
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting the enzymatic activity of CxxC motif-containing PDI family members†
Tsubura
Kuramochi‡
ab,
Yukino
Yamashita‡
c,
Kenta
Arai
 de,
Shingo
Kanemura
a,
Takahiro
Muraoka
*cf and
Masaki
Okumura
*ab
de,
Shingo
Kanemura
a,
Takahiro
Muraoka
*cf and
Masaki
Okumura
*ab
aFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, 6-3 Aramaki-Aza-Aoba, Aoba-ku, Sendai, Miyagi 980-8578, Japan. E-mail: okmasaki@tohoku.ac.jp
bDepartment of Molecular and Chemical Life Sciences, Graduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan. E-mail: muraoka@go.tuat.ac.jp
dDepartment of Chemistry, School of Science, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan
eInstitute of Advanced Biosciences, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan
fKanagawa Institute of Industrial Science and Technology, 3-2-1 Sakato, Takatsu-ku, Kawasaki, Kanagawa 213-0012, Japan
First published on 13th May 2024
Abstract
Compounds harboring high acidity and oxidizability of thiol groups permit tuning the redox equilibrium constants of CxxC sites of members of the protein disulphide isomerase (PDI) family and thus can be used to accelerate folding processes and increase the production of native proteins by minimal loading in comparison to glutathione.
Chemical boosters of enzymatic activity provide us with a better understanding of enzymatic function and allow us to combat enzyme-related pathologies. Members of the protein disulphide isomerase (PDI) family are responsible for catalyzing oxidative protein folding in the endoplasmic reticulum (ER).1–3 Several studies have reported that PDIs play important roles in protein misfolding-related pathologies, such as amyotrophic lateral sclerosis, Alzheimer's disease (AD), Parkinson's disease, and type-II diabetes.4,5 Loss-of-function of PDIs can thus result in severe diseases, such as neurodegeneration and diabetes. Notably, redox active site dysfunction in PDIA1 and PDIA6 CxxC motifs induced by posttranslational chemical modifications has been observed in AD,6 suggesting that misfolding-related pathologies can be ameliorated using redox chemistry.7
Extracellular PDIs are promising targets for thrombosis-related and tumor diseases owing to the upregulation of the PDI expression level, and several PDI inhibitors, such as 16F16, bisphenol A, PACMA31, LOC14, and piericone, have been reported.8–12 Many of these antagonists abolish the PDI catalytic activity by acting on cysteines within CxxC motifs or by competitively inhibiting its function by binding to its substrate-binding pocket. However, in the case of PDIA1, an allosteric switch of its substrate-binding pocket increases its catalytic activity;13 yet, only a few studies were reported about an enhancer of PDIs activity.16 Regarding thiol compound-mediated catalytic activity,17 developing novel thiol compounds to target cysteines within CxxC motifs could be extremely effective.
In the ER, glutathione has a central role in thiol–disulphide exchange reactions, where both its reduced (GSH) and its oxidized (GSSG) form catalyze oxidative protein folding18 and control the PDIs redox status.19 Regarding thiol compounds that can replace glutathione (GSH; pKa = 9.17, E°′ = −256 mV), we previously reported that para-substituted N-methylated pyridinylmethanethiol (pMePySH; pKa = 7.34, E°′ = −211 mV) enhances both the acidity and oxidizability of the thiol groups, which allows several clients to accelerate oxidative folding by 1-equivalent loading as semi-enzymatic activity (Fig. 1A).20 Such a highly redox-reactive function of pMePySH by minimum loading prompted us to explore how the PDIs enzymatic activity can be controlled via the CxxC motif.
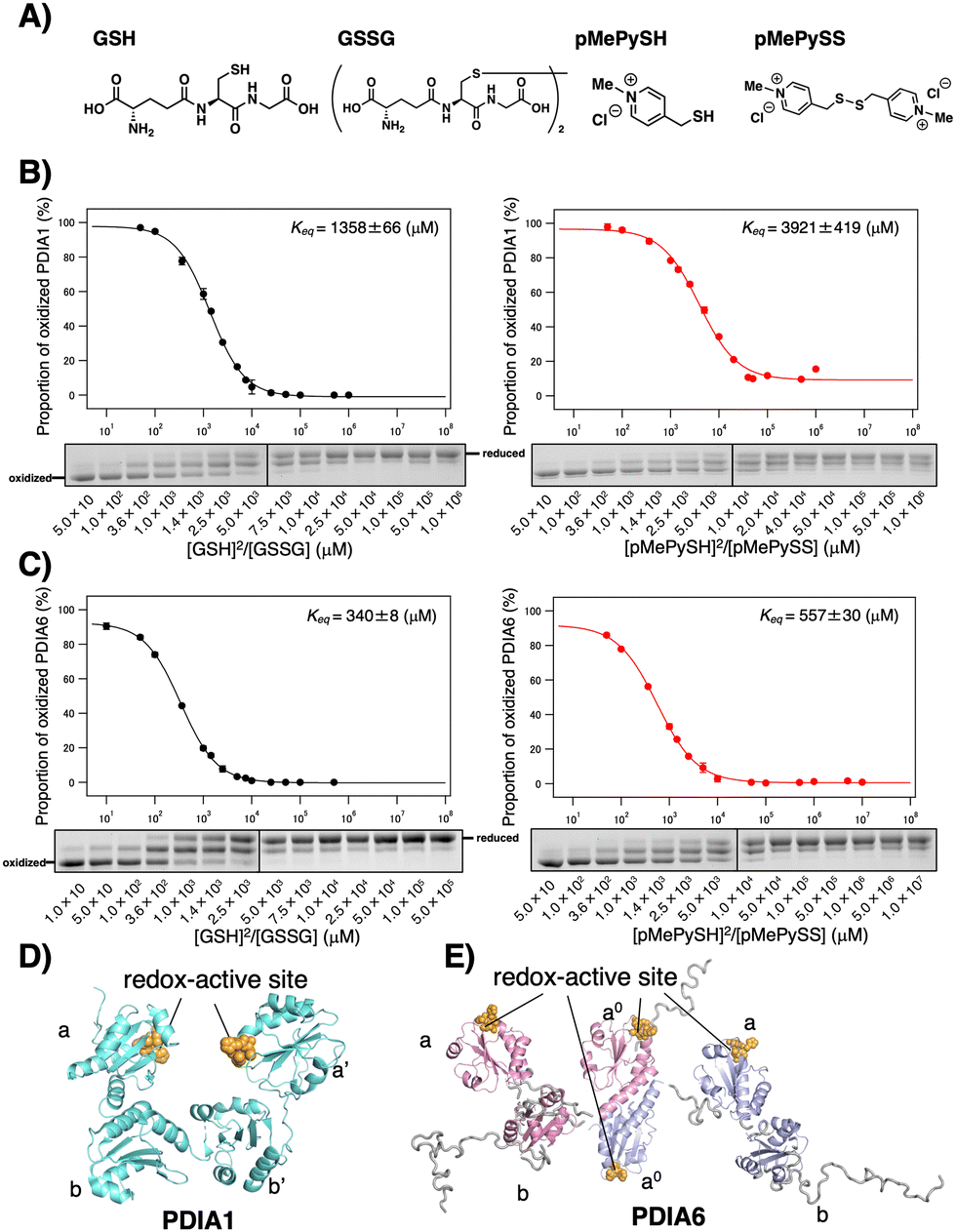 | ||
Fig. 1 Redox equilibrium constants of PDIA1 and PDIA6 using different GSH/GSSG or pMePySH/pMePySS ratios. (A) Chemical structures of GSH, GSSG, pMePySH, and pMePySS. Redox equilibrium constants of (B) PDIA1 and (C) PDIA6 using different GSH/GSSG or pMePySH/pMePySS ratios. Free PDI thiol groups (0.3 μM) were modified with malPEG2000 after incubation with different [GSH]2/[GSSG] or [pMePySH]2/[pMePySS] ratios in a degassed buffer containing 90 μM GSSG or pMePySS and various concentrations of GSH or pMePySH (30–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μM) at 30 °C. Keq values were determined from at least three independent experiments. (D) PDIA1 consists of four Trx-like domains, the first and last of which contain redox-active CxxC motifs. PDIA1 (PDB code; 4EKZ) crystal structure revealing four domains, which form an overall U-shaped structure.14 (E) PDIA6 consists of three Trx-like domains, the first and second of which contain redox-active CxxC motifs. PDIA6 dimerizes in solution via a unique dimeric motif.15 The dimeric motif is comprised of a Leu- and Val-rich region in the first redox active a0 domain. Of note, in contrast to PDIA1, the solvent-exposed redox active sites in each PDIA6 Trx-like domain rapidly introduce disulphide bond(s) into the client proteins. This structural feature is quite different from that of PDIA1. 000 μM) at 30 °C. Keq values were determined from at least three independent experiments. (D) PDIA1 consists of four Trx-like domains, the first and last of which contain redox-active CxxC motifs. PDIA1 (PDB code; 4EKZ) crystal structure revealing four domains, which form an overall U-shaped structure.14 (E) PDIA6 consists of three Trx-like domains, the first and second of which contain redox-active CxxC motifs. PDIA6 dimerizes in solution via a unique dimeric motif.15 The dimeric motif is comprised of a Leu- and Val-rich region in the first redox active a0 domain. Of note, in contrast to PDIA1, the solvent-exposed redox active sites in each PDIA6 Trx-like domain rapidly introduce disulphide bond(s) into the client proteins. This structural feature is quite different from that of PDIA1. | ||
Among PDIs, PDIA1, PDIA6, and PDIA15 are physiologically involved in the oxidative folding of proinsulin and insulin secretion,21 and PDIA3 is also essential for the efficient folding of glycoproteins in cells.22 We herein validated a strategy via the CxxC motif of PDIA1, PDIA3, PDIA6, and PDIA15 due to the redox-related pathological significance. To evaluate the “redox” molecule that enhances enzymatic activity by minimal loading, we investigated its effect on the PDI active sites when its concentration was in the μM rather than in the mM range.19 Although the thiol–disulphide catalytic CxxC motif is highly conserved in the PDI family (Fig. S1 in ESI†), the reactivity of each active site with glutathione differs depending on the structural characteristics of the PDIs (Fig. 1B, C and Fig. S2, ESI†). The redox equilibrium constant of each PDI member was determined by incubating purified PDIs in redox buffers containing different ratios of [GSH] to [GSSG] or [pMePySH] to [pMePySS]. To monitor the number of free thiol(s) of Cys residue(s), the reaction mixtures were modified by malPEG2000 (average Mn = 2000) and subsequently separated by SDS-PAGE (Fig. S2C, ESI†). The apparent redox equilibrium constants of four representative PDIs (PDIA1, PDIA3, PDIA6, and PDIA15) were calculated from the dose–response curve for redox agents obtained from the stained gel intensity (Fig. 1B, C and Fig. S3, ESI†). Consistent with a previous report,19 PDIA1 exhibited the highest redox equilibrium constant (Keq = 1358 μM), whereas PDIA6 exhibited the lowest constant (Keq = 340 μM). The PDIA1 redox active sites face the interior of the U-shaped structure,1,14 whereas the same sites in PDIA6 are exposed to the solvent,15 resulting in significantly different reactivity between the PDIs and glutathione active sites (Fig. 1D and E). As for pMePySH/pMePySS, the ranking of the redox equilibrium constants among PDIs was the same as those of GSH/GSSG (Fig. 2). However, among four representative PDI family members, the redox equilibrium constants using pMePySH/pMePySS were higher than those using GSH/GSSG, indicating higher reactivity with the redox active CxxC sites within the PDI family (Fig. 2).
 | ||
| Fig. 2 Redox equilibrium constant (Keq) of PDI family members using different (A) GSH/GSSG or (B) pMePySH/pMePySS ratios. The Keq values of PDIA1, PDIA3, PDIA6, and PDIA15 using pMePySH/pMePySS were determined as 3921, 2809, 1441, and 557 μM, respectively. | ||
To validate the ability to catalyze the oxidative folding against a reduced and denatured client, the oxidative folding of bovine pancreatic trypsin inhibitor (BPTI) was monitored in the presence of disulphide/thiol compounds ([BPTI] = 30 μM; [disulphides] = 90 μM; [thiols] = 360 μM). Native BPTI has three pairs of cysteine residues that can form disulphide bonds, i.e., Cys5–Cys55, Cys14–Cys38, and Cys30–Cys51 (Fig. 3A); therefore, since pMePySS accelerates the oxidative folding most efficiently by 1-equivalent loading as semi-enzymatic activity,20 we used a 90 μM disulphide concentration. Considering that the optimal condition for PDIA1 activity is [GSH]:[GSSG] = 5:117 and that a [GSH]:[GSSG] = 3:1 may be present in the ER,23 [thiols] = 360 μM and [disulphides] = 90 μM were used. Regarding on-path BPTI folding (Fig. 3B), quasi-native intermediates such as N′ and N* fold into native-like structures with two disulphide bonds and form native structures (N) via NSHSH. In the absence of PDIs, BPTI folds relatively slowly, with yields of 34.7% for GSH/GSSG and 72.6% for pMePySH/pMePySS in 120 min (Fig. S4, ESI†). Consistent with previous reports,1,2 BPTI folding was accelerated by PDIA1 or PDIA6 under GSH/GSSG, with yields of 90.4% for PDIA1 and 58.7% for PDIA6 in 120 min (Fig. 3C, F and Table 1). In comparison to GSH/GSSG, pMePySH/pMePySS enhanced the BPTI folding efficiency, which resulted in an N yield of 96.5% for PDIA1 and 80.7% for PDIA6 after 120 min (Fig. 3D and G). To further calculate the BPTI folding kinetics, the rate constants for BPTI folding were determined to be kGSH-PDIA1 = 2.3 × 10−2 min−1, kpMePySH-PDIA1 = 9.0 × 10−2 min−1, kGSH-PDIA6 = 1.6 × 10−2 min−1, and kpMePySH-PDIA6 = 4.1 × 10−2 min-1, i.e., the PDI-catalyzed reaction rate using pMePySH/pMePySS was indeed 2.5–4.0 times faster than that using the typical GSH/GSSG system (Fig. 3E, H and Fig. S5, ESI†). Of note, PDIA1 and PDIA6 significantly accelerated the production of N in BPTI under the redox environment using pMePySH/pMePySS (Fig. 3).
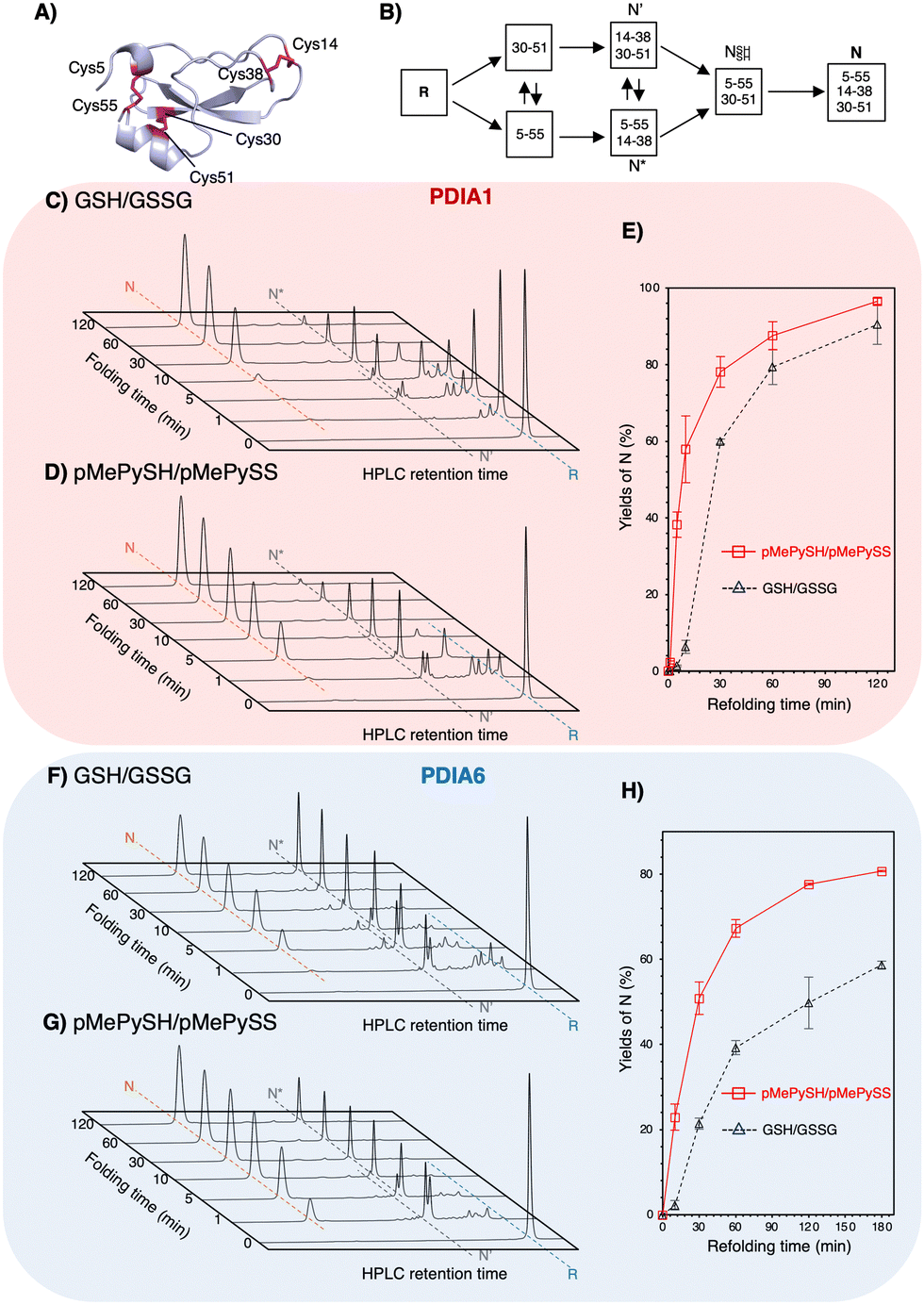 | ||
| Fig. 3 Both PDIA1 and PDIA6 under the pMePySH/pMePySS redox system accelerated the oxidative folding rates. (A) The crystal structure of BPTI (PDB code; 6PTI). The natively folded BPTI has three disulphide bonds (Cys5–Cys55, Cys14–Cys38, and Cys30–Cys51). (B) Oxidative folding pathway of BPTI. Time-course reverse-phase HPLC analyses of oxidative folding of BPTI (30 μM) in the presence of: (C) PDIA1 (0.3 μM) under a GSH (360 μM)/GSSG (90 μM) redox system, (D) PDIA1 (0.3 μM) the pMePySH (360 μM)/pMePySS (90 μM) redox system, (F) PDIA6 (0.3 μM) under a GSH (360 μM)/GSSG (90 μM) redox system, and (G) PDIA6 (0.3 μM) under the pMePySH (360 μM)/pMePySS (90 μM) redox system, respectively. (E) and (H) Time-course plots of native BPTI yields in (C), (D), (F) and (G). N, N′, N*, and R represent native, folding intermediates with 2-disulfide pairings (30–51 and 5–55 for N′, and 5–55 and 14–38 for N*), and reduced forms of BPTI. Eluent buffers, water (containing 0.1% TFA) and CH3CN (containing 0.1% TFA) with a linear gradient; flow rate, 1.0 mL min−1; detection wavelength, 229 nm; temperature, 30 °C. | ||
| Combination of enzymes and redox agents | Native BPTI yieldsa (%) |
|---|---|
| a Yields were calculated from the area of RP-HPLC. Errors indicate the mean ± SEM of three independent experiments. | |
| GSH/GSSG | 34.7 ± 1.0 |
| pMePySH/pMePySS | 72.6 ± 4.3 |
| PDIA1+GSH/GSSG | 90.4 ± 5.1 |
| PDIA1+pMePySH/pMePySS | 96.5 ± 0.8 |
| PDIA6+GSH/GSSG | 58.7 ± 0.9 |
| PDIA6+pMePySH/pMePySS | 80.7 ± 0.2 |
To further ensure the generality of the substrate catalyst, we investigated its ability to promote oxidative folding of human proinsulin, a peptide of biopharmaceutical interest for diabetes treatment. Human proinsulin consists of three chains, A, B, and the so-called C peptide, which connects A and B, with two disulphide bonds (CysB7–CysA7 and CysB19–CysA20) residing between chains A and B and one (CysA6–CysA11) constituting an intra-A-chain bond (Fig. 4A). The three disulphide bonds are crucial for the native folding of mature insulin, but we observed that its refolding yield in the μM range of GSH/GSSG was grossly impaired at neutral pH (Fig. 4B). Therefore, PDIs are thought to be involved in proinsulin oxidative folding under physiological conditions,21 and PDI-mediated catalytic activity enhancement is extremely important for insulin production. Compared with the GSH/GSSG system, both PDIA1 and PDIA6 with pMePySH/pMePySS increased the efficiency of native formation of human proinsulin by approximately 10%. Remarkably, GSH/GSSG alone failed to increase the native form of proinsulin, but PDIA1 in the μM range of pMePySH/pMePySS increased the yield by up to 81.6% (Fig. 4B). Thus, PDIA1 and PDIA6 markedly increased the yield of the protein native structures by minimal loading of pMePySH in comparison to glutathione.
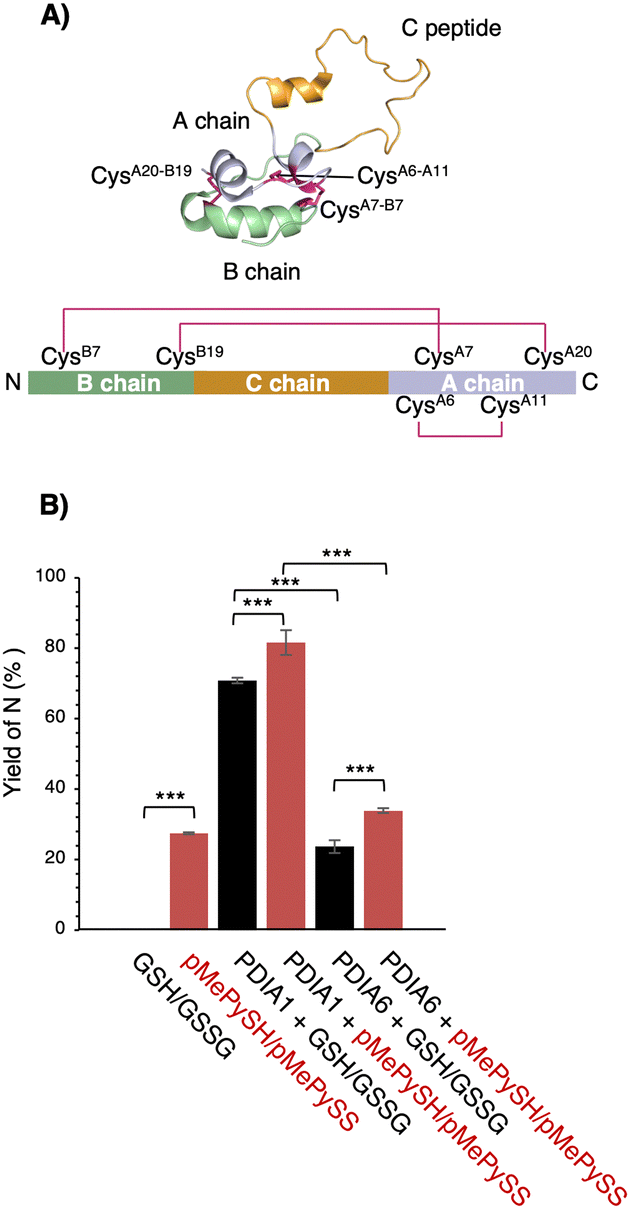 | ||
| Fig. 4 Refolding yields of proinsulin in the presence of GSH/GSSG or pMePySH/pMePySS with/without PDIA1 or PDIA6. (A) Solution structure of proinsulin (PDB code; 2KQP). The natively folded proinsulin has three disulphide bonds (CysB7–CysA7, CysB19–CysA20 and CysA6–CysA11). (B) The yields of native proinsulin (5 μM) in the presence of GSH/GSSG or pMePySH/pMePySS with/without PDIA1 (0.5 μM) or PDIA6 (0.5 μM) after 5 min are shown. Oxidative folding was performed at pH 7.5. Before HPLC analyses, aliquots of the folding solutions were pre-treated with aqueous 2-aminoethyl methanethiosulfonate (AEMTS) to quench the reaction by blocking the free SH groups into –SSCH2CH2NH3+. Error bars indicate the mean ± SEM of three independent experiments. The statistical significance of differences was examined by one-way analysis of variance with Tukey honestly significant difference (HSD) post hoc testing. All statistical tests were performed using KaleidaGraph statistical software (Synergy Software) at a significance level of α = 0.05. | ||
Differences in redox equilibrium constants among individual PDI family members are believed to sustain the complicated but elaborate redox network in the ER that controls protein homeostasis.19 Therefore, pathological redox imbalances can be caused by uncontrolled oxidation (e.g., excessive amount of reactive oxygen species) or reduction (e.g., hypo-oxidation in the ER contributing to the etiology of misfolding diseases such as diabetes and AD24). Further, dysfunction of PDI family members is known to cause pathological diseases such as neurodegeneration and diabetes.4,7 Therefore, chemically controlling the PDI family as ER-resident redox enzymes is crucial.
Here, we found that pMePySH/pMePySS redox system-mediated catalysis accelerated the folding kinetics of PDIA1 (with a relatively high Keq) and PDIA6 (with a relatively low Keq). This work could offer a proof of concept regarding the existence of redox crosstalk in the ER, i.e., thiol compounds targeting the cysteines of PDI family CxxC motifs and increasing the PDI family member enzymatic activity more efficiently than glutathione, a typical redox agent of universal use. Regarding the chemical booster of PDI activity, an allosteric switch operating via the substrate-binding b′ domain increases PDI activity,13 but a similar allosteric effect can also lead to decreased activity.11 One reason for this apparent paradox is that regulation of the both function and structure is beyond the proof-of-concept for chemical design due to the conformational dynamic nature of the PDI family.1,25 Therefore, chemical boosters of the PDI family activity via CxxC motif(s) may further provide clues to new approaches for combating PDI family-related misfolding pathologies.
MO conceived the idea of this study. TK, YY, SK, and KA performed the experiments. MO and TM supervised the implementation of this study. MO wrote the manuscript. All authors critically reviewed and revised the manuscript draft and approved the final version for submission.
This research was funded by a JSPS KAKENHI grant (grant numbers 22H02205 (to MO)), a Fund for the Promotion of Joint International Research (grant number 23KK0105 (to MO)), JSPS Grants-in-Aid for Transformative Research Areas (B) (grant numbers JP21H05095 (to MO) and JP21H05096 (TM)), the Japan Science and Technology Agency FOREST Program (grant numbers JPMJFR201F (to MO) and JPMJFR2122 (to TM)), a grant from the Takeda Science Foundation (to MO and TM), a grant from the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to MO), a grant from the Naito Foundation (to MO), a grant from the Uehara Memorial Foundation (to MO), a grant from the Terumo Life Science Foundation (to MO), a grant from the Astellas Foundation for Research on Metabolic Disorders (to MO), a grant from The Asahi Glass Foundation (to MO and TM), Mitsui Sumitomo Insurance Welfare Foundation (to MO), a grant from the Daiichi Sankyo Foundation of Life Science (to MO), and a grant from THE Sumitomo FOUNDATION (to MO). This work was supported by JST SPRING, Grant number JPMJSP2114. Generous support was received from the FRIS CoRE, which is a shared research environment at Tohoku University. We thank Prof. Kenji Inaba (Kyushu University) for the kind gift of PDI family plasmids.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- M. Okumura, K. Noi, S. Kanemura, M. Kinoshita, T. Saio, Y. Inoue, T. Hikima, S. Akiyama, T. Ogura and K. Inaba, Nat. Chem. Biol., 2019, 15, 499–509 CrossRef CAS PubMed.
- M. Okumura, S. Kanemura, M. Matsusaki, M. Kinoshita, T. Saio, D. Ito, C. Hirayama, H. Kumeta, M. Watabe, Y. Amagai, Y. H. Lee, S. Akiyama and K. Inaba, Structure, 2021, 29, 1357–1370 CrossRef CAS PubMed.
- R. Kojima, M. Okumura, S. Masui, S. Kanemura, M. Inoue, M. Saiki, H. Yamaguchi, T. Hikima, M. Suzuki, S. Akiyama and K. Inaba, Structure, 2014, 22, 431–443 CrossRef CAS PubMed.
- M. Matsusaki, S. Kanemura, M. Kinoshita, Y. H. Lee, K. Inaba and M. Okumura, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864(2), 129338 CrossRef CAS PubMed.
- U. Woehlbier, A. Colombo, M. J. Saaranen, V. Pérez, J. Ojeda, F. J. Bustos, C. I. Andreu, M. Torres, V. Valenzuela, D. B. Medinas, P. Rozas, R. L. Vidal, R. Lopez-Gonzalez, J. Salameh, S. Fernandez-Collemann, N. Muñoz, S. Matus, R. Armisen, A. Sagredo, K. Palma, T. Irrazabal, S. Almeida, P. Gonzalez-Perez, M. Campero, F. B. Gao, P. Henny, B. van Zundert, L. W. Ruddock, M. L. Concha, J. P. Henriquez, R. H. Brown and C. Hetz, EMBO J., 2016, 35, 845–865 CrossRef CAS PubMed.
- T. Uehara, T. Nakamura, D. Yao, Z. Q. Shi, Z. Gu, Y. Ma, E. Masliah, Y. Nomura and S. A. Lipton, Nature, 2006, 441, 513–517 CrossRef CAS PubMed.
- T. Muraoka, M. Okumura and T. Saio, Chem. Sci., 2024, 15, 2282–2299 RSC.
- B. G. Hoffstrom, A. Kaplan, R. Letso, R. S. Schmid, G. J. Turmel, D. C. Lo and B. R. Stockwell, Nat. Chem. Biol., 2010, 6, 900–906 CrossRef CAS PubMed.
- S. Xu, A. N. Butkevich, R. Yamada, Y. Zhou, B. Debnath, R. Duncan, E. Zandi, N. A. Petasis and N. Neamati, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16348–16353 CrossRef CAS PubMed.
- A. Kaplan, M. M. Gaschler, D. E. Dunn, R. Colligan, L. M. Brown, A. G. Palmer, 3rd, D. C. Lo and B. R. Stockwell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2245–2252 CrossRef CAS PubMed.
- M. Okumura, H. Kadokura, S. Hashimoto, K. Yutani, S. Kanemura, T. Hikima, Y. Hidaka, L. Ito, K. Shiba, S. Masui, D. Imai, S. Imaoka, H. Yamaguchi and K. Inaba, J. Biol. Chem., 2014, 289, 27004–27018 CrossRef CAS PubMed.
- G. Zheng, K. Lv, H. Wang, L. Huang, Y. Feng, B. Gao, Y. Sun, Y. Li, J. Huang, P. Jin, X. Xu, F. D. Horgen, C. Fang and G. Yao, J. Am. Chem. Soc., 2023, 145, 3196–3203 CrossRef CAS PubMed.
- R. H. Bekendam, P. K. Bendapudi, L. Lin, P. P. Nag, J. Pu, D. R. Kennedy, A. Feldenzer, J. Chiu, K. M. Cook, B. Furie, M. Huang, P. J. Hogg and R. Flaumenhaft, Nat. Commun., 2016, 7, 12579 CrossRef CAS PubMed.
- C. Wang, W. Li, J. Ren, J. Fang, H. Ke, W. Gong, W. Feng and C. C. Wang, Antioxid. Redox Signaling, 2013, 19, 36–45 CrossRef PubMed.
- M. Okumura, S. Kanemura, M. Matsusaki, M. Kinoshita, T. Saio, D. Ito, C. Hirayama, H. Kumeta, M. Watabe, Y. Amagai, Y. H. Lee, S. Akiyama and K. Inaba, Structure, 2021, 29, 1357–1370 CrossRef CAS PubMed.
- A. B. Khan, N. Gupta, Q. Rashid, I. Ahmad, S. Bano, U. Siddiqui, M. Abid and M. A. Jairajpuri, Med. Drug Discovery, 2020, 6, 100029 CrossRef.
- M. M. Lyles and H. F. Gilbert, Biochemistry, 1991, 30, 613–619 CrossRef CAS PubMed.
- J. L. Arolas, F. X. Aviles, J. Y. Chang and S. Ventura, Trends Biochem. Sci., 2006, 31, 292–301 CrossRef CAS PubMed.
- K. Araki, S. Iemura, Y. Kamiya, D. Ron, K. Kato, T. Natsume and K. Nagata, J. Cell Biol., 2013, 202, 861–874 CrossRef CAS PubMed.
- S. Okada, Y. Matsumoto, R. Takahashi, K. Arai, S. Kanemura, M. Okumura and T. Muraoka, Chem. Sci., 2023, 14, 7630–7636 RSC.
- Y. Tsuchiya, M. Saito, H. Kadokura, J. I. Miyazaki, F. Tashiro, Y. Imagawa, T. Iwawaki and K. Kohno, J. Cell Biol., 2018, 217, 1287–1301 CrossRef CAS PubMed.
- C. E. Jessop, S. Chakravarthi, N. Garbi, G. J. Hämmerling, S. Lovell and N. J. Bulleid, EMBO J., 2007, 26, 28–40 CrossRef CAS PubMed.
- C. Hwang, A. J. Sinskey and H. F. Lodish, Science, 1992, 257, 1496–1502 CrossRef CAS PubMed.
- J. D. Watson, Lancet, 2014, 383, 841–843 CrossRef PubMed.
- M. Okumura, K. Noi and K. Inaba, Curr. Opin. Struct. Biol., 2021, 66, 49–57 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01712a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |