid dilution strategy where the ligation mixture was added directly to a redox buffer11b,30 (final protein conc., ∼0.3 mg mL−1) was adopted, and the refolding was allowed for 24 h at 4 °C (Fig. 4C-e). The folded protein 24a was finally obtained by a single HPLC purification step with an isolated yield of 31.2% (over three steps). The proper folding of 24a was confirmed by ESI-MS (∼–2 Da vs.24) (Fig. 4D and Table S5†) and fluorescence emission spectra (Fig. 4F and S61†). Furthermore, the presence of a disulfide bond Cys3–Cys26 was confirmed via trypsin digestion and ESI-MS (Fig. S62†).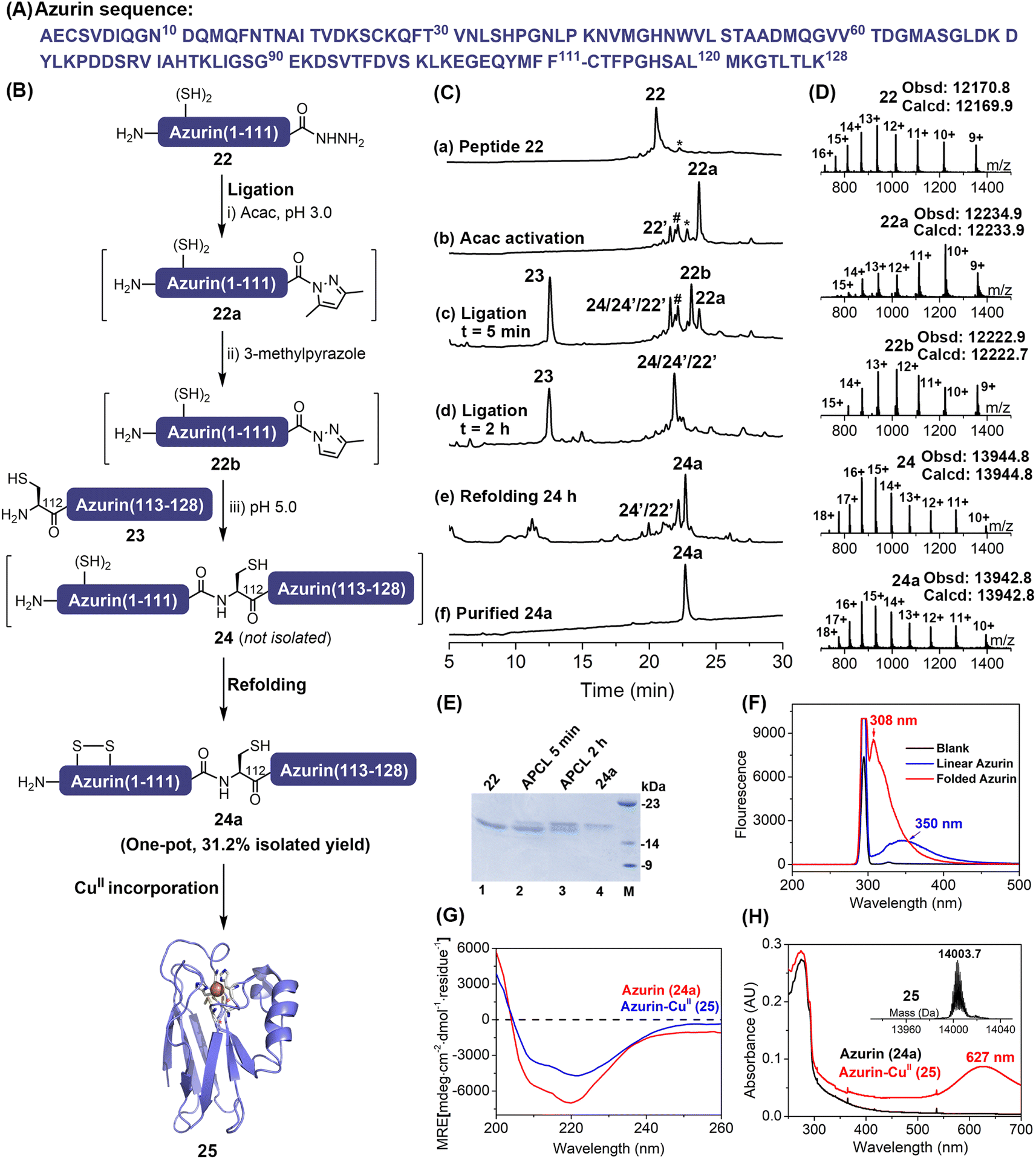 | ||
| Fig. 4 (A) Sequence of wild type azurin; the ligation site is indicated with a dash. (B) Scheme for the semi-synthesis of azurin 25via one-pot expressed protein ligation–refolding with APCL. (C) HPLC traces (214 nm) of major reaction steps. (a) The purified 22; (b) acac activation, 22a is the acyl pyrazole product, 22′ is the hydrolysis by-product, the peak # denotes the hydrazone intermediate and peak * denotes column contaminants; (c and d) APCL at 5 min and 2 h, 22b is the 3-methylpyrazole substituted intermediate and 24′ is the truncated versions of 24 at Met13; (e) the refolding at 24 h; (f) the purified folded protein 24a. (D) The ESI-MS analysis of the major reaction steps. (E) SDS-PAGE analysis of ligation traces and purified folded protein 24a. (F) Fluorescence emission spectra of the linear azurin (24) and folded azurin (24a). (G) Circular dichroism spectra of the synthetic azurin (24a) and azurin-CuII (25). (H) The UV-visible absorption spectra of the apo-protein 24a and azurin-CuII25, inset: the deconvoluted ESI-MS of azurin-CuII25. | ||
Lastly, the folded protein 24a was reconstituted with CuII, exhibiting a characteristic absorption band at 627 nm for the Cys112—CuII coordination (Fig. 4H and S63†). Moreover, ESI-MS data (Fig. 4H and S63†) also demonstrated the successful incorporation of the CuII centre, and the CD spectrum (Fig. 4G and Table S6†) verified that the protein structure was maintained after the incorporation of CuII, agreeing well with the literature.28a,31 Therefore, our reductant-free APCL strategy enabled by 3-methylpyrazole provides a one-pot expressed protein ligation and refolding protocol, streamlining the access of the blue-copper protein azurin.
Total synthesis of the sulfated hirudin variant 1 (sY63-HV1) through the APCL strategy
Tyrosine-O-sulfation (sY) is a widespread post-translational modification; especially in salivary proteins like hirudins, the exact role of this modification has, however, been difficult to elucidate, due partially to its instability during purification from natural extract.32 CPS has recently evolved as an important tool in accessing sulfated proteins.32c,33 Herein, we document our effort in applying the APCL strategy in the synthesis of the hirudin variant 1 from leech bearing the labile sY modification at Tyr63, sY63-HV1. For this purpose, the target protein was disconnected into two segments at Lys27–Cys28, and both were obtained by Fmoc-SPPS. The neopentyl (nP) protecting group of the sulfate was reported to be stable under typical Fmoc-SPPS and TFA cleavage conditions,34 hence Fmoc-Tyr(SO3nP)-OH was chosen as the monomer. According to initial observations, the HV1(1–27) segment containing four internal Cys residues could form thiolactone almost immediately when the corresponding acyl pyrazole was activated with 3-methylpyrazole, imidazole, or MPAA.33 A higher ligation pH (at 6.5) was therefore chosen and imidazole was selected as the catalyst to enable the ligation of any thiolactone, if formed. As shown in Fig. 5, HV1(1–27) segment 26 was readily converted to its corresponding acyl pyrazole form (26a) within 15 min, as judged by HPLC-MS. Without purification, the HV1(28–65)-sY63 fragment 27, imidazole and TCEP were added, and the pH was adjusted to 6.5, after which 26a rapidly transformed to its thiolactone 26b. Nevertheless, the ligation between 26b and 27 went smoothly to afford the full-length protein 28 (Fig. S66 and S67†), albeit a bit slower (i.e., completed within 9 h), which agrees well with the reported method.33 Interestingly, the HV1(1–27) segment did not undergo C-terminal lactamization (at Lys27) during the ligation, unlike the previous cases for peptides 9 and 16, which suggests that this side-reaction is rather sequence specific or partially suppressed by the formation of thiolactone. It's worth mentioning that, similar to reported observations,33,34b the nP protecting group was removed automatically during the ligation process in the presence of imidazole. In an effort to improve the overall yield, we consulted our previous work to carry out the ligation and refolding without intermediate HPLC purification.11b,30 For this purpose, the ligation mixture was exchanged into a buffer containing 6 M Gdn·HCl and 0.2 M Na2HPO4 (pH 6.0) by ultracentrifugation (3 kDa cut-off) and was directly added dropwise into the refolding buffer (final conc. 0.25 mg mL−1). The refolding was allowed for 16 h at RT, and the refolded sY63-HV1 29 was obtained by a single HPLC purification step with an isolated yield of 39% (over three steps). The proper folding of 29 was confirmed by ESI-MS (∼–6 Da vs.28) (Fig. 5C). For comparison, the same protein was also synthesized by a conventional MPAA method (Fig. S68 and S69†), and more ultracentrifugation washes were needed to remove all MPAA (8 times vs. 3 times for the APCL strategy), resulting in lower isolated yield (27.5%). Overall, the efficient synthesis of the sulfated sY63-HV1 demonstrates that the mild APCL strategy could tolerate some of the most labile protein post-translational modifications.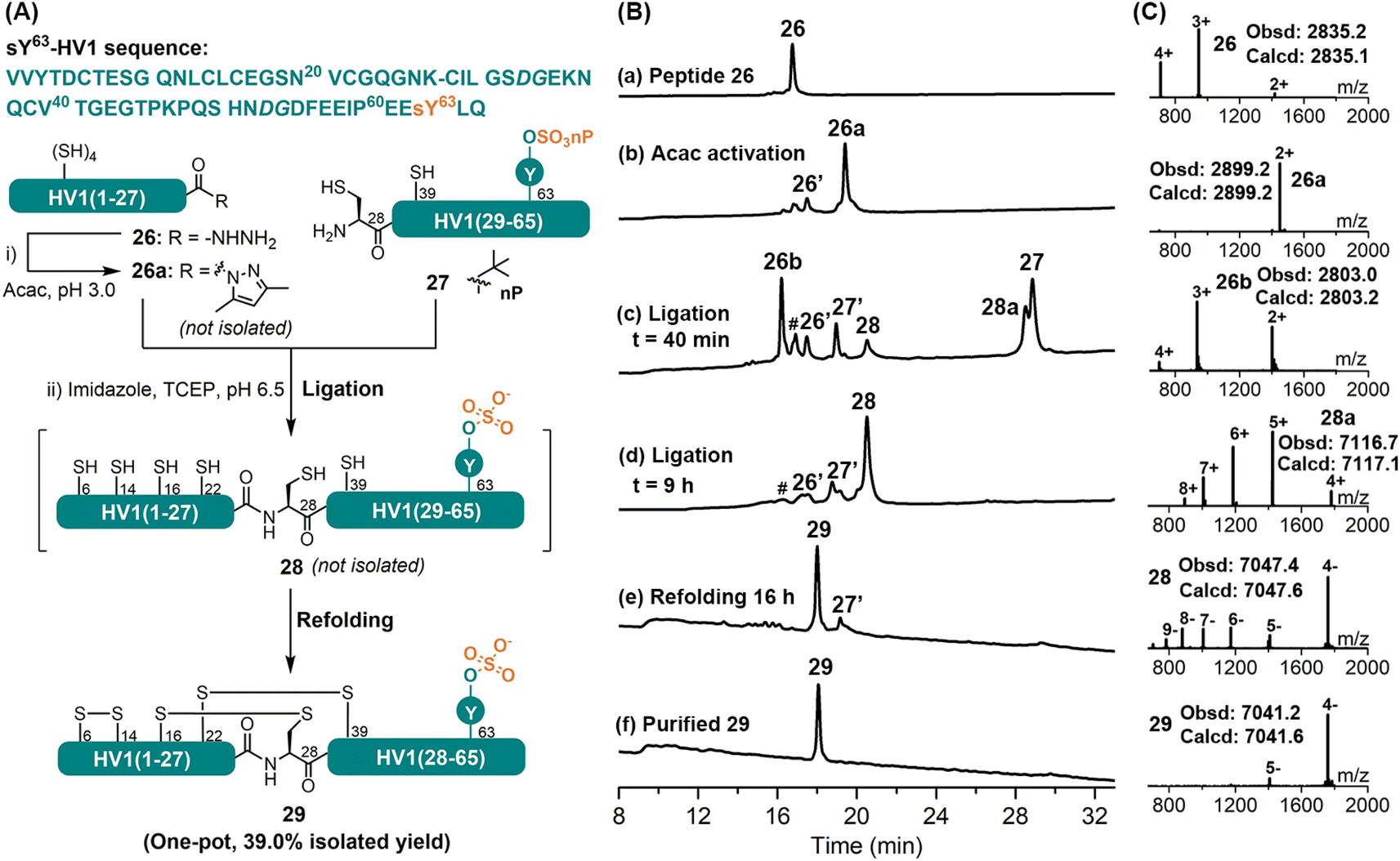 | ||
| Fig. 5 (A) Sequence and synthetic scheme for the sY63-HV1 29. The ligation site is indicated with a dash and any dipeptides used are in italic. (B) HPLC traces (214 nm) of major reaction steps. (a) The purified peptide 26; (b) acac activation, 26a is an acyl pyrazole product and 26′ is a hydrolysis by-product; (c and d) APCL at 40 min and 9 h, 26b corresponds to the thiolactone derivative of 26a and 27′ corresponds to the nP removed derivative of 27. 28a corresponds to the ligation product, 28 corresponds to the nP removed product and peak # denotes unknown by-products; (e) the refolding at 16 h; (f) the purified folded product 29. (C) ESI-MS characterization of key intermediates and products. | ||
Synthesis of the cyclic peptide kalata B1 through the APCL strategy
Disulfide-rich cyclic peptides have high structural rigidity and are resistant to chemical, enzymatic or thermal attacks, making them promising candidates for the development of peptide-based therapeutic drugs.35 We were interested to explore the viability of the APCL strategy in preparing cyclic peptides. To this end, cyclization of a model peptide, CGYLKFKSLA-NHNH2 (15), was tested. As shown in Fig. S70 and S71,† both 3-methylpyrazole and imidazole gave the cyclic peptide 15c with excellent isolated yields (76–77.4%) through APCL.Encouraged by these positive results, we moved to synthesize a large cyclotide, kalata B1 (kB1), which is from the African plant Oldenlandia affinis. kB1 is a head-to-tail cyclic peptide with a knotted arrangement of three disulfide bonds, which makes it an extremely stable and versatile drug lead or scaffold for pharmaceutical development.36 Aiming to achieve a highly efficient synthetic route for kB1, the reductant-free 3-methylpyrazole-assisted APCL strategy that allows one-pot cyclization and refolding was adopted (Fig. S73 and S74†). In this case, the linear kB1 hydrazide 30 was treated with acac to form the corresponding acyl pyrazole fragment (30a) within 2 h. The cyclization was then initiated by direct addition of 3-methylpyrazole and adjustment of pH to 5.0. Again, the 3-methylpyrazole exchanged intermediate 30b could be clearly observed, and within 3 h an extremely clean conversion to the targeted cyclization product 31 was achieved (Fig. 6B-e and S73 and S74†). Without any purification and extraction steps, the resulting mixture was directly added dropwise into the folding buffer containing 2-propanol (i-PrOH)36a (final conc. 0.2–0.4 mg mL−1). To our delight, the oxidative folding was accomplished within 15 h, giving the folded kB1 32 as a single peak in an excellent isolated yield (68.5% in one pot over three steps). The proper folding of 32 was confirmed by ESI-MS (∼–6 Da vs.31) and the CD spectrum (see the left inset in Fig. 6B-f), which agrees well with the reported literature.36a In addition, we have synthesized kB1 with the conventional MPAA method (Fig. S75 and S76†), where two HPLC purification steps were required (one for the removal of MPAA and TCEP and another for final purification), and the overall isolated yield (27.5%) was much lower than with the current strategy. Collectively, these results demonstrate that the APCL strategy is highly viable for the synthesis of cyclic peptides, especially those bearing one or more disulfides, featuring fewer purification steps and higher synthetic yield.
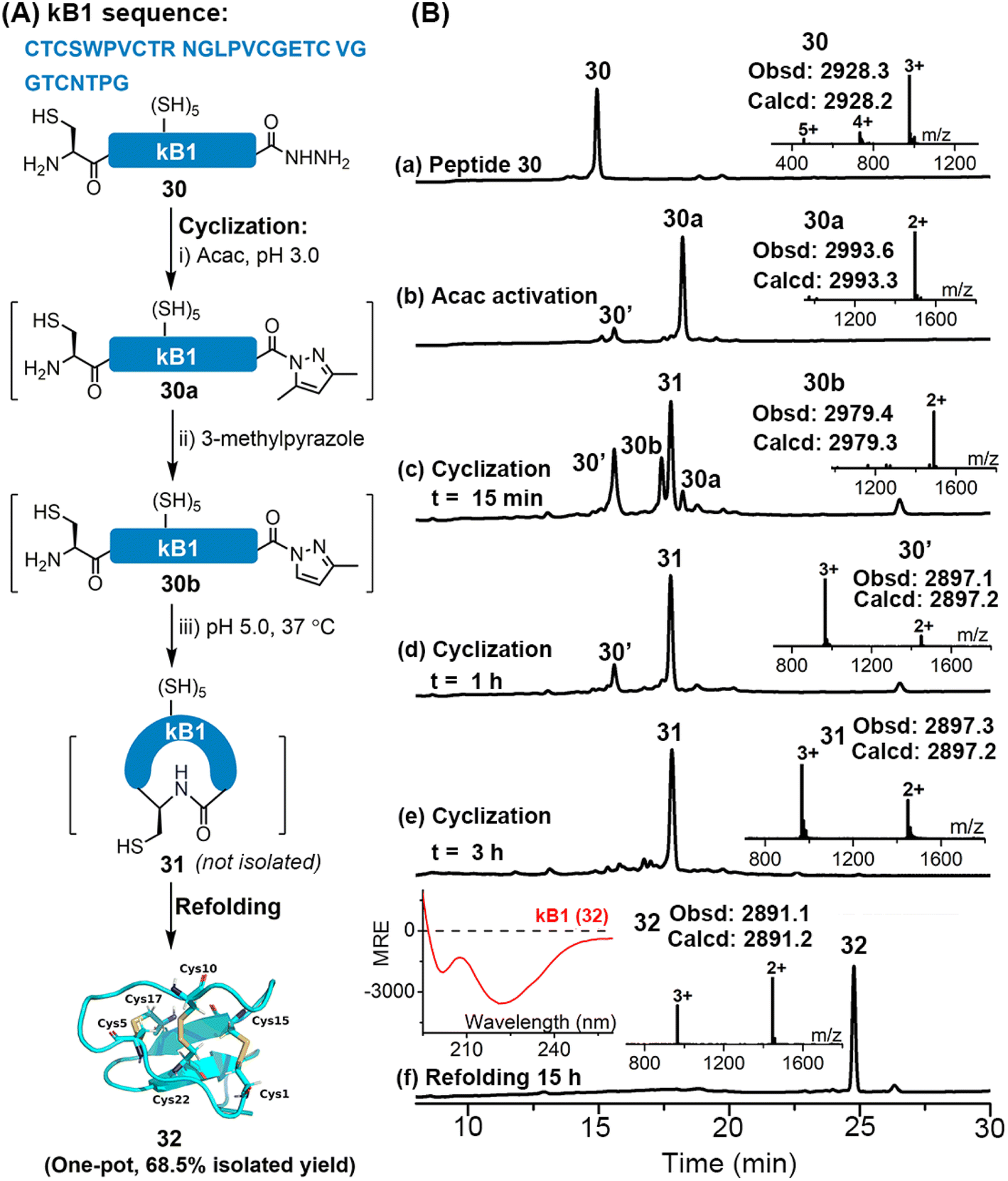 | ||
| Fig. 6 (A) Sequence and synthetic scheme for the cyclic peptide kB1 32via one-pot cyclization–refolding with APCL. (B) HPLC traces (214 nm) and ESI-MS characterization of the major reaction steps. (a) The purified 30; (b) acac activation, 30a is the acyl pyrazole and 30′ is the thiolactone; (c–e) cyclization at 15 min, 1 h and 3 h, respectively. 30b is a 3-methylpyrazole substituted intermediate and 31 is the cyclization product; (f) the refolding at 15 h, 32 is the folded product. Inset: CD spectrum (left) of the synthetic 32. | ||
Conclusion
In summary, we demonstrated that 3-methylpyrazole and imidazole can activate the weak acyl donor peptidyl N-acyl pyrazole to enable its direct ligation with an N-terminal cysteine peptide efficiently at pH 5.0 and 6.5, respectively. Building upon the easily accessible peptidyl hydrazides (through SPPS or recombinant expression) and their facile conversion to acyl pyrazoles, the current APCL strategy is viable for both protein total and semi-synthesis. Importantly, since no peptidyl thioester or thiol additive is needed for the ligation, the APCL strategy can be conveniently combined with one-pot MFD or oxidative protein folding, which greatly improves the synthetic efficiency by avoiding the preparation of a peptidyl thioester and its side-reactions (e.g., hydrolysis), reducing intermediary purification steps, and shortening overall operation time. The practicality and generality of the strategy are showcased by the total synthesis of ubiquitin via an N-to-C sequential ligation–MFD strategy and the ligation–folding of the semi-synthetic copper protein azurin, a sulfated hirudin variant, and the cyclotide kB1, all in a one-pot fashion. Given the efficiency and simplicity of APCL enabled by the commercially available and inexpensive azole reagents, we expect it to be widely utilized to improve the efficiency of CPS, complementing the current synthetic toolbox.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
PL performed the experiments and wrote the manuscript. PL and CH conceptualized the project and revised the manuscript.Conflicts of interest
A Chinese patent is filed covering the strategy described in this manuscript (ZL 202310576757.4).Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22277029 and 22077036). The technical assistance in fluorescence emission spectroscopy from Ms Jindi Luo is gratefully acknowledged. We also thank Ms Qi Zhang, Ms Xinyue Zhang, Ms Qin He, Mr Ye Yang, Mr Pengpeng Li and Mr Lan Xu for assistance with peptide synthesis.References
- (a) Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed; (b) S. Dong, J.-S. Zheng, Y. Li, H. Wang, G. Chen, Y. Chen, G. Fang, J. Guo, C. He, H. Hu, X. Li, Y. Li, Z. Li, M. Pan, S. Tang, C. Tian, P. Wang, B. Wu, C. Wu, J. Zhao and L. Liu, Sci. China: Chem., 2024, 67, 1060–1096 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- (a) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC; (b) V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- (a) S. Futaki, K. Sogawa, J. Maruyama, T. Asahara, M. Niwa and H. Hojo, Tetrahedron Lett., 1997, 38, 6237–6240 CrossRef CAS; (b) J. Alsina, T. S. Yokum, F. Albericio and G. Barany, J. Org. Chem., 1999, 64, 8761–8769 CrossRef CAS PubMed; (c) H. Hojo, Y. Kwon, Y. Kakuta, S. Tsuda, I. Tanaka, K. Hikichi and S. Aimoto, Bull. Chem. Soc. Jpn., 1993, 66, 2700–2706 CrossRef CAS; (d) R. von Eggelkraut-Gottanka, A. Klose, A. G. Beck-Sickinger and M. Beyermann, Tetrahedron Lett., 2003, 44, 3551–3554 CrossRef CAS.
- (a) E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed; (b) V. Diemer, O. Firstova, V. Agouridas and O. Melnyk, Chem.–Eur. J., 2022, 28, e202104229 CrossRef CAS PubMed.
- (a) Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed; (b) C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed; (c) J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS PubMed; (d) K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS PubMed; (e) L. R. Malins and R. J. Payne, Aust. J. Chem., 2015, 68, 521–537 CrossRef CAS; (f) H. M. Burke, L. McSweeney and E. M. Scanlan, Nat. Commun., 2017, 8, 15655 CrossRef PubMed; (g) Y. Gui, L. Qiu, Y. Li, H. Li and S. Dong, J. Am. Chem. Soc., 2016, 138, 4890–4899 CrossRef CAS PubMed.
- H. Rohde, J. Schmalisch, Z. Harpaz, F. Diezmann and O. Seitz, ChemBioChem, 2011, 12, 1396–1400 CrossRef CAS PubMed.
- T. Moyal, H. P. Hemantha, P. Siman, M. Refua and A. Brik, Chem. Sci., 2013, 4, 2496–2501 RSC.
- (a) P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS PubMed; (b) R. E. Thompson, X. Liu, N. Alonso-García, P. J. B. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS PubMed; (c) M. Haj-Yahya and H. A. Lashuel, J. Am. Chem. Soc., 2018, 140, 6611–6621 CrossRef CAS PubMed; (d) Y. C. Huang, C. C. Chen, S. Gao, Y. H. Wang, H. Xiao, F. Wang, C. L. Tian and Y. M. Li, Chem.–Eur. J., 2016, 22, 7623–7628 CrossRef CAS PubMed; (e) P. W. Erickson, J. M. Fulcher, P. Spaltenstein and M. S. Kay, Bioconjugate Chem., 2021, 32, 2233–2244 CrossRef CAS PubMed; (f) K. Ohkawachi, D. Kobayashi, K. Morimoto, A. Shigenaga, M. Denda, K. Yamatsugu, M. Kanai and A. Otaka, Org. Lett., 2020, 22, 5289–5293 CrossRef CAS PubMed; (g) S. Suzuki, Y. Nakajima, N. Kamo, A. Osakabe, A. Okamoto, G. Hayashi and H. Murakami, Molecules, 2023, 28, 3655 CrossRef CAS PubMed.
- (a) D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed; (b) T. Durek, V. Y. Torbeev and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4846–4851 CrossRef CAS PubMed.
- (a) K. Sakamoto, S. Tsuda, M. Mochizuki, Y. Nohara, H. Nishio and T. Yoshiya, Chem.–Eur. J., 2016, 22, 17940–17944 CrossRef CAS PubMed; (b) P. Liao and C. He, Front. Chem., 2021, 9, 735149 CrossRef CAS PubMed.
- T. S. Chisholm, D. Clayton, L. J. Dowman, J. Sayers and R. J. Payne, J. Am. Chem. Soc., 2018, 140, 9020–9024 CrossRef CAS PubMed.
- (a) K. Kitagawa, H. Adachi, Y. Sekigawa, T. Yagami, S. Futaki, Y. J. Gu and K. Inoue, Tetrahedron, 2004, 60, 907–918 CrossRef CAS; (b) J. Alsina, S. A. Kates, G. Bárány and F. Albericio, Methods Mol. Biol., 2005, 298, 195–208 CAS; (c) J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed; (d) J. A. Camarero, B. J. Hackel, J. J. de Yoreo and A. R. Mitchell, J. Org. Chem., 2004, 69, 4145–4151 CrossRef CAS PubMed; (e) M. Yanase, K. Nakatsu, C. J. Cardos, Y. Konda, G. Hayashi and A. Okamoto, Chem. Sci., 2019, 10, 5967–5975 RSC; (f) J. Dheur, N. Ollivier and O. Melnyk, Org. Lett., 2011, 13, 1560–1563 CrossRef CAS PubMed; (g) H. E. Elashal, Y. E. Sim and M. Raj, Chem. Sci., 2017, 8, 117–123 RSC; (h) B. Cowper, T. M. Sze, B. Premdjee, A. F. Bongat White, A. Hacking and D. Macmillan, Chem. Commun., 2015, 51, 3208–3210 RSC; (i) B. Yan, W. Shi, L. Ye and L. Liu, Curr. Opin. Chem. Biol., 2018, 46, 33–40 CrossRef CAS PubMed; (j) G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed; (k) J. X. Wang, G. M. Fang, Y. He, D. L. Qu, M. Yu, Z. Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- K. Sakamoto, S. Tsuda, H. Nishio and T. Yoshiya, Chem. Commun., 2017, 53, 12236–12239 RSC.
- (a) Y. Wang, L. Han, N. Yuan, H. Wang, H. Li, J. Liu, H. Chen, Q. Zhang and S. Dong, Chem. Sci., 2018, 9, 1940–1946 RSC; (b) Y. Dao, B. Wang, W. Dong, J. Zhang, C. Zhong, Z. Zhang and S. Dong, Chin. J. Chem., 2021, 39, 2509–2516 CrossRef CAS.
- J.-S. Zheng, S. Tang, Y.-K. Qi, Z.-P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- (a) J. Thom, D. Anderson, J. McGregor and G. Cotton, Bioconjugate Chem., 2011, 22, 1017–1020 CrossRef CAS PubMed; (b) D. Zhang, Z. Wang, S. Hu, N.-Y. Chan, H. T. Liew, J. Lescar, J. P. Tam and C.-F. Liu, Bioconjugate Chem., 2022, 33, 238–247 CrossRef CAS PubMed; (c) A. L. Adams, B. Cowper, R. E. Morgan, B. Premdjee, S. Caddick and D. Macmillan, Angew. Chem., Int. Ed., 2013, 52, 13062–13066 CrossRef CAS PubMed; (d) Y. Kajihara, Y. Kanemitsu, M. Nishihara, R. Okamoto and M. Izumi, J. Pept. Sci., 2014, 20, 958–963 CrossRef CAS PubMed; (e) Y. Qiao, G. Yu, K. C. Kratch, X. A. Wang, W. W. Wang, S. Z. Leeuwon, S. Xu, J. S. Morse and W. R. Liu, J. Am. Chem. Soc., 2020, 142, 7047–7054 CrossRef CAS PubMed; (f) L. Shi, H. Chen, S.-Y. Zhang, T.-T. Chu, Y.-F. Zhao, Y.-X. Chen and Y.-M. Li, J. Pept. Sci., 2017, 23, 438–444 CrossRef CAS PubMed; (g) Z. Mo, S. Lin, W. Chen and C. He, Angew. Chem., Int. Ed., 2022, 61, e202115377 CrossRef CAS PubMed; (h) Y.-M. Li, Y.-T. Li, M. Pan, X.-Q. Kong, Y.-C. Huang, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 2198–2202 CrossRef CAS PubMed.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS PubMed.
- (a) T. E. Creighton, BioEssays, 1988, 8, 57–63 CrossRef CAS PubMed; (b) J. M. Thornton, J. Mol. Biol., 1981, 151, 261–287 CrossRef CAS PubMed.
- Y. Li, J. Liu, Q. Zhou, J. Zhao and P. Wang, Chin. J. Chem., 2021, 39, 1861–1866 CrossRef CAS.
- (a) H. A. Staab and A. Mannschreck, Chem. Ber., 1962, 95, 1284–1297 CrossRef CAS; (b) A. R. Katritzky, S. Hoffmann and K. Suzuki, ARKIVOC, 2004, 14–22 Search PubMed; (c) T. Mino, S. Sakamoto and I. Hamachi, Biosci., Biotechnol., Biochem., 2021, 85, 53–60 CrossRef PubMed.
- (a) Y. Li, A. Yongye, M. Giulianotti, K. Martinez-Mayorga, Y. Yu and R. A. Houghten, J. Comb. Chem., 2009, 11, 1066–1072 CrossRef CAS PubMed; (b) G.-M. Fang, H.-K. Cui, J.-S. Zheng and L. Liu, ChemBioChem, 2010, 11, 1061–1065 CrossRef CAS PubMed.
- (a) P. Siman, S. V. Karthikeyan, M. Nikolov, W. Fischle and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 8059–8063 CrossRef CAS PubMed; (b) F. K. Deng, L. Zhang, Y. T. Wang, O. Schneewind and S. B. H. Kent, Angew. Chem., Int. Ed., 2014, 53, 4662–4666 CrossRef CAS PubMed; (c) H. Sun and A. Brik, Acc. Chem. Res., 2019, 52, 3361–3371 CrossRef CAS PubMed.
- D. Bang, G. I. Makhatadze, V. Tereshko, A. A. Kossiakoff and S. B. Kent, Angew. Chem., Int. Ed., 2005, 44, 3852–3856 CrossRef CAS PubMed.
- (a) K. S. A. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2010, 49, 9126–9131 CrossRef CAS PubMed; (b) L. A. Erlich, K. S. A. Kumar, M. Haj-Yahya, P. E. Dawson and A. Brik, Org. Biomol. Chem., 2010, 8, 2392–2396 RSC.
- (a) R. Ramage, J. Green, T. W. Muir, O. M. Ogunjobi, S. Love and K. Shaw, Biochem. J., 1994, 299, 151–158 CrossRef CAS PubMed; (b) F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS PubMed; (c) S. N. Bavikar, L. Spasser, M. Haj-Yahya, S. V. Karthikeyan, T. Moyal, K. S. Ajish Kumar and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 758–763 CrossRef CAS PubMed.
- J. Schmalisch and O. Seitz, Chem. Commun., 2015, 51, 7554–7557 RSC.
- (a) D. K. Garner, M. D. Vaughan, H. J. Hwang, M. G. Savelieff, S. M. Berry, J. F. Honek and Y. Lu, J. Am. Chem. Soc., 2006, 128, 15608–15617 CrossRef CAS PubMed; (b) T. D. Wilson, Y. Yu and Y. Lu, Coord. Chem. Rev., 2013, 257, 260–276 CrossRef CAS.
- J. Liu, O. Ekanayake, D. Santoleri, K. Walker and S. Rozovsky, ChemBioChem, 2020, 21, 346–352 CrossRef CAS PubMed.
- P. Liao, H. Liu and C. He, Chem. Sci., 2022, 13, 6322–6327 RSC.
- (a) Y. Yu, N. M. Marshall, D. K. Garner, M. J. Nilges and Y. Lu, J. Inorg. Biochem., 2022, 234, 111863 CrossRef CAS PubMed; (b) N. Bonander, B. Göran Karlsson and T. Vänngård, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1251, 48–54 CrossRef PubMed.
- (a) W. B. Huttner, Nature, 1982, 299, 273–276 CrossRef CAS PubMed; (b) M. J. Stone and R. J. Payne, Acc. Chem. Res., 2015, 48, 2251–2261 CrossRef CAS PubMed; (c) J. W. C. Maxwell and R. J. Payne, Curr. Opin. Chem. Biol., 2020, 58, 72–85 CrossRef CAS PubMed.
- Y. Yang, M. Liang, R. Wang and C. He, Chin. Chem. Lett., 2023, 34, 107806 CrossRef CAS.
- (a) L. S. Simpson, J. Z. Zhu, T. S. Widlanski and M. J. Stone, Chem. Biol., 2009, 16, 153–161 CrossRef CAS PubMed; (b) C. Li and C. He, Org. Biomol. Chem., 2020, 18, 7559–7564 RSC.
- (a) M. L. Colgrave and D. J. Craik, Biochemistry, 2004, 43, 5965–5975 CrossRef CAS PubMed; (b) D. J. Craik, J. S. Mylne and N. L. Daly, Cell. Mol. Life Sci., 2010, 67, 9–16 CrossRef CAS PubMed.
- (a) N. L. Daly, S. Love, P. F. Alewood and D. J. Craik, Biochemistry, 1999, 38, 10606–10614 CrossRef CAS PubMed; (b) J. S. Zheng, S. Tang, Y. Guo, H. N. Chang and L. Liu, ChemBioChem, 2012, 13, 542–546 CrossRef CAS PubMed; (c) X. Jia, S. Kwon, C. I. A. Wang, Y. H. Huang, L. Y. Chan, C. C. Tan, K. J. Rosengren, J. P. Mulvenna, C. I. Schroeder and D. J. Craik, J. Biol. Chem., 2014, 289, 6627–6638 CrossRef CAS PubMed; (d) S. J. de Veer, M.-W. Kan and D. J. Craik, Chem. Rev., 2019, 119, 12375–12421 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06697e |
| This journal is © The Royal Society of Chemistry 2024 |