DOI:
10.1039/D4DT00183D
(Paper)
Dalton Trans., 2024,
53, 6731-6746
Tuning of surface oxygen vacancies for enhancing photocatalytic performance under visible light irradiation in Sb2WO6 nanostructures†
Received
20th January 2024
, Accepted 14th March 2024
First published on 26th March 2024
Abstract
Tuning of vacancies in photocatalytic materials has emerged as a versatile strategy to enhance visible light absorption and photocatalytic activity. In this study, surface oxygen vacancies (defects) were incorporated on antimony tungstate to boost its photocatalytic activity, which was examined by studying the degradation of model pollutants under visible light irradiation. Specifically, a two-to-three-fold increase in photocatalytic activity was observed for oxygen vacancy-rich antimony tungstate in comparison to its pristine counterpart. This improvement in the photocatalytic performance can be attributed to the presence of oxygen vacancies in the material, which leads to an enhanced absorption of light, decrease in the recombination of charge carriers, and increase in the number of active sites. In addition, owing to the nature of the surface charge present, the photocatalysts were found to be selective for the degradation of cationic pollutants in comparison to anionic and neutral pollutants, and can thus be used for the separation of a mixture of pollutants. Furthermore, scavenger studies illustrate that holes play a major role in the photocatalytic degradation of pollutants. Moreover, the excellent photostability of oxygen vacancy-rich antimony tungstate over three consecutive cycles demonstrates its potential as a good photocatalyst for the degradation of pollutants. Overall, this study demonstrates that the engineering of surface vacancies on perovskite oxide materials can render them as efficient single component photocatalysts for environmental remediation applications.
Introduction
Sustainable environmental clean-up has become one of the major global challenges for the human race.1 Specifically, the treatment of wastewater or polluted water is an important task for researchers all over the world. According to the World Health Organization (WHO), more than 2000 million people all over the world are using polluted water every day for various activities.2 Therefore, the study of emerging water pollutants such as dyes and pharmaceutical compounds has become an important research area. These compounds remain in wastewater and water bodies as micro-pollutants for a long time and pose a threat to marine life.3 Many toxic organic pollutants, such as dyes, fertilizers, phenols, and pharmaceuticals, are difficult to remove using conventional treatment methods. This is because each class of pollutant contains vast varieties. These organic pollutants are non-biodegradable in water because of their chemically stable nature. Conventional methods such as coagulation and adsorption do not completely remove pollutants from water and mineralize them.4 Other wastewater treatment methods such as reverse osmosis or chlorination require high power consumption or generate secondary waste.5 Thus, the wastewater treatment problem requires a solution that is cost-efficient and offers high efficiency.6
Nanostructured semiconductors have shown tremendous potential for environmental remediation using photocatalysis.7 In recent years, photocatalysis has emerged as a green technique to deal with environmental remediation-related issues. This approach comes with various merits such as the use of renewable sources, low cost, mild conditions, and reusability over other conventional techniques.8,9 Over the past few years, various nitride-based, metal oxide, metal sulfide, and metal–organic frameworks (MOF)-based semiconductors have been reported for organic pollutant degradation.10–14 However, because of the rapid recombination of charge carriers, large band gap, and low light absorption, these photocatalysts generally show sluggish activity.15 Various strategies, including the formation of a nanocomposite and heterojunction and band gap engineering using defect-rich materials, are employed to improve photocatalytic activity.16,17 Among these, defect-rich materials have specifically caught the attention of researchers because of their ability to significantly enhance the surface active sites, as well as light absorption.18
Perovskite oxide-based semiconductors are very popular because of their unique structural properties. Their structural formula is ABO3, where A and B are transition metals that are in the +2 and +4 oxidation states, respectively.19 Different numbers of transition metals can be adjusted to A and B sites, making them a huge class of materials.20 Various types of vacancies or doping can be done in these materials. Many vacancy-rich perovskite materials, such as Bi2MoO6, Bi2WO6, SrTiO3, CaTiO3, etc., have been reported for many photocatalytic applications, such as pollutant degradation, nitrogen fixation and H2 production.15,18,21 Defects can be of various types based on their size, synthesis method or doping level.22 They can be categorized into point defects, line defects, planer defects and volume defects. Among all, point defects (vacancies and doping) are often discussed because of their facile synthesis routes.23,24 There have been a few materials previously reported in literature studies based on oxygen vacancies for pollutant degradation, such as CuFe2O4, TiO2, Bi2WO6etc.25–27 These literature reports show the enhancement in photocatalytic activity after the introduction of oxygen vacancies. The characterization techniques for oxygen vacancies are well established, but the reaction mechanism involving the oxygen vacancies is not very clear. There is also a dearth in the information available on the controllable synthesis of oxygen vacancies in the photocatalyst. So, it can be said that there is still a need to perform further research to gain more insights related to the structural changes, as well as reaction mechanisms, after incorporation of oxygen vacancies in the photocatalyst. One such material in the perovskite family having the Aurivillius phase is Sb2WO6, where Sb, W and O are present in the +3, +6 and −2 oxidation states, respectively. Sb2WO6 has a band gap of approximately 2.62 eV, which makes this material suitable for various photocatalytic applications.28 In 2014, Hu et al.29 demonstrated the photocatalytic ability of Sb2WO6 structures for the degradation of rhodamine B dye. These hierarchical structures were prepared by varying ratios of ethylene glycol and water. In other work, the photocatalytic activity of the nanocomposite of Sb2WO6 with carbon quantum dots has been explored. This report also demonstrated the degradation of rhodamine B dye up to 83%.30 Zhang et al.31 demonstrated the photocatalytic ability of the S-scheme Sb2WO6/Bi4O5I2 heterojunction for the degradation of tetracycline and doxycycline with the removal rate of 88.2% and 83.9%, respectively. Furthermore, oxygen vacancies in Sb2WO6 microspheres have been introduced using low-temperature plasma by Peng et al.32 This work also demonstrated the degradation of rhodamine B with the removal efficiency of 72% in 6 h. However, there are very few reports available on defect-rich Sb2WO6 nanostructures that demonstrate their effect on photocatalytic activities.
In this work, we have synthesized pristine Sb2WO6 (SWO) and its oxygen vacancy-rich counterparts, DSWO1, DSWO2, DSWO3, using ascorbic acid in different amounts (100 mg, 200 mg, 300 mg) by employing a facile solvothermal method. The evaluation of their photocatalytic performance has been done by studying the degradation of organic dyes, crystal violet (CRV) and rhodamine B (RhB), and a colorless pharmaceutical pollutant, levofloxacin (LFX), under visible light irradiation. In addition, the selectivity of the photocatalyst was studied using a mixture of dyes. Furthermore, the photocatalytic degradation pathways of CRV and RhB were investigated using high-resolution mass spectrometry. Additionally, the adsorption studies of RhB over the surface of the photocatalyst was performed. The defected photocatalyst DSWO2 with oxygen vacancies showed better activity than pristine SWO. The generation of oxygen-vacancy rich centers helps in reducing the recombination of charge carriers, which is the main reason for the enhanced activity. Thus, this work offers a deeper level understanding into the structural properties of defect-engineered materials, as well as their application in photocatalytic pollutant degradation.
Experimental section
Chemicals
Antimony chloride (SbCl3) ≥98% and sodium tungstate dihydrate (Na2WO4·2H2O) ≥99% were purchased from Sigma Aldrich, India. Ethanol (C2H5OH) was purchased from Analytical CSS Reagent, India. The organic dyes, crystal violet and rhodamine B, were obtained from Sigma Aldrich, India, and levofloxacin was purchased from Alkem Laboratory, India. Deionized (DI) water (18 MΩ cm) was used from ELGA PURELAB option-R7.
Synthesis of antimony tungstate nanostructures
The synthesis of antimony tungstate (SWO) nanostructures was performed via hydrothermal method, as reported in earlier literature with a slight difference.33 In brief, 2 mmol of antimony trichloride (SbCl3) was dissolved in 8 mL ethanol. This was stirred until a homogeneous mixture was obtained. After that, 1 mmol sodium tungstate dihydrate (Na2WO4·2H2O) was dissolved in 8 mL deionized water. Both solutions were mixed and stirred for at least 15 min. The pH of the solution was then adjusted to 2 using 1 M NaOH solution. The resultant solution was then transferred to a stainless-steel autoclave with Teflon lining, and heated for 24 h at 180 °C. After completion of the reaction, the autoclave was cooled at room temperature, and green-colored precipitates were collected for SWO. The collected precipitates were washed multiple times with deionized water and ethanol. Finally, the product was oven dried at 80 °C overnight.
Synthesis of the oxygen-vacant antimony tungstate
Oxygen vacancy-rich antimony tungstate samples were prepared using a one-step hydrothermal method. In a typical procedure, 2 mmol of antimony trichloride (SbCl3) was dissolved in 8 mL ethanol. The solution was stirred until SbCl3 was completely dissolved. Afterwards, different amounts of ascorbic acid (100, 200 and 300 mg) were added to the above mixture, while continuous stirring was maintained. Then, 1 mmol of sodium tungstate dihydrate (Na2WO4·2H2O) was dissolved in 8 mL deionized water in another beaker and was subsequently added to the above solution. The pH of the resulting solution was adjusted to 2 using 1 M NaOH solution. The resultant solution was then transferred to a stainless-steel autoclave with Teflon lining and heated for 24 h at 180 °C. After completion of the reaction, the autoclave was cooled at room temperature, and brown-colored precipitates were collected. At last, the collected precipitates were washed multiple times with deionized water and ethanol. Finally, the product was oven dried at 80 °C overnight. These samples prepared with 100, 200, 300 mg of ascorbic acid were named DSWO1, DSWO2, and DSWO3, respectively.
Photocatalytic pollutant degradation
The activity of pure SWO and the oxygen vacancy-rich (DSWO1, DSWO2, DSWO3) samples was examined by photocatalytic degradation of two organic dyes, viz., crystal violet (CRV) and rhodamine B dye (RhB). In brief, 10 mg of the photocatalyst was dispersed into 20 mL of 6 × 10−5 M aqueous solution of CRV and into 20 mL of 4 × 10−5 M aqueous solution of RhB. These solutions were kept under dark conditions for 60 min to achieve pollutant and photocatalyst adsorption–desorption equilibrium. For the photocatalytic reaction, the reaction mixture was kept in a photoreactor under visible light irradiation consisting of three 45 W white light-emitting compact fluorescence light (CFL) bulbs. After every 30 min, 1 mL of suspension was taken out during the reaction and centrifuged to separate the photocatalyst. The absorbance of the CRV and RhB dyes can be seen at 592 nm and 556 nm, respectively, to examine their degradation. Additionally, reaction samples collected at different time intervals were inspected using high-resolution mass spectroscopy (HRMS) to determine the breakdown route of the dye molecules. The degradation percentage of RhB and CRV was estimated by using the equation below:15
| Degradation (%) = 1 − C/C0 × 100 |
Here, C0 is the absorbance of CFX and RhB before visible light irradiation, and C is the absorbance of CRV and RhB over different time intervals under visible light irradiation.
Results and discussion
Synthesis and structural studies
The synthesis of oxygen vacancy-rich Sb2WO6 was done using a one-step solvothermal approach. Ascorbic acid was added to the reaction mixture during the synthesis process. A schematic illustration of the synthesis process is shown in Scheme 1. As shown in Fig. 1a, the powder X-ray diffraction pattern (PXRD) of the as-synthesized samples is consistent with the JCPDS card no. 00-047-1680, suggesting the triclinic phase of the samples. The PXRD characteristic peaks of SWO are observed at 2θ = 20.38, 26.99, 29.30, 32.85, 36.59, 40.32, 47.44, 49.93, 53.31 and 55.44°. These peaks are assigned to the (01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), (11), (003), (20), (020), (202), (02
), (11), (003), (20), (020), (202), (02![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), (22), (310) and (222) diffraction planes, respectively.34 Similar peak patterns were observed for DSWO1, DSWO2 and DSWO3. However, the peak corresponding to the (20) lattice plane is shifted to a lower value for the oxygen vacancy-rich DSWO1 and DSWO3 (Fig. 1b). The shifting of the peak for DSWO3 is much lower as compared to DSWO1. Such shifting of peaks indicates the lattice expansion of the oxygen vacancy-rich samples. On the other hand, the peak was seen shifting to a higher value for DSWO2, suggesting the lattice contraction. The formation of an oxygen vacancy (Ov) is strongly related to the lattice distortions. Oxygen vacancies are a very common type of defect in metal oxides, and they can be present in charged (+2) or neutral (0) forms. Neutral vacancies lead to lattice expansion, whereas charged vacancies lead to lattice contraction.35 The metal cation (M+) that is closest to the neutral oxygen vacancy moves along the direction of the M–O bond. So, the distance between metal and neutral oxygen vacancy (M–Ov) is elongated as compared to the M–O bond. This elongation of the bond leads to lattice expansion.36 However, charged oxygen vacancies have high electronegativity. Due to the high electronegativity of the oxygen vacancy centers, a contraction of the lattice takes place. The lattice expansion in DSWO1 and DSWO3 indicate the presence of neutral vacancies, whereas the lattice contraction in DSWO2 indicates the presence of charged vacancies. Although the oxygen vacancy-rich samples are synthesized using the same method, it is extremely difficult to control the nature of the oxygen vacancies formed in the samples. The crystallite size, lattice strain and lattice parameters of all of the samples are calculated (Table S1†). The crystallite size and lattice strain have been calculated using the following equations:
), (22), (310) and (222) diffraction planes, respectively.34 Similar peak patterns were observed for DSWO1, DSWO2 and DSWO3. However, the peak corresponding to the (20) lattice plane is shifted to a lower value for the oxygen vacancy-rich DSWO1 and DSWO3 (Fig. 1b). The shifting of the peak for DSWO3 is much lower as compared to DSWO1. Such shifting of peaks indicates the lattice expansion of the oxygen vacancy-rich samples. On the other hand, the peak was seen shifting to a higher value for DSWO2, suggesting the lattice contraction. The formation of an oxygen vacancy (Ov) is strongly related to the lattice distortions. Oxygen vacancies are a very common type of defect in metal oxides, and they can be present in charged (+2) or neutral (0) forms. Neutral vacancies lead to lattice expansion, whereas charged vacancies lead to lattice contraction.35 The metal cation (M+) that is closest to the neutral oxygen vacancy moves along the direction of the M–O bond. So, the distance between metal and neutral oxygen vacancy (M–Ov) is elongated as compared to the M–O bond. This elongation of the bond leads to lattice expansion.36 However, charged oxygen vacancies have high electronegativity. Due to the high electronegativity of the oxygen vacancy centers, a contraction of the lattice takes place. The lattice expansion in DSWO1 and DSWO3 indicate the presence of neutral vacancies, whereas the lattice contraction in DSWO2 indicates the presence of charged vacancies. Although the oxygen vacancy-rich samples are synthesized using the same method, it is extremely difficult to control the nature of the oxygen vacancies formed in the samples. The crystallite size, lattice strain and lattice parameters of all of the samples are calculated (Table S1†). The crystallite size and lattice strain have been calculated using the following equations:| | kλ = β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ | (1) |
| | | ε = β/tanθ | (2) |
 |
| | Scheme 1 Schematic representation for the synthesis of pristine sample (SWO) and oxygen vacancy-rich samples (DSWO1, DSWO2, and DSWO3). | |
 |
| | Fig. 1 (a and b) PXRD patterns, (c) Raman spectra and (d) FTIR spectra of SWO, DSW01, DSWO2, and DSWO3. | |
Here, k is Scherrer constant, λ is the wavelength of the X-ray (1.54 Å), β is the full width at half maximum (FWHM), θ is the angle of diffraction, and ε corresponds to the lattice strain. The change in crystallite size, lattice strain, and lattice parameters suggest the lattice distortion after the insertion of oxygen vacancies in the samples (DSWO1, DSWO2, DSWO3). The maximum lattice strain has been observed in DSWO3, which suggests the maximum amount of oxygen vacancies in it.
Raman spectra of SWO, DSWO1, DSWO2 and DSWO3 are displayed in Fig. 1c. Normal modes of SWO have no effect after the creation of oxygen vacancies. The normal modes of SWO, DSWO1, DSWO2 and DSWO3 can be seen at 78 cm−1, 208 cm−1, 280 cm−1 and 859 cm−1.37 The stretching and bending modes of the Sb–O bonds in the (Sb2O2)2+ layers are merged with the WO6 bending modes ranging from 100 to 600 cm−1. The stretching modes of the W–O bonds in the WO6 octahedra can be evidenced in the region from 600 cm−1 to 1000 cm−1. The peak around 859 cm−1 in Fig. 1c corresponds to the stretching of W–O bonds. The drop in the intensity of peaks in the oxygen vacancy-rich samples, corresponding to the W–O bond stretching modes, indicates the increase in the concentration of oxygen vacancies.38 In addition, Fourier transform infrared (FTIR) spectroscopic measurements were performed for SWO, DSWO1, DSWO2 and DSWO3 samples, as shown in Fig. 1d. The peak at 712 cm−1 is the characteristic peak of the W–O bond. The peak around 520 cm−1 corresponds to the W–O–W bonds.39
Morphological and compositional studies
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) studies were carried out to investigate the surface morphology of the control (SWO) and a representative (DSWO2) sample. The morphologies of the SWO and DSWO2 samples are shown in Fig. 2a, b and Fig. 2c, d, respectively. The morphology of SWO was observed as nanostructured spheres, whereas for the oxygen vacancy-rich DSWO2 sample, an aggregation of particles composed of small nanosheets can be seen. It is clear from Fig. 2d that oxygen vacancies have changed the morphology of the sample. The incorporation of oxygen vacancies in DSWO2 inhibits the growth of the particles and thereby alters its morphology.18 Nevertheless, the nanosheet-like morphology provides extra exposure of edge sites, which increases the accessibility of surface active sites and can be helpful in increasing the photocatalytic activity.40 The TEM images of DSWO2 are shown in Fig. 2e and f. The TEM images also show the nanosheet-like morphology of the DSWO2 sample. In addition, the high-resolution transmission electron microscopy (HRTEM) image of DSWO2 (Fig. 2g) reveals the lattice fringes with a d-spacing equal to 0.34 nm, corresponding to the (11) lattice plane.41 Furthermore, the inverse fast Fourier transform (IFFT) and line profile of this lattice plane are shown in Fig. 2h and i. The energy-dispersive analysis (EDAX) spectra and elemental mapping of SWO (Fig. S1†) and DSWO2 verify the presence of Sb, W and O as the constituent elements (Fig. 2j and k–n). According to the mapping data, all of the elements in the sample are uniformly distributed.
 |
| | Fig. 2 SEM images of (a and b) SWO and (c and d) DSWO2; (e and f) TEM, (g) HRTEM images, (h) IFFT, and (i) line analysis of DSWO2; (j) EDAX spectra and (k–n) elemental spectra of DSWO2. | |
To understand the composition and oxidation state of the elements in the samples, X-ray photoelectron spectroscopy (XPS) measurements were performed on SWO and DSWO2. The survey spectrum (Fig. S2) (ESI†) shows the presence of all of the elements (Sb, O, W) in both samples. In Fig. 3a, Sb 3d and O 1s high-resolution spectra are shown as their peaks merge together. The peaks for 3d5/2 and 3d3/2 have been observed at 530.2 and 539.7 eV for SWO. However, the same peaks for DSWO2 have been observed with a small shift at 530.4 and 539.9 eV. These peaks correspond to the +3 oxidation state of Sb.42 This slight increase in binding energy can be attributed to the presence of oxygen vacancies in the DSWO2 sample. As oxygen vacancies are more electronegative in nature, they tend to increase the binding energy of the surrounding atoms.43 For SWO, the peak for O 1s is split into two peaks at 530.2 and 530.7 eV, which correspond to the Sb–O/Sb 3d5/2 and W–O bonds, respectively. Similar peaks have been observed for DSWO2 at 530.4 eV and 530.9 eV.41 The peak at 532.0 eV corresponds to the adsorbed water molecules on the surface of DSWO2. In addition, the peaks for W 4f7/2 and W 4f5/2 spectra were recorded at 35.68 eV, 37.85 eV and 35.94 eV, and at 37.98 eV for SWO and DSWO2, respectively (Fig. 3b). The binding energy of the peaks indicates the +6-oxidation state of W in both samples. Peaks for the +5 oxidation state of W were also recorded, which were seen at 34.87 eV, 36.88 eV and 35.43 eV, and at 37.45 eV for SWO and DSWO2, respectively. Moreover, the Sb 4d peaks were observed in the W 4f spectra of both samples at lower binding energies.
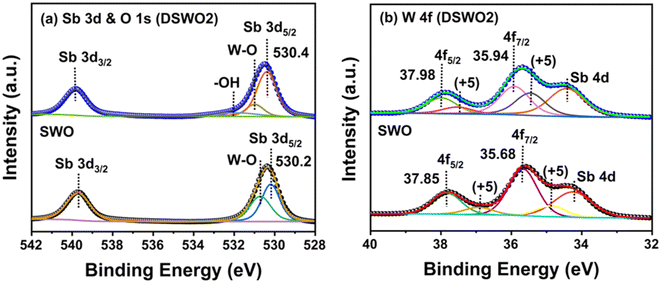 |
| | Fig. 3 (a and b) High-resolution XPS of SWO and DSWO2. | |
Optical and band gap studies
The optical properties and band gap measurements of the as-synthesized samples (SWO, DSWO1, DSWO2 and DSWO3) were obtained. The band gap of all of the samples was measured using diffuse reflectance spectroscopy (DRS). In Fig. 4a, the absorption spectra of all of the samples are shown. It can be clearly seen that with the increase in oxygen vacancy concentration, the absorption edge of samples with oxygen vacancies, i.e., DSWO1, DSWO2 and DSWO3, has shifted to a higher wavelength (red shift). The absorption edge of SWO, DSWO1, DSWO2 and DSWO3 is observed at 556.8, 560.4, 581.7 and 586.5 nm, respectively. The shifting of the absorption edge indicates a greater absorption of light in the oxygen vacancy-rich samples in comparison to pristine SWO. The energy vs. Kubelka–Munk function plot was used to determine the band gap of all of the samples (Fig. S3†). The band gaps were found to be 2.13, 2.09, 1.96 and 1.88 eV for SWO, DSWO1, DSWO2 and DSWO3, respectively. This decrease in the band gap can be ascribed to the presence of oxygen vacancies.
 |
| | Fig. 4 (a) UV absorption spectra, (b) VB XPS spectra, and (c) band diagram of SWO, DSWO1, DSWO2 and DSWO3 (inset: photograph of samples). | |
Furthermore, to investigate the band positions of samples, VB XPS measurements were performed. It can be seen in Fig. 4b that the position of the valence band (VB = 2.00 eV) remained constant after the creation of oxygen vacancies in all of the samples. This suggests the emergence of defect energy levels below the CB, rather than above VB. The CB positions for all of the samples were determined by using the following equation:
Here, ECB and EVB are the conduction band and valence band positions, respectively. Eg corresponds to the band gap of the photocatalyst. Based on this equation, the CB values were determined to be −0.13, −0.09, 0.04 and 0.12 eV for SWO, DSWO1, DSWO2 and DSWO3, respectively. The lower part of the conduction band of the Sb2WO6 is dominated by W 5d, and the upper part is dominated by W 5d and Sb 5p orbitals. This leads to the shifting of the conduction band to negative potentials. In addition, the valence band is dominated by Sb 5s and O 2p orbitals. Due to the high electronegativity of Sb, the valence band of Sb2WO6 formed near the lower valence band potentials. This leads to the formation of a band gap of 2.13 eV for Sb2WO6. However, due to the formation of oxygen vacancies in Sb2WO6 nanostructures, the conduction band shifts to the lower potentials and decreases the band gap.44 The shifting of CB can be understood by the formation of donor energy levels with an increase in oxygen vacancies. With an adequate amount of oxygen vacancies in the sample, the formation of a defect energy level takes place and tends to overlap with CB. This overlapping of the defect energy level and CB generates an energy band which shifts the CB band position towards a more positive value; hence, the band gap decreases. The color of all of the samples (Fig. 4c) also suggests a change in the band gap and the respective positions of VB and CB for SWO, DSWO1, DSWO2, DSWO3, respectively, as shown in Fig. 4c.
Surface area, thermogravimetric and temperature-programmed desorption studies
The specific surface area of the as-synthesized samples was calculated using Brunauer–Emmett–Teller (BET) analysis. The N2 adsorption desorption isotherm, multi-point BET, and pore size distribution curves corresponding to SWO, DSWO1, DSWO2 and DSWO3 are shown in Fig. S4 (ESI†). These isotherms corresponding to all of the samples are type VI with H3 hysteresis loop, indicating their mesoporous nature.41,45 The specific surface area of SWO, DSWO1, DSWO2 and DSWO3 was found to be 31.9, 34.7, 36.6 and 36.9 m2 g−1, respectively. The pore diameter in all of the samples was found in the range of 100–150 nm. The samples with oxygen vacancies have shown larger surface areas, as compared to pristine SWO. This increase in surface area can be understood by the change in the morphology of the samples after incorporation of oxygen vacancies, as observed in SEM images (Fig. 2c). The maximum surface area for DSWO3 has been observed, which can be attributed to the amount of ascorbic acid used in the hydrothermal synthesis of the sample. Thus, it can be inferred that the amount of ascorbic acid used in the synthesis is directly proportional to the increase in the surface area, and the oxygen vacancy-rich DSWO samples have high surface area as compared to the pristine sample SWO.
In addition, the thermal stability of SWO and DSWO2 samples was investigated using thermogravimetric analysis (TGA), as shown in Fig. 5a. It can be seen from the graph that both of the samples are stable up to 350 °C. For SWO, a mass loss of 3% could be evidenced from 350 °C to 700 °C. This mass loss can be ascribed to the evaporation of water molecules from the sample surface. However, a more significant loss of up to 20% can be seen after 700 °C. This decline in weight is due to pyrolysis of SWO, which further tends to release lattice oxygen.46 An overall mass loss of 23% could be noticed in SWO up to 800 °C. Whereas, in the case of DSWO2, 6% of loss up to 550 °C is due to surface-adsorbed water molecules and other hydrocarbons. Furthermore, a mass loss of 52% is observed up to 800 °C, which is due to decomposition of DSWO2, leading to the release of lattice oxygen. It can be inferred from the data that the pristine SWO sample has higher thermal stability than its oxygen vacancy counterpart, DSWO2.
 |
| | Fig. 5 (a) TGA curves and (b) NH3-TPD curves of SWO and DSWO2. | |
In order to confirm the presence of oxygen vacancies in the samples, temperature-programmed desorption (TPD) studies were performed on SWO and DSWO2 using NH3 gas (Fig. 5b). As oxygen vacancies are electron attracting in nature and behave as Lewis acid sites, NH3 gas can easily get chemisorbed on oxygen vacancy sites due to the presence of the lone pair of electrons on the N-atom.47 The adsorption of NH3 was done over the surface of the catalysts using the NH3 probe at 50 °C. Then, desorption of NH3 was checked on the surface of SWO and DSWO2 up to 800 °C. The peaks at 389 °C and 597 °C were observed for pristine SWO, whereas peaks at 423 °C and 654 °C were observed for DSWO2. The peaks observed for the samples signify the chemisorption of the NH3 gas over the catalyst surface, which generally takes place above 300 °C.48 However, the peaks observed for DSWO2 at higher temperatures infer the stronger chemisorption of NH3 as compared to the SWO surface. Also, the amount of NH3 chemisorbed on the SWO and DSWO2 surfaces was found to be 0.579 mmol g−1 and 2.66 mmol g−1, respectively. The higher amount of NH3 chemisorbed on the DSWO2 surface also signifies the increased amount of oxygen vacancies in it compared to that in SWO.
Photocatalytic activity studies
The photocatalytic activity studies were performed on CRV and RhB dyes as model pollutants under visible light irradiation. The absorption peaks of CRV and RhB dyes were observed at 592 nm and 556 nm, respectively. The initial concentrations of CRV and RhB were 6 × 10−5 M and 4 × 10−5 M, respectively. For the photocatalysis studies, the sample was taken every 30 min and was examined using UV-vis spectroscopy. The time-dependent UV absorption spectra of CRV taken at various time intervals in controlled conditions, like without a catalyst (WC) and dark, as well as in the presence of SWO, DSWO1, DSWO2, DSWO3, is shown in Fig. S5 (ESI†). The reaction mixture was first kept in the dark for 60 min with different catalysts, and an initial decrease in the absorbance up to 60 min can be attributed to the adsorption of the pollutant over the catalyst surface. In dark conditions, the adsorption of CRV over DSWO2 was 20%. In the absence of the photocatalyst, neither adsorption nor degradation was observed. The adsorption of CRV was found to be 14%, 17%, 19%, and 10%, and the photocatalytic degradation was determined to be 23%, 57%, 71%, and 52% in the presence of SWO, DSWO1, DSWO2, and DSWO3, respectively. It is clear from the absorption spectra that the degradation of CRV has increased in the case of the oxygen vacancy-rich samples (DSWO1, DSWO2, DSWO3), and DSWO2 is the best performing photocatalyst. The Langmuir-Hanselwood model for pseudo-first-order kinetics studies, as shown in eqn (4), was employed:49| | | lnC/C0 = −kt | (4) |
Here, C is the initial concentration and C0 is the concentration of CRV corresponding to the initial absorbances, A and A0, of the reaction mixture taken during different time intervals. The kinetics plots of time vs. C/C0 and −lnC/C0 are shown in Fig. 6a and b, respectively. The adsorption and degradation in controlled conditions, as well as in the presence of photocatalysts, are shown in Fig. 6c. The rate constant was found to be 0.0009 min−1, 0.0031 min−1, 0.0045 min−1, and 0.0029 min−1 for SWO, DSWO1, DSWO2, and DSWO3, respectively, as shown in Fig. 6d.
 |
| | Fig. 6 (a and b) Kinetic curves of photocatalytic CRV degradation, (c) histogram of comparative adsorption and degradation of CRV, and (d) histogram for rate constants for all of the photocatalysts under visible light irradiation. | |
Furthermore, the degradation of RhB was examined and the UV-vis spectra corresponding to WC, dark condition, and in the presence of all of the photocatalysts are shown in Fig. S6 (ESI†). The degradation of RhB without any photocatalyst did not show any degradation. However, under dark conditions, it has shown some amount of adsorption on the catalyst surface. These results indicate the importance of the photocatalyst and light source for the photo-assisted degradation studies. In addition, kinetics studies were done to gain better insights on the degradation of RhB on the DSWO2 surface. The plot of time (min) vs. C/C0 and −ln(C/C0) are shown in Fig. 7a and b. It can be evidenced that all of the samples show a good linear fit along with the highest activity photocatalyst, DSWO2. The adsorption of RhB over SWO, DSWO1, DSWO2, and DSWO3 was calculated to be 17%, 18%, 13%, and 16%, respectively. Furthermore, the degradation of RhB over these catalysts’ surface was determined to be 45%, 63%, 86%, and 67%, respectively, under visible light irradiation (Fig. 7c). The rate constant (k) values were determined to be 0.0022 min−1, 0.0036 min−1, 0.0080 min−1, and 0.0041 min−1 for SWO, DSWO1, DSWO2, and DSWO3, respectively (Fig. 7d).
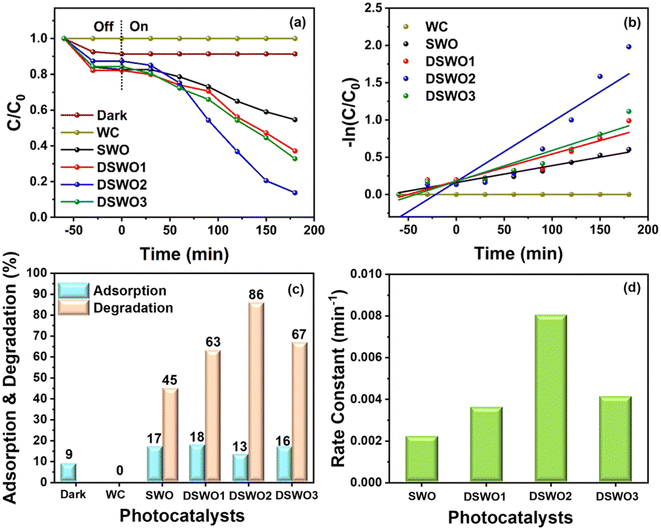 |
| | Fig. 7 (a and b) Kinetic curves of photocatalytic RhB degradation, (c) histogram of comparative adsorption and degradation of RhB, and (d) histogram for rate constants for all of the photocatalysts under visible light irradiation. | |
As both CRV and RhB are colored dyes, they have the tendency to undergo photosensitization and self-degradation under light irradiation. To eradicate this doubt, the photocatalytic degradation of a colorless pharmaceutical pollutant, levofloxacin (LFX), was also performed under identical experimental conditions and the obtained results are shown in Fig. S7 (ESI†). It was found that 52% of LFX was degraded using the DSWO2 photocatalyst. This result clearly shows the importance of the photocatalyst towards the degradation of pollutants.
Intermediates of RhB and CRV photocatalytic degradation
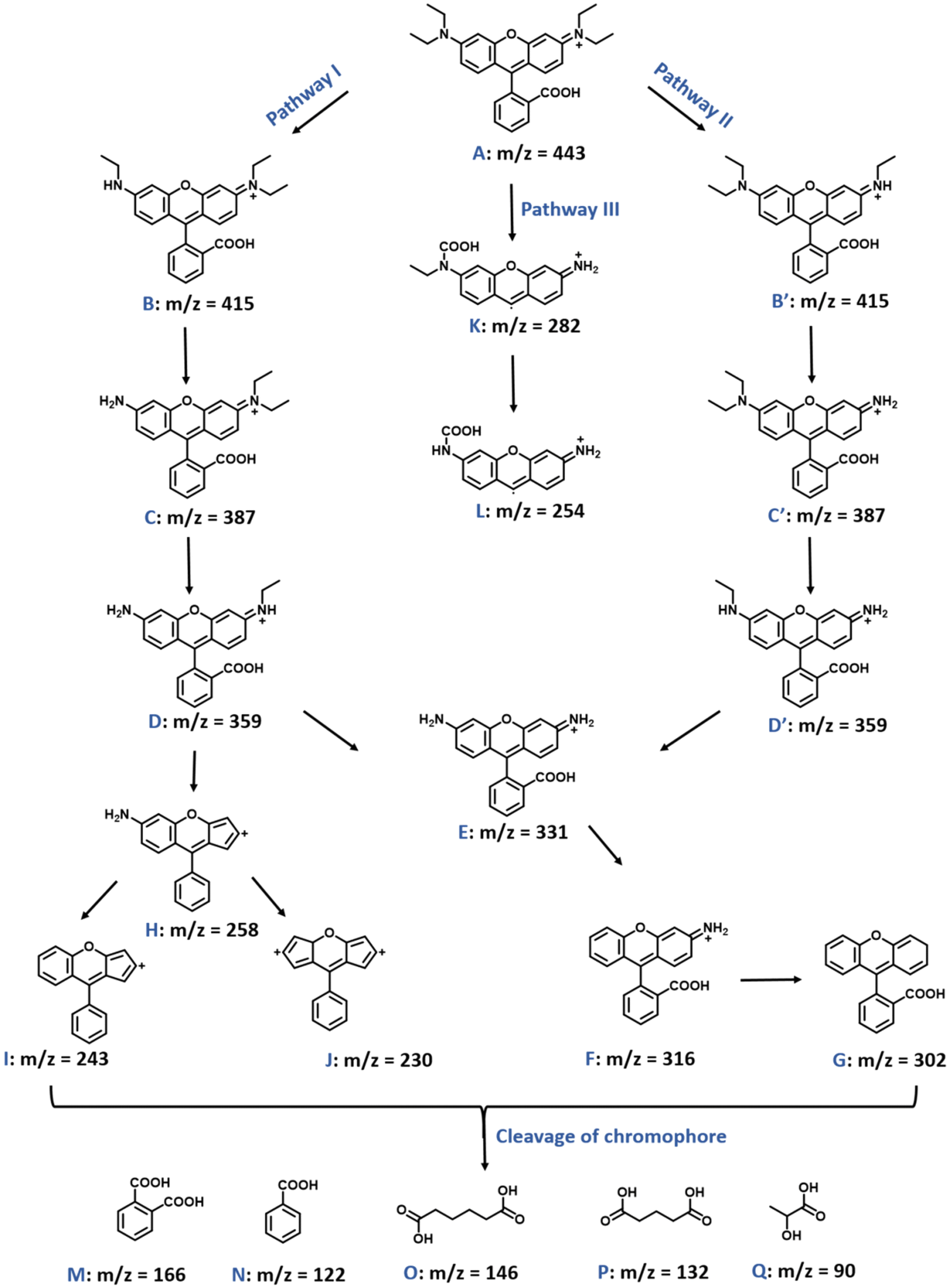
The degraded samples of both pollutants, CRV and RhB (under visible light irradiation using DSWO2 as photocatalyst), were subjected to mass spectrometry to analyze the degradation fragments. The reaction mixtures taken at different time intervals were analyzed. For CRV, the peak at A (m/z = 372) corresponds to pure CRV dye. The degradation of CRV proceeds in two pathways, as shown in Fig. 8. The first pathway involves the N-demethylation of CRV molecules that can lead to the formation of fragments B (m/z = 358) and C (m/z = 344). Furthermore, complete N-demethylation of CRV molecules leads to the formation of D (m/z = 288). Subsequently, the removal of the benzene ring along with the –NH2 group attack of the *OH radical on the carbon atom leads to the formation of fragment E (m/z = 212). The second pathway involves the degradation of CRV molecules via oxidation by *OH radicals. The attack of the *OH radicals at the central carbon atom leads to the formation of the benzophenone derivative of the CRV molecules. Such benzophenone intermediate can be observed in the intermediate F (m/z = 268). The intermediate F can further dissociate into smaller molecules via three different sub-pathways. In sub-pathway I, the removal of one N-methyl group forms an intermediate H (m/z = 254). The removal of one methyl group from H results in the formation of intermediate E (m/z = 212). In sub-pathway II, the removal of the –N(CH3)2 group leads to the formation of G (m/z = 225). In sub-pathway III, the attack of the *OH radical on the carbon atom of the benzophenone intermediate (F) results in the formation of I (m/z = 165) and its decarbonylation forms the fragment J (m/z = 137). Further loss of the –OH and N-methyl groups results in the formation of aniline, which leads to different intermediates, such as K (m/z = 121), L (m/z = 107) and M (m/z = 93). The intermediate M (m/z = 93) can further undergo the opening of the benzene ring and leads to the formation of smaller molecules, which are expected to finally mineralize to even smaller molecules, like CO2, H2O and NO2.50–52
 |
| | Fig. 8 Various fragments formed during the photocatalytic degradation of CRV over DSWO2 in the presence of visible light. | |
Also, the degradation pathway of RhB was analyzed using various degradation samples collected at different time intervals using mass spectrometry (Fig. 9). The mass peak obtained at A (m/z = 443) was observed for pure RhB solution. The deethylation of RhB molecules can happen in either one of the N-atoms via two pathways I and II, and can lead to the formation of intermediates B, B′ (m/z = 415). Furthermore, removal of the other three ethyl molecules from N-atoms leads to the formation of intermediates like C, C′ (m/z = 387), D, D′ (m/z = 359) and E (m/z = 331). The intermediate D (m/z = 359) undergoes decarboxylation and rearrangement after removal of the –C2H5NH2 group, resulting in the formation of H (m/z = 258). The intermediate H (m/z = 258) can also undergo further degradation to result in two other intermediates, I (m/z = 243) and J (m/z = 230). Also, the removal of only one –NH2 group from E (m/z = 331) leads to the formation of F (m/z = 316). This intermediate is further degraded to G (m/z = 302) by losing another –NH2 group. In addition, the pure RhB molecule (m/z = 443) can result in the formation of intermediates K (m/z = 282) and L (m/z = 254) after deethylation and carboxylation at one N-atom (Pathway III). After cleavage of the chromophore, some smaller molecules were also identified, which includes intermediates M, N, O, P and Q (m/z = 166, 122, 146, 132 and 90). These smaller intermediate molecules will further undergo benzene ring opening, leading to mineralization of the RhB molecule into CO2, H2O and NO2.53,54
 |
| | Fig. 9 Various fragments formed during the photocatalytic degradation of RhB over DSWO2 in the presence of visible light. | |
Selectivity of the photocatalyst
In order to check the selectivity of the DSWO2 photocatalyst, the degradation of anionic dye (brilliant yellow: BY) was investigated. A 5 × 10−5 M concentration of the BY solution was taken. No degradation of BY was observed even after 180 min, as can be seen in Fig. S8a (ESI†). In addition, a mixture of dyes (RhB and BY, CRV and BY), mixed in 1:1 ratio, was taken. These mixtures were held under dark conditions for 60 min to achieve adsorption–desorption equilibrium before being subjected to photocatalytic degradation for 180 min. The absorption spectra of CRV and BY, RhB and BY are presented in Fig. S8b and c (ESI†). It was seen that in the mixture of CRV and BY, CRV was degraded up to 75% in 180 min, whereas the degradation of BY was not evidenced. However, a shift in the absorption spectra of BY could be seen with an increase in the degradation of CRV, which could be due to interaction between the degraded smaller fragments of CRV and BY dye. Similarly, the mixture of RhB and BY showed degradation of only RhB (59%). In this case as well, no degradation of BY could be evidenced. It can be inferred from the above results that the photocatalyst, DSWO2, was selective for cationic pollutants like RhB and CRV. This selectivity of the DSWO2 catalyst can also be understood by zeta potential values. Table S2† shows the zeta potential values of SWO, DSWO1, DSWO2 and DSWO3, respectively. It can be inferred from the zeta potential values that with an increase in oxygen vacancies, the values were less negative (−23.1, −18.6, −18.2 mV for DSWO1, DSWO2, DSWO3, respectively) as compared to pristine SWO (−41.3 mV). This change can be ascribed to the electronegative nature of the oxygen vacancies, which increases the overall positive charge on the surface of the catalysts.55
Adsorption studies of the RhB dye
The adsorption of cationic dye molecules like RhB and CRV is the result of electrostatic interactions due to the negatively charged surface of the photocatalyst (DSWO2). The experimental equilibrium adsorption of DSWO2 is determined by varying the concentration of the RhB solution from 1 × 10−5 to 4 × 10−5 M. The adsorption capacity of catalyst was determined by dispersing the catalyst over a fixed amount of time (60 min). The resulting data were fitted into Langmuir and Freundlich isotherms, as given in eqn (5) and (6):56–58| | | Ce/qe = 1/KLqm + Ce/qm | (5) |
| | | logqe = logKF + logCe/n | (6) |
Here, Ce and qe correspond to the equilibrium concentration (mg L−1) and equilibrium adsorption capacity (mg g−1), respectively, qm is the maximum theoretical amount of dye adsorbed on the DSWO2 surface (mg g−1), kL and kF are bonding energy of adsorption (L mg−1) and adsorption capacity of the adsorbent (mg1−n Ln g−1), respectively, and n is the constant related to the adsorption intensity and adsorption capacity.
It was observed that DSWO2 exhibits the maximum adsorption capacity of 13.46 mg g−1 for a 4 × 10−5 M solution of RhB dye (Fig. S9a, ESI†). Furthermore, the adsorption data were well-fitted into the pseudo-second order kinetics model, which was confirmed by the correlation factor (R2 = 0.99) for all of the concentrations. The resultant data fitted into the Langmuir and Freundlich isotherms is shown in Fig. S9c and d (ESI†). The correlation factors for both models are 0.96 and 0.94, respectively. The fitting of data clearly shows that the adsorption of RhB over the surface of DSWO2 follows the Langmuir model. Furthermore, the slope and intercept of Fig. S9c† produced the values of qm and kL, which were found to be 21.0 mg g−1 and 0.144 L mg−1, respectively.
Mechanism of photocatalytic degradation
The light irradiated on the photocatalyst surface leads to the generation of electron–hole pairs (e−–h+). These electron–hole pairs take part in the redox reaction. The photogenerated charge carriers can react with water molecules to form superoxide anions (O2*−) and hydroxyl radical (*OH). These reactive oxygen species are accountable for the redox reaction that occurs on the surface of the photocatalyst. To check the role of the active species, scavenger studies were performed for the photocatalytic degradation of both RhB and CRV dyes, as shown in Fig. 10a and b. Chloroform (CHCl3), ethylenediamine (EDTA) and isopropyl alcohol (IPA) were used as scavengers for O2*−, h+ and *OH radicals, respectively. For CRV, the degradation was decreased to 32%, 4% and 37% using CHCl3, EDTA and IPA, respectively, as compared to the situation without the scavenger (71%). Based on these results, the role of the active species in the degradation of CRV can be described as follows: h+ > O2*− > *OH. Also, the photocatalytic degradation of RhB in the presence of CHCl3, EDTA and IPA decreased to 48%, 2% and 22%, respectively; whereas, it was 86% without the use of any scavenger. In a similar way, based on the obtained results, the role of the active species in the degradation of RhB can be described as follows: h+ > *OH > O2*−. In the degradation process of CRV and RhB, h+ acted as the most active species as compared to *OH and O2*−.
 |
| | Fig. 10 (a and b) Scavenger study results for CRV and RhB and (c and d) recyclability of DSWO2 for up to 5 cycles for CRV and RhB. | |
Recyclability and photostability are two important aspects of a photocatalyst. The recyclability of DSWO2 was checked for up to five cycles for CRV and RhB (Fig. 10c and d). It can be seen from Fig. 10c that the degradation of CRV was decreased from 71% to 50%. Also, a decrease in RhB degradation was seen from 86% to 69% after 5 cycles. This decrease in CRV and RhB degradation can be assigned to the photocatalyst loss during repeated recycling steps. Furthermore, XPS analysis of the recovered DSWO2 sample was performed to examine its stability after repeated use for five consecutive cycles (Fig. S10, ESI†). It was found that the survey spectrum showed all of the peaks of the constituent elements (Sb 3d, O 1s, W 4f). Furthermore, there were no changes seen in the individual high-resolution spectra of the elements, which indicates the structural stability of the catalyst after multiple cycles. Furthermore, the PXRD pattern of the DSWO2 catalyst was obtained after the recycling experiments, and was compared with that of the fresh DSWPO2 catalyst. The PXRD pattern of the DSWO2 catalyst matches well before and after the photocatalytic experiments, which suggests its structural stability even after five cycles. Also, the original color of DSWO2 was regained after multiple washing (Fig. S11†).
Based on the experimental data, a suitable reaction mechanism for the degradation of pollutants on the surface of DSWO2 is shown in Scheme 2. The VB and CB positions of DSWO2 are at 2.00 and 0.04 eV, respectively. The irradiated light on the surface of DSWO2 generated electron (e−) and hole (h+) pairs. These photo-generated e− and h+ pairs react with H2O and O2 to form active oxygen species. The formation of O2*− on the DSWO2 surface is negligible because of the less negative potential of DSWO2 CB (0.04 eV), as compared to the standard redox potential of O2/O2*− (−0.33 eV). Also, the redox potential of *OH/–OH and *OH/H2O are 1.99 eV and 2.68 eV, respectively.59 So, the direct formation of *OH radicals from H2O is not possible because of the less positive potential of DSWO2 VB (2.00 eV). However, *OH radicals can be formed by OH− anions with photo-generated h+. So, the photo-generated *OH radicals can take part in the degradation process of CRV and RhB dyes. Furthermore, the h+ in the VB of DSWO2 can be utilized directly for the degradation of CRV and RhB dyes. In addition, the oxygen vacancies present in the photocatalyst can act as trapping sites for the photo-generated electrons. These electrons can also generate O2*− radicals by reacting with O2 molecules, and can further help in the degradation of RhB and CRV dyes. Based on the above points, the degradation mechanism has been given below:
| h+VB + RhB/CRV → Degraded products |
| *OH + RhB/CRV → Degraded products |
| O2*− + RhB/CRV → Degraded products |
 |
| | Scheme 2 Schematic illustration of CRV and RhB degradation over the surface of DSWO2. | |
All of the photo-generated reactive species (h+, *OH, O2*−) oxidise RhB and CRV molecules to smaller fragments, and are further converted to CO2 and H2O.
Photoluminescence studies
In order to investigate the effect of oxygen vacancies on the photo-generated charge carriers, photoluminescence (PL) studies were done for pristine SWO and the oxygen vacancy-rich samples DSWO1, DSWO2, and DSWO3. These studies were carried out at room temperature with an excitation wavelength of 350 nm. A high charge recombination rate corresponds to high PL intensity. As per the PL spectra obtained (Fig. S11) (ESI†), SWO shows the maximum PL emission, which shows its high charge recombination rate and thereby accounts for its low photocatalytic activity. The decrease in PL emission intensity was observed for the oxygen vacancy-rich samples. This can be assigned to the presence of oxygen vacancies. As the photo-generated electrons get trapped on the oxygen vacancy sites, the recombination rate decreases and hence the photocatalytic activity increases. As the oxygen vacancies increase, the PL intensity decreases. Among all the photocatalysts, DSWO3 shows the lowest recombination rate, but the photocatalytic activity is higher for DSWO2. That is because in the case of DSWO3, defects are created on the surface as well as in the bulk. So, the defects that are in bulk will also trap electrons, but those electrons will not be able to effectively participate in the reaction. The PL studies reveal the recombination behavior of the photo-generated charge carriers, and support the observed photocatalytic activity of the samples.
Conclusions
In summary, we synthesized pristine sample (SWO) and defect rich samples (DSWO1, DSWO2 and DSWO3) with different concentrations of oxygen vacancies using ascorbic acid in varying amounts. Their structure, morphology, optical properties, and photocatalytic activities have been investigated in detail. Specifically, the differences in the morphology after incorporation of oxygen vacancies were examined by SEM and TEM analysis. Furthermore, the increase in the binding energy of the oxygen vacancy-rich DSWO2 sample was confirmed by XPS studies. In addition, the effect of varying oxygen vacancies was seen on the band gap and CB values. These samples were then utilized for the degradation of colored pollutants like CRV and RhB dyes, and a colorless pollutant, LFX. It was found that one of the catalysts having the optimal amount of oxygen vacancies (DSWO2) was the best photocatalyst, showing a degradation of 71% and 86% for CRV and RhB dyes, respectively. Based on the zeta potential values of DSWO2, it was found to be more selective for cationic pollutants than anionic and neutral pollutants. Also, the degradation of only cationic dyes in the mixture shows its selectivity. The increase in photocatalytic activity of DSWO2 can be ascribed to its increased surface area, decreased band gap, and increased light absorption. Furthermore, the oxygen vacancies behave as electron trapping sites, which can further decrease the recombination rate of e− and h+. Overall, this work provides insight into the defect (vacancy)-engineered materials and its effect on the structural properties, as well as the photocatalytic activities. This work also provides a good example of perovskite oxide-based band gap engineered materials as efficient single component photocatalysts for environmental remediation applications.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We are thankful to the Advanced Material Research Centre (AMRC), IIT Mandi, for their laboratory and the characterization facilities. The financial support obtained from the Department of Science and Technology – Science and Engineering Research Board (DST-SERB) under the core research grant (CRG/2022/003559) is gratefully acknowledged. Manisha Sharma and Anitya Sharma thank the Ministry of Education (MoE), Government of India, for their respective doctoral research fellowships.
References
- T. Kalhorizadeh, B. Dahrazma, R. Zarghami, S. Mirzababaei, A. M. Kirillov and R. Abazari, New J. Chem., 2022, 46, 9440–9450 RSC
 .
.
- M. Borges, H. de Paz Carmona, M. Gutiérrez and P. Esparza, Catalysts, 2023, 13, 1023 CrossRef CAS .
- C. Sivaraman, S. Vijayalakshmi, E. Leonard, S. Sagadevan and R. Jambulingam, Catalysts, 2022, 12, 544 CrossRef CAS .
- C.-C. Wang, J.-R. Li, X.-L. Lv, Y.-Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 RSC .
- Y. Vasseghian, A. Khataee, E.-N. Dragoi, M. Moradi, S. Nabavifard, G. O. Conti and A. M. Khaneghah, Arabian J. Chem., 2020, 13, 8458–8480 CrossRef CAS .
- T. Nunes, M. Teodoro, M. Bomio and F. Motta, Dalton Trans., 2022, 51, 18234–18247 RSC .
- A. Kumar, S. Kumar, A. Bahuguna, A. Kumar, V. Sharma and V. Krishnan, Mater. Chem. Front., 2017, 1, 2391–2404 RSC .
- A. Kumar, S. Kashyap, M. Sharma and V. Krishnan, Chemosphere, 2022, 287, 131988 CrossRef CAS PubMed .
- Z. Wang, W. Li, J. Wang, Y. Li and G. Zhang, Chemosphere, 2023, 311, 137000 CrossRef CAS PubMed .
- C. Díaz, M. Segovia and M. L. Valenzuela, Photochem, 2022, 2, 609–627 CrossRef .
- V. Gadore, S. R. Mishra and M. Ahmaruzzaman, Environ. Sci. Pollut. Res., 2023, 1–48 Search PubMed .
- F.-X. Wang, Z.-C. Zhang, X.-H. Yi, C.-C. Wang, P. Wang, C.-Y. Wang and B. Yu, CrystEngComm, 2022, 24, 5557–5561 RSC .
- H. Jindal, D. Kumar, M. Sillanpaa and M. Nemiwal, Inorg. Chem. Commun., 2021, 131, 108786 CrossRef CAS .
- R. P. Gaikwad, D. R. Naikwadi, A. V. Biradar and M. B. Gawande, ACS Appl. Nano Mater., 2023, 6, 1859–1869 CrossRef CAS .
- M. Sharma, A. Kumar and V. Krishnan, Nanotechnology, 2022, 33, 275702 CrossRef PubMed .
- J. Zhang, Y. Fang, Y. Zhang, Y. Lin, Y. Gui and L. Liu, ACS Appl. Nano Mater., 2023, 6, 22301–22310 CrossRef CAS .
- X. Liang, G. Wang, X. Dong, G. Wang, H. Ma and X. Zhang, ACS Appl. Nano Mater., 2018, 2, 517–524 CrossRef .
- A. Kumar, M. Sharma, S. Sheoran, S. Jaiswal, A. Patra, S. Bhattacharya and V. Krishnan, Nanoscale, 2023, 15, 11667–11680 RSC .
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS .
- J. P. Attfield, P. Lightfoot and R. E. Morris, Dalton Trans., 2015, 44, 10541–10542 RSC .
- A. Kumar, M. Kumar, V. N. Rao, M. V. Shankar, S. Bhattacharya and V. Krishnan, J. Mater. Chem. A, 2021, 9, 17006–17018 RSC .
- R. Shi, Y. Zhao, G. I. Waterhouse, S. Zhang and T. Zhang, ACS Catal., 2019, 9, 9739–9750 CrossRef CAS .
- F. Liu and Z. Fan, Chem. Soc. Rev., 2023, 52, 1723–1772 RSC .
- A. Kumar and V. Krishnan, Adv. Funct. Mater., 2021, 31, 2009807 CrossRef CAS .
- R.-R. Ding, W.-Q. Li, C.-S. He, Y.-R. Wang, X.-C. Liu, G.-N. Zhou and Y. Mu, Appl. Catal., B, 2021, 291, 120069 CrossRef CAS .
- C. Zhao, Y. Yang, L. Luo, S. Shao, Y. Zhou, Y. Shao, F. Zhan, J. Yang and Y. Zhou, Sci. Total Environ., 2020, 747, 141533 CrossRef CAS PubMed .
- L. Chen, B. Xu, M. Jin, L. Chen, G. Yi, B. Xing, Y. Zhang, Y. Wu and Z. Li, J. Mol. Struct., 2023, 1278, 134911 CrossRef CAS .
- D. Karmakar, S. Datta and D. Jana, J. Alloys Compd., 2021, 881, 160586 CrossRef CAS .
- S.-P. Hu, C.-Y. Xu, F.-X. Ma, L. Cao and L. Zhen, Dalton Trans., 2014, 43, 8439–8445 RSC .
- W. Li, Z. Wang, Y. Li, J. B. Ghasemi, J. Li and G. Zhang, J. Hazard. Mater., 2022, 424, 127595 CrossRef CAS PubMed .
- Y. Zhang, H. Liu, J. Peng, J. Guo, L. Ding, X. Cao, Y. Chang, W. B. Soltan and G. Liu, J. Environ. Chem. Eng., 2024, 12, 111548 CrossRef CAS .
- Q. Peng, Q. Ruan, B. Wang, J. Liu, C. Huang, X. Zhu, D. Li, L. Liu, Y. Wang and X. Zhang, Colloids Surf., A, 2024, 681, 132724 CrossRef CAS .
- L. Zhang, T. Xu, X. Zhao and Y. Zhu, Appl. Catal., B, 2010, 98, 138–146 CrossRef CAS .
- X. Yang, H. Wu and C. Feng, J. Mater. Sci.: Mater. Electron., 2020, 31, 4761–4768 CrossRef CAS .
- D. S. Aidhy, B. Liu, Y. Zhang and W. J. Weber, Comput. Mater. Sci., 2015, 99, 298–305 CrossRef CAS .
- M. Tyunina, O. Pacherova, T. Kocourek and A. Dejneka, Sci. Rep., 2021, 11, 15247 CrossRef CAS PubMed .
- C. Huang, S. Ma, Y. Zong, J. Gu, J. Xue and M. Wang, Photochem. Photobiol. Sci., 2020, 19, 1697–1706 CrossRef CAS PubMed .
- R. Wang, Z. Jiang, L. Xu and C. Liu, J. Mater. Sci.: Mater. Electron., 2021, 32, 6931–6941 CrossRef CAS .
- U. Rafiq and K. Majid, J. Mater. Sci.: Mater. Electron., 2019, 30, 5965–5977 CrossRef CAS .
- K. Chu, Y.-h. Cheng, Q.-q. Li, Y.-p. Liu and Y. Tian, J. Mater. Chem. A, 2020, 8, 5865–5873 RSC .
- B. Miao, Y. Chu, X. Zheng and H. Su, Mater. Sci. Semicond. Process., 2021, 125, 105636 CrossRef CAS .
- Y. Wang, K. Wang, J. Wang, X. Wu and G. Zhang, J. Mater. Sci. Technol., 2020, 56, 236–243 CrossRef CAS .
- Y. Lv, W. Yao, R. Zong and Y. Zhu, Sci. Rep., 2016, 6, 19347 CrossRef CAS PubMed .
- B. Huang and J. N. Hart, Phys. Chem. Chem. Phys., 2020, 22, 1727–1737 RSC .
- L. Yang, Y. Xin, C. Yao and Y. Miao, J. Mater. Sci.: Mater. Electron., 2021, 32, 13382–13395 CrossRef CAS .
- J. Jin, J. Sun, K. Lv, X. Guo, Q. Hou, J. Liu, J. Wang, Y. Bai and X. Huang, J. Colloid Interface Sci., 2022, 605, 342–353 CrossRef CAS PubMed .
- X. Ren, Z. Zhang, Y. Wang, J. Lu, J. An, J. Zhang, M. Wang, X. Wang and Y. Luo, RSC Adv., 2019, 9, 15229–15237 RSC .
- Y. Zhang, L. Guo, Y. Wang, T. Wang, T. Ma, Z. Zhang, D. Wang, B. Xu and F. Fu, J. Mater. Sci. Technol., 2022, 110, 152–160 CrossRef CAS .
- A. Kumar, Y. Singla, M. Sharma, A. Bhardwaj and V. Krishnan, Chemosphere, 2022, 308, 136212 CrossRef CAS PubMed .
- Y.-R. Huang, Y. Kong, H.-Z. Li and X.-M. Wei, Environ. Technol. Innovation, 2020, 18, 100780 CrossRef .
- H.-J. Fan, S.-T. Huang, W.-H. Chung, J.-L. Jan, W.-Y. Lin and C.-C. Chen, J. Hazard. Mater., 2009, 171, 1032–1044 CrossRef CAS PubMed .
- P. A. Ajibade and A. E. Oluwalana, Nanomaterials, 2021, 11, 2000 CrossRef CAS PubMed .
- R. Xie, K. Fang, Y. Liu, W. Chen, J. Fan, X. Wang, Y. Ren and Y. Song, J. Mater. Sci., 2020, 55, 11919–11937 CrossRef CAS .
- T. S. Natarajan, M. Thomas, K. Natarajan, H. C. Bajaj and R. J. Tayade, Chem. Eng. J., 2011, 169, 126–134 CrossRef CAS .
- D. Cui, L. Wang, K. Xu, L. Ren, L. Wang, Y. Yu, Y. Du and W. Hao, J. Mater. Chem. A, 2018, 6, 2193–2199 RSC .
- H. Kaur, S. Walia, A. Karmakar, V. Krishnan and R. R. Koner, J. Environ. Chem. Eng., 2022, 10, 106667 CrossRef CAS .
- H. Kaur, R. Kumar, A. Kumar, V. Krishnan and R. R. Koner, Dalton Trans., 2019, 48, 915–927 RSC .
- S. Mohanty, M. Sharma, A. Kumar and V. Krishnan, J. Phys. Chem. C, 2023, 127, 17711–17722 CrossRef CAS .
- A. Kumar, V. Navakoteswara Rao, A. Kumar, M. Venkatakrishnan Shankar and V. Krishnan, ChemPhotoChem, 2020, 4, 427–444 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Material characterization; calculated crystallite size, lattice strain and lattice parameters; EDAX and elemental mapping of SWO; XPS survey spectra of SWO and DSWO2; Tauc plots for SWO, DSWO1, DSWO2, and DSWO3; BET plots for SWO, DSWO1, DSWO2, and DSWO3; UV-vis absorption spectra of CRV, RhB, LFX, and the mixture of dyes; Zeta potential values for SWO, DSWO1, DSWO2, and DSWO3; Langmuir adsorption isotherm and Freundlich adsorption isotherm of RhB on DSWO2; XPS spectra of the recycled DSWO2 photocatalyst; PXRD of the recycled sample; PL spectra of SWO, DSWO1, DSWO2, and DSWO3; Comparison table of different material activities. See DOI: https://doi.org/10.1039/d4dt00183d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*