
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective fluoride sensing by a novel series of lanthanide-based one-dimensional coordination polymers through intramolecular proton transfer†
Vaibhav
Singh
 a,
Lakshmi Thachanadan
Suresh
a,
Jean-Pascal
Sutter
*b and
Arun Kumar
Bar
*a
a,
Lakshmi Thachanadan
Suresh
a,
Jean-Pascal
Sutter
*b and
Arun Kumar
Bar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Tirupati, Tirupati – 517507, Andhra Pradesh, India. E-mail: a.bar@iisertirupati.ac.in
bLaboratoire de Chimie de Coordination du CNRS (LCC-CNRS), Université de Toulouse, CNRS, Toulouse, France. E-mail: jean-pascal.sutter@lcc-toulouse.fr
First published on 21st March 2024
Abstract
A novel series of one-dimensional coordination polymers (CPs) is achieved via a facile one-pot synthesis strategy employing the nitrate salts of trivalent lanthanides, a pentadentate chelating ligand, and triphenylphosphine oxide at a controlled stoichiometry under ambient conditions. All the CPs are characterized comprehensively using spectroscopic, X-ray crystallographic and magnetometric studies. The CPs are found to be thermally stable up to a significantly high temperature and resistant to water for an indefinite time. They are photoactive and exhibit selective fluoride ion (F−) sensing with excellent efficiency both colorimetrically and fluorimetrically in the solid-state as well as in solution. The presence of F− concomitantly sensitizes the photoluminescence enhancement and visual decolourization of the CPs in solution owing to the ground-state intra-molecular proton transfer. The photophysical response of the CPs to F− in solution was found to be instantaneous (<30 s). The sensitivity of detection is observed to be significantly high over a wide range of F− concentrations, covering the beneficial and detrimental domains of F− concentrations in drinking water. The limit of detection (LoD) under ambient conditions was found to be in the micromolar (μM) range—the best being 0.22 μM found using UV-vis spectrometry and 7.5 μM using fluorimetry. In comparison, the USEPA standard cut-off for the upper limit of F− concentration in drinking water is 211 μM, and the LoD of measuring F− concentration using the USEPA standard method using a fluoride-selective electrode is 26.3 μM. The CPs display markedly high selectivity toward F− with negligible-to-no interference from the commonly abundant ions (Cl−, Br−, I−, CH3CO2−, CO32−, SO42−, HPO42−, NH4+, Na+, K+, Mg2+, and Ca2+) in terms of UV-vis spectral change. Moreover, they also exhibit solid-state IR-spectrometric sensitivity towards F− under ambient conditions.
Introduction
Fluoride ions (F−) exhibit well-documented dichotomous behaviour spanning over beneficial as well as detrimental domains.1 The beneficial domain includes dental health care,1g,j,k,2 osteoporosis therapy,3 medicinal cleaning agents, environmental pesticides,1c,g,h positron emission tomography,4 and material industries.1a,k Obviously, all of these useful applications have an upper limit, exceeding which will lead to detrimental effects, which include osteoporosis and related diseases,3 fluorosis,1g urolithiasis,5 neurological and metabolic disorders,1i,k gastric and kidney malfunctions,1k cancer,6 and low intelligent quotient in child.7 Notably, due to the small size and high charge density of F− and the high polarity of water molecules, there is an easy association of F− with water molecules through strong H-bonding interactions, thereby making water the most efficient carrier for F−.8 The penetration and accumulation of F− in different organs of the human body is way higher compared to other common anions, mainly, because of the smaller size and higher charge density of F−.1i The common sources of F− in the environment are chemical industry wastes, medical processes and discharge, degradation of nerve agents, corrosion of naturally occurring F− containing ores, etc.1a,9 Therefore, the detection of F− ions both in the solid-state and solution, especially in aqueous/protic solutions, is of utmost importance. Ironically, the beneficial-detrimental dichotomy is very narrowly spaced in several aspects of the human body. For example, as per the United States Environmental Protection Agency (USEPA), the upper cut-off for the F− concentration in drinking water stands at 211 μM to safeguard osteofluorosis and related diseases, while the requisite fluoride concentration is 105 μM for better dental health.1d,2a,10 Therefore, the efficient and accurate detection of F− is extremely important. Moreover, there are plenty of other anions that are required for a healthy human body at a relatively higher concentration level than F−.11 Therefore, selective F− sensing with negligible-to-no interference from commonly abundant ions is equally important. In view of such a crucial analytical requirement, great advancements have already been made broadly in two categories – one being the physical analytical methods and the other being the chemosensors. The most commonly used physical analytical methods for F− detection are 19F nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry, atomic absorption spectrophotometry (AAS), standard Willard and Winter methods, F− selective electrodes, ion chromatography, etc.12However, the essence of developing efficient chemosensors originates from various limitations and inconveniences pertaining to these physical methods. For example, high cost, complicated and time-consuming procedures, incompatibility in real-time onsite analysis, highly skilled operators and vulnerability of such high-end equipment to the environments are among the major concerns. On the other hand, chemosensors could address most of the aforementioned disadvantages and challenges. The desired characteristics for a potential F− chemosensor include high performance in aqueous/alcoholic media, high selectivity towards F− with negligible-to-no interference from other common anions, and a detection limit covering the deficiency. High efficiency, selectivity and application-wise suitability for the chemosensors to be portable for the applications are still the areas under advancement. Nevertheless, several optical F− sensors have been developed that exhibit fluorescent and colorimetric responses upon interactions with F− through different ways. The chemosensors that act as Lewis acids, hydrogen bond donors, chemodosimeters and through-space charge transfer are the most prominent ones.1a,f,9a,13 It is worth pointing out that polymeric chemosensors are proven to be better chemosensors than mono/oligomeric analogues due to the better cooperativity for the formers. Several biomolecular and supramolecular systems are found to be promising in this regard.14 The advantages of inorganic coordination polymers (CPs) over organic/bio-organic polymers lie in the easy synthesis, high purity and better tunability with various functionalities. Lanthanide (Ln) ions-based coordination complexes possess huge prospects in this regard. It is mainly because they are generally optically active, they exhibit narrow absorbance and sharp emission that are essential for high-precision sensing and imaging, their optical activity can be greatly tailored by antenna effects, and Ln(III) ions have extremely high affinity towards F− owing to favourable hard-acid–hard-base interactions.11,15 Moreover, most of the Ln ions inherit strong unquenched orbital angular momentum that results in high magnetic anisotropy via spin–orbit coupling.16 Consequently, Ln-based coordination complexes can also be equipped with large dielectric constant and large magnetization ground states to facilitate high-resolution sensing and imaging.11a,13a,17 Hierarchical lanthanide-based discrete molecular complexes are explored towards various anion sensing, including F−.11,17,18 However, lanthanide-based polymeric systems with the desired functionality and topology are less known primarily due to the difficulty in controlling the coordination geometry around the Ln ions. One requires aesthetic synthetic strategies to equip the Ln-complexes with desired topology, dimensionality and functionality. Nevertheless, several metal–organic frameworks (MOF's) incorporating lanthanides and mixed-lanthanides are reported to demonstrate promises in the arena of anion and small molecule sensing and imaging.19 To the best of our knowledge, the Ln-based one-dimensional (1D) CPs exhibiting selective fluoride sensing are rare.
Herein we report syntheses, characterization, chemosensing behaviour and the magnetic property of a series of novel, air stable trivalent lanthanide-based 1D CPs, [(κ5,μ2-H2L)Ln(TPPO)(κ2-NO3)]n (Ln = Y (1·Y); Gd (1·Gd); and Dy (1·Dy)), employing a rigid Schiff base ligand with κ5-N3O2 ligating sites, monodentate TPPO and κ2-NO3 – in conjunction with Ln(III) ions (Scheme 1, where H4L = N′,N′′′-((1E,1′E)-pyridine-2,6-diylbis(ethan-1-yl-1-ylidene))bis(2-hydroxybenzohydrazide), TPPO = triphenylphosphine oxide). Similar reactions in the absence of TPPO are known to result in discrete molecular complexes.20 The ancillary TPPO ligand renders the stability of 1D CPs. Though the CPs could be achieved in the presence of TPPO with lower equivalence, the optimum yield is found to be in the presence of approximately 2 equivalents of TPPO. The presence of a large excess of TPPO leads to the formation of mononuclear complexes, [(κ5-H2L)Ln(TPPO)2(κ2-NO3)] (Ln = Y (2·Y); Gd (2·Gd); and Dy (2·Dy)), alongside the formation of the 1D CPs, 1·Ln (Scheme 1, see the Experimental section for details). Though we could not isolate the mononuclear complexes, 2·Ln, in pure forms in their bulks, herein, we report their single-crystal X-ray diffraction structures. However, all the complexes were synthesized under aerobic conditions. The 1D CPs were characterized comprehensively by spectroscopic and X-ray crystallographic techniques both at their molecular levels and in bulks. Magnetic property investigations were carried out on the polycrystalline solid samples of 1·Ln (Ln = Gd and Dy). We also report herein their colorimetric and fluorimetric chemosensing behaviour.
 | ||
| Scheme 1 Schematic representation for the syntheses of the 1D coordination polymers, 1·Ln, and the mononuclear complexes, 2·Ln (Ln = Y, Gd and Dy). | ||
Experimental section
Materials and methods
All the reagents and solvents used for the syntheses were used as received from the commercial suppliers. The Schiff base ligand, H4L, was synthesized following the reported procedure.21 Triphenylphosphine oxide (TPPO) was synthesized starting from triphenylphosphine following the reported procedure.22 Syntheses and manipulations of samples for all the experiments were carried out under aerobic conditions. All the spectroscopic studies were carried out under ambient conditions. UV-vis spectral studies were carried out using μ(M) methanol solutions of the complexes and an Agilent Hewlett-Packard 8453 diode array UV-vis spectrometer. 1H and 31P NMR spectral studies were carried out on the Y analogues of the complexes in deuterated solvents (CDCl3/CD3OD) for 1·Y using a Bruker model Ascend 400 FT-NMR spectrometer. Fourier transform infrared (FT-IR) spectroscopic studies were performed on the thin layers of neat samples with a Bruker-Alpha Eco-ATR FT-IR spectrometer.X-ray crystallographic studies
Suitable single crystals for X-ray diffraction were coated with paratone oil and mounted onto the goniometer. The X-ray crystallographic data were obtained from Bruker (D8 Advance, Da Vinci) diffractometer using the Mo-Kα radiation source and equipped with an Oxford Cryo-system. The structures were solved by direct methods using SIR92 or Superflip and refined by means of least-square procedures on F using the PC version of the program Olex2. The scattering factors for all the atoms were used as listed in the International Tables for X-ray crystallography.23 Absorption correction was performed using a multi-scan procedure. The H atoms were repositioned geometrically. The H atoms were initially refined with soft restraints on the bond lengths and angles to regularize their geometry and U ∼ iso ∼ (H) (in the range 1.2–1.5 times U ∼ eq ∼ of the parent atom), after which the positions were refined with riding constraints. All non-hydrogen atoms were refined anisotropically. Crystallographic data and refinement parameters for the single-crystal X-ray data analyses for all the complexes are summarized in Table S2.† The selected bond parameters are provided in Table S3.† CIF has been deposited at CCDC (https://www.ccdc.cam.ac.uk) with references 2311694–2311699† for the complexes 1·Y, 1·Dy, 1·Gd, 2·Dy, 2·Gd and 2·Y, respectively. The solid-state phase purity of the isolated polycrystalline solid samples of the complexes was confirmed by powder X-ray diffraction (PXRD) using a Rigaku (SmartLab) diffractometer with Cu(Kα) radiation source radiation, λ = 1.5406 Å. The PXRD studies were carried out with 2θ ranging from 5 to 50° with a step size of 0.01.Magnetic property measurements
Magnetic measurements for all the samples were carried out using a Quantum Design MPMS 5S SQUID magnetometer in the temperature range of 2–300 K. The measurements were performed on polycrystalline samples. The freshly isolated crystalline powders of the complexes were mixed with grease (except for the Gd complex) and placed in gelatin capsules. The temperature dependences of the magnetization were measured in an applied field of 1 kOe and the isothermal field dependence of the magnetizations was collected up to 5 T. The molar susceptibility (χM) was corrected for the sample holder, grease and the diamagnetic contribution of all the atoms by using Pascal's tables.24 Magnetic data for 1·Gd were fitted using the spin Hamiltonians as coded in the PHI software.25 AC susceptibility was collected using an AC field of 3 Oe.Computational studies
Single point energy and gas-phase energy optimization were performed using DFT on GAUSSIAN-09 platform26 utilizing hybrid B3LYP basis functions27 with mixed basis sets: 6-311G*28 for all elements except for Y, and the small-core Hay–Wadt pseudopotential (indicated in the Gaussian code as LANL2DZ)29 for Y. The crystallographic coordinates of the repeat unit of 1·Y were used as it is for the truncation to obtain the model complex NaH2LY, except that the neighbouring Y ions were replaced with Na+ ion. The UV-vis spectroscopic calculations were performed using TD-DFT on the GAUSSIAN-09 platform utilizing hybrid B3LYP basis functions with mixed basis sets: 6-311G* for all the elements. The isosurface maps were plotted with isovalue 0.02 using GassView4.1 software and are presented in the 4 × 10−4μB Å3 isosurface.Synthesis procedure
1·Y: Isolated yield 65% (128 mg). UV-Vis: λmax = 349 nm, ε = 3.86 × 104 M−1 cm−1; λmax = 396 nm, ε = 1.97 × 104 M−1 cm−1. NMR (CDCl3/CD3OD (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v)): δ ppm: 1H: 2.49 (s, 6H, –CH3) 6.90–7.00 (m, 4H, aromatic-CH) 7.37–7.43 (m, 8H, aromatic –CH) 7.46–7.58 (m, 9H, aromatic) 7.67–7.74 (m, 2H, aromatic –CH) 8.04–8.10 (t, 1H, aromatic –CH). 8.15–8.21 (d, 2H, aromatic –CH). 31P: 36.62 IR (cm−1): υC
:1 v/v)): δ ppm: 1H: 2.49 (s, 6H, –CH3) 6.90–7.00 (m, 4H, aromatic-CH) 7.37–7.43 (m, 8H, aromatic –CH) 7.46–7.58 (m, 9H, aromatic) 7.67–7.74 (m, 2H, aromatic –CH) 8.04–8.10 (t, 1H, aromatic –CH). 8.15–8.21 (d, 2H, aromatic –CH). 31P: 36.62 IR (cm−1): υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1557(w), 1542(m); υCN = 1612(w), 1591(m). Elemental analysis (%) calculated for C41H34YN6O8P (FW = 858.62 g mol−1): C 57.35; H 3.99; N 9.79; observed: C, 57.27; 4.03; N, 9.57.
O = 1557(w), 1542(m); υCN = 1612(w), 1591(m). Elemental analysis (%) calculated for C41H34YN6O8P (FW = 858.62 g mol−1): C 57.35; H 3.99; N 9.79; observed: C, 57.27; 4.03; N, 9.57.
1·Gd: Isolated yield 70% (148 mg). UV-Vis: λmax = 347 nm; ε = 3.61 × 104 M−1 cm−1; λmax = 397 nm, ε = 1.82 × 104 M−1 cm−1 IR (cm−1): υCO = 1556(w); υCN = 1612(w), 1588(m). Elemental analysis (%) calculated for C41H34GdN6O8P (FW = 926.96 g mol−1): C 53.12; H 3.70; N 9.07; observed: C, 53.27; H, 3.76; N, 8.99.
1·Dy: Isolated yield 60% (130 mg). UV-Vis: λmax = 349 nm; ε = 3.34 × 104 M−1 cm−1. IR (cm−1): υCO = 1555(w); υCN = 1612(w), 1590(m) Elemental analysis (%) calculated for C41H34DyN6O8P (FW = 932.21 g mol−1): C 52.82; H 3.68; N 9.02; observed: C, 52.97; H, 3.72; N, 8.97.
Results and discussion
Syntheses, spectroscopic characterization and physical properties
The complexes were synthesized by refluxing the ligand (H4L) with corresponding hydrated Ln(III) nitrate salt in 1:1 molar ratio under vigorous stirring conditions in ethanol medium followed by treatment with two equivalents of triethylamine as the base at room temperature (Scheme 1, see the Experimental section for details). Upon addition of a white slurry of H4L in ethanol into the transparent solution of lanthanide salt under stirring conditions at room temperature, the reaction mixture turned immediately into dark yellow with concomitant consumption of the white suspended particles of H4L. A sharp change from white slurry to dark yellow during the reactions indicated the complexation of the ligands with the metal ions. The whole reaction mixture appeared translucent orange solution upon refluxing for an hour. The colour of the reaction solution darkened further upon the addition of two equivalents of ethanolic solution (9:1 v/v EtOH/Et3N) of Et3N at room temperature. The further colour change could be ascribed to the deprotonation of the amide N of the ligands upon the addition of the base. Solid TPPO (2 equivalents) was added in a portion into the reaction mixture under stirring conditions at room temperature after 15 minutes of the base addition. The colour of the whole reaction solution faded into light yellow and fluorescent lemon-yellow crystalline precipitate started to appear within a few minutes – implying ligation of TPPO molecules and the formation of the CPs. The precipitate was isolated by filtration followed by washing with diethyl ether, consequently, aerial drying to obtain the products as lemon-yellow solids with excellent isolated yields (60–70%). A distinct change from a white slurry to a dark yellow solution during the progress of the reactions indicated the complexation of the ligands with the metal ions. The ligation of the TPPO molecules and the formation of the coordination polymers were indicated by the change in colour from dark yellow to light yellow, and the precipitations of the lemon-yellow solids. Though the 1D CPs were isolated from the reactions in the presence of TPPO with less equivalence, the yield was found to be optimum for the reactions with 2 equivalents of TPPO. The absorbance in the visible range with the λmax near 400 nm results in the exhibition of the characteristic yellow colour of the complexes (Fig. S1, see ESI†). The solid-state infra-red (IR) spectroscopic studies displayed a significant decrease in carbonyl stretching frequencies and an increase in imine stretching frequencies in the complexes compared to the free Schiff base ligands (Table S1 and Fig. S2, see ESI†). The increment of the imine stretching frequencies directly implies coordination of the ligands through imine nitrogen centres. However, electronic resonance between the amide nitrogen and the carbonyl group is expected to enhance the deprotonated form of the ligands, thereby, decreasing the bond order of the carbonyl group significantly. Consequently, the carbonyl stretching frequencies of the complexes are expected to decrease in spite of the coordination through the carbonyl oxygen.30 The room temperature solution 1H NMR spectrum of 1·Y confirmed the TPPO and the Schiff base ligand ratio to be 1:1 (Fig. S3, see ESI†). The 31P NMR spectra of 1·Y displayed downfield chemical shifts (δ = 36.62 ppm, sharp in CD3OD/CDCl3) compared to the free TPPO ligand (δ = 35.67 ppm in CD3OD/CDCl3), thereby indicating the ligation of TPPO to the metal centres (Fig. S4, see ESI†). The appearance of a sharp singlet 31P signal indicates the presence of a single product in the solution. Elemental analyses (see the Experimental section) on the polycrystalline solid samples of the complexes revealed the chemical compositions as described in Scheme 1. The X-ray diffraction quality single crystals were grown from the slow evaporation of the CHCl3 solutions of the complexes at ambient conditions. Single crystal X-ray diffraction analyses unambiguously confirmed the formation of the molecular complexes for both the 1D CPs, 1·Ln, and the mononuclear complexes, 2·Ln. The powder X-ray diffraction analyses on the isolated polycrystalline samples of the 1D CPs confirmed their phase purity in the solid-state bulk (Fig. S5, see ESI†). The variable temperature solid-state FT-IR spectral analyses revealed high thermal stability of the CPs (Fig. S6, see ESI†). All these complexes are significantly soluble in common alcohols and chlorinated solvents, but sparingly soluble in water. However, the solubility of these complexes is fairly high in aqueous alcoholic media. Notably, the time-dependent FT-IR analyses on the polycrystalline solid samples of the polymers soaked under water at room temperature indicated that these polymers are indefinitely waterproof (Fig. S7, see ESI†).
X-ray crystallographic characterization
The analyses of the single crystal X-ray diffraction data revealed the molecular structures of both the 1D CPs, 1·Ln, and the mononuclear complexes, 2·Ln. In the cases of the formers, the building units consist of one nonacoordinate Ln(III) metal centre, one doubly deprotonated organic ligand (H2L2−) with κ5 binding mode, one monodentate TPPO and one κ2 NO3− (Fig. 1). Two neighbouring building blocks are tethered with one deprotonated phenolic hydroxyl group of the organic ligand establishing the 1D coordination backbone, as shown in Scheme 1. On the other hand, a mononuclear complex is composed of a nonacoordinate Ln(III) centre incorporating a κ5 H2L2−, two monodentate TPPO, and a κ2 NO3− (Scheme 1). The structure of the mononuclear complexes could be realized upon replacing the tethering phenolic hydroxyl group of the ligand in the repeat units of the 1D CPs with a TPPO molecule – keeping the topology and environment of the inner coordination sphere pretty much similar. The solid-state molecular structures for the Dy analogues (1·Dy and 2·Dy) are portrayed in Fig. 1 as the representative structures for their respective series. The molecular structures for the rest of the complexes are displayed in ESI (Fig. S8–S13, see ESI†). | ||
| Fig. 1 The ball-and-stick models of the solid-state single-crystal X-ray molecular structures of the 1·Dy (top, displayed up to three consecutive repeat units) and 2·Dy (bottom). The coordinating atoms are labelled. Color codes: cyan, Dy; orange, P; red, O; blue, N; grey, C. The H atoms, co-crystallized solvent molecules, and the lower occupancy C atoms of the phenyl rings that are split due to thermal disorder in 1·Dy are omitted for clarity. | ||
All the complexes of the series 1·Ln crystallize in a monoclinic crystal system with a P21/n space group incorporating four repeat units per unit cell, while all the mononuclear analogues, 2·Ln, crystallize in a monoclinic crystal system with a P21/c space group (Fig. S14–19†). Crystallographic data and refinement parameters for the complexes are provided in Table S2 (see ESI†). The selected bond lengths and bond angles associated with the coordination environments of the complexes are summarized in Table S3.† All the Ln complexes in their respective series are isostructural and isomorphic. Therefore, herein, the solid-state molecular structures are described only for the Dy analogues as their representative structures. A molecule of the ligand chelates the Ln ion through two carbonyl oxygen (O1 and O2), two imine nitrogen (N2 and N4) and the pyridyl nitrogen (N3) atoms completing its N3O2 pentacoordination leading to a pseudo pentagonal basal plane around the Ln centre, as shown in Fig. 1. In 1·Ln the central [N3O2] cavity is singly deprotonated. The hydrazide N–N–C(O) angles found one above and one below the 111° threshold angle – distinguishing the protonated hydrazide group from the deprotonated one.30 In 2.Ln, both N–N–C(O) angles are very close to the threshold (in the range of 110.5–111.5°) – supporting the deprotonation of each hydrazide moiety. Interestingly, the [N3O2] coordinating sites are almost coplanar – dihedral angles between the planes of pyridine moiety and the amide functional groups of the ligand ranging from 0° to 8° for 1·Ln, and from 8° to 10° for 2·Ln. However, the Ln centre is significantly out of the N2–N3–N4 planes (0.651 Å for 1·Dy, and 0.623 Å for 2·Dy). Such an out-of-planarity of the Ln centres renders the basal ligand resemble umbrella-shaped geometry where a TPPO molecule coordinates the metal centre from the concave side and a NO3− anion chelates the metal ions from the convex side. Therefore, the coordination sites of the metal centre sterically open up to accommodate further ancillary ligand(s) from the convex side, facilitating the formation of 1D CPs upon binding another building-block through the phenolic hydroxyl group (through O3), or the formation of the mononuclear complex upon coordinating with another TPPO molecule (through O9). Apparently, the formation of the 1D CPs is kinetically controlled. They precipitate out of the reaction medium once formed due to low solubility. The solid-state phase purity for all the 1D CPs was confirmed by powder X-ray diffraction (PXRD) analyses on the polycrystalline solid samples. Excellent agreement between the experimental and simulated PXRD patterns revealed the phase purity of the isolated products (Fig. S5, see ESI†).
Magnetic property
The temperature dependence of the molar magnetic susceptibilities, χM, for 1·Ln (Ln = Gd and Dy) were investigated over the temperature range 2–300 K. The results are plotted as χMT vs. T in Fig. 2. The experimentally observed χMT values (cm3 mol−1 K) at 300 K are found to be 7.88 (1·Gd) and 13.73 (1·Dy) – agreeing well with the theoretical values considering the magnetically isolated Ln(III) ion (7.88 for Gd with S = 7/2, giso = 2; and 14.17 for Dy with J = 15/2, gJ = 4/3). Upon lowering the temperature, the χMT values hardly change for 1·Gd, reaching 7.71 cm3 mol−1 K for 2 K. Such a behaviour agrees with mainly a Curie behaviour. The isothermal field dependence of the magnetizations for 1·Gd reached 7.0 μB at 2 K and 50 kOe (Fig. 2). Modelling of this behaviour was performed for an S = 7/2 spin and possible inter-Gd exchange interactions were considered within the mean-field approximation (zJ parameter). The simultaneous analyses of χMT vs. T and M vs. H at 2, 3, and 4 K gave best-fit parameters g = 1.999 ± 0.001 and zJ = −0.0036 ± 0.0007 cm−1, confirming practically the absence of the exchange interaction between the Ln enters within the 1D arrangements. For 1·Dy the χMT vs. T behaviour is characterized by a steady decrease with lower T (Fig. 2), to reach 9.65 cm3 mol−1 K at 2 K. Such a decrease can be ascribed to the crystal-field effects and the subsequent depopulation of the magnetic states upon lowering the temperature.31 Field dependence of the magnetization at 2, 3, and 4 K reaches 5.1 μB (Fig. S20†), close to 5 μB expected for a ground state with a pure MJ = 15/2 term. However, it can be noticed that after the rapid increase of M for low fields (<10 kOe), a smooth augmentation was observed with increasing H, this suggests close-lying excited states or admixed terms. Notably, it is well-documented that Ln complexes can exhibit magnetization blocking below a critical temperature giving rise to slow relaxation of magnetization – widely known as single-molecule magnet (SMM) behaviour.32 The SMM behaviour is closely related to the magnetic anisotropy exhibited by certain Ln ions, especially Dy(III) ion that exhibits fascinating SMM behaviour once stabilized under appropriate coordination geometries and crystal fields.16a,33 We have also reported previously that similar coordination environments around the Dy(III) ion similar to those found in 1·Ln and 2·Ln induce SMM behavior.34 In order to have an insight into a possible magnetization blocking and slow relaxation of the magnetization, AC susceptibility was measured for 1·Dy both in the absence of and under the applied DC magnetic fields (Fig. S21†). Nothing or just the onset of an out-of-phase AC susceptibility (χ′′) components was observed above 2 K, ruling out any SMM-type behaviour. | ||
| Fig. 2 The experimental χMT vs. T plots for 1·Gd (blue open circles) and 1·Dy (black open circles); and the M vs. H plots (inset) for 1·Gd along with the best fits (red solid lines). The best-fit parameters are discussed in the text. | ||
Chemosensing behaviour
As mentioned above, there has been noticeable progress with hierarchical Ln-based discrete molecular complexes as the chemosensors for various anions including F−. However, F− sensing by Ln-based polymeric systems is rare perhaps due to the synthetic difficulties in controlling the coordination geometry and structural dimensionality. Zeng, X. et al. employed a series of mixed Ln(III)-based metalorganic frameworks (MOFs) as the ratiometric chemosensors for the visual detection of F− in water with the detection limits in the range of ppb levels.35 Notably, rational synthesis for the mixed ratiometric Ln complexes possesses synthetic challenges. Zhou, J.-M. et al. reported Eu(III) and Tb(III) based two isostructural MOFs that exhibit efficient sensing of F− and various organic small molecules including common alcohols via fluorescence quenching.19c However, potential chemosensors with high efficiency and selectivity for F− are yet to appear for suitable and convenient applications. The 1D CPs reported herein (1·Ln; Ln = Y, Gd and Dy) are extremely facile to synthesize and isolate in pure forms with excellent yields. They are stable under ambient conditions both in solid states and in solutions. They are waterproof and resistant to heat up to significantly high temperatures (>300 °C, Fig. S6, see ESI†). They exhibit instantaneous real-time response in the presence of F− at a concentration level as low as nanomolar (nM), which is way lower than the upper cut-off for the F− concentration in drinking water. They can sense F− in solutions of common alcohols, chlorinated solvents, and in aqueous media as well as in the solid-state. Their use as sensors appears to be highly economical as they exhibit large molar extinction coefficients (in the range of 16000–20000 M−1 cm−1) for the absorptions in the visible domain (λmax ∼ 400 nm). Thus, a micromolar solution of the complexes suffices the detection of F−. All of these characteristics of 1·Ln prompted us to investigate their detailed sensing behaviour and the mechanistic aspects.
The characteristic f–f transitions corresponding to the ground states of Gd(III) (8S7/2 → 6P5/2, 6P7/2; 308 nm) and Dy(III) (6H15/2 → 4F9/2, 475, 570, 660 and 750 nm; 6H15/2 → 4I9/2, 455, 540, 615 and 695 nm) free ions in the UV-vis range are well known to be very weak and sharp, while the Y(III) is a diamagnetic ion and its ground state transitions lie beyond the UV-vis range.36 Though the ligand employed herein (H4L) is sparingly soluble in common organic solvents in its pristine form, all the complexes (1·Ln and 2·Ln) are fairly soluble in most of the common organic solvents including alcohols.
The room temperature UV-vis spectrum of 1·Ln in aqueous methanol exhibits five distinct bands with absorption maxima (λabs.max; nm) at ∼401, 351, 315, 255, 220 and 204 having full widths at the half-maxima of ∼52, 37, 42, 50, 25 and 16 nm, respectively (Fig. S1, see ESI†). They are also characterized by markedly high molar extinction coefficients in the range of 1.66 × 104–9.21 × 104 M−1 cm−1. Notably, various heavy element chemical analytes strongly absorb below 250 nm in the UV-vis spectral domain.37 Thus, detailed UV-vis spectrometric studies were restricted above 250 nm. However, all the 1D CPs fairly fluoresce. The excitation (PL) spectra for the methanolic solution of 1·Dy reveal the exhibition of fluorescence over a wide range of wavelengths (200–500 nm, Fig. S22, see ESI†). Though the maximum photon counts were observed at the excitation of 250 nm, the maximum change in photon counts upon titration with F− was observed at the excitation of 349 nm (Fig. S23 and S24, see ESI†). Thus, the latter excitation wavelength was the obvious choice for the detailed PL spectral analyses. The room temperature steady-state PL spectra of the methanolic solutions of 1·Ln exhibit three distinct emission maxima (λabs.max; nm) at ∼435, 412 and 388 (Fig. S25, see ESI†). The investigation for the time-dependent spectral changes for 1·Ln revealed almost instantaneous (<30 seconds) responses and rapid virtual saturation (within ∼2 minutes for fluorimetry and ∼10 minutes for UV-vis spectrometry (Fig. 3(i) and S26, S27, see ESI†)) after the additional treatment with NH4F. All of the 1D CPs exhibited similar spectral profiles and similar sensing behaviour – implying the photophysical and sensing behaviours of these complexes are more of the ligand characteristics than the metalions. Therefore, the systematic studies are detailed for the Dy analogue. The Y and Gd analogues were studied for comparative and conclusive investigations. It is worth mentioning that the spectral dynamics at different absorption and emission maxima at different concentration domains of F−are found to be non-linear. The UV-Vis spectroscopic studies for 1·Dy displayed that the absorptions near λabs.max (nm) = 349 and 397 diminish, and near 322 and 280 enhance upon titration with F− (Fig. 3(ii)). The spectral change was prominent up to around 500 equivalents of F−. The relative magnitude of spectral changes at different absorption and emission maxima for 1·Dy upon treatment with F− were investigated by both PL and UV-vis, spectrometrically. The best responses were observed near λabs.max = 280 nm and λemi.max = 435 nm in the UV-vis and emission spectra, respectively (Fig. 3(iii)). However, the visual yellow colour of the solution of 1·Dy turns colourless upon treatment with <100 equivalents of F− with respect to the Dy ion in 1·Dy (Fig. 3(v)). On the other hand, the photon counts are found to increase across the PL spectrum for 1·Dy upon treatment with F−, and the spectral change was prominent up to around 50 equivalents of F− (Fig. S28, see ESI†). In order to determine the standard calibration curves and the limit of detections (LoD), detailed photoluminescence (PL) and UV-vis spectrometric readings were monitored. The micromolar (μM; with respect to Ln centre in the 1D CPs) stock solutions in methanol were titrated with millimolar (mM) aqueous methanolic (1:1 v/v) solution of NH4F such that the 1D CPs are at 12.5 μM strengths, and the concentrations of F− are varied between 0–1250 nM in the study-solutions, which correspond to the fluoride–lanthanide ion ratios (F:Ln) varying from 0 to 0.1 (see the Experimental section for details). Delay times of 2 min and 10 min were maintained between the addition of NH4F solution into the complex solution and the measurement of the spectral responses for PL and UV-vis spectrometric studies, respectively. The relative response ratios, X0 − Xc/X0, were plotted against the F− concentration (c), where X0 and Xc stand for the response reads (X = photon counts, I, for PL, and absorbances, A, for the UV-vis spectral analyses) of the complexes in the absence of F− and in the presence of F− with concentration c to obtain the calibration curves (Fig. S28–S31, see ESI†). Both the PL and UV-vis spectrometric calibration curves for all the 1D CPs displayed detectable responses with F− concentration <50 nM, which corresponds to <0.005 equivalents of F− per Ln centre of the 1D CPs. Notably, magnitudes of responses are found to be non-monotonous across the concentration domains of F− even for a particular absorption or emission maximum of a particular Ln analogue – implying different sensitivity and limit of detection (LoD) towards F− at different domains of its concentrations. The standard LoD was calculated following the 3σ/m formalism where m stands for the slope of the linear regime of a calibration curve, and σ stands for the standard deviation of blank samples. The detailed LoDs are tabulated in the ESI (Tables S4–S7†). The standard LoDs determined in the nM concentration domains of F− under ambient conditions are found to be in the micromolar range – the best being at λemi.max = 388 nm (7.5 μM for 1·Y, 14.8 μM for 1·Gd and 16.1 μM for 1·Dy) via fluorimetry, and at λabs.max = 280 nm (0.22 μM for 1·Y, 2.28 μM for 1·Gd and 2.3 μM for 1·Dy) via UV-vis spectrometry, which is 10 to 1000 times lower than the USEPA standard cut-off for the upper limit of F− concentration (211 μM) in drinking water, and comparable to 100 times more sensitive than the USEPA standard method of measuring of F− concentration (26.3 μM) using a fluoride-selective electrode. Comparatively, the Gd analogue is found to be the best candidate through the PL probe, while the Y analogue is the best candidate through the UV-Vis probe (Fig. S32, see ESI†). In view of the practical applications, a sensor must exhibit a no-to-negligible response to the other commonly abundant anions (such as Cl−, Br−, I−, CO32−, CH3CO3−, NO3−, SO42− and HPO42−) and cations (such as NH4+, Na+, K+, Ca2+ and Mg2+). The fluorescence responses of the complexes were investigated upon treatment with ammonium salts of the aforementioned anions and the NO3− salts of the aforementioned cations. Interestingly, the change in the UV-vis absorbance is negligibly small for all other common ions compared to F−. The change in fluorescence is also found to be negligibly small for all other common ions except for SO42− and HPO42− compared to the F− (Fig. 3 and S33, see ESI†).
 | ||
| Fig. 3 (i): The time-dependent relative UV-vis absorption changes for the 12.5 μM (with respect to Ln center) solutions of 1·Dy (green), 1·Gd (red) and 1·Y (blue) at λabs.max (nm) = 277 (stars), 320 (up-triangles), 348 (circles), and 402 (squares) upon treatment with 25 equivalents of NH4F. The solid lines are for better visualisation only. (ii): The gradual change in UV-vis spectra of 12.5 μM solution of 1·Dy upon titration with NH4F solution between 0 and 500 equivalents at room temperature. (iii): The change in absorbances (|A − A0|, yellow bars) at λabs.max (nm) = 277, 320, 348, and 402 in the UV-vis spectra, and the change in the photon counts (|I − I0|, green bars) at λemi.max (nm) = 435, 412, and 388 in the steady-state PL spectra of 2 mL of 12.5 μM methanolic solutions of 1·Dy upon treatment with NH4F (30 s after the addition of 5 equivalents for PL, and 30 min after the addition of 25 equivalents for UV-vis) at the room temperature. (iv): The absorbance changes of the 12.5 μM solution of 1·Dy upon treatment with 100 equivalents of different ions as indicated in the abscissa at an absorption maximum of 397 nm. (v): The visual colour of the 500 μM methanolic solution of 1·Dy alongside the colour of the same 500 μM methanolic solutions of 1·Dy upon treatment with 100 equivalents of different ions including F−. A0(I0) corresponds to the readings in the absence of F− ion, while A(I) corresponds to the readings after treatment with the analyte. See the Experimental section for more details. | ||
A significant blue shift in the UV-vis spectra of the complexes in the presence of F− compared to their pristine forms in aqueous methanolic solutions indicates the static (via the sensor-analyte complexation) sensing over the dynamic (via senor-analyte collisions) sensing. One of the most intriguing aspects is the concomitant fluorescence enhancement and the visual decolorization of the complexes upon treatment with F−. The as-synthesized sample of H4L exists as an off-white solid at STP, and it is sparingly soluble in most of the common solvents at ambient conditions. Obviously, such a Schiff base ligand molecule can assume various intramolecular H-bond driven rotamers and keto-iminol tautomers which are separated by low-lying, thermally accessible energy barriers. A set of such obvious rotamers and tautomers (c1–c8) are portrayed in Scheme S1 (see ESI†). The dominant rotamers/tautomers, and consequently, the steady-state spectroscopic behaviour (especially the UV-vis absorbance, photo-luminescence and FT-IR stretching frequencies) of the ligand molecules are dictated by the media of their presence and the other chemical associates. The keto form (one or all of c1, c2, c5, c6; Scheme S1, see ESI†) of the ligand molecules is well-known to dominate over the iminol form (one or all of c3, c4, c7, c8; Scheme S1, see ESI†) in the solid-states. It is evident from the solid-state FT-IR spectrum of the as-synthesized solid sample that it exhibits sharp and strong characteristic amide carbonyl stretching frequencies (νCO = 1640 cm−1). Notably, an H4L molecule consists of three aromatic moieties – one pyridine-imine moiety and two benzoic moieties, tethered together by the amide N atoms. The amide N atoms are at the sp2 hybridized state in iminol forms where the aromatic moieties are unified through extended π-conjugation. On the other hand, the amide N atoms are at sp3 hybridized state in keto forms, keeping all the aromatic moieties independent with respect to π-conjugation. Therefore, the absorption maxima are expected to appear at higher wavelengths for the iminol forms compared to their keto congeners. Considering the intramolecular H-bonding interactions and charge density distribution, the ligand molecules are shifted to the iminolate forms upon deprotonation, which is regularly observed for such ligands by displaying intense coloration upon deprotonation and complexation with the metal ions.34,38 Notably, the chelate effects render the ligand molecules into all cis conformations (c5–c8; Scheme S1, see ESI†), especially for the mononuclear complexes. To be specific, considering the reactions described herein, treatment of H4L with Ln(NO3)3 in the presence of two equivalents of Et3N as a base could lead to the formation of mononuclear building blocks (c9–c11), as depicted in the Scheme S2 (see ESI†) notwithstanding the coordinated neutral ligands/solvents for clarity. The doubly deprotonated form of the ligand molecule, H2L2−, could assume one of the following three states in the building blocks: all-iminolate (c9), keto-iminolate (c10) and all-keto (c11). One can anticipate the proton transfer from the phenolic –OH group to the amide N centre in the keto forms for more favourable charge density distributions. Such a proton transfer is on-demand where the phenolic hydroxyl O coordinates to a Lewis acid centre, as is the case in the 1D CPs, 1·Ln, to avoid electron density depletion at the more electronegative O atom (phenolic O centre) versus N atom (amide N centre), as depicted in Scheme S2 (c12, see the ESI†). The phenomenon could well be supported by the comparative single crystal X-ray data of the 1D CPs, 1·Ln, and their mononuclear congeners, 2·Ln. Both the amide C–N bond distances in 2·Ln are similar to each other (1.334 Å and 1.349 Å for 2·Y; 1.340 Å and 1.341 Å for 2·Gd; 1.342 Å and 1.337 Å for 2·Dy), while those in 1·Ln are significantly inequal (1.328 Å and 1.353 Å for 1·Y; 1.331 Å and 1.360 Å for 1·Gd; 1.327 Å and 1.353 Å for 1·Dy). At the same time, both the amide C–O bond distances in 2·Ln are also similar to each other (1.271 Å and 1.270 Å for 2·Y; 1.260 Å and 1.272 Å for 2·Gd; 1.260 Å and 1.271 Å for 2·Dy), while the corresponding bond parameters in 1·Ln differ significantly (1.278 Å and 1.248 Å for 1·Y; 1.279 Å and 1.246 Å for 1·Gd; 1.244 Å and 1.281 Å for 1·Dy). It implies more of a keto form of the amide functionality at both sides of the ligand molecule for the mononuclear complexes. While in the case of the 1D CPs, the amide functionality at the side where the phenolic hydroxyl O coordinates to the Ln centre of the neighbouring building-block assumes the iminolate form and the other one assumes keto form. Upon coordination with F−, the Lewis acidity of the Ln(III) centre diminishes significantly toward H2L2−, which renders the proton transfer from the non-coordinated phenolic hydroxyl group to its neighbouring amide N centre, and consequently establishing the all-keto form of H2L2− (c13). The phenomenon is strongly evidenced by the solid-state FT-IR spectral studies. The stretching frequencies that are characteristic of the amide carbonyl group (νCO) and the iminol imine group (νCN) for the pristine ligand (H4L) appear in the range 1650–1640 cm−1 with a very strong absorbance, and 1590–1610 cm−1 with a medium absorbance (Fig. S2 and Table S3†). The frequencies corresponding to the amide bond (νC–N) along with the aromatic ring stretching appear near 1550 cm−1 with a very strong absorbance, implying the dominance of the all-keto forms (c1/c2/c5/c6; Scheme S1†). In the solid-state FT-IR spectrum of the 1D CPs (1·Ln), the absorbance bands in the range 1650–1640 cm−1 vanish. The bands near 1550 cm−1 diminish markedly and the bands corresponding to the imine group enhance significantly, implying the dominance of the all iminolate (c9)/keto iminolate (c10) forms of the ligand in the 1D CPs (Scheme S2, see ESI†). Upon treatment of the 1D CPs with F− in the solid states, the resultant FT-IR spectrum displayed a re-appearance of the band corresponding to the amide carbonyl group (νCO; 1645–1700 cm−1; Fig. S34, see ESI†), implying the presence of the all-keto form (c13, Scheme S2, see ESI†).
Computational investigation
To obtain further insights into the electronic structures, detailed computational studies were carried out on the different isomers of H4L, and on their doubly deprotonated forms (H2L2−) as well as on the model compounds of Y analogue of the 1D CPs using density functional theory (DFT, see the Experimental section, ESI†).26 The coordinates of the energy-optimized geometries are tabulated in ESI (Tables S9 and S10†). The gas phase energy optimization studies revealed that a pristine ligand molecule can indeed exist in different conformations (Chart S1–S8, see ESI†) – c2 being the most stable one where the molecule exists in the all keto-all trans form with the phenolic H pointing towards the carbonyl O (Table S11 and Chart S2, see ESI†). However, the energy differences between c2 and the other conformers lie in the range of 5–250 kcal M−1 (Table S11, see ESI†). Interestingly, upon double deprotonation, the dianionic form (H2L2−) is found to exist only as the all-keto-all trans phenolate form with the phenolate O atoms pointing towards the amide H (Chart S9, see ESI†). The absence of imaginary frequency and the preservation of spin multiplicity affirmed the energy-optimized geometries resembling the global minima in their potential surfaces. Moreover, the UV-Vis spectrum of c2 computed by employing the time-dependent density functional theory (TD-DFT) was found to be in excellent agreement with the UV-Vis spectrum of the as-synthesized sample of H4L (Fig. S36, see ESI†). However, the isosurface corresponding to the HOMO was found to spread across the molecular skeleton for c2, while it is dominantly located on the phenolate moieties for H2L2− (Charts S1–9, ESI†). The HOMO–LUMO energy gap in H2L2− (73.98 kCal M−1) was found to be significantly lower than that in c2 (93.70 kCal M−1; Table S11, see ESI†). The computed UV-vis spectroscopic investigation revealed that H2L2− in gas phase absorbs lights covering the visible domain (λabs.max = 232–447 nm), while the absorbance for c2 in gas phase is restricted in the UV region (λabs.max = 183–344 nm Fig. S36–S38, ESI†). The 1D CPs are truncated into mononuclear model complexes owing to realistic computations. As the structural properties and the photophysical behaviours are analogous to each other for the 1·Ln series, computational studies are carried out only on the Y analogue as the representative. As described above in the X-ray crystallographic section, a repeat unit of the 1D CPs is a mononuclear building block composed of a doubly deprotonated form of the ligand (H2L2−) chelating an Ln(III) ion via κ5-N3O2 fashion, a chelating nitrate anion (κ2 NO3−), a triphenyl phosphine oxide molecule coordinating the same Ln(III) ion via the phosphine oxide ligating site (Fig. 1). Such a building block is tethered with a neighbouring building block via the coordination between the Ln(III) centre of the building block and one of the phenolic O ligating sites of the neighbouring building block to develop the 1D CPs. In order for the closest resemblance, the mononuclear model complex is assumed where a monodentate phenolate ligand is introduced in the inner coordination environment via O–Ln coordination, and the neighbouring building block is replaced with Na+ ion making it a neutral unit (the model complex will be presented as NaH2LY here onwards, Chart S10, ESI†). The fluorinated model complexes are hypothesized as the following two monomeric complexes – H4LYF and Na2H2YF (see the Experimental section for details). The initial guesses for both of them were derived from NaH2LY upon coordinating an F− to the Y centre. In addition, all the amide-N and phenoxide-O atoms are protonated for H4LYF (Chart S11, ESI†), while both the amide-N atoms are protonated and both the phenoxide-O are coordinated with Na+ ions for Na2H2YF (Chart S12, see ESI†). Notably, though the energy optimization resulted in analogous structures for both H4LYF and Na2H2YF, they distinctly differ from the repeating unit of the single crystal X-ray molecular structure of 1·Y in terms of the following three aspects – (i) the NO3− being weakly interacting monodentate ligand, (ii) the phenyl moiety of the monodentate phenoxide ligand being far apart from the pyridyl moiety of the pentadentate chelating ligand, and (ii) the ethaneylylidenehydrazide (CN–NH–CO) functional groups on both the sides of the pyridyl moiety being equalized for the formers (Fig. 4 and Table S8, see ESI†). Such deviations could be attributed to the following reasons – (i) significant reduction in electron affinity and/or development of steric crowding at the Y centre after coordination with F−, (ii) the phenoxide ligand being freed from an integral part of the pentadentate chelating ligand as in the 1D CP, and (iii) re-establishment of the keto form in the ethaneylylidenehydrazide functional group upon protonation. Nevertheless, the spatial arrangement of the pentadentate chelating ligand and the topology of the coordination geometry around the Y centre remain practically the same (Fig. 4 and Table S8, see ESI†). The continuous shape measures (CShM's) analyses employing the SHAPE program39 based on the Pinsky–Avnir algorithm,40 the definitions of minimal distortion paths,41 and generalized interconversion coordinates,42 revealed that the coordination polyhedra for all the complexes in 1·Ln series are closest to the muffin topology with Cs symmetry, and for all the complexes in 2·Ln series are closest to the spherical capped square antiprism topology with C4v symmetry (Table S12, see 22 ESI†). On the other hand, the Y centres in the energy-optimized geometries of H4LYF and Na2H2YF are found to assume tricapped trigonal prism J51 with D3h symmetry, which is very narrowly deviated (the difference in the minimal distortion paths value = 0.9 for the former and 1.2 for the latter) from the topology of the inner coordination environments for 1·Ln series (Table S12, see ESI†). The single-point energy calculation on NaH2LY displayed the isosurface corresponding to its HOMO dominantly locating on the bridging phenolate moiety, and the isosurface corresponding to its LUMO being mainly metal centric (Fig. 5). Its HOMO–LUMO energy gap (25.2 kCal M−1) was found to be remarkably redshifted compared to H2L2− (74 kCal M−1) or compared to c2 (93.7 kCal M−1, Fig. 5 and Table S11, ESI†), supporting the experimental observation of immediate coloration upon complex formations. Surprisingly, the isosurfaces corresponding to the HOMOs for H4LYF and Na2H2YF are still dominantly locate on the bridging phenolate moieties, alike NaH2LY, while the isosurfaces corresponding to their LUMOs are completely shifted from the
metal centres and are distributed over the skeletons of the pentadentate chelating ligands (Fig. 5). Thus, an electronic transition associated with HOMO–LUMO could be ascribed to the ligand-to-metal charge transfer for NaH2LY, while it is a ligand centric n/π-to-π* charge transfer for H4LYF and Na2H2YF.43 The HOMO–LUMO energy gaps for the fluorinated model complexes are found to be significantly higher than those for the non-fluorinated complexes (Fig. 5 and Table S11†), revealing the root for the experimentally observed phenomenon of decolorization of the 1D CPs upon treatment with F−. Moreover, the lanthanide ions are known to inherit low quantum yields in photoluminescence.44 Therefore, ligand-to-metal charge transfer in 1·Ln is expected to lead to weak photoluminescence. On the other hand, the isosurfaces corresponding to the LUMOs of the fluorinated model complexes are significantly inflated and symmetrical, which effectively enhances the transition probability. Consequently, a ligand-centric radiative decay is non-surprising for a HOMO–LUMO excitation in the fluorinated complexes.
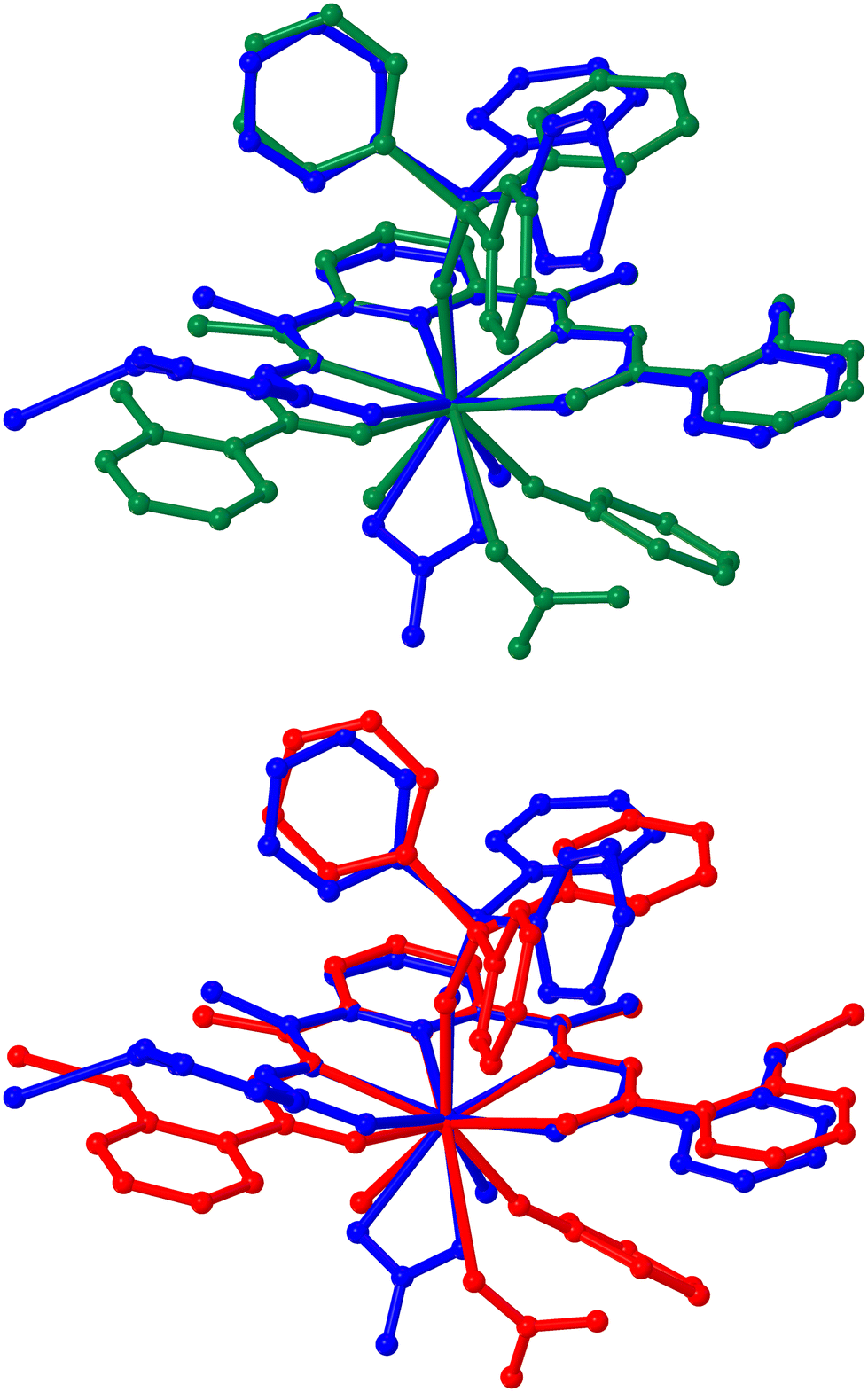 | ||
| Fig. 4 Comparative views between the ball-and-sticks models of a repeat unit of the single-crystal X-ray molecular structure of 1·Y (blue) and the optimized geometries of its hypothetical fluorinated model complexes H4LYF (green), and Na2H2LYF (red). For each pair of structures, atomic sites are projected with respect to a common local coordinate system overlapping the imine-N, pyridyl-N and Y atoms. The H atoms are omitted for clarity. | ||
 | ||
| Fig. 5 The isosurface distributions (isovalue = 0.02; the green and brown coloured surfaces representing the mutually opposite phases of the corresponding wave function) for the HOMO (left) and the LUMO (right) over the energy-optimized structures of c2, H2L2−, H4LYF, Na2H2LYF and over the structure of NaH2LY as truncated from the single crystal X-ray structure of the 1D coordination polymer 1·Y. The atomic color codes: cyan, Y; yellow, P; purple, Na; red, O; blue, N; dark grey, C; light grey, H. | ||
Conclusions
The 1D CPs, 1·Ln, open up a huge scope for exploring hierarchical Ln-based complexes in search of portable and suitable chemosensors for fluoride ions that play immense roles in human society covering both the beneficial and detrimental domains. As the heart of the sensing by these 1D CPs lies in the intramolecular proton transfer between the closest possible atomic centres via one of the most favourable chemical phenomena, one probably could not think of a better alternative for the faster chemical response. Moreover, in view of the practical convenience, an ideal chemosensor could be the one that is ready for as-collected analyte samples. These 1D CPs provide a very wide prospect, especially considering the F− sensing, by exhibiting sensitivity in solid states and in solutions without being interfered with by the other commonly occurring ions. The visual detection could be regarded as the most economical and convenient method of sensing. Decolorization of the solutions of 1·Ln upon treatment with F− strengthens their capability as sensors. The limits of F− detection are found to be orders of magnitude higher than those required for distinguishing the beneficial and detrimental domains of F−. However, it is worth pointing out that all the sensing studies reported herein are carried out with laboratory chemical analytes. No field samples were investigated. Nevertheless, these series of complexes can be synthesized from commercially readily available metal salts (hydrated lanthanide nitrates), and easily synthesizable organic ligands (Schiff bases) under ambient conditions using ethanol as the solvents. The facile synthesis, excellent yield, highest purity in crystalline form, high thermal resistivity and prolonged stability in water endow these CPs with potential applicability. As the Schiff base ligands are quite abundant with versatile functionality, one can achieve a library of analogous 1D CPs with more fascinating chromophores and luminophores to tailor chemosensitivity. These CPs also provide the scopes for further chemical decoration using hierarchical ancillary ligands. In addition, the lanthanide ions also provide the opportunity to endow such complexes with enthralling magnetic behaviour to tune high-resolution imaging and sensing at the molecular levels as well as in the bulks.Author contributions
The manuscript was written through the contributions of all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AKB thanks the Ministry of Education, Government of India for financial aid through the Scheme for Transformational and Advanced Research in Sciences (STARS, MoE/STARS-1/Apr2019/333) to carry out the investigation.References
-
(a) Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed
; (b) C. McDonagh, C. S. Burke and B. D. MacCraith, Chem. Rev., 2008, 108, 400–422 CrossRef CAS PubMed
-
(a) P. Briss, W. Bailey, L. K. Barker, L. T. Beker, E. Beltrán-Aguilar, M. B. Bigley, L. Birnbaum, A. Chattopadhyay, J. Donohue, E. Doyle, I. Garcia, B. Gooch, J. Goodman, J. N. Gracia, S. O. Griffin, L. Grummer-Strawn, J. Hirschman, F. Hyman, T. Iafolla, W. Kohn, A. M. Lester, N. S. Makrides, R. Manski, A. M. Osorio, B. Silverman and T. Sinks, Public Health Rep., 2015, 130(4), 318–331 CrossRef PubMed
- D. Briancon, Rev. Rhum. Engl. Ed., 1997, 64, 78–81 CAS
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed
- E. V. Yusenko, F. P. Kapsargin and P. N. Nesterenko, J. Anal. Chem., 2014, 69, 474–479 CrossRef CAS
-
D. W. Morry and C. Steinmaus, Evidence of the carcinogenicity of fluoride and its salts, Reproductive and Cancer Hazard Assessment Branch, Office of Environmental Health Hazard Assessment, California Environmental Protection Agency, USA, 2011, p. 22 Search PubMed
- Y. Lu, Z. Sun, L. Wu, X. Wang, W. Lu and S. Liu, Fluoride, 2000, 33, 74–78 CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
-
(a) P. Chen, W. Bai and Y. Bao, J. Mater. Chem. C, 2019, 7, 11731–11746 RSC
- R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915–4918 CrossRef CAS PubMed
-
(a) S. Shinoda and H. Tsukube, Analyst, 2011, 136, 431–435 RSC
-
(a) H. Yahyavi, M. Kaykhaii and M. Mirmoghaddam, Crit. Rev. Anal. Chem., 2016, 46, 106–121 CrossRef CAS PubMed
-
(a) P. Hou, S. Chen, H. Wang, J. Wang, K. Voitchovsky and X. Song, Chem. Commun., 2014, 50, 320–322 RSC
-
(a) Y. Ma, Q. Mou, P. Yan, Z. Yang, Y. Xiong, D. Yan, C. Zhang, X. Zhu and Y. Lu, Chem. Sci., 2021, 12, 11740–11747 RSC
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS
-
(a) A. K. Bar, P. Kalita, M. K. Singh, G. Rajaraman and V. Chandrasekhar, Coord. Chem. Rev., 2018, 367, 163–216 CrossRef CAS
- L. Lima and R. Tripier, Curr. Inorg. Chem., 2011, 1, 36 CrossRef CAS
-
(a) X. Liu, J. Xu, Y. Lv, W. Wu, W. Liu and Y. Tang, Dalton Trans., 2013, 42, 9840–9846 RSC
-
(a) Y. Su, J. Yu, Y. Li, S. F. Z. Phua, G. Liu, W. Q. Lim, X. Yang, R. Ganguly, C. Dang, C. Yang and Y. Zhao, Commun. Chem., 2018, 1, 12 CrossRef
- A. K. Mondal, S. Goswami and S. Konar, Dalton Trans., 2015, 44, 5086–5094 RSC
- C. Pelizzi and G. Pelizzi, J. Chem. Soc., Dalton Trans., 1980, 10, 1970 RSC
- L. Dubován, A. Pöllnitz and C. Silvestru, Eur. J. Inorg. Chem., 2016, 2016, 1521–1527 CrossRef
- International Tables for Crystallography. DOI:10.1107/97809553602060000001.
-
O. Kahn, Molecular Magnetism, VCH, Weinheim, 1993 Search PubMed
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. B. Rendell, J. C., S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009. 15 Search PubMed
- A. D. J. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS
-
T. H. J. Dunning and P. J. Hay, in Modern Theoretical Chem. Eur. J, ed.H. F. Schaefer III, Plenum, New York, 1976, vol. 3 Search PubMed
- P. J. Hay and R. J. Wadt, Chem. Phys. Chem., 1985, 82, 299 CAS
- J.-P. Sutter, V. Béreau, V. Jubault, K. Bretosh, C. Pichon and C. Duhayon, Chem. Soc. Rev., 2022, 51, 3280–3313 RSC
- M. L. Kahn, J.-P. Sutter, S. Golhen, P. Guionneau, L. Ouahab, O. Kahn and D. Chasseau, J. Am. Chem. Soc., 2000, 122, 3413–3421 CrossRef CAS
-
(a) Z. Zhu and J. Tang, Natl. Sci. Rev., 2022, 9, nwac194 CrossRef PubMed
-
(a) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed
- V. Singh, D. Das, S. Anga, J.-P. Sutter, V. Chandrasekhar and A. K. Bar, ACS Omega, 2022, 7, 25881–25890 CrossRef CAS PubMed
- X. Zeng, J. Hu, M. Zhang, F. Wang, L. Wu and X. Hou, Anal. Chem., 2020, 92, 2097–2102 CrossRef CAS PubMed
-
J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed
- T. J. Cooper, R. A. Simcoe, K. L. Cooksey, R. Bordoloi, D. R. Miller, G. Furesz, M. L. Turner and E. Bañados, Astrophys. J., 2019, 882, 77 CrossRef CAS
-
(a) P. Kalita, N. Ahmed, A. K. Bar, S. Dey, A. Jana, G. Rajaraman, J.-P. Sutter and V. Chandrasekhar, Inorg. Chem., 2020, 59, 6603–6612 CrossRef CAS PubMed
-
(a) S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem.
Rev., 2005, 249, 1693–1708 CrossRef CAS
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed
- D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed
- J. Cirera, E. Ruiz and S. Alvarez, Chem. – Eur. J., 2006, 12, 3162–3167 CrossRef CAS PubMed
-
(a)
A. Piccone, Probing pyrolysis on the surface of thermal protection systems, Scilight, 2022, p. 2022 Search PubMed
- Y. Hasegawa, Y. Wada and S. Yanagida, J. Photochem. Photobiol., C, 2004, 5, 183–202 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: The experimental and the computational details, synthetic procedures, UV-Vis, IR and NMR spectra, single crystal X-ray structures and the coordination polyhedra, powder X-ray diffractograms, crystallographic data and refinement parameters, crystallographic bond parameters, SHAPE analysis data and additional magnetic properties analyses plots. CCDC 2311694–2311699. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00598h |
| This journal is © The Royal Society of Chemistry 2024 |