
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aminophosphonium organocatalysts for the ring-opening copolymerisation of epoxide and cyclic anhydride†
Ella F.
Clark
 a,
Estelle
Dunstan
a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*c
a,
Estelle
Dunstan
a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*c
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
bMaterials and Chemical Characterisation Facility, University of Bath, UK
cDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: antoine.buchard@york.ac.uk
First published on 22nd October 2024
Abstract
The Kirsanov reaction has been used to synthesise air stable, efficient and selective bifunctional aminophosphonium catalysts for the alternating ring-opening copolymerisation of cyclohexene oxide and phthalic anhydride without the need for a co-initiator.
Ring-Opening Copolymerisation (ROCOP) is a living polymerisation technique, which produces polyesters by selectively incorporating epoxides and cyclic anhydrides in an alternating fashion.1,2 Control over the rate and alternating selectivity is determined by the catalyst, with the first example reported by Fischer in 1960.3 Since then, numerous highly active catalysts have been developed, including metal complexes based on porphyrin (Al(III), Cr(III), Co(III), Mn(III)),4–6 salen (Al(III), Cr(III), Co(III), Mn(III)),7–10 β-diiminate (Zn(II))11 and dinucleating Schiff base (e.g., Al(III)/K(I))12,13 ligands.14 More recently, organocatalysts have emerged as effective catalysts for ROCOP.15 The first metal free catalysis for ROCOP was reported by Gnanou, Feng and coworkers; Et3B, in combination with an onium halide or onium alkoxide initiator, was shown to selectively catalyse the ROCOP of propylene oxide (and later other epoxides) and CO2.16,17 This has inspired other organocatalytic systems based on the combination of a Lewis acid (e.g., thio(urea)) and nucleophilic initiator (e.g., bis(triphenylphosphoranylidene)ammonium chloride (PPNCl)).18–23 Phosphazene bases and benzyl alcohol have also been used in combination with thiourea moieties.21,24,25In situ, this creates an aminophosphonium cation, which can act as a Lewis acid catalyst, and an alkoxide initiator (a system also efficient for ROP24).
Aminophosphonium salts can also be directly produced through the Kirsanov reaction, via the oxidation of a phosphine with Br2, followed by nucleophilic addition of an amine to form an aminophosphonium bromide salt, although anion exchange is possible.26 Furthermore, such strategy relies on a wide pool of commercially available primary amines. We therefore envisioned that the Kirsanov reaction could be used to readily synthesise and tailor simple and air/moisture stable aminophosphonium salts towards ROCOP.
We hypothesised that the combination of the aminophosphonium moiety with a tertiary amine would weaken the N–H bond, improving epoxide activation as well as facilitating proton shuttling towards chain end stabilisation, with the bromide anion initiating the polymerisation. 1 was thus generated in one pot by reaction of readily available triphenyl phosphine (PPh3) with commercial, bifunctional N,N-dimethylethylenediamine (Fig. 1), isolated in near quantitative yield (99%) as an air stable white powder, and characterised by multinuclear (31P{1H}, 13C{1H}, 1H) NMR spectroscopy (Fig. S1 and S2, ESI†). Mass spectrometry detected a singly positively charged species with an m/z = 349.18 (Fig. S8, ESI†), and X-ray diffraction analysis of single crystals also confirmed a monocationic structure (Fig. 1). Efforts to protonate the tertiary amine by addition of HCl (2 mol L−1 in Et2O) affected no change in the 31P{1H} NMR spectrum (Fig. S22, ESI†).
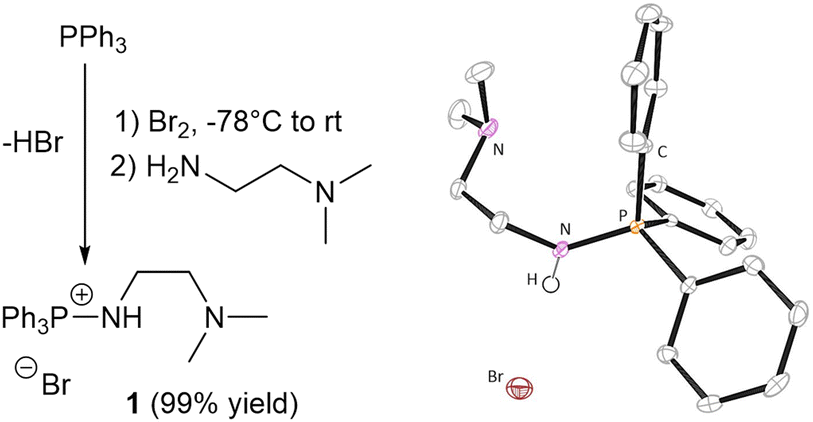 | ||
| Fig. 1 Left: Synthesis of 1. Right: Molecular structure of 1 obtained by X-ray diffraction analysis. Thermal ellipsoids are drawn to the 50% probability level. Hydrogen atoms are omitted for clarity, except for the aminophosphonium proton. | ||
Contrary to previous syntheses of aminophophonium salts using the Kirsanov reaction, no additional base is required to trap side-product HBr, which we postulate is removed during work-up. It is worth noting that when an additional base is used to trap HBr (e.g., DABCO), 1 is also unequivocally obtained, as evidenced by identical characterisation data.
1 was next tested for the benchmark ROCOP of cyclohexene oxide (CHO) and phthalic anhydride (PA), with no solvent and at 120 °C. At [CHO]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PA]0:[1]0 loadings of 400:200:1, 1 was found to catalyse the reaction, producing polymers with good activity (TOF = 78–80 h−1) and good selectivity for ester vs. ether linkages (86–91%) (Table 1, entries 1 and 2; Fig. S23, ESI†). At full conversion of PA, the resulting polymer had an Mn,SEC of 14100 g mol−1 as measured by SEC (against narrow polystyrene standards) (Table 1, entry 2). The alternating nature of the resultant polymers was further evidenced by MALDI-ToF mass spectrometry analysis which detected polymeric series with a repeat unit of 246 g mol−1, consistent with an alternating PA/CHO polymer sequence (Fig. S24, ESI†). The living/controlled polymerization character of the ROCOP was illustrated by the linear increase of the Mn,SEC with PA conversion whilst maintaining low dispersities (Fig. S25, ESI†). SEC traces showed a bimodal distribution which were attributed to residual diol impurities in CHO and diacid impurities in PA, limiting the obtention of high molecular mass (Fig. S26, ESI†).
:[PA]0:[1]0 loadings of 400:200:1, 1 was found to catalyse the reaction, producing polymers with good activity (TOF = 78–80 h−1) and good selectivity for ester vs. ether linkages (86–91%) (Table 1, entries 1 and 2; Fig. S23, ESI†). At full conversion of PA, the resulting polymer had an Mn,SEC of 14100 g mol−1 as measured by SEC (against narrow polystyrene standards) (Table 1, entry 2). The alternating nature of the resultant polymers was further evidenced by MALDI-ToF mass spectrometry analysis which detected polymeric series with a repeat unit of 246 g mol−1, consistent with an alternating PA/CHO polymer sequence (Fig. S24, ESI†). The living/controlled polymerization character of the ROCOP was illustrated by the linear increase of the Mn,SEC with PA conversion whilst maintaining low dispersities (Fig. S25, ESI†). SEC traces showed a bimodal distribution which were attributed to residual diol impurities in CHO and diacid impurities in PA, limiting the obtention of high molecular mass (Fig. S26, ESI†).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Cat | [CHO]0:[PA]0:[cat]0 |
Temp. (°C) | Time (min) | PA Conv.b (%) | TONc | TOFd (h−1) | CHO Conv.e (%) | % Select.f | M n,SEC (ĐM) |
| a Reactions carried out at [CHO]0 = 5.8 mol L−1, neat at 120 °C unless otherwise stated. b Conversion of PA determined by 1H NMR spectroscopy by relative integration of the aromatic protons in PA (CDCl3, δ 7.95–7.80 ppm) and poly(CHO-alt-PA) (CDCl3, δ 7.52–7.33 ppm). c TON = (moles of PA consumed) × (moles of catalyst)−1. d TOF = (moles of PA consumed) × (moles of catalyst)−1 × (time of reaction)−1. e Conversion of CHO determined by 1H NMR spectroscopy by relative integration of protons singlets in CHO (CDCl3, δ 3.02 ppm) and poly(CHO-alt-PA) (CDCl3, δ 5.06 ppm). f Selectivity of ester vs. ether links determined by 1H NMR spectroscopy using the relative integration of the ester and ether linkages polyester (CDCl3, δ = 5.13 ppm) and polyether (CDCl3, δ = 3.61–3.30 ppm). g M n,SEC in g mol−1, calculated by SEC relative to polystyrene standards in tetrahydrofuran (THF) eluent; ĐM = Mw/Mn. h 1 was acidified with 1 equivalent of HCl prior use in catalysis (see ESI). | ||||||||||
| 1 | 1 | 400:200:1 |
120 | 60 | 39 | 78 | 78 | 24 | 86 | 6100 (1.20) |
| 2 | 1 | 400:200:1 |
120 | 150 | >99 | 200 | 80 | 63 | 91 | 14100 (1.23) |
| 3 | 2 | 400:200:1 |
120 | 60 | 9 | 18 | 18 | 5 | 83 | <1000 |
| 4 | 3 | 400:200:1 |
120 | 40 | 93 | 186 | 279 | 49 | 96 | 14900 (1.14) |
| 5 | 4 | 400:200:1 |
120 | 60 | 14 | 28 | 28 | 7 | 66 | <1000 |
| 6 | 1 | 200:200:1 |
120 | 30 | 36 | 72 | 144 | 37 | 93 | 3900 (1.16) |
| 7 | 1 | 200:200:1 |
120 | 60 | 46 | 92 | 92 | 47 | 92 | 6800 (1.20) |
| 8 | 1 | 400:200:1 |
140 | 15 | 29 | 58 | 232 | 16 | 89 | 3600 (1.14) |
| 9 | 1 | 400:200:1 |
160 | 15 | 18 | 36 | 144 | 15 | 83 | <1000 |
| 10 | 1 | 400:0:1 |
120 | 60 | — | — | — | — | — | <1000 |
| 11 | 1 | 400:200:1 |
120 | 120 | 28 | 56 | 28 | 19 | 76 | 2000 (1.20) |
| 12 | PPNCl | 400:200:1 |
120 | 30 | 100 | 200 | >400 | 55 | 62 | <1000 |
| 13 | Ph3PO | 400:200:1 |
120 | 60 | <1 | — | — | <1 | — | <1000 |
This was corroborated using MALDI-ToF mass spectrometry which detected several polymer distributions, including higher Mn,SEC species initiated from residual cyclohexane diol (Fig. S24, ESI†). Lower Mn,SEC species showed polymers with a 2-bromo-cyclohexanol end group, confirming that the bromide anion of 1 can act as an initiator via nucleophilic attack onto CHO.
Catalytic activity could be further increased to 232 h−1 by performing the reaction at 140 °C (Table 1, entry 8). However, further increasing the temperature to 160 °C led to a decrease in activity (TOF = 144 h−1), suggesting side reactions or degradation (Table 1, entry 9). Lowering the reaction temperature to 60 °C significantly reduced the TOF, although ROCOP was possible (TOF = 5 h−1). ROCOP of CHO and PA at a [CHO]0:[PA]0:[1]0 ratio of 200:200:1 resulted in non-significant improvement of the selectivity towards ester linkages (Table 1, entries 6 and 7). Control experiments with [CHO]0:[PA]0:[1]0 ratios of 400:2:1 and 400:0:1 showed no conversion after 90 minutes, indicating 1 does not catalyse CHO ROP (Table 1, entry 10). Acidified 1 (1·HCl) was also tested in catalysis, and showed significantly reduced activity compared to its non-acidified counterpart (TOF = 28 vs. 78 h−1) with notably reduced selectivity (76% of ester linkages) (Table 1, entry 11 vs. 1). This points towards the monocationic species 1 being the active catalytic species. 1 is more selective towards polyesters than previous iminophosphorane organocatalysts.25 However, where comparisons are possible, 1 is less active than the ammonium- or phosphonium-containing organoboron systems designed by Wu and team (TOF = 80 vs. 258 vs. 1024 h−1; respectively).27,28
Monofunctional aminophosphonium salt 2 was synthesised using benzyl amine and tested in catalysis (Table 1, entry 4), but showed significantly lower activity compared to 1, with 9% PA conversion after 60 min. Commercial bifunctional phosphonium/tertiary amine salt, 3, was also tested. After 40 minutes the reaction had solidified with a PA conversion of 93%, producing poly(CHO-alt-PA) with an Mn,SEC of 14900 g mol−1 and ĐM of 1.14 (Table 1, entry 3). Collectively, these results suggest the key role of the tertiary amine group for catalysis, likely through the facilitation of proton shuttling towards the activation of monomers and the stabilisation of transition states and growing chain ends. Finally, a diaminophosphonium salt, 4, was synthesised and tested for the ROCOP of CHO. 13% conversion was achieved after 60 minutes, showing limited activity, consistent with the results obtained with aminophosphonium salts that do not feature a tertiary amine (2) or have an additional ammonium group (1·HCl). Catalysis with commercial PPNCl achieved a high TOF (>400 h−1) but SEC analysis showed that only oligomers were produced (Mn,SEC < 1000 g mol−1) (Table 1, entry 12). Previous studies have found PPNCl capable of catalysing CHO/PA ROCOP, at 110 °C in toluene.29 Triphenyl phosphine oxide was shown to be catalytically inactive (Table 1, entry 13).
To gather insights into the catalytically active form of 1, in situ31P{1H} NMR reaction monitoring was carried out. Polymerisations were performed at a [PA]0:[1]0 ratio of 50:1 in an excess of CHO at 120 °C under argon in a J Young NMR tube. After 60 minutes, all PA had reacted according to 1H NMR analysis. No change in the signal of 1 was observed in the 31P{1H} NMR spectra throughout the experiment, indicating either that the phosphorus remains remote from the active catalytic site or that the resting state of the catalyst is structurally similar to 1 in CHO. However, no degradation of the catalyst was observed by 31P{1H} NMR analysis, and coupled with the inactivity of the Ph3PO, this points towards 1 being a true catalyst which remains intact throughout polymerisation (Fig. S27, ESI†).
The kinetic profile of the ROCOP of CHO/PA, catalysed by 1, was monitored with in situ FTIR spectroscopy. Polymerisations were performed at a [CHO]0:[PA]0:[1]0 loading of 800:400:1 at 120 °C without solvent and followed by analysis of the absorption intensity at 1783 cm−1, corresponding to the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration in PA. A zero-order dependence on [PA] was determined from linear plots of [PA] against time (Fig. S28, ESI†). The order with respect to CHO was next considered, CHO consumption being inferred by measuring the growth of the νCO vibration at 1722 cm−1 in poly(CHO-alt-PA). Plots of ln([CHO]t/[CHO]0) against time showed a linear correlation, revealing a first order dependence on [CHO] (kCHO = 4.42 × 10−5 s−1; Fig. S29, ESI†). We next demonstrated that 1 was catalytically active when handled in air (Fig. S28, ESI†). While the kinetic profile unexpectedly deviated from linearity, which could be due to side reactions including progressive catalyst deactivation, ROCOP proceeded faster in air than under argon. Such rate acceleration has also been observed for metal-catalysed ROCOP,30 when chain transfer agents (CTAs) are used or in the presence of air/water, but is yet to be fully rationalised. The likely moisture in the air functioning as CTA was also reflected in the final molar masses, with polymers with higher Mn,SEC produced under argon (16300 g mol−1, ĐM = 1.23) and lower Mn,SEC, monomodal polymers produced in air (4400 g mol−1, ĐM = 1.10). Only a small number of air tolerant catalysts for the ROCOP of CHO/PA have been reported so far.13,30,31
O vibration in PA. A zero-order dependence on [PA] was determined from linear plots of [PA] against time (Fig. S28, ESI†). The order with respect to CHO was next considered, CHO consumption being inferred by measuring the growth of the νCO vibration at 1722 cm−1 in poly(CHO-alt-PA). Plots of ln([CHO]t/[CHO]0) against time showed a linear correlation, revealing a first order dependence on [CHO] (kCHO = 4.42 × 10−5 s−1; Fig. S29, ESI†). We next demonstrated that 1 was catalytically active when handled in air (Fig. S28, ESI†). While the kinetic profile unexpectedly deviated from linearity, which could be due to side reactions including progressive catalyst deactivation, ROCOP proceeded faster in air than under argon. Such rate acceleration has also been observed for metal-catalysed ROCOP,30 when chain transfer agents (CTAs) are used or in the presence of air/water, but is yet to be fully rationalised. The likely moisture in the air functioning as CTA was also reflected in the final molar masses, with polymers with higher Mn,SEC produced under argon (16300 g mol−1, ĐM = 1.23) and lower Mn,SEC, monomodal polymers produced in air (4400 g mol−1, ĐM = 1.10). Only a small number of air tolerant catalysts for the ROCOP of CHO/PA have been reported so far.13,30,31
To shed further light on the catalytic active species, modelling of the ring-opening of CHO was carried out using DFT calculations (see ESI†), having been identified by kinetics study as a key catalytic step. For simplicity, only the opening by the bromide anion (initiation step) was modelled (Fig. 2). The propagation step will be analogous but for a carboxylate growing chain as the nucleophile. Consistent with pKa values and the molecular structure inferred from X-ray diffraction, 1's most stable isomer (by 8.2 kcal mol−1) is the aminophosphonium species (P–NH) and not the quaternary ammonium salt. Activation of CHO by hydrogen bonding was found to happen preferentially at the aminophosphonium, but to be overall endergonic (+8.8 kcal mol−1). Next, the lowest energy barrier for CHO ring-opening was calculated to be the nucleophilic attack of the bromide away from the PPh3 group, which had an energy barrier of 32.5 kcal mol−1. ROCOP being quantitative at 120 °C within a few hours, circa 30 kcal mol−1 is an acceptable energy barrier. The resulting alkoxide was shown to be stabilised by the aminophosphonium moiety. The reactivity of a putative doubly protonated 1 (1·HBr) was also examined by DFT calculations (Fig. S31, ESI†). While the concurrent activation of CHO by both the ammonium and aminophosphonium moiety remained endergonic (+10.0 kcal mol−1), the barrier to ring-opening was found to be 29.3 kcal mol−1, lower but within error to what is calculated for the monocationic species at this cursory stage. This preliminary study clearly calls for further mechanistic investigation, including a full conformation search on the propagation step.
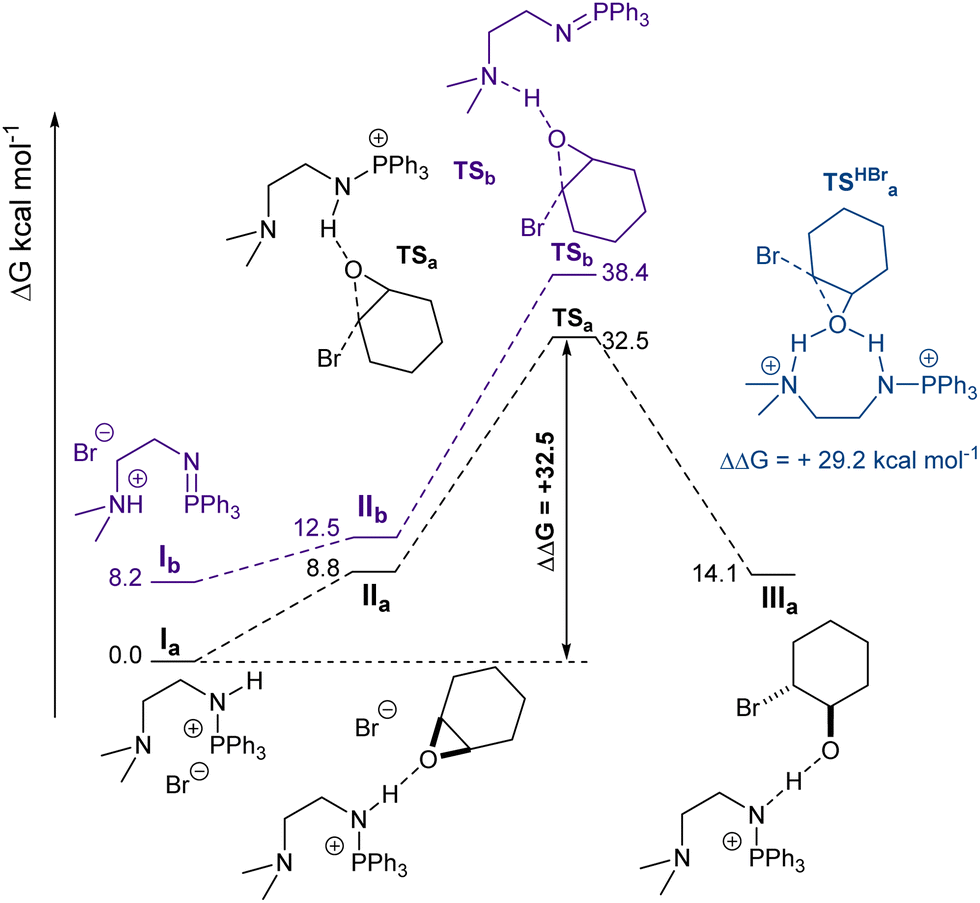 | ||
| Fig. 2 DFT computed reaction profiles for the ring-opening of CHO by 1 (initiation step). | ||
To conclude, the Kirsanov reaction has been used to synthesise a simple, novel organocatalyst which has been shown to be active in the ROCOP of CHO and PA, including in air. Our results demonstrate the potential of aminophosphonium salts, made from abundant phosphines and primary amines, as simple organocatalysts for ROCOP, and a promising and tuneable alternative to widely used PPNCl.
Research funding from the Royal Society (UF/160021 and URF\R\221027: fellowship to AB, RGF\R1\180036 studentship to EFC) is acknowledged in support of this study.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- R. F. Fischer, J. Polym. Sci., 1960, 44, 155–172 CrossRef CAS.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS.
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS.
- W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef CAS PubMed.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- J. Zhang, L. Wang, S. Liu and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202111197 CrossRef CAS PubMed.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- X. Liang, W. Wang, D. Zhao, H. Liu and Y. Zhu, Polym. Chem., 2023, 14, 4918–4926 RSC.
- J. Wang, Y. Zhu, M. Li, Y. Wang, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 61, e202208525 CrossRef CAS PubMed.
- H. Li, H. Luo, J. Zhao and G. Zhang, ACS Macro Lett., 2018, 7, 1420–1425 CrossRef CAS PubMed.
- J. Xu, X. Wang and N. Hadjichristidis, Nat. Commun., 2021, 12, 7124 CrossRef CAS PubMed.
- H. Li, J. Zhao and G. Zhang, ACS Macro Lett., 2017, 6, 1094–1098 CrossRef CAS PubMed.
- A. M. Goldys and D. J. Dixon, Macromolecules, 2014, 47, 1277–1284 CrossRef CAS.
- M. Hirschmann, R. Zunino, S. Meninno, L. Falivene and T. Fuoco, Catal. Sci. Technol., 2023, 13, 7011–7021 RSC.
- A. Picot, H. Dyer, A. Buchard, A. Auffrant, L. Vendier, P. Le Floch and S. Sabo-Etienne, Inorg. Chem., 2010, 49, 1310–1312 CrossRef CAS PubMed.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 Search PubMed.
- R. Xie, Y.-Y. Zhang, G.-W. Yang, X.-F. Zhu, B. Li and G.-P. Wu, Angew. Chem., Int. Ed., 2021, 60, 19253–19261 CrossRef CAS PubMed.
- Z. Hošťálek, O. Trhlíková, Z. Walterová, T. Martinez, F. Peruch, H. Cramail and J. Merna, Eur. Polym. J., 2017, 88, 433–447 CrossRef.
- E. J. K. Shellard, W. T. Diment, D. A. Resendiz-Lara, F. Fiorentini, G. L. Gregory and C. K. Williams, ACS Catal., 2024, 14, 1363–1374 CrossRef CAS PubMed.
- Y. Manjarrez, A. M. Clark and M. E. Fieser, ChemCatChem, 2023, 15, e202300319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; spectroscopic, mass spectrometry and crystallographic data for 1, 3 and 4; SEC traces, spectroscopic and MALDI-ToF mass spectrometry data for the polymers; DFT calculations data and associated digital repositories. CCDC 2374662 and 2374926. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03947e |
| This journal is © The Royal Society of Chemistry 2024 |