
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dinuclear platinum(II) complexes featuring rigidly linked Pt(NCN)X units: the effect of X = SCN− in favouring low-energy, excimer-like luminescence†
Rebecca J.
Salthouse
 *a,
Yana M.
Dikova
a,
Marc K.
Etherington
b and
J. A. Gareth
Williams
*a
*a,
Yana M.
Dikova
a,
Marc K.
Etherington
b and
J. A. Gareth
Williams
*a
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: rebecca.salthouse@durham.ac.uk; j.a.g.williams@durham.ac.uk
bDepartment of Mathematics, Physics and Electrical Engineering, Northumbria University, Ellison Building, Newcastle upon Tyne, NE1 8ST, UK
First published on 30th October 2024
Abstract
Interfacial intermolecular interactions between phosphorescent, square-planar, cyclometallated platinum(II) complexes may lead to the formation of bimolecular excited states that emit at lower energy than the isolated complexes in dilute solution. We study compounds in which two Pt(NCN)Cl units are appended onto a rigid xanthene scaffold to favour the intramolecular formation of such states and thus promote low-energy emission even at high dilution {where NCN represents a cyclometallated tridentate ligand based on 2,6-di(2-pyridyl)benzene}. Here, we show how the metathesis of the monodentate Cl− ligand to thiocyanate SCN− has a profound effect on the emissive properties of such compounds in solution and in polymer-doped and neat films. Intramolecular Pt⋯Pt interactions are promoted by the change to SCN− (as evident by a short Pt⋯Pt distance of 3.253(4) Å in the crystal, determined by X-ray diffraction). This increased propensity for the Pt(NCN) units to interact, induced by the thiocyanate, is also manifest in the emission spectra: the spectra show only the low-energy, excimer-like bands in solution, even at very low concentrations. That contrasts with the appearance of emission bands typical both of isolated Pt(NCN) units and of excimers for the chloro parent compound. Nevertheless, data at low temperature and in dilute polymer-doped films suggest that some degree of conformational change is still required to form the low-energy emitting states. Meanwhile, the change of the monodentate ligand from chloride to iodide suppresses the formation of the low-energy-emitting states and lowers the emission efficiency. Taken together, the results offer new insight into strategies for obtaining efficient NIR-emitting phosphors based on dinuclear PtII2 excited states.
Introduction
Materials that emit light efficiently within the deep-red and near-infrared (NIR) regions of the electromagnetic spectrum are in demand for various applications. For example, NIR-emitting lanthanides are widely used in modern fibre-optic communications,1 and visible light communication strategies based on optical intensity modulation are being expanded into the NIR region.2 Meanwhile, the use of light in medicine – both diagnostically in bioimaging and biosensing, and therapeutically in photodynamic therapy (PDT) – is optimal in the near-NIR region (700–1400 nm), matching the so-called “window of transparency” of biological tissue.3 Whilst the most well-established NIR-emitting materials are ionic, there is huge interest in the potential of molecular materials as emitters for NIR-OLEDs (organic light-emitting devices).4Nevertheless, the availability of efficient, molecular, deep-red and NIR emitters remains limited. Several factors conspire to reduce the efficiency of low-energy emitters compared to, say, those that emit in the green region, where quantum yields approaching unity are now commonplace. Firstly, when the energy gap between the excited and ground states is reduced to shift emission to longer wavelength, non-radiative vibrational decay is promoted through the so-called “energy gap law”.5,6 Secondly, when the strategy of using extended planar chromophores is used to attain the necessary small HOMO–LUMO gap, energy-depletive intermolecular interactions often come into play, particularly for fluorescent organic emitters.7 Thirdly, for systems that exploit a heavy metal such as Ir(III) or Pt(II) to promote the harvesting of triplet states in phosphorescent OLEDs through spin–orbit coupling (SOC), the extent of metal character in the excited state (and hence the efficiency of SOC) falls off as the ligands become more extended, due to a poorer energy match between filled metal and ligand orbitals.8
We and others have been exploring the formation of low-energy-emitting excimers and aggregates of organometallic platinum(II) complexes as a strategy for generating deep red/NIR light.9–14 Pt(II) complexes featuring the tridentate, NCN-coordinating ligand 2,6-di(2-pyridyl)benzene (dpyb), and derivatives thereof, have a high propensity to form strongly emissive excimers at elevated concentrations in solution.15 For Pt(dpyb)Cl, for example (Fig. 1), the broad excimer emission band centred at around 700 nm in CH2Cl2 grows in with increasing concentration (>10−5 M) at the expense of the structured, unimolecular emission band (λ0,0 = 491 nm).15b Similar behaviour is observed in films comprising the complex doped into a suitable host material, where the combination of emission bands from unimolecular and bimolecular excited states can be used to generate white light from only one emissive component.16 Meanwhile, neat films lead to exclusively excimeric photoluminescence; this behaviour also translates to electroluminescence (EL) and hence to deep-red-emitting phosphorescent OLEDs.17
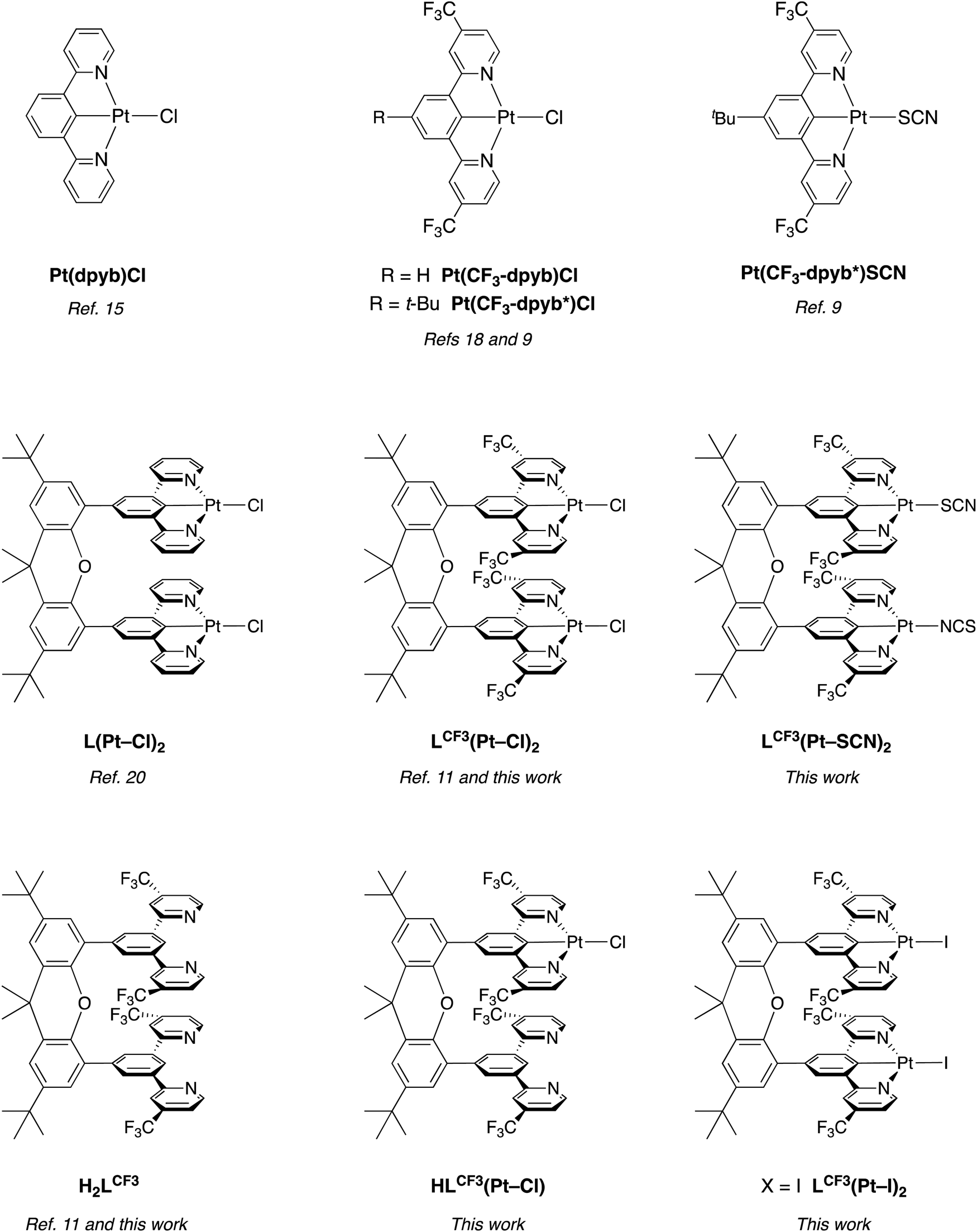 | ||
| Fig. 1 Structures of the dinuclear complexes LCF3(Pt–SCN)2 and LCF3(Pt–I)2 reported in this work; the related examples L(Pt–Cl)2 and LCF3(Pt–Cl)2 with chloride as the monodentate ligand; reference mononuclear complexes (top row); the proligand H2LCF3 and the mononuclear compound HLCF3(Pt–Cl). See text for discussion of the binding mode of SCN. | ||
In work to shift the emission further into the NIR, three relevant strategies have emerged:
(1) Electron-withdrawing substituents in the pyridine rings stabilise and hence red-shift the excimer (conversely, electron-donating substituents induce a blue shift).18 These trends mirror those on the unimolecular emission,15d rationalised in terms of the broadly similar localisation of the HOMO and LUMO in dimers to those in isolated molecules.9 The excimer bands of Pt(CF3-dpyb)Cl and its 4-tert-butyl derivative Pt(CF3-dpyb*)Cl (Fig. 1) are thus shifted to around λmax = 750 nm, such that almost all of the emission lies in the NIR region.9
(2) The metathesis of the monodentate chloride in such Pt(NCN)Cl complexes to isothiocyanate (–NCS) or thiocyanate (–SCN) promotes the formation of aggregates in the ground state, as originally demonstrated by Roberto and co-workers,19 and further studied recently.10 The resulting dimers, trimers and higher oligomers emit more deeply into the NIR; for example, for thermally evaporated neat films of Pt(CF3-dpyb*)SCN, the emission λmax is around 960 nm.10
(3) In an attempt to allow lower concentrations of Pt(II) materials to be used in doped films – as opposed to the high concentrations/neat films required for the intermolecular excimer-like emission bands to dominate over unimolecular emission – we have explored the strategy of covalently linking two such Pt(NCN)Cl units to a rigid xanthene scaffold, e.g. L(Pt–Cl)2 and LCF3(Pt–Cl)2 (Fig. 1).11,20 The two organoplatinum units are oriented in such a way that face-to-face interactions between them may be facilitated, thereby potentially enhancing the proportion of low-energy emission from excimer- or aggregate-like states, relative to that from independent Pt(NCN)Cl units. This design strategy has been found to be successful in that, in solution, emission from “intramolecular excimers” does now dominate even under very dilute conditions (compared to the higher concentrations necessary for intermolecular excimers to form in mononuclear Pt(NCN)Cl complexes). In doped polymer films, on the other hand, these molecules display little such excimeric emission (at low loadings), apparently owing to the need for a significant geometrical rearrangement to move the Pt(NCN) units close enough for the necessary interfacial interactions to occur.11
In this contribution, we describe work aimed at combining all three of the above strategies to target efficient NIR emission, including in doped films. The new dinuclear compound LCF3(Pt–SCN)2 (Fig. 1) has thus been prepared, with a view to profiting from the influence of the SCN ligands that had previously been found to promote ground-state interfacial interactions in mononuclear complexes, while simultaneously exploiting the anticipated red shift associated with the CF3 substituents and the rigid interconnecting scaffold. The photoluminescence properties of this new compound are reported in solution and thin films, along with those of the iodo derivative LCF3(Pt–I)2, which is found to behave very differently. The properties are compared with those of LCF3(Pt–Cl)2 and its mononuclear analogue HLCF3(Pt–Cl) (Fig. 1).
Results and discussion
Synthesis and characterisation of the mono- and dinuclear complexes
Xanthene has proved to be an effective rigid backbone to hold units of interest together, both metal-containing and purely organic.21,22 The presence of tert-butyl substituents in the 2 and 9 positions should aid the solubility of resulting compounds. The requisite ditopic bis-tridentate ligand H2LCF3 was prepared by Suzuki coupling of 4-borylated-2,6-bis(4-trifluoromethylpyrid-2-yl)-benzene with 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethyl-xanthene as described previously.11 In the diplatination of H2LCF3 with K2PtCl4 to form LCF3(Pt–Cl)2, some of the mono-platinated compound HLCF3(Pt–Cl) was also formed, which was separated by preparative column chromatography (synthetic details are given in the ESI†). The metathesis of the chloride ligand in mononuclear Pt(NCN)Cl complexes has previously been accomplished by pre-treatment with silver trifluoromethanesulfonate (AgOTf) in acetone, whereby the chloride ligand is displaced (as AgCl) by a weakly bound acetone molecule for subsequent displacement by the desired ligand under mild conditions.9,19 The same strategy was used here using 2 molar equivalents of AgOTf. The desired thiocyanate and iodo complexes LCF3(Pt–SCN)2 and LCF3(Pt–I)2 were then generated by addition of KSCN or KI, as bright red and orange solids respectively. They were purified by a series of washings and recrystallizations (see ESI†). The metathesis of Cl was clearly reflected in the 1H NMR spectra, which showed a substantial shift (Δδ > 0.5 ppm) of the pyridine H6 resonance (adjacent to the Pt–X bond) to high frequency in the thiocyanate case, and conversely a shift of similar magnitude to low frequency for the iodo complex (Fig. S1, ESI†).The NCS ligand is well-known as an ambidentate ligand that can bind either through the nitrogen or through the sulfur atom. The former is typically observed for harder, 1st-row metal ions such as Ni2+, while the latter is expected to be more likely with softer metals. That the binding mode is finely balanced for Pt(II) is evident from the few previously reported examples of Pt(NCN)–NCS complexes, of which two feature the N-bound ligand and the third S-bound.23 Similarly, in complexes with related pyridyl-based ligands such as Pt(N,N-Me2bpy)(SCN)2 (Me2bpy = 4,4′-dimethylbipyridine), results suggest that the S-bound form is the kinetic product while the N-isomer is thermodynamically the more stable, and mixtures may thus ensue according to the conditions.24 In the case of LCF3(Pt–SCN)2, an X-ray diffraction study on crystals obtained from DMF solution reveals one N-bound and one S-bound ligand per molecule (Fig. 2). This single crystal is, of course, not necessarily representative of the bulk material. In principle, IR spectroscopy offers a means of distinguishing between N- and S-binding, as the relevant NCS stretches conveniently appear in the otherwise featureless 2000–2200 cm−1 region.25 Here, different batches of LCF3(Pt–SCN)2 consistently showed a broad, slightly split peak centred at 2080 cm−1. This value is more akin to that for S-binding in the previous studies on mononuclear Pt(NCN)X complexes [N-bound = 2108 cm−1; S-bound 2074 cm−1],9 but the broadness of the peak and the splitting could be consistent with both forms being present throughout the material. This may simply reflect a lack of a clear preference for one form over the other and thus the presence of a mixture of three species {i.e., LCF3(Pt–SCN)2 and LCF3(Pt–NCS)2, as well as the LCF3(Pt–SCN)(Pt–NCS) found in the X-ray study} or that there exists some kind of complementarity that thermodynamically favours the adoption of one N-bound and one S-bound ligand within a given molecule. For concision, we shall continue to refer to the compound as “LCF3(Pt–SCN)2.”
 | ||
| Fig. 2 The structure of LCF3(Pt–SCN)2 in crystals determined by X-ray diffraction. Left: Molecular structure highlighting the –NCS and –SCN binding modes of the ancillary ligand within the molecule and the short intramolecular Pt⋯Pt distance. Right: The packing in the crystal, with shortest intermolecular interplanar distance of 3.445(15) Å. | ||
In the crystal (Fig. 2), the Pt(NCN) units are twisted relative to the xanthene scaffold, with a torsion angle of 53.3(7)°. The Pt⋯Pt distance of 3.253(4) Å is shorter than the sum of the van der Waals radii of two Pt atoms (2 × 1.75 Å = 3.5 Å), indicative of intramolecular metallophillic interactions. There are also intermolecular π–π interactions at play, with the shortest intermolecular interplanar distance at 3.445(15) Å. The shortest intermolecular Pt⋯Pt distance is 4.127(2) Å.
Solution-state photophysics
The absorption and emission spectra of the binuclear complexes LCF3(Pt–X)2 (X = Cl, I, SCN) in dilute CH2Cl2 solution at 295 K are shown in Fig. 3, together with those of the mononuclear HLCF3(Pt–Cl) for comparison; numerical data are compiled in Table 1. The absorption spectra are typical of cyclometallated Pt(II) complexes,26 with intense bands at λ < 330 nm attributed to aromatic π → π* transitions, and somewhat weaker bands at longer wavelengths assigned to charge-transfer transitions associated with the introduction of the metal. In LCF3(Pt–Cl)2, the molar absorptivity in the latter region is roughly double that of mononuclear HLCF3(Pt–Cl) (Table 1), consistent with these bands arising from the metalated units, whereas ε values at shorter wavelengths are increased to a much lesser extent, as the shorter-wavelength bands arise from transitions within the aromatic rings that are common to both the mono- and dinuclear complexes. There is no evidence of any additional low-energy absorption band(s) in the dinuclear complex that would be expected if MMLCT states formed through intramolecular interactions;27,28 the absence of such bands is consistent with earlier observations on L(Pt–Cl)2.11 The metathesis of X from Cl to I or SCN has little effect on the absorption spectra (Fig. 3), other than the absorption tailing out to longer wavelengths for the iodo derivative, again consistent with previous work on mononuclear complexes.9 | ||
| Fig. 3 Normalised absorption and emission spectra of LCF3(Pt–X)2 in dilute deoxygenated CH2Cl2 solution (approx. 5 × 10−6 M) at 295 K; X = Cl, SCN, and I denoted by solid black, red, and blue lines, respectively. The mononuclear HLCF3PtCl is shown by the black dashed line. | ||
| Complex | Absorption at 295 Kaλmax/nm (ε/M−1 cm−1) | Emission at 295 Kb | Emission at 77 Kef | ||||
|---|---|---|---|---|---|---|---|
| λ max/nm short-λ bandc | λ max/nm long-λ bandc | Φ lum | τ/μse | λ max/nm | τ/μs | ||
a In CH2Cl2.
b In deoxygenated CH2Cl2 solution.
c LCF3(Pt–Cl)2 displays two sets of bands in dilute solution (see Fig. 3), one at higher energy (“short-λ” column) and one at lower-energy (“long-λ” column) while LCF3(Pt–SCN)2 and LCF3(Pt–I)2/HLCF3(Pt–Cl) show only the latter and former respectively, with entries in the according columns. Excitation wavelength employed, λex = λmax of the lowest-energy absorption band.
d Photo-luminescence quantum yield in dilute solution measured relative to aqueous, air-equilibrated [Ru(bpy)3]Cl2, for which Φlum = 0.04.
e Where two sets of bands are observed, * indicates the lifetime or wavelength of the long-λ band. Excitation wavelength employed, λex = 405 nm.
f In diethyl ether/isopentane/ethanol glass (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 v/v). :2:1 v/v).
|
|||||||
| LCF3(Pt–Cl)2 | 304 (56700), 357 (10300), 380 (8690), 412 (13700), 425 (13000) |
542, 582 | 762 | 0.12 | 7.0 0.45* | 540, 580 | 9.4 |
| LCF3(Pt–SCN)2 | 310 (43900), 360 (8490), 425 (10200) |
— | 714 | 0.11 | 1.2 | 539, 573 663, 773* | 11 |
| LCF3(Pt–I)2 | 308 (39000), 393 (5820), 435 (8390) |
551, 584 | — | 0.01 | 6.8 | 546, 584 | 9.6 |
| HLCF3Pt–Cl | 300 (45200), 396 (4970), 413 (7360), 430 (6120) |
543, 582 | — | 0.22 | 7.2 | 532, 572 | 10 |
All the complexes are luminescent in deoxygenated solution at 295 K (Fig. 3) with lifetimes of the order 1–10 μs, a range comparable to those typical of mononuclear Pt(NCN)Cl complexes. The mononuclear HLCF3(Pt–Cl) displays green emission (λmax = 543 nm) similar to that of Pt(CF3dpyb)Cl.9 There is no evidence of luminescence at lower energy attributable to an excimer, nor does any such emission appear at higher concentrations (Fig. S2, ESI†). Meanwhile, time-resolved data reveal barely any self-quenching of the excited state: lifetimes are only slightly attenuated with increasing concentration. The Stern–Volmer quenching constant is estimated to be approximately 108 M−1 s−1, namely around 50× smaller than for Pt(dpyb)Cl, for example. Intermolecular interactions are thus clearly minimal in the case of HLCF3(Pt–Cl).
In contrast, in our earlier work, LCF3(Pt–Cl)2 showed two bands: one of λmax 542 nm attributed to excited states localised on one Pt(NCN) unit, and the second at low energy centred at 762 nm and attributable to an excimer.11 In dilute solutions (up to about 4 × 10−6 M), the intensity of the latter relative to the former remained constant with concentration, supportive of an intramolecular origin to the excimer band (i.e., involving interactions between two Pt(NCN) units within the same molecule, as we reported previously). But, the relative intensity of red emission then increased at higher concentrations, interpreted in terms of excimeric species then being formed intermolecularly under those conditions. The Stern–Volmer quenching constant estimated from the unimolecular excited-state lifetimes was around 6× higher than for HLCF3(Pt–Cl). Thus, the diplatinated structure apparently promotes intermolecular interactions compared to its monoplatinated analogue.
The iodinated derivative LCF3(Pt–I)2 differs from the chloro LCF3(Pt–Cl)2 in that the emission is much weaker (Φlum < 0.01), with no low-energy band growing in with concentration (Fig. 3; see also Fig. S3 (ESI†) at varying concentration). This latter observation mirrors what was previously observed for mononuclear Pt(CF3dpyb*)I versus Pt(CF3dpyb*)Cl,11 perhaps due to the larger iodide ligands disfavouring the interfacial approach of the Pt(NCN) units. No detrimental effect of iodide on the quantum yield was observed in that case, however.
The new, dinuclear thiocyanate complex LCF3(Pt–SCN)2 behaves quite differently from either of the halogenated analogues. Here, the introduction of the thiocyanate ligands leads exclusively to an excimer-like, low-energy emission band for LCF3(Pt–SCN)2, λmax = 715 nm (Fig. 3), irrespective of the concentration (the range 10−7–10−4 M was investigated). The excitation spectrum matches closely the absorption spectrum, consistent with assignment to an excimer (Fig. S4, ESI†). There is no evidence of any emission at higher energy due to excited states confined to a single Pt(NCN) unit, as there was in the chloro analogue. The original hypothesis – namely that metathesis from Cl to SCN would lead to excited states spanning both Pt(NCN) units even under dilute conditions – is thus vindicated. The broad emission band is blue-shifted by around 50 nm compared to that of LCF3(Pt–Cl)2, in line with the earlier observations on the mononuclear complexes Pt(CF3-dpyb*)X, where the excimer for X = SCN was blue-shifted relative to that for X = Cl.11 The emission maximum of 715 nm is still, however, somewhat red-shifted relative to the corresponding value of 690 nm for L(Pt–Cl)2 through the influence of the CF3 substituents, and the quantum yield of 0.11 is respectable for a molecular phosphor emitting at λ > 700 nm. Despite the persistence of the low-energy band at all concentrations investigated (representative spectra are shown in Fig. S5, ESI†), the complex does show some concentration quenching with a Stern–Volmer constant of 1.6 × 109 M−1 s−1, indicating that intermolecular interactions do also come into play to some extent as the concentration increases.
Excimer or aggregate?
Our previous experimental work on L(Pt–Cl)2 and LCF3(Pt–Cl)2 concluded that the excited state spanning the two Pt(NCN) units responsible for the low-energy emission in solution was excimer-like in character; i.e., one involving a molecular configuration that forms in the excited state, rather than a pre-existing ground-state interaction (e.g., as would be found in an aggregate).11 TD-DFT calculations indicated that a substantial geometric distortion is required relative to the ground state, to move the metal centres of the Pt(NCN) units to a distance short enough to form excimer-like states. In the present instance, the ground-state Pt⋯Pt distance of 3.253(4) Å in the crystal is, in principle, already potentially short enough for a ground-state interaction to be present, at least in the solid state. However, as noted above, no low-energy absorption band was visible in solution, which would be the typical hallmark of an MMLCT state.27,28 Further insight comes from the emission spectra in a rigid glass at 77 K (Fig. 4). The spectrum of LCF3(Pt–SCN)2 under these conditions – and likewise for the chloro parent – is dominated by structured emission bands in the green region of the spectrum, λ0,0 = 539 nm, typical of the mononuclear parent complexes {i.e., of isolated, unimolecular Pt(NCN) units}. Apparently, then, the rigid environment inhibits the attainment of the excited state responsible for the low-energy band observed at room temperature, supporting the notion that it is excimer-like in origin. For LCF3(Pt–SCN)2, however, two additional, weaker, broad bands are observed, centred at 663 and 773 nm. It is likely that these are due to species with intramolecular and intermolecular ground-state interactions between Pt(NCN) units, respectively, that have been frozen out at 77 K (analogous to the behaviour in films, discussed further below). | ||
| Fig. 4 Normalised emission spectra of LCF3(Pt–X)2 at 77 K in diethyl ether/isopentane/ethanol (2:2:1 v/v); X = Cl, SCN, and I denoted by solid black, red, and blue lines, respectively. The mononuclear complex HLCF3(Pt–Cl) is shown as the black dashed line. | ||
Emission in films
The emission of the new complexes was subsequently investigated in films of doped poly(methylmethacrylate) (PMMA), at loadings of 0.1, 1 and 10% by mass, as well as in neat (100%) films. Details of the preparation of the films are given in the ESI.† We first consider LCF3(Pt–SCN)2. At the low 0.1% loading at 295 K, the complex shows two bands of roughly comparable intensity, with λmax 540 and 700 nm (Fig. 5). This behaviour can be rationalised by reference to the behaviour in solution at room temperature – where only the long wavelength band was observed (around 714 nm) – and the behaviour at 77 K, where the spectrum was dominated by the shorter-wavelength emission (λmax = 546 nm). In the polymer host – a more rigid environment than in solution – not all molecules will have Pt(NCN) units in the conformation required to form the intramolecular excimer, and so some emission then appears from excited states confined to “isolated” Pt(NCN) units. Upon increasing the loading to 1% and then 10%, the lower-energy band red shifts and increases in intensity relative to the shorter wavelength band. We attribute this behaviour to intermolecular interactions coming into play at these higher loadings, which deplete the excited states localised on isolated Pt(NCN) units. The red-shift with increased loadings – which terminates with a λmax of 830 nm for the neat film – probably reflects the increasing contribution of oligomers (e.g., trimers and tetramers) with slightly lower-energy excited states, as was observed in our previous work on mononuclear Pt(CF3dpyb*)SCN. These oligomers are likely ground-state species formed through aggregation: the absorption spectrum of the neat film shows quite an intense low-energy shoulder tailing to > 600 nm (Fig. S6, ESI†), not present in solution, and such bands are typical of, for example, metal–metal-to-ligand charge-transfer states.27,28 Table S3 (ESI†) summarises the decay times of the emission bands in the films. The high-energy unimolecular emission in the dilute films (0.1 and 10%) has a lifetime of around 6 μs, whilst the low-energy band is much shorter lived, around 200 ns. These two values are comparable to the unimolecular and excimer bands of mononuclear Pt(dpyb)Cl and many of its derivatives.15b–d An overlay of the solution, 77 K, and neat film spectra is given in the ESI,† highlighting the contrasting behaviour under the different conditions (Fig. S7, ESI†). | ||
| Fig. 5 Emission spectra of LCF3(Pt–SCN)2 doped into PMMA films at the loadings indicated (% by weight) and in neat film, at 295 K. The emission spectrum in CH2Cl2 at 295 K from Fig. 3 is also shown again here for comparison (black dashed line). | ||
It is pertinent to compare the behaviour of LCF3(Pt–SCN)2 in films both with (i) the chloro analogue LCF3(Pt–Cl)2 and (ii) the mononuclear Pt(CF3dpyb*)SCN investigated previously:
(i) In our previous study, we examined LCF3(Pt–Cl)2 in polystyrene (PS) films. Only a very small contribution from the low-energy band was observed at doping levels of 1 and 20% (Fig. S8, ESI†). Indeed, a neat film was required to reach a roughly 1:1 ratio of the short- and long-wavelength bands, yet such a ratio is almost attained even by 0.1% in the case of LCF3(Pt–SCN)2. As the different polymer used in that study (polystyrene as opposed to PMMA) might influence the behaviour, we have now also examined LCF3(Pt–Cl)2 in PMMA at 0.1, 1 and 10% loadings (Fig. S9, ESI†). A somewhat higher proportion of the lower-energy band is found in PMMA than in PS at corresponding concentrations. Nevertheless, the low-energy band is again proportionally much weaker for LCF3(Pt–Cl)2 than for LCF3(Pt–SCN)2 at a given concentration (e.g., a 1:1 ratio requires roughly 10% doping in PMMA for the chloro parent, almost 100× higher than for the thiocyanate derivative). The comparison highlights the profound influence of the change from X = Cl to SCN in favouring the formation of the low-energy-emitting excited states that span two (or more) Pt(NCN) units.
(ii) Although the neat films of both the dinuclear LCF3(Pt–SCN)2 and its mononuclear analogue Pt(CF3dpyb*)SCN both show only the long wavelength band, the band was further red-shifted in the mononuclear case to around λmax = 940 nm. This difference – albeit a relatively small one in energy terms (1300 cm−1 based on λmax values) – is consistent with a more significant contribution, in the mononuclear case, from the oligomeric species that were calculated to have slightly lower-lying excited states than dimers. The quantum yield of the neat film of LCF3(Pt–SCN)2 is measured at 0.05, superior to the value of 0.01 for Pt(CF3dpyb*)SCN under the same conditions, the difference likely being due, at least in part, to the faster non-radiative decay expected for the lower-energy excited states in the latter.
Finally, we note that the iodo derivative LCF3(Pt–I)2 does not show the behaviour of the thiocyanate complex in film. On the contrary, even in neat film, the emission spectrum is dominated by the higher-energy emission band from excited states associated with isolated Pt(NCN) units, with only a rather weak band at low-energy around 740 nm (Fig. S10, ESI†). Apparently then, the iodo ligands either hinder the formation of excited states spanning two Pt(NCN) units, or the subsequent emission from such states is suppressed through enhanced non-radiative decay, or indeed both. Close inspection of the low-energy edge of the absorption spectrum in the neat film does reveal a weak tail, implying some weak aggregation (Fig. S10, ESI†).
Concluding remarks
It is increasingly clear from the present and earlier studies that the emission properties of “tweezer-like” dinuclear platinum(II) complexes – comprising two rigidly connected Pt(NCN)X units – are complicated, owing to the possibility of both intramolecular and intermolecular interactions between the Pt(NCN)X units, both of which may occur either in the excited state or ground state or both. From the point of view of practical applications, however, it is the resulting emission range and efficiency under prevailing conditions that are important. The key conclusion drawn here is that the thiocyanate ligand promotes these interfacial interactions, enhancing low-energy emission from excited states that span two Pt(NCN)X units. Indeed, both in dilute solution at room temperature and in neat films, the spectra essentially feature only low-energy bands arising from such states, but their nature differs. Ground-state interactions are important in the neat films, leading to a further red shift of around 100 nm compared to solution. In contrast to thiocyanate, iodide ligands are found to inhibit the low-energy emission, leading to emission being dominated – both in solution and films – by excited states associated with isolated Pt(NCN)X units, similar to that displayed by the corresponding mononuclear complexes in solution. The results described are likely to provide insight and inspiration for the future design of NIR-emitting phosphors for diverse applications, including both doped and neat-film OLEDs.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for LCF3(Pt–SCN)2 have been deposited at the CCDC, no. 2370430.†Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank Dr Dmitry Yufit for determining the structure of LCF3(Pt–SCN)2 through X-ray diffraction and Dr Piotr Pander for helpful discussions. Part funding of PhD studentships to R. J. S. and Y. M. D. from Durham University Chemistry Department is gratefully acknowledged.References
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- A. Minotto, P. A. Haigh, L. G. Lukasiewicz, E. Lunedei, D. T. Gryko, I. Darwazeh and F. Cacialli, Light: Sci. Appl., 2020, 9, 70 CrossRef PubMed.
- (a) P. Liu, X. Mu, X.-D. Zhang and D. Ming, Bioconjugate Chem., 2020, 31, 260–275 CrossRef CAS PubMed; (b) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; (c) Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Coord. Chem. Rev., 2016, 310, 16–40 CrossRef CAS; (d) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bond., 2015, 165, 205–256 CrossRef CAS; (e) L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front, 2024, 8, 1731–1766 RSC.
- (a) R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS; (b) J. V. Caspar and T. J. Meyer, Inorg. Chem., 1983, 22, 2444–2453 CrossRef CAS.
- Y.-C. Wei, K.-H. Kuo, Y. Chi and P.-T. Chou, Acc. Chem. Res., 2023, 56, 689–699 CrossRef CAS PubMed.
- For a discussion of mechanisms of such quenching in hosts relevant to OLEDs, see: A. P. Green and A. R. Buckley, Phys. Chem. Chem. Phys., 2015, 17, 1435–1440 RSC.
- (a) P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS; (b) D. W. Kozhevnikov, V. N. Kozhevnikov, M. S. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed.
- R. J. Salthouse, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, Chem. Sci., 2022, 13, 13600–13610 RSC.
- P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- P. Pander, M. T. Walden, R. J. Salthouse, A. Sil, D. S. Yufit, F. B. Dias and J. A. G. Williams, J. Mater. Chem. C, 2023, 10, 15335–15346 RSC.
- (a) K. T. Ly, R. W. Chen-Cheng, H. W. Lin, Y. J. Shiau, S. H. Liu, P. T. Chou, C. S. Tsao, Y. C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63–68 CrossRef; (b) S. F. Wang, L.-W. Fu, Y.-C. Wei, S.-H. Liu, J.-A. Lin, G.-H. Lee, P.-T. Chou, J.-Z. Huang, C.-I. Wu, Y. Yuan, C.-S. Lee and Y. Chi, Inorg. Chem., 2019, 58, 13892–13901 CrossRef CAS PubMed; (c) G. Ni, J. Yan, Y. Wu, F. Zhou, P.-T. Chou and Y. Chi, Inorg. Chem. Front., 2023, 10, 1395–1401 RSC; (d) Ref. 6 provides a concise summary of the strategies employed in the work of Chi, Chou and co-workers.
- J. Kang, X. Zhang, H. Zhou, X. Gai, T. Jia, L. Xu, J. Zhang, Y. Li and J. Ni, Inorg. Chem., 2016, 55, 10208–10217 CrossRef CAS PubMed.
- Z. Wen, Y. Xu, X. Song, J. Miao, Y. Zhang, K. Li and C. Yang, Adv. Opt. Mater., 2013, 11, 2300201 CrossRef.
- (a) D. J. Cardenas, A. M. Echavarren and M. C. Ramirez de Arellano, Organometallics, 1999, 18, 3337–3341 CrossRef CAS; (b) J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed; (c) S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed; (d) D. L. Rochester, S. Develay, S. Zalis and J. A. G. Williams, Dalton Trans., 2009, 1728–1741 RSC; (e) E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- (a) M. Cocchi, J. Kalinowski, D. Virgili, V. Fattori, S. Develay and J. A. G. Williams, Appl. Phys. Lett., 2007, 90, 163508 CrossRef; (b) W. Mroz, C. Botta, U. Giovanella, E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore and J. A. G. Williams, J. Mater. Chem., 2011, 21, 8653–8661 RSC.
- (a) M. Cocchi, D. Virgili, V. Fattori, J. A. G. Williams and J. Kalinowski, Appl. Phys. Lett., 2007, 90, 023506 CrossRef; (b) M. Cocchi, J. Kalinowksi, V. Fattori, J. A. G. Williams and L. Murphy, Appl. Phys. Lett., 2009, 94, 073309 CrossRef.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501–15510 RSC.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, F. Demartin, M. Cocchi, P. Brulatti, V. Fattori and J. A. G. Williams, Chem. Commun., 2012, 48, 3182–3184 RSC.
- S. Develay and J. A. G. Williams, Dalton Trans., 2008, 4562–4564 RSC.
- (a) R. Munoz-Rodriguez, E. Bunuel, J. A. G. Williams and D. J. Cardenas, Chem. Commun., 2012, 48, 5980–5982 RSC; (b) Z. Guo, S. M. Yiu and M. C. W. Chan, Chem. – Eur. J., 2013, 19, 8937–8947 CrossRef CAS PubMed; (c) R. Okamura, T. Wada, K. Aikawa, T. Nagata and K. Tanaka, Inorg. Chem., 2004, 43, 7210–7217 CrossRef CAS PubMed.
- (a) H. Liu, L. Shen, Z. Cao and X. Li, Phys. Chem. Chem. Phys., 2014, 16, 16399–16406 RSC; (b) H. Tsujimoto, D. G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed; (c) M. Chen, Y. J. Bae, C. M. Mauck, A. Mandal, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 9184–9192 CrossRef CAS PubMed.
- N-bound NCS was confirmed by crystallography in ref. 19, and it was implicitly assumed in: A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. De Cola, Dalton Trans. 2015, 44, 8478–8487. An S-bound SCN was confirmed by crystallography in ref. 9.
- M. J. Coyer, R. H. Herber, J. Chen, M. Croft and S. P. Szu, Inorg. Chem. 1994, 33, 716–721 Search PubMed.
- (a) G. P. Mcquillan and I. A. Oxton, Dalton Trans., 1978, 1460–1464 RSC; (b) M. Kabesova and J. Gazo, Chem. Zvesti, 1980, 34, 800–841 CAS.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- (a) V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145 CrossRef CAS; (b) W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261–6265 CrossRef CAS; (c) W. Lu, M. C.-W. Chan, N. Y. Zhu, C. M. Che, C. N. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed; (d) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed; (e) E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- For an accessible recent review of the importance of MMLCT states in Pt(II) systems, see: M. Walesa-Chorab, J. Photochem. Photobiol. C, 2024, 59, 100664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterisation of new materials; X-ray diffraction and crystal data; details of photophysical instrumentation and sample preparation; additional absorption and emission spectra as referred to in the main text. CCDC 2370430. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj03357d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |