
Cell or cell derivative-laden hydrogels for myocardial infarction therapy: from the perspective of cell types
Ziqing
Xiong
a,
Qi
An
a,
Liqiang
Chen
b,
Yucheng
Xiang
b,
Lian
Li
*b and
Yaxian
Zheng
 *cd
*cd
aDepartment of Cardiovascular Surgery, West China Hospital, Sichuan University, Chengdu, Sichuan, China
bKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, Sichuan Engineering Laboratory for Plant-Sourced Drug and Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu, Sichuan, China. E-mail: liliantripple@163.com
cDepartment of Pharmacy, Affiliated Hospital of Southwest Jiaotong University, The Third People's Hospital of Chengdu, Chengdu, Sichuan, China. E-mail: ZhengYX95@163.com
dInstitute of Biomedical Engineering, College of Medicine, Southwest Jiaotong University, Chengdu, Sichuan, China
First published on 21st September 2023
Abstract
Myocardial infarction (MI) is a global cardiovascular disease with high mortality and morbidity. To treat acute MI, various therapeutic approaches have been developed, including cells, extracellular vesicles, and biomimetic nanoparticles. However, the clinical application of these therapies is limited due to low cell viability, inadequate targetability, and rapid elimination from cardiac sites. Injectable hydrogels, with their three-dimensional porous structure, can maintain the biomechanical stabilization of hearts and the transplantation activity of cells. However, they cannot regenerate cardiomyocytes or repair broken hearts. A better understanding of the collaborative relationship between hydrogel delivery systems and cell or cell-inspired therapy will facilitate advancing innovative therapeutic strategies against MI. Following that, from the perspective of cell types, MI progression and recent studies on using hydrogel to deliver cell or cell-derived preparations for MI treatment are discussed. Finally, current challenges and future prospects of cell or cell derivative-laden hydrogels for MI therapy are proposed.
1. Introduction
According to the World Health Organization (WHO), more than 17.9 million people die each year from cardiovascular disease (CVD), and the number is steadily rising.1 By 2030, the global economic burden of CVD is anticipated to reach $104.4 billion.2 Among CVD-related deaths, myocardial infarction (MI) and associated heart failure are the most common causes.3 MI is triggered by the sudden narrowing or blockage of coronary arteries, which leads to a prolonged reduction or cessation of blood supply to the heart.4 Myocardial cells undergo necrotic apoptosis during ischemia due to the lack of oxygen or nutrients.5 The process of fibrous remodeling leads to the formation of scars, which can impede normal cardiac function and ultimately result in severe heart failure.6 While current therapeutic methods, such as prompt reperfusion, pharmacological thrombolysis, and surgical intervention, can alleviate the process of MI, they do not address the urgent challenge of regenerating and repairing myocardial tissue. This presents a significant obstacle in the field of MI therapy.7–9Researchers have developed various cell-based MI therapeutic strategies over the years to restore cardiac tissue and reverse cardiac function.10–13 These strategies have evolved from the initial cell therapy to extracellular vesicles (EVs). (1) Cell therapy. Source cells include mesenchymal stem cells (MSCs), endothelial progenitor cells (EPCs), and cardiac-derived stem cells.14,15 Mechanisms involved in this therapy are pro-angiogenic factor releasement and cardiomyocyte differentiation.16 (2) Cell-derived extracellular vesicle therapy (e.g., MSCs, EPCs, and macrophage-derived exosomes).17,18 Mechanisms include reduced monocyte invasion and macrophage polarization control.19,20 However, the clinical application of cell or cell-inspired therapy is hindered by low cell survival rates, inefficient heart targeting, and rapid clearance from cardiac sites.21,22
Encapsulating cell or cell-derived preparations in hydrogels or patches is a promising resolution.23,24 While cardiac patches require surgical implantation and result in low patient compliance, the injectable hydrogel is less intrusive and traumatic, and has preferable biocompatibility due to its structural similarity to the myocardium's extracellular matrix.25–28 The porous structure of swelling hydrogel also helps maintain cell activity and reduce blood elimination in vivo, making it an excellent choice for encapsulation.29,30 Previous studies have demonstrated that injecting hydrogel into myocardial infarction sites can attenuate biomechanical remodeling of the heart, leading to improved 5 year survival rates for MI patients.31 As such, utilizing an injectable hydrogel delivery strategy in conjunction with cell or cell-derived therapy may be a promising approach to further improve outcomes for MI patients.32 Moreover, biomimetic nanoparticle (e.g., erythrocyte, neutrophil, and platelet cell membrane-camouflaged nanoparticles) has emerged as a new tool for MI therapy. These delivery systems have the potential to target the site of myocardial infarction and repair endogenous heart tissue by releasing loaded active cytokines or other drugs.33–36
In light of the foregoing, this review first presented an overview of the pathological process of myocardial infarction, focusing on the role of each cell type in heart recovery post MI. Then, we discussed the advantages and drawbacks of hydrogel delivery and cell-based therapy, as well as their cooperative relationship in MI treatment. Notably, we reviewed the current studies on using hydrogel delivery technologies to deliver cell or cell-derived preparations for MI treatment. Finally, current challenges and future prospects of cell or cell derivative-laden hydrogels for MI therapy are proposed.
2. The role of different cells in heart recovery post MI
Myocardial infarction is closely linked to the interactions and collaboration among various cells in the heart, blood, and bone marrow.37 The heart, being the primary site of MI, comprises several cell types such as cardiomyocytes, cardiac fibroblasts, endothelium, immune cells, and the myocardium extracellular matrix.38 During the progression of MI, neutrophils and monocytes, which are the major functional blood or bone marrow cells, also play a critical role.39 Each cell type has a specific function in the physiological condition, and their interaction and collaboration are essential for the heart to function normally. In the pathological stage of MI, several cascade reactions occur in the specific cells of the heart, blood circulation, and bone marrow, leading to cardiac remodeling and subsequent heart failure.40–42 This process is mainly divided into four phases: inflammation, proliferation, maturation, and remodeling.43 This review outlines the biological activity and function of different cells in heart recovery post MI (Fig. 1).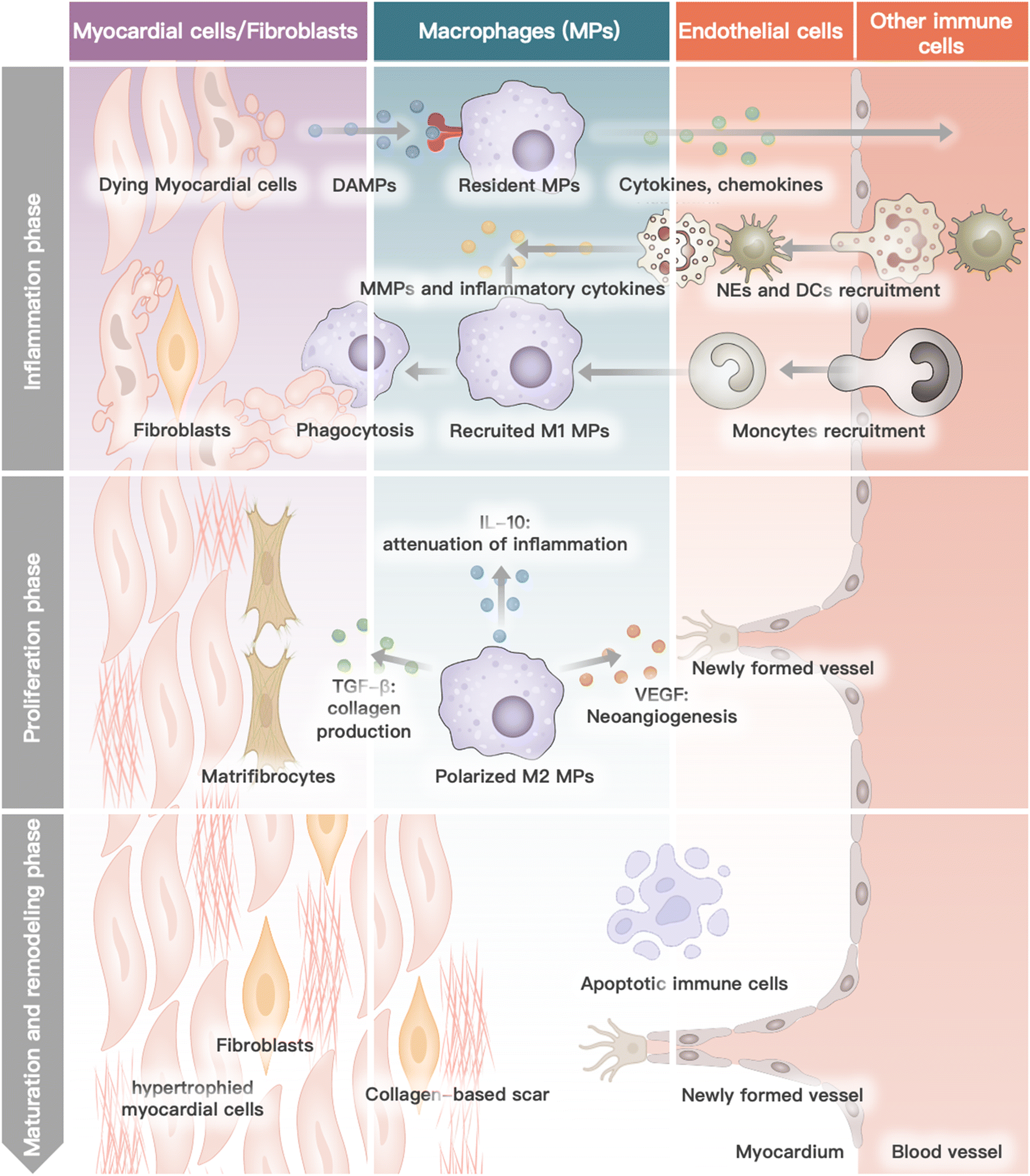 | ||
| Fig. 1 The role of different cells in heart recovery post MI. Dying myocardial cells release damage-associated molecular patterns (DAMPs). Resident macrophages recognize DAMPs and initialize inflammatory cascade by releasing cytokines and chemokines, leading to the recruitment of neutrophils (NEs), dendritic cells (DCs) and monocytes. Monocytes are differentiated into M1-phenotype macrophages (MPs) and, together with NEs, secrete matrix metalloproteinases (MMPs) and inflammatory cytokines. Resident and recruited MPs are responsible for phagocytosis and degradation of necrotic tissue. In subsequent proliferation phage, MPs are polarized into pro-inflammatory M2-phenotype MPs, releasing transforming growth factor-β (TGF-β), interleukin-10 (IL-10), and vascular endothelial growth factor (VEGF). Three cytokines can respectively enhance the collagen production ability of matrificrocytes, attenuate inflammation, and promote neovascularization. In the maturation and remodeling phase, monocyte recruitment ends. Eventually, the scar tissue is formed by hypertrophied cardiomyocytes and cross-linked extracellular. | ||
2.1 Cardiomyocytes
Myocardial ischemia causes irreparable damage to cardiomyocytes during the inflammatory phase.44 This process mediates cardiomyocyte death and the release of damage-related molecular patterns through multiple pathways such as necrosis, regulation of death, and autophagy.45 Cardiomyocytes will hypertrophy with the addition of new myofibrils during the ensuing MI process to adjust to the physical demands of the injured heart.462.2 Macrophages
During the inflammatory phase, resident macrophages receive death signals from cardiomyocytes via the pattern recognition receptor.47 Subsequently, macrophages release pro-inflammatory factors and chemokines into the circulation system, including the chemokine (C–C motif) ligand 2 (CCL2) and interleukin-1β (IL-1β). In response to cytokines, monocytes are attracted to the ischemic region and then transform into M1-phenotype macrophages with a pro-inflammatory character.48 At this stage, the inflammatory effect will be amplified in cascades. In the proliferative phase, macrophages are polarized into M2-phenotype macrophages with anti-inflammatory property.48,49 M2-phenotype macrophages secrete a variety of cytokines, such as TGF-β, IL-10, and VEGF, to promote tissue repair and angiogenesis.50 At the maturation stage, the recruitment of macrophages ends. Macrophages remaining in the ischemic region will undergo apoptosis.512.3 Cardiac fibroblasts
In normal cardiac tissue, cardiac fibroblasts are mainly responsible for the secretion of extracellular matrix, facilitating the infiltration of immune cells.51 During the proliferative phase, macrophages activate cardiac fibroblasts by releasing TGF-β.51 In the maturation phase, activated fibroblasts transform into matrifibrocytes.52 The cardiac extracellular matrix is cross-linked to form scar tissue. During the remodeling phase, chronic immune system overactivation will lead to fibrosis and increase tissue stiffness in infracted and remote myocardium.532.4 Endothelial
Physiologically, the principal function of endothelial cells is to provide a barrier between blood and cardiac tissue by regulating vascular permeability.54 The initial inflammatory response of MI causes the expression of surface adhesion molecules by endothelial cells and increases the intercellular space between endothelial cells.55 This action substantially increases vascular permeability and aids immune cell recruitment and infiltration. In the proliferative phase, endothelial cells proliferate in response to cytokines such as VEGF, which mediates vascular regeneration and remodeling at the site of ischemia.56 Endothelial cells further proliferate during the maturation phase to produce vessel lumens, which are subsequently stabilized by recruited pericytes. During the remodeling phase, as scar tissue continues to form, microvascular rarefied and vascular density is reduced in the heart.572.5 Other immune cells
During heart recovery period post MI, except for macrophages mentioned above, other immune cells (e.g., monocytes, neutrophils, dendritic cells) rapidly coordinate their function to contain inflammation by removing dying cells and promoting cardiomyocyte replenishment. Monocytes and neutrophils from the bone marrow or spleen are recruited to the site of ischemia by endothelial cell surface adhesion molecules and inflammatory chemokines.58 Among them, induced by pro-inflammatory cytokines, monocytes transform into M1-phenotype macrophages with a pro-inflammatory character. During the proliferative phase, macrophages polarize into anti-inflammatory M2-phenotype macrophages. The re-emergence of immune cells during the remodeling phase causes inflammation in remote and non-infarcted myocardium.59 Dendritic cells play a significant role in post-MI antigen presentation, immune activation, and other processes.42 Initially, immature DCs migrate through the vascular endothelium into damaged heart tissue, where they are activated by DAMPs. These activated DCs then engulf antigens released by necrotic cardiomyocytes. Subsequently, the DCs migrate to lymph nodes and stimulate the production of CD8+ T cells, triggering specific immunity. On the other hand, EVs released by DCs can activate regulatory T cells (Tregs), which suppress the activity of CD8+ T cells and regulate the differentiation of monocytes and macrophages. CD8+ T cells, Tregs, and other immune cells work together to maintain the equilibrium of the heart's immune environment post MI.3. Collaborative effects of hydrogel with cell-based therapy
3.1 Necessities and drawbacks of hydrogel in MI therapy
Due to the function of supplying blood to the entire body as a mechanical pump, the homeostasis of the biomechanical environment of the heart matters a lot.60 The elasticity of each cardiac cell and the extracellular matrix are particularly important. During cardiac systole and diastole, the elastic modulus of the normal heart varies cyclically.61 The modulus of elasticity in the infarcted region tends to decrease and then increase within 24 hours of the onset of MI.62 This pattern is associated with the degradation and then increase of extracellular matrix (ECM) during the progression of MI.63 It has been reported that hydrogels could replace the degraded ECM after being injected into the ischemic region, compensating for the loss of contractility in the acute ischemic region and providing good mechanical support for cardiomyocyte function.64 Another study found that by increasing the thickness of the ventricular wall in the infarcted region, hydrogel could improve the ejection fraction of the left ventricle, effectively delaying the remodeling process of the myocardium after the onset of MI.65Furthermore, several natural and synthetic hydrogels (e.g., fibrin hydrogels, alginate hydrogels, hyaluronic acid hydrogels, and the cell-interacting peptide RAD16-II hydrogels) have been shown to have bioactivities that can improve the therapeutic efficacy of MI.33,66 These bioactivities include immune cell recruitment, inflammatory microenvironment modulation, and pro-angiogenic effects. A clinical phase II trial of an alginate hydrogel (Algisyl-LVRTM) for intracardiac injection has been completed.67 Although the hydrogel is able to maintain the biomechanical environment of the heart after the onset of MI and mitigate the MI process, the inability to regenerate cardiomyocytes remains an unresolved issue. In a clinical trial called PRESERVATION-1 (NCT01226563), a total of 231 patients were administered IK-5001 and compared with 102 healthy controls.68 However, no visible effect was observed after a period of 6 months. It was noted that serious adverse events were more common in the treatment group (5%) than in the control group (2.9%). Additionally, the treatment group also exhibited higher rates of mortality and myocardial infarction compared to the control group. In conclusion, these findings suggest that severe MI patients did not experience significant anatomical changes or improvements in their quality of life but were more prone to severe adverse reactions. This is because there are still many problems with hydrogel alone. On the one hand, the lack of cells makes this therapy only “passive”, unable to regenerate new myocardium. On the other hand, since the hydrogel is mainly relied on to maintain the cardiac biomechanical environment after MI, the injection volume of the hydrogel is relatively large. It may be more likely to cause adverse reactions, such as ventricular ectopy or ventricular arrhythmia.
3.2 Necessities and drawbacks of cell-based therapy
Due to the superior MI therapeutic effects, stem cell therapy has received much attention.69 In 2011, the first MSCs preparation in the world for myocardial infarction therapy (Hearticellgram-AMI) was launched in Korea.70 It is prepared by extracting blood from the autologous sciatic bone marrow of patients. MSCs are cultured for three to four weeks before being intracoronary infused. Clinical trials have shown that stem cell preparation injection improved heartbeats in patients with MI. Several autologous bone marrow MSCs administrated by intracoronary infusion for MI therapy are also in phase III clinical trials.71–73 These findings highlight the significance of stem cell therapy in the treatment of heart diseases such as MI. They stimulate endogenous myocardial activation by secreting a variety of repair and regenerative cytokines. It is characterized by decreased myocardial cell death and hypertrophy, increased neovascularization, and improved function of existing myocardial cells.74 However, stem cells survive in the heart only for a short time after being infused.Surprisingly, it is estimated that more than 90% of stem cells infused into the heart do not reach the heart.75 The situation occurs for two reasons. (1) Because of the abundant blood flow in the heart, stem cells infused are quickly eliminated and cannot be retained for an extended period of time to exert therapeutic effects.76 (2) Stem cell therapy should be started as soon as an acute injury occurs. However, during acute injury, the infarcted area of the heart has a robust immune and inflammatory response.77 Therefore, stem cells injected into this area are subjected to a hostile environment, reducing cell viability. As a result, selecting an appropriate delivery system to increase intracardiac retention and cell survival rate is critical to promoting the efficacy of cell-based MI therapy.
Injectable hydrogels, among various biological scaffolds, use the properties of polymeric materials that undergo a sol–gel transition. These hydrogels can incorporate cells and retain them in the infarct site. Furthermore, in the early stages of MI, the hydrogel could effectively separate loading cells from the robust inflammatory and immune environments.78,79 Additionally, a more suitable in vivo microenvironment for stem cell differentiation can be created by adding pro-stem cell differentiation siRNA (e.g., miR-1a) or cytokines to the hydrogel.80,81 Moreover, it has been reported that the elasticity of the matrix influences the differentiation efficiency of stem cells.82 Human mesenchymal stem cells differentiate more readily in hydrogels with a higher elastic modulus. Based on this, it is reasonable to speculate that stem cell delivery via a hydrogel similar to the elasticity of cardiac tissue is more conducive to stem cell differentiation to cardiomyocytes.83 In one study, pluripotent stem cells were cultured in a polyacrylamide hydrogel with an elastic modulus of 55 kPa rather than a traditional tissue culture medium.35 The stem cells were able to differentiate into cardiomyocytes more efficiently.
Notably, some researchers believe that the potential mechanism of MI cell therapy is primarily related to their paracrine function.84 The paracrine function of stem cells refers to the release of extracellular vesicles from these stem cells into the stroma. Active substances found in extracellular vesicles include stem cell-associated proteins, miRNAs, and DNA fragments.85 By fusing with the cell membrane of damaged or surviving cardiomyocytes, stem cell-derived exosomes release their active contents into the recipient cells, enhancing cardiomyocyte viability and function at the infarct site. Current research has focused on using extracellular vesicles as drug delivery systems for miRNAs or active proteins that promote angiogenesis for myocardial regeneration.86,87 However, to date, there is no clinical trial on using extracellular vesicle-loaded hydrogels for MI therapy. Facing the same problem as cell therapy, extracellular vesicles are still rapidly eliminated by the rich blood flow of the heart after intravenous or intracoronary administration.88 As a result, extracellular vesicle delivery to the infarct site is inefficient. Similarly, sparse drug-loading space combined with the long-term retention ability of hydrogel at the infarct site makes it an ideal vehicle for extracellular vesicle MI therapy.89
In conclusion, researches indicate that the use of injectable hydrogels and cell or cell-derived agents can be mutually beneficial and complementary in treating MI. While the cardiomyocyte differentiation ability and paracrine function of cell or cell-derived agents can effectively improve cardiac function after MI, compensating for the inability of hydrogels to reverse cardiac fibrosis, the ECM-like three-dimensional structure of the injectable hydrogel provides mechanical support for the heart. Additionally, the abundant cell-loading space makes hydrogel an ideal microenvironment for cell proliferation and differentiation.90 The residency of the hydrogel at the site of post-injection infarct provides targeting properties for cell-derived agents, which compensates for the drawbacks of cell or cell-derived preparation. These drawbacks include low cell survival, rapid clearance, and poor targeting properties.91 In this article, we summarize studies about hydrogel delivery systems that encapsulate cell or cell-derived preparations for myocardial infarction therapy.
4. Cell or cell derivative-laden hydrogels for MI therapy
In order to meet the cells or extracellular vesicles delivery requirements of hydrogels, the physicochemical properties of hydrogels need to be carefully considered, including porosity, stiffness, swelling ability, complex mechanical properties, and degradation ability.92 These physicochemical properties dictate the fate of cells or EVs after being encapsulated and delivered. For example, it has been reported that in order to deliver human embryonic stem cells (hESCs), the stiffness of the hydrogel needs to be around 10 kPa. This is because only hydrogels with this stiffness can maintain hESCs proliferation and pluripotency.93 Degradability provides the space for cell stretching and proliferation. According to another study, it has been found that MSCs only spread in a degradable hydrogel made from RGD-modified HA, as opposed to a non-degradable matrix.93 Therefore, it is necessary to adjust these physicochemical properties and complex mechanical properties of hydrogels to meet the delivery or treatment need for cells or cell derivatives-based MI therapy.Materials and preparation methods affect the physicochemical properties of hydrogels. Based on the biological material used, hydrogels can be classified as natural or synthetic. Fibrin glue, alginate, hyaluronic acid, and collagen hydrogels are the most commonly used natural hydrogels.30,92 The majority of synthetic hydrogels are homopolymers and copolymers of acrylamide (AAM) and its derivatives, homopolymers and copolymers of acrylic acid (AA) and its derivatives, followed by polyvinyl alcohol (PVA) and polyethylene glycol (PEG) (Table 1).92 The preparation methods of hydrogels include physical methods (ultraviolet light, γ-ray irradiation, heating, freeze-drying), chemical methods (modification of functional groups such as carboxyl and amino groups, and addition of crosslinking agents such as glutaraldehyde, etc.), and enzymatic methods (glutamine transaminase, etc.).94 Physical crosslinking methods may decrease cell activity, while chemical crosslinking and enzymatic crosslinking require complex modifications or additional reagents. The interactions between the cell or cell-derived agents and the hydrogel matrix must be considered during the preparation of the hydrogels. On the one hand, hydrogels must meet the fundamental requirements for in vitro culture and in vivo transplantation, such as the absence of small molecule cross-linking agents, moderate cross-linking conditions, and the avoidance of toxic by-product generation.33,95 As reported, gelation and cryo-gelation hydrogels are less toxic to adipose-derived mesenchymal cells than conventional covalent cross-linked and photopolymerized hydrogels, indicating they are a more promising tool in vivo application.96,97 On the other hand, the physical and mechanical properties of hydrogels are crucial for their application in stem cell tissue engineering.98 Although the hydrogels have a stable three-dimensional porous structure for encapsulating cells or cell-derived agents, the biotherapeutic adding will inevitably affect the macroscopic properties of hydrogels.99,100 Herein, we introduced two cell-based approaches for MI therapy from the perspective of cell types.
| Materials | Advantages | Disadvantages | |
|---|---|---|---|
| Natural hydrogels | Fibrin | • Good biocompatibility | • Poor mechanical stability |
| • Good biodegradability | • Easily agglomerate at high concentrations | ||
| • Promotes angiogenesis | |||
| • Fast gelation | |||
| Alginate | • Bio-inert | • Modifications needed for cell binding | |
| • Fast gelation | • Poor cell adhesion | ||
| • Good stability | • Easily agglomerate at high concentrations | ||
| Hyaluronic acid | • Good biocompatibility | • Rapid degradation | |
| • Promotes proliferation and angiogenesis | • Poor mechanical stability | ||
| • Fast gelation | |||
| Collagen | • Cell–matrix interactions | • Poor mechanical stability | |
| • Promotes cell attachment | • Slow gelation | ||
| • Easily agglomerate at high concentrations | |||
| Synthetic hydrogels | AAM derivatives | • Good hydrolytic stability | • Weak degradability |
| • Similar to the extracellular matrix | • Cell toxicity | ||
| AA derivatives | • Good biocompatibility | • Weak degradability | |
| • Easily functionalized | |||
| PVA derivatives | • Promoting cell survival and proliferation | • Weak ability to adsorb proteins | |
| • Degradability | • Poor cell adhesion | ||
| PEG derivatives | • Good hydrophilicity | • Low cell proliferation and adhesion | |
| • Good biocompatibility | • Weak biodegradation | ||
| • Easily functionalized | • Intrinsic toxicity of synthetic schemes | ||
4.1 Cell-laden hydrogels for MI therapy
Cells used in cell therapy primarily consist of bone marrow- or adipose-derived mesenchymal stem cells (ADSCs), blood- or bone marrow-derived endothelial progenitor cells, blood- or bone marrow-derived monocytes, cardiac-derived stem cells, skeletal myoblasts (SkMs), and multi-source-derived cardiomyocytes (Fig. 2). The therapeutic mechanism of cell-based MI therapy is summarized in Table 2.101–103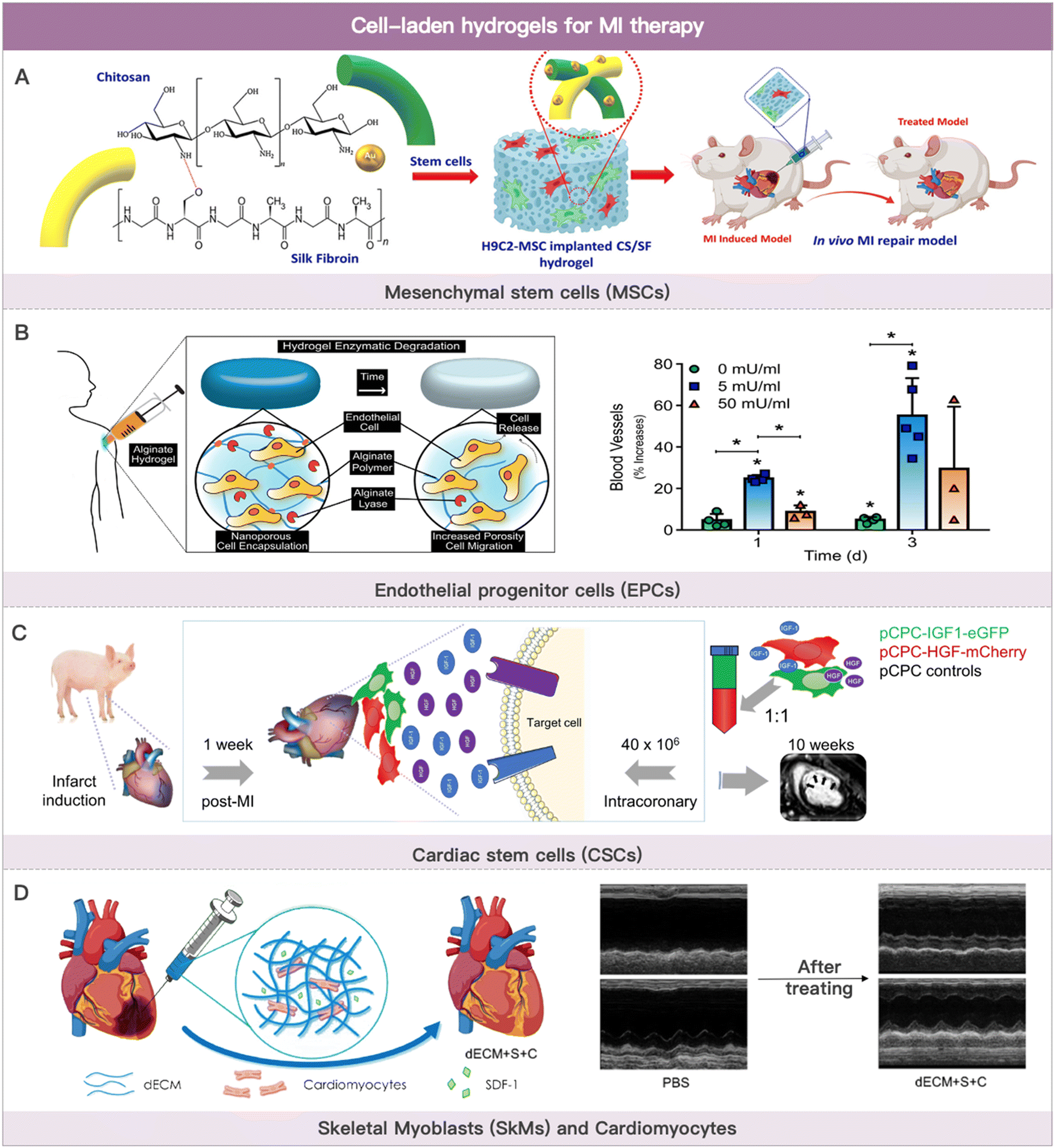 | ||
| Fig. 2 Cell-laden hydrogels for MI therapy. (A) Schematic illustration of the composited hydrogel formation loaded with MSCs for myocardial functional recovery therapy. Reproduced with permission from ref. 113, Copyright Elsevier 2023. (B) Schematic illustration of hydrogels with various alginate lyase concentrations over the course of three weeks and angiogenesis effect of EPCs-loaded hydrogel. Reproduced with permission from ref. 123, Copyright Elsevier 2018. (C) Safety evaluation of coronary artery injection of porcine cardiac progenitor cells in an acute MI porcine model. Reproduced from ref. 142 under the open access policy of MDPI 2021. (D) Schematic illustration of decellularized porcine extracellular matrix to deliver exogenous cardiomyocytes for MI treatment. Reproduced with permission from ref. 159, Copyright American Chemical Society 2023. | ||
| Cell sources | Therapeutic mechanism |
|---|---|
| Mesenchymal stem cells | • Extensive paracrine effect |
| • Inducement of angiogenesis by secreting angiogenic factors | |
| • Differentiation into cardiomyocyte | |
| Endothelial progenitor cells | • Incorporation into neovascularization |
| • Releasement of pro-angiogenic factors | |
| Monocytes | • Differentiation into macrophages and dendritic cells |
| • New collateral blood vessel formation | |
| Cardiac-derived stem cells | • Extensive paracrine effect |
| • Modulation of ischemic microenvironment | |
| Cardiomyocytes | • Differentiate into cardiomyocyte |
| • Electromechanical integration with host cardiomyocytes |
Wang et al. discovered that a cyclodextrin/PEG–PCL–PEG hydrogel could promote bone marrow-derived MSC cell colonization and differentiation in the infarcted area. This hydrogel was absorbed four weeks after administration. Thereby, MSCs were mostly retained in the infarcted tissue.108 This effectively increased vessel density while inhibiting infarct dilatation and left ventricular remodeling. The pore size of the hydrogel can also influence the paracrine effect of MSCs. Qazi et al. reported that medium pore size hydrogels (500 m) promoted pro-angiogenic factor secretion by MSCs more effectively compared with small pore size (400 m) and large pore size (600 m) hydrogels.109 This class of drug delivery systems has anti-inflammatory properties in addition to cardiac tissue repair. By converting adenosine monophosphate (AMP) to adenosine via CD73, bone marrow-derived MSCs encapsulated in hydrogels effectively suppressed acute inflammatory cascade in MI mice model.110 MSCs are more abundant in the stromal fraction of adipose tissue than in bone marrow. Adipose tissue is also easily accessible via liposuction. As a result, adipose-derived MSCs are a good source of MSCs. Chen et al. cultured ADSCs in cross-linked transglutaminase gelatin (Col-Tgel). Col-Tgel created an adaptive microenvironment for ADSCs at the infarct site that is conducive to differentiation and secretion, thereby improving ADSCs cardioprotection.111 In addition to the commonly used fibrin hydrogels, alginate hydrogels, and hyaluronic acid hydrogels, researchers have developed various other hydrogel systems for MSC cardiac delivery. Koivunotko et al. discovered nano-fibrillated cellulose (NFC) as a superior biomaterial scaffold capable of delivering MSCs. NFC maintained the high viability and angiogenic properties of ADSCs in vitro. As a result, NFC was able to deliver MSCs for MI therapy.112 Wu et al. co-loaded MSCs and cardioblast cells (H9C2 cells) into chitosan-silk fibroin hydrogel. This co-culture strategy reduced the electrophysiological barriers of native MSCs and released heart-friendly cytokines, which slowed down the acute MI process.113 Ding et al. designed a novel injectable hydrogel to encapsulate MSCs, achieving better therapeutic results in MI treatment. This hydrogel has a dual function of reactive oxygen removal and oxygen generation.114 The lack of electromechanical coupling of the hydrogel to the host myocardial tissue and the inability to monitor the implantation may compromise optimal treatment outcomes. Zhu et al. prepared an injectable conductive nanocomposite hydrogel. The hydrogel consisted of gold nanorods (GNRs), synthetic silicate nanoplatelets (SNs), and PLGA–PEG–PLGA. Among them, SNs supported the proliferation of MSCs.115 Carbon nanotubes modified poly-(N-isopropylacrylamide) (PNIPAAm) hydrogel was prepared by Li et al. The hydrogel has good electrical conductivity and mechanical support properties. It can effectively deliver ADSCs to the infarct site and maintain their activity.116
Various materials such as fibrin hydrogels, hyaluronic acid, alginate, self-assembling peptide (SAP), and ECM for hydrogel delivery of EPCs have been reported.121–125 For example, Campbell et al. reported an alginate hydrogel that can be degraded by lytic enzymes. In this work, alginate lyase and outgrowth endothelial cells (OECs) (a subpopulation of EPCs) were co-loaded in a hydrogel. After injection into the heart, this hydrogel is progressively degraded by enzymatic action, with an increase in pore size and subsequent release of the encapsulated OECs.123 After MI occurs, adverse remodeling of the ventricles leads to an abnormal biomechanical environment of the heart. For this reason, Gaffey et al. designed a novel injectable shear-thinning hyaluronic acid hydrogel. This hydrogel significantly increased the efficiency of intracardiac transplantation and retention of EPCs. In addition, the hydrogel adhered to the infarct site, stabilized the border zone myocardium, and slowed down the adverse myocardial remodeling process. The hyaluronic acid hydrogel, together with encapsulated EPCs reduced left ventricular dilatation, thus improving cardiac function.126 Cardiac lymphatic vessel damage leads to cardiac lymphedema. Recent studies have shown that stimulation of lymphatic angiogenesis reduces cardiac lymphedema and slows down the MI process. However, little is known about the effect of lymphatic endothelial progenitor cells (LEPCs) on the formation of cardiac lymph nodes. Zhang et al. evaluated the safety and efficacy of a self-assembling peptide hydrogel encapsulating LEPCs and VEGF-C for treating MI. The results showed that the cell hydrogel delivery system promoted the survival of transplanted cells and cardiac lymphatic angiogenesis. Notably, cardiac lymph-angiogenesis facilitated the clearance of inflammatory cells from the infarct site, thereby alleviating cardiac edema and ventricular remodeling.125 Engineered heart tissue transplantation requires a good vascular network to provide oxygen and nutrients to the living cells. Based on this, Yang et al. proposed spheroids of early vascular cells (EVCs) derived from hESCs rather than single-cell forms as transplant “seeds” for reconstructing microvascular networks. EVCs are capable of forming vascular networks in extracellular matrix hydrogels. After being injected into the ischemic site of the heart in MI mice, this delivery system effectively reduced infarct size and improved cardiac function.124
Fewer studies used hydrogel to deliver MNCs for MI therapy. Li et al. discovered that hydrogels enhanced BM-MNC colonization in the infarct site. Hydrogel injection with BM-MNCs promoted angiogenesis and prevented scar expansion in MI rabbits, facilitating cardiac function restoration.134 In a large animal model, Chen et al. assessed the safety and efficacy of BM-MNCs-loaded hyaluronic acid hydrogel. According to the findings, this strategy significantly increased left ventricular ejection fraction, systolic function, and neovascular differentiation in the hearts of the porcine MI model. Notably, the hyaluronic acid hydrogel effectively promoted the differentiation of BM-MNCs at the infarct site.131
In addition, studies have reported a population of progenitor cells derived from resident cardiac stem cells, termed cardiac progenitor cells (CPCs).137 In contrast to terminally differentiated cardiomyocytes, CPCs are highly proliferative and can theoretically differentiate into all types of cardiomyocytes to efficiently reconstruct damaged cardiac tissue and promote angiogenesis. Cardiospheres are self-adhesive heterogeneous cell populations obtained from in vitro cardiac tissue cultures.138 It typically contains several different types, including endothelial cells, mesenchymal cells, and c-kit, Sca-1, and CD34 expressed stem cells.139 Because of their feeblish differentiation capacity, the therapeutic potential of CDCs for MI basically relies on their paracrine function. Although CDCs do not significantly improve LV (left ventricle) ejection fraction and LV volume, they reduce scar tissue and increase local ventricular wall contractility after MI.
Phase I clinical trials have been completed with injectable gelatin hydrogels containing human cardiac-derived stem cells and fibroblast growth factor (bFGF).140 Takehara et al. demonstrated that hydrogels co-loaded with bFGF and CDCs could improve post-infarction cardiac function by modulating the ischemic microenvironment and enhancing cardiomyocyte viability.141 Prat-Vidal et al. evaluated the safety of coronary artery injection of porcine cardiac progenitor cells (pCPCs) in an acute MI porcine model.142 A clinical trial further validated the technical feasibility and short-term safety of using fibrin hydrogel delivery of CPCs.143 In addition, Masato Kanda reported a three-dimensional hydrogel scaffold (CPC-PRGmx) for culturing CPCs. CPC-PRGmx consisted of RGD peptide-conjugated self-assembling peptide and insulin-like growth factor-1 (IGF-1). This cell hydrogel delivery strategy was potential in MI therapy.144 The hydrophilic hydrogel environment is more conducive to improving cell viability and achieving uniform cell distribution in engineered cardiac tissues. Chang et al. prepared N-hydroxysuccinimide-functionalized hyaluronic acid hydrogels to encapsulate CDCs and enhance cell viability. The NHS-activated HA hydrogels reacted with primary amines in the myocardium to form a more stable three-dimensional structure with the infarcted tissue, allowing the hydrogel to remain at the injection site for over 4 weeks. The encapsulated CDCs effectively improved left ventricular ejection fraction after myocardial infarction.145
The hydrogel delivery system is vital to increase cell retention and ameliorate the cell transplantation microenvironment. Oligo [poly(ethylene glycol) fumarate] (OPF) hydrogels were used to encapsulate mouse embryonic stem cells (mESCs) by Wang et al. In vivo ascorbic acid induces mESCs to differentiate into cardiomyocytes and other cell types in all three lineages. OPF hydrogels effectively increased cell retention and reduced infarct size together with ESC-CMs.155 Nakajima et al. validated that water-soluble gelatin hydrogel was a suitable biomaterial for delivering ESC-CMs.156 Erythropoietin (EPO) could promote cell survival. Chow et al. found that treatment with polyethylene glycol hydrogels encapsulating iPSC-CMs with EPO improved cardiac function and increased infarct thickness and muscle content after MI.157 Delivery of iPSC-CMs using conventional hydrogels suffered from poor attachment capacity, single function, and inability to control the direction of differentiation of iPSCs. To address these problems, Khazaei et al. designed an iPSC-CMs-integrated conductive hydrogel microneedle. The anisotropic structure of the encapsulated parallel-aligned carbon nanotube induced the directional alignment of iPSCs and endowed it with preferable conductivity. MSCs differentiated into cardiomyocytes under the stimulation of MSCs-related genes (e.g., microRNA-1) or differentiation factors (e.g., Myocardin). In addition, hydrogels with three-dimensional structures used for cell culture also improved the induction efficiency of MSC-derived cardiomyocytes (MSC-CMs).158 Wu et al. used a decellularized porcine extracellular matrix to deliver exogenous cardiomyocytes into the infarcted region. The co-loaded stromal cell-derived factor 1 (SDF-1) alleviated the ischemic microenvironment and improved the engraftment environment of cardiomyocytes.159
4.2 Cell-derived extracellular vesicle-laden hydrogels for MI therapy
EVs are natural lipid nanoparticles released by cells that can transport various macromolecular active substances (e.g., cytokines and non-coding RNA).164 Apoptotic bodies (size between 1 and 4 μm), microvesicles (size between 50 and 1000 nm), and exosomes (size between 30 and 100 nm) are the three main types of EVs.165–168 Apoptotic bodies are the remains of cells undergoing apoptosis or programmed cell death. Microvesicles are membrane-bound sacs that are shed from cell surfaces into the extracellular environment in a highly regulated process. Exosomes are generated in the endosomal compartment, which contains the proteins, DNA, and RNA of the cells that secrete them. EVs contain a wide range of biological activities and participate in many physiological or pathological cardiovascular diseases processes, such as angiogenic regulation, blood pressure regulation, cardiomyocyte hypertrophy, and cardiac fibrosis.169,170Most mechanisms of MI therapy with stem cells involve the paracrine release of extracellular vesicles containing bioactive substances. As a result, some researchers have extracted extracellular vesicles from stem cells in vitro for MI therapy.171 Preclinical studies have confirmed the therapeutic potential of EVs in alleviating ischemic heart injury.172 The therapeutic mechanisms primarily include reduced monocyte infiltration in ischemic cardiac tissue, angiogenesis promotion, and macrophage polarization regulation. Compared with cell therapy, the therapeutic effect of EVs by releasing active contents (e.g., miRNAs and cytokines) in MI is more direct. Furthermore, EVs are only nanometers in size. Thus, EVs are able to penetrate tissue and cells deeper.173 It should be noted that EVs (particularly stem cell-derived EVs) are typically less effective due to a lack of targeting ability in the heart and in the infarct region.174 The recently developed injectable hydrogels show the potential to conquer this issue. EVs-containing hydrogels can be prepared through “sponge absorption,” mix and cross-linking, and in situ hydrogel formation.175 This section summarizes the research on using EVs-loaded hydrogels for MI therapy according to the type of source cells (Fig. 3).
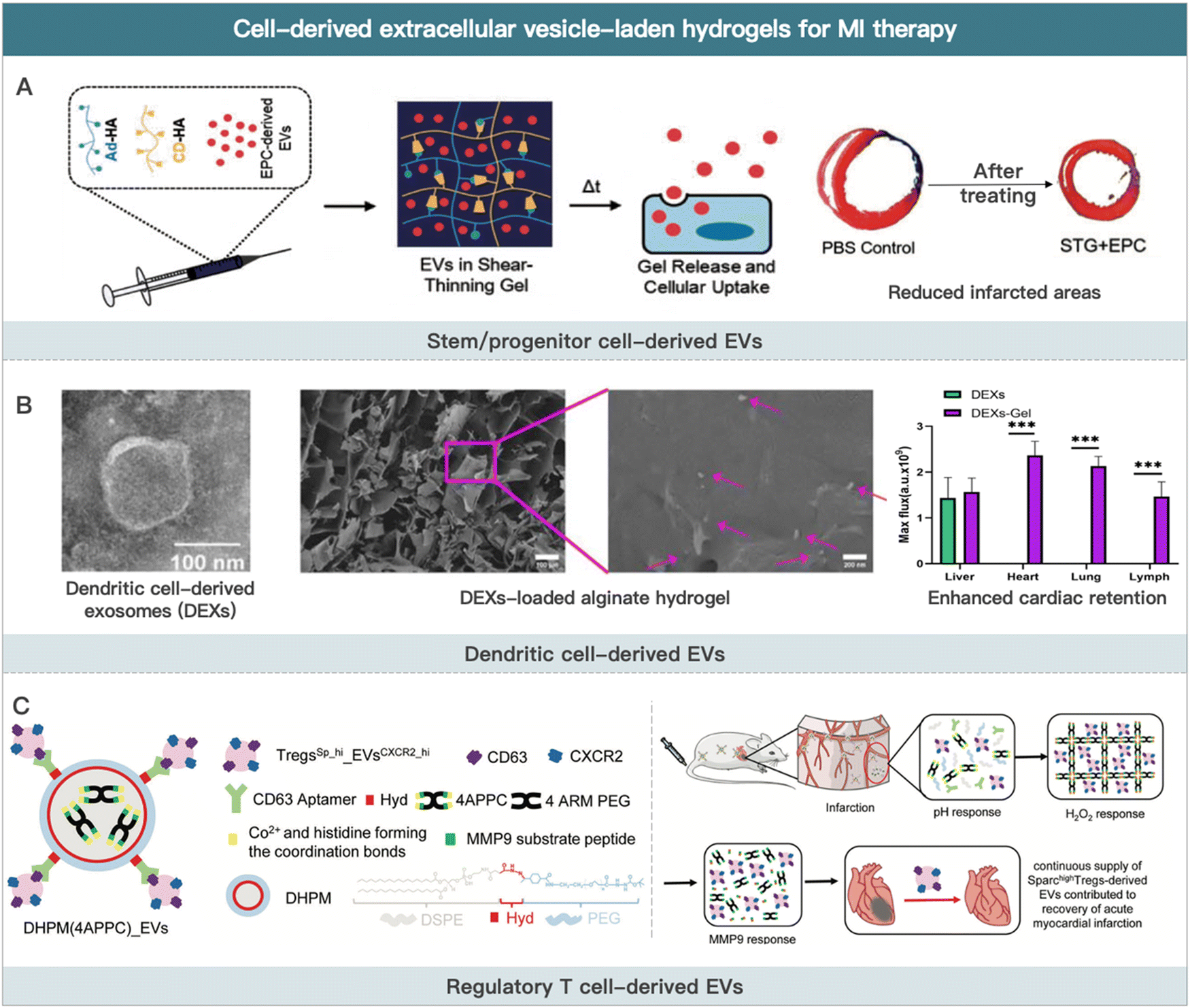 | ||
| Fig. 3 Cell-derived extracellular vesicle-laden hydrogels for MI therapy. (A) Schematic illustration of EPCs-derived EVs for improving border zone remodeling post-MI. Reproduced with permission from ref. 19, Copyright Oxford University Press 2018. (B) Schematic illustration of dendritic cell-derived EVs loaded alginate hydrogel for improving cardiac function post-MI. Reproduced from ref. 190 under the open access policy of Springer Nature 2021. (C) Schematic illustration of Sparchigh Tregs derived-EVs loaded hydrogel for promoting the recovery of cardiac function. Reproduced with permission from ref. 192, Copyright Wiley-VCH GmbH 2022. | ||
However, one of the critical limitations of their clinical application is the inefficient production rate of highly active EVs. It has been reported that using three-dimensional hydrogels as the culture medium could significantly enhance the yield of MSC-derived EVs compared to the two-dimensional liquid medium. EVs obtained via this method were more potential in terms of cardio-protection due to the similar culture medium structure with the physiological environment. It allowed stem cells to respond positively to exogenous stimuli (e.g., temperature, pH, and nutrition) and produce higher-quality EVs.185 The expression of ISL1 could enhance the paracrine function of MSCs. Hu et al. used a gene transfection technique to generate MSCs with high ISL1 expression. This strategy made it possible to prepare MSC-derived EVs (ISL1-MSCs-Exo) efficiently. Using angiogenin-1 hydrogel (Ang-1 hydrogel) to encapsulate ISL1-MSCs-Exo increased EVs retention at the ischemic site and improved the anti-apoptotic and pro-angiogenic ability of EVs.186 In addition, researchers demonstrated an approach to prepare high amounts of extracellular vesicles with high bioactivity from endothelial progenitor cells by stimulation with silicate ions derived from silicate ceramics. The engineered EVs encapsulated in hydrogels significantly improved angiogenesis at the post-MI infarct site.187
5. Challenges and future prospects
5.1 Challenges
Injectable hydrogels have been highlighted to be the ideal heart tissue analogs. They served as functional myocardial tissue after efficient encapsulation and release of various cells or cell-derived EVs. However, several critical issues need to be addressed and clarified on the road to the commercialization of cell or cell derivative-laden hydrogels for MI treatment.5.2 Prospective future of MI therapy using cell-derived nanoparticles
Plenty of biomimetic targeted drug delivery systems have been developed via cell membrane-camouflaged technology.200 Numerous studies have shown that encapsulating cell membranes onto inner cores can endow nanoparticles with multiple biological functions. For example, erythrocyte membrane-camouflaged nanoparticles have a prolonged in vivo half-life due to the high expression of CD47 signaling molecules on the membrane surface.201 Neutrophil membrane-camouflaged nanoparticles accumulate at MI inflammation sites due to inflammatory chemotactic effects of vascular endothelial adhesion molecules (e.g., P-selectin and chemokine receptors).202 Compared with cell therapy, cell membrane-camouflaged biomimetic nanoparticles have following several advantages. (1) Multiple adhesion molecules expressed on the cell membrane endow the inner core with prolonged circulation time and infarct/inflammation targetablity.203 As a result, cell membrane-camouflaged nanoparticles exhibited a lower demand for hydrogel delivery compared to cell therapy. Few studies used hydrogel to deliver cell membrane-camouflaged nanoparticles for MI therapy. (2) Cell membrane-camouflaged nanoparticles do not have survival issues, which is unique to cell therapy. Besides, cell membrane-coated camouflaged nanoparticle formulation is more stable during storage. (3) Cell membrane-camouflaged nanoparticles are commonly nanometer in size. As a result, they can efficiently cross the tissue and cell barrier and penetrate deeper.204 (4) Cell membrane-camouflaged nanoparticles show lower T-cell immune response levels than cell preparations.205 Additionally, in contrast to cell-derived EVs, cell membrane-camouflaged nanoparticles have the advantages of various inner core selections, superior drug loading capacity, and high preparation efficiency.The preparation of cell membrane-camouflaged nanoparticles is primarily divided into four steps.206 (1) Cell membrane extraction and separation. Freezing or hypotonic are usually employed to lyse cells. Differential centrifugation is used to separate the cell membrane from the rest of the cell contents. (2) Cell membrane vesicle preparation. The lipid co-extrusion method is used to generate nanometer-sized cell membrane vesicles. (3) Inner core preparation. PLGA is the most commonly used core material, prepared using a solvent substitution method. (4) Cell membrane-camouflaged nanoparticles preparation. Lipid co-extrusion, microfluidics, or ultrasonication are commonly used to wrap the cell membrane onto the inner core to fabricate core–shell nanoparticles. Although the preparation process is time-consuming, it has a higher yield than that of cell-derived EVs.
Stem cell membranes express and attach with pro-cardiac or vascular regenerative cytokines.207 Neutrophil membranes bind inflammatory factors at the site of infarction, reducing inflammation. However, the therapeutic effect of cell membrane-coated nanoparticles for MI is extremely limited due to the lack of paracrine effect and release of endogenous active substances. To compensate for the diminished MI therapeutic activity of cell membrane-camouflaged nanoparticles, researchers usually load drugs in the inner core nanoparticles, including small molecule drugs (e.g., cyclosporine A and celecoxib) and active biomacromolecules (e.g., pro-angiogenic factors and miRNAs).208,209 Although cell membrane-camouflaged nanoparticles are not in high demand for hydrogel delivery as an emerging cell-derived MI therapeutic strategy, we still summarize the relevant studies in this section due to their potential MI therapeutic effect and intimate connection with cell therapy (Fig. 4).
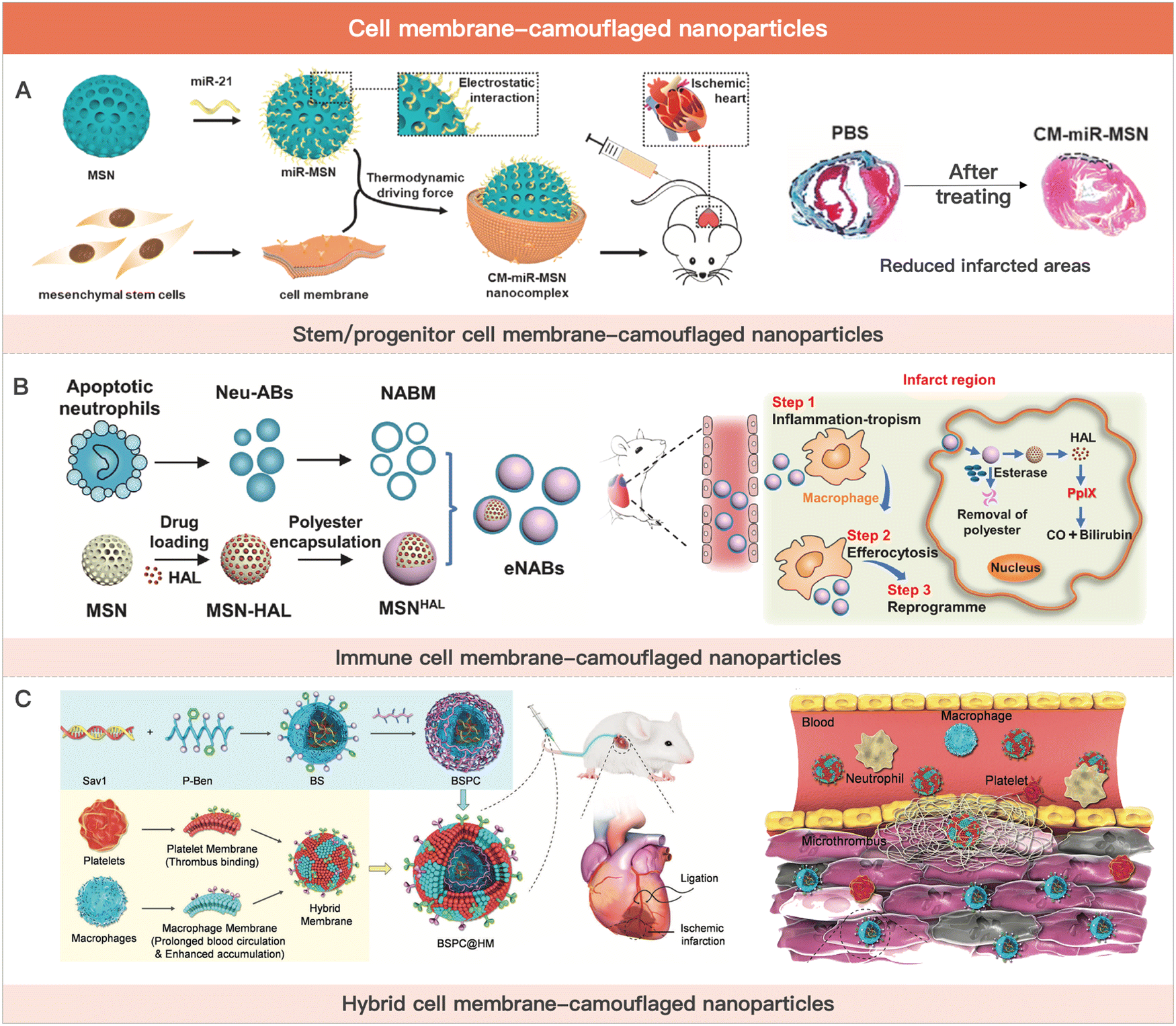 | ||
| Fig. 4 Cell membrane-camouflaged nanoparticles for MI therapy. (A) Schematic illustration of stem cell membrane-camouflaged exosome-mimicking nano-complex for the miRNA-mediated repair of MI injury. Reproduced with permission from ref. 223, Copyright Elsevier 2020. (B) Schematic of engineered neutrophil apoptotic bodies (eNABs) for MI treatment. Reproduced from ref. 229 under the open access policy of Elsevier 2021. (C) Schematic illustration of the platelet–macrophage hybrid membrane-coated nano-complexes for the myocardial siSav1 delivery and MI/IR injury management. Reproduced with permission from ref. 227, Copyright Wiley-VCH GmbH 2023. | ||
Platelets inhibit cardiovascular bleeding injury by forming clots through aggregation.217 Platelets have a variety of membrane-bound molecules on their membranes, such as GPIIb/IIIa, CD62P, immunomodulatory proteins, CD47, and CD55.184 During the inflammatory phase of infarction, CD62P on the platelet surface binds to P-selectin glycoprotein ligand-1 (PSGL-1) on monocytes, forming monocyte-platelet aggregates (MPA).218 Thus, platelet membrane-camouflaged nanoparticles can target the ischemic and inflammatory milieu after MI. On the one hand, the damage factors released early in MI recruit platelets/platelet-derived nanoparticles. On the other hand, platelet membrane-camouflaged nanoparticles can bind to monocytes in blood circulation. Monocytes are further recruited to the site of MI inflammation induced by chemokines. These processes enhance the post-MI cardiac targeting ability of platelet-derived nanoparticles. Zhang et al. reported a platelet membrane-camouflaged silica nanoparticle to deliver H-D-Arg-Dmt-Ly-Phe-NH2 (SS31) peptide with antioxidant capacity. This strategy effectively delivered SS31 peptide to the damaged heart site to exert antioxidant function.219 In addition, researchers have delivered non-selective β-blockers (Carvedilol), tissue fibrinogen activator (tPA), and CSCs-derived secretome by platelet membrane-coated nanoparticles for treating MI.220
Tang et al. demonstrated the safety and efficacy of CSCs cell membrane-camouflaged nanoparticles (CMMPs), which carry secreted and membrane proteins similar to those of CSCs. In a mouse model of myocardial infarction, CMMPs enhanced cardiac function without causing infiltration of T cells in mice.198 Yao et al. used this drug delivery system for the cardiac delivery of miRNAs. The nanoparticles effectively encapsulated miRNAs and protected them from in vivo protease degradation. miRNAs delivered to the site of ischemic injury inhibited the translation of apoptosis-related proteins, effectively improving cardiac function in MI model mice.223
Monocytes/macrophages mediate the innate immune response in vivo. After MI occurs, monocytes are recruited to the infarct site and differentiate into macrophages.224 Together with the cardiac resident macrophages, the recruited macrophages regulate the inflammatory balance at all phases of MI. They are able to bind to vascular cell adhesion molecule-1 (VCAM-1) on chemically injured vascular endothelial cells.225 By inheriting the inherent targeting and migration ability of macrophages, the nanoparticle delivery system was able to deliver polydopamine with antioxidant capacity to the infarcted myocardium. It has shown great potential in myocardial ischemia/reperfusion injury (MI/RI) therapy.226 Based on this, Zhou et al. used macrophage cell membrane-camouflaged nanoparticles to deliver Sav1 siRNA. By down-regulating Sav1 expression levels in injured myocardium, the nano complex significantly inhibited cardiomyocyte apoptosis and restored cardiac function.227
Neutrophils infiltrate the inflammatory regions of myocardial infarction through the interaction of surface adhesion molecules with endothelial cells. Based on this, Han et al. used neutrophil membrane-camouflaged nanoparticles to alleviate ischemic myocardial injury-mediated inflammation and promote angiogenesis. The mechanism involved the pro-angiogenic effect of IL-5 and the inflammatory factor absorption effect of the neutrophil membrane.195 During the inflammatory and proliferative phases of MI, neutrophils apoptosis and are phagocytosed by macrophages.228 This leads to a polarization of macrophages from pro-inflammatory M1-phenotype macrophages to anti-inflammatory M2-phenotype. This process mediates the transition of MI from the inflammatory phase to the proliferative phase. Administration of neutrophil-derived apoptotic vesicles early in MI to suppress the inflammatory microenvironment in the infarcted region may be an effective strategy for MI treatment. In addition, neutrophils are recruited to the infarcted region during the inflammatory phase. Thus, intravenous infusion of neutrophil-derived apoptotic vesicles is able to target the site of ischemic inflammation in the heart, which greatly reduces the need for hydrogel delivery. Bao et al. used neutrophil apoptotic vesicle membranes wrapped in silica nanoparticles to mimic neutrophil apoptosis in the MI state. The functionalized neutrophil apoptotic vesicles had excellent inflammatory targeting ability and enhanced macrophage efferocytosis and reprogramming for inflammation resolution. This strategy could promote cardiac tissue regeneration after MI.229
Regulatory T lymphocytes express the Forkhead box P3 (Foxp3) transcription factor, which plays an important role in maintaining immune system homeostasis and suppressing the pro-inflammatory immune responses.230 Saxena et al. reported that Treg depletion increased inflammatory response and dilated left ventricular in mice after myocardial infarction.231 Another study reported similar results. The adoptive transfer of Tregs attenuated the post-infarction inflammatory response with poor cardiac remodeling.232 Inspired by this, Li et al. prepared Treg biomimetic nanoparticles that mimic Treg to perform various functions, such as targeting ischemic myocardium, anti-inflammation, anti-apoptosis, and scavenging reactive oxygen species. This strategy significantly alleviated left ventricular remodeling and enhanced cardiac function and has the potential to treat MI/RI.233
6. Conclusion
In conclusion, from the perspective of cell types, we introduced cell or cell derivative-laden hydrogels for MI therapy and the prospective future by using cell-derived nanoparticles (Fig. 5). These therapies have been developed to address the limitations of traditional cell therapy and to move towards engineered cell therapy. However, each therapy modality has its own advantages and disadvantages (Table 3). Cell or cell-inspired MI therapy requires hydrogel delivery, especially cell therapy and cell-derived EVs, as they are rapidly eliminated after in vivo infusion and are unable to remain in the ischemic region for long periods of time.234 Currently, several stem cell intracardiac infusions are in clinical phase III trials for MI treatment.132 However, there have been limited clinical trials on the use of cell-derived EVs for MI treatment. A single study on the use of EVs derived from cardiovascular progenitor cells has entered clinical trials for non-ischemic myocardiopathy therapy.177 The challenges associated with traditional drug delivery systems include low yield, difficulty in targeting specific areas in vivo, and challenges in quality assessment. However, cell membrane-camouflaged biomimetic drug delivery systems, such as those utilizing red blood cells or immune cell membrane-camouflaged nanoparticles, offer potential advantages such as long-term circulation and targeted delivery to inflammatory areas. As a result, cell membrane-camouflaged nanoparticles have a lower demand for hydrogel administration when compared to cell treatment. However, researchers need to address several issues (e.g., stability, large-scale production, animal model selection, and administration timing) before the commercialization of cell or cell derivative-laden hydrogels for MI treatment.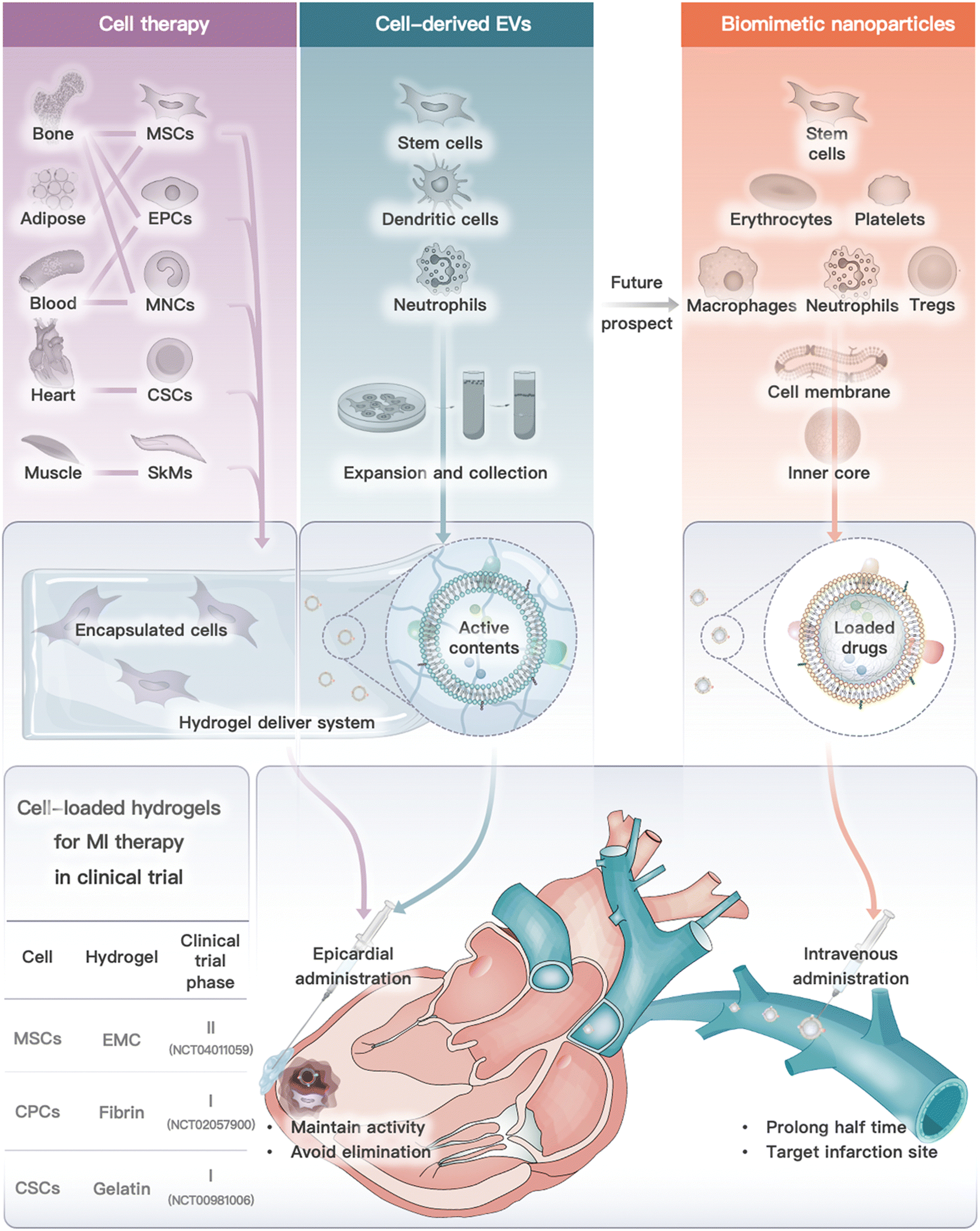 | ||
| Fig. 5 Cell or cell derivative-laden hydrogels for MI therapy and future prospect on biomimetic nanoparticles. Cell or cell-derived EVs need to be encapsulated into hydrogels and delivered by epicardial administration (or other feasible routes), while cell membrane camouflaged nanoparticles exhibited lower demand for hydrogel delivery, which are commonly administrated by intravenous injection. Notably, three clinical trials of cell-loaded hydrogel for MI therapy are also summarized.141,143,153 | ||
| Cell sources | Advantages | Disadvantages | |
|---|---|---|---|
| Cell therapy | • Mesenchymal stem cells | • High differentiation and proliferation activity | • Low cell viability |
| • Endothelial progenitor cells | • Paracrine effect | • Poor formulation storage stability | |
| • Monocytes | • Fast clearance | ||
| • Cardiac-derived stem cells | • Activates local T-cell response | ||
| • Skeletal myoblasts | |||
| • Cardiomyocytes | |||
| Cell-derived EVs | • Stem/progenitor cells (CPCs, EPCs, MSCs) | • No viability issues | • Low yield |
| • Dendritic cells | • Relatively stable formulation storage | • Fast clearance, poor targeting | |
| • Regulatory T cells | • High tissue penetration ability | • No differentiation and proliferation ability | |
| • Can release active substances | |||
| • Weak local T-cell response | |||
Despite the hurdles and concerns, cell or cell derivative-laden hydrogel formulations for MI are expected to be available soon, owing to the significant potential of cell or cell-inspired techniques in MI therapy, as well as the rapid progress of injectable hydrogels in clinical trials.
Author contributions
Z. X. mainly contributed to this work, and the manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to many scientists whose research results are cited here. This work was financially supported by the National Natural Science Foundation of China (82104069) and Sichuan Science and Technology Program (2022089).References
- M. S. Nawaz, B. Shoaib and M. A. Ashraf, Heliyon, 2021, 7, e06948 CrossRef PubMed.
- D. Mozaffarian, E. J. Benjamin, A. S. Go, D. K. Arnett, M. J. Blaha, M. Cushman, S. de Ferranti, J.-P. Després, H. J. Fullerton, V. J. Howard, M. D. Huffman, S. E. Judd, B. M. Kissela, D. T. Lackland, J. H. Lichtman, L. D. Lisabeth, S. Liu, R. H. Mackey, D. B. Matchar, D. K. McGuire, E. R. MohlerIII, C. S. Moy, P. Muntner, M. E. Mussolino, K. Nasir, R. W. Neumar, G. Nichol, L. Palaniappan, D. K. Pandey, M. J. Reeves, C. J. Rodriguez, P. D. Sorlie, J. Stein, A. Towfighi, T. N. Turan, S. S. Virani, J. Z. Willey, D. Woo, R. W. Yeh and M. B. Turner, Circulation, 2015, 131, e29–e322 Search PubMed.
- A. R. Roudkoli, S. Behrooj, H. Farshidi, F. Khorrami and S. S. Korani, J. Preventive Epidemiol., 2022, 7, e26167–e26167 Search PubMed.
- G. Ren, O. Dewald and N. G. Frangogiannis, Curr. Drug Targets: Inflammation Allergy, 2003, 2, 242–256 CAS.
- X. Chang, R. Liu, R. Li, Y. Peng, P. Zhu and H. Zhou., Int. J. Biol. Sci., 2023, 19, 426 CrossRef CAS PubMed.
- R. G. Gourdie, S. Dimmeler and P. Kohl, Nat. Rev. Drug Discovery, 2016, 15, 620–638 CrossRef CAS PubMed.
- S. P. D'Souza, M. A. Mamas, D. G. Fraser and F. Fath-Ordoubadi, Eur. Heart J., 2011, 32, 972–982 CrossRef PubMed.
- Y. Zhu, S. Chen, X. Zhao, S. Qiao, Q. Yang, R. Gao and Y. Wu, Br. J. Clin. Pharmacol., 2022, 88, 490–499 CrossRef PubMed.
- R. Sui, X. Liao, X. Zhou and Q. Tan, Stem Cell Rev. Rep., 2011, 7, 172–180 CrossRef PubMed.
- M. Li, H. Wu, Y. Yuan, B. Hu and N. Gu, View, 2022, 3, 20200153 CrossRef CAS.
- A. E. S. Shafei, M. A. Ali, H. G. Ghanem, A. I. Shehata, A. A. Abdelgawad, H. R. Handal, K. A. Talaat, A. E. Ashaal and A. S. El-Shal, J. Gene Med., 2017, 19, e2995 CrossRef PubMed.
- K. Huang, S. Hu and K. Cheng, Adv. Healthcare Mater., 2019, 8, 1801011 CrossRef PubMed.
- N. Dib, H. Khawaja, S. Varner, M. McCarthy and A. Campbell, J. Cardiovascular Trans. Res., 2011, 4, 177–181 CrossRef PubMed.
- J. Ankrum and J. M. Karp, Trends Mol. Med., 2010, 16, 203–209 CrossRef PubMed.
- R. Alonaizan and C. Carr, Biochem. Soc. Trans., 2022, 50, 269–281 CrossRef CAS PubMed.
- S. C. Suresh, V. Selvaraju, M. Thirunavukkarasu, J. W. Goldman, A. Husain, J. A. Palesty, J. A. Sanchez, D. W. McFadden and N. Maulik, Int. J. Cardiol., 2015, 201, 517–528 CrossRef PubMed.
- L. Yang, J. Zhu, C. Zhang, J. Wang, F. Yue, X. Jia and H. Liu, Aging, 2019, 11, 1129 CrossRef CAS PubMed.
- S. Bian, L. Zhang, L. Duan, X. Wang, Y. Min and H. Yu, J. Mol. Med., 2014, 92, 387–397 CrossRef CAS PubMed.
- C. W. Chen, L. L. Wang, S. Zaman, J. Gordon, M. F. Arisi, C. M. Venkataraman, J. J. Chung, G. Hung, A. C. Gaffey, L. A. Spruce, H. Fazelinia, R. C. Gorman, S. H. Seeholzer, J. A. Burdick and P. Atluri, Cardiovasc. Res., 2018, 114, 1029–1040 CrossRef CAS PubMed.
- N. Akbar, J. E. Digby, T. J. Cahill, A. N. Tavare, A. L. Corbin, S. Saluja, S. Dawkins, L. Edgar, N. Rawlings, K. Ziberna, E. McNeill, E. Johnson, A. A. Aljabali, R. A. Dragovic, M. Rohling, T. G. Belgard, I. A. Udalova, D. R. Greaves, K. M. Channon, P. R. Riley, D. C. Anthony and R. P. Choudhury, JCI Insight, 2017, 2, e93344 CrossRef PubMed.
- Y. Yamada, S. Minatoguchi, H. Kanamori, A. Mikami, H. Okura, M. Dezawa and S. Minatoguchi, J. Cardiol., 2022, 80, 80–87 CrossRef PubMed.
- S. M. Parizadeh, R. Jafarzadeh-Esfehani, M. Ghandehari, M. R. Parizadeh, G. A. Ferns, A. Avan and S. M. Hassanian, J. Cell. Physiol., 2019, 234, 16904–16912 CrossRef CAS PubMed.
- M. Zhou, Q. Zuo, Y. Huang and L. Li, Acta Pharm. Sin. B, 2022, 12, 3383–3397 CrossRef CAS PubMed.
- J. Li, P. Zhang, M. Zhou, C. Liu, Y. Huang and L. Li, ACS Nano, 2022, 16, 6064–6079 CrossRef CAS PubMed.
- T. Chang, C. Liu, K. Lu, Y. Wu, M. Xu, Q. Yu, Z. Shen, T. Jiang and Y. Zhang, J. Mater. Sci. Technol., 2021, 94, 77–89 CrossRef CAS.
- T. D. Johnson and K. L. Christman, Expert Opin. Drug Delivery, 2013, 10, 59–72 CrossRef CAS PubMed.
- J. Li, Y. Yan, P. Zhang, J. Ding, Y. Huang, Y. Jin and L. Li, J. Controlled Release, 2022, 351, 231–244 CrossRef CAS PubMed.
- Y. Yan, J. Li, X. Yi, C. Liu, Z. Zhou, Y. Huang and L. Li, J. Controlled Release, 2022, 352, 747–758 CrossRef CAS PubMed.
- A. Hasan, A. Khattab, M. A. Islam, K. A. Hweij, J. Zeitouny, R. Waters, M. Sayegh, M. M. Hossain and A. Paul, Adv. Sci., 2015, 2, 1500122 CrossRef PubMed.
- L. Wang, M. Neumann, T. Fu, W. Li, X. Cheng and B. Su, Curr. Opin. Colloid Interface Sci., 2018, 38, 135–157 CrossRef CAS.
- Z. Zheng, Y. Tan, Y. Li, Y. Liu, G. Yi, C. Yu and H. Wei, J. Controlled Release, 2021, 335, 216–236 CrossRef CAS PubMed.
- J. L. Ungerleider and K. L. Christman, Stem Cells Transl. Med., 2014, 3, 1090–1099 CrossRef CAS PubMed.
- C. Yao, W. Wu, H. Tang, X. Jia, J. Tang, X. Ruan, F. Li, D. Leong, D. Luo and D. Yang, Biomaterials, 2020, 257, 120256 CrossRef CAS PubMed.
- R. Zhang, S. Wu, Q. Ding, Q. Fan, Y. Dai, S. Guo, Y. Ye, C. Li and M. Zhou, Drug Delivery, 2021, 28, 1109–1119 CrossRef CAS PubMed.
- X. Liu, X. Zhong and C. Li, Chin. Chem. Lett., 2021, 32, 2347–2358 CrossRef CAS.
- X. Li, G. Hong, G. Zhao, H. Pei, J. Qu, C. Chun, Z. Huang and Z. Lu, Front. Pharmacol., 2022, 13, 1274 Search PubMed.
- A. M. Lodrini and M. J. Goumans, Front. Cardiovasc. Med., 2021, 8, 750510 CrossRef CAS PubMed.
- G. A. Gray, I. S. Toor, R. F. P. Castellan, M. Crisan and M. Meloni, Curr. Opin. Physiol., 2018, 1, 46–51 CrossRef CAS PubMed.
- U. Chalise, M. Becirovic-Agic and M. L. Lindsey, WIREs Mech. Dis., 2023, 15, e1584 CrossRef CAS PubMed.
- P. Christia and N. G. Frangogiannis, Eur. J. Clin. Invest., 2013, 43, 986–995 CrossRef CAS PubMed.
- K. A. Mateesku, P. A. Lebedev and N. F. Scherbakova, Aspirantskiy Vestnik Povolzhiya, 2016, 16, 190–195 Search PubMed.
- Y. Zaidi, E. G. Aguilar, M. Troncoso, D. V. Ilatovskaya and K. Y. DeLeon-Pennell, Cell. Signalling, 2021, 77, 109837 CrossRef CAS PubMed.
- S. D. Francis Stuart, N. M. De Jesus, M. L. Lindsey and C. M. Ripplinger, J. Mol. Cell. Cardiol., 2016, 91, 114–122 CrossRef CAS PubMed.
- D. Cheng, J. Zheng, F. Hu, W. Lv and C. Lu, Front. Physiol., 2021, 12, 717187 CrossRef PubMed.
- X. Wang, Z. Guo, Z. Ding and J. Mehta, J. Am. Heart Assoc., 2018, 7, e008024 CrossRef PubMed.
- M. Hori and K. Nishida, Cardiovasc. Res., 2009, 81, 457–464 CrossRef CAS PubMed.
- L. Timmers, J. P. G. Sluijter, J. K. van Keulen, I. E. Hoefer, M. G. J. Nederhoff, M. J. Goumans, P. A. Doevendans, C. J. A. van Echteld, J. A. Joles, P. H. Quax, J. J. Piek, G. Pasterkamp and D. P. V. de Kleijn, Circ. Res., 2008, 102, 257–264 CrossRef CAS PubMed.
- V. Dayan, G. Yannarelli, F. Billia, P. Filomeno, X. Wang, J. E. Davies and A. Keating, Basic Res. Cardiol., 2011, 106, 1299–1310 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, J. Li, C. Li, R. Zhao, C. Shen, W. Liu, J. Rong, Z. Wang, J. Ge and B. Shi, Theranostics, 2023, 13, 2192 CrossRef CAS PubMed.
- C. Tian, M. Li, X. Shuai, F. Jiang, Y. Dong, Y. Gui, Z. Zhang, R. Qin, Z. Kang, L. Lin, A. Sarapultsev, B. Wu, S. Luo and D. Hu, Phytother. Res., 2023, 37, 50–61 CrossRef CAS PubMed.
- M. D. Tallquist and J. D. Molkentin, Nat. Rev. Cardiol., 2017, 14, 484–491 CrossRef PubMed.
- H. Venugopal, A. Hanna, C. Humeres and N. G. Frangogiannis, Cells, 2022, 11, 1386 CrossRef CAS PubMed.
- Q. Feng, Q. Li, H. Zhou, L. Sun, C. Lin, Y. Jin, D. Wang and G. Guo, Front. Immunol., 2023, 13, 1084460 CrossRef PubMed.
- A. Schmidt, D. Ladage, C. Steingen, K. Brixius, T. Schinköthe, F. Klinz, R. H. G. Schwinger, U. Mehlhorn and W. Bloch, Eur. J. Cell Biol., 2006, 85, 1179–1188 CrossRef CAS PubMed.
- C. G. Manole, V. Cismaşiu, M. Gherghiceanu and L. M. Popescu, J. Cell. Mol. Med., 2011, 15, 2284–2296 CrossRef CAS PubMed.
- Y. L. Tang, Q. Zhao, X. Qin, L. Shen, L. Cheng, J. Ge and M. L. Phillips, Ann. Thorac. Surg., 2005, 80, 229–237 CrossRef PubMed.
- M. J. Schotman and P. Y. Dankers, Adv. Mater. Interfaces, 2022, 9, 2100942 CrossRef CAS.
- D. Y. Nah and M. Y. Rhee, Korean Circ. J., 2009, 39, 393–398 CrossRef CAS PubMed.
- S. D. Prabhu, Circ. Res., 2014, 114, 1558–1560 CrossRef CAS PubMed.
- V. Talman and H. Ruskoaho, Cell Tissue Res., 2016, 365, 563–581 CrossRef CAS PubMed.
- X. Lin, Y. Liu, A. Bai, H. Cai, Y. Bai, W. Jiang, H. Yang, X. Wang, L. Yang, N. Sun and H. Gao, Nat. Biomed. Eng., 2019, 3, 632–643 CrossRef CAS PubMed.
- A. I. Hassaballah, M. A. Hassan, A. N. Mardi and M. Hamdi, PLoS One, 2013, 8, e82703 CrossRef PubMed.
- Z. Fan, M. Fu, Z. Xu, B. Zhang, Z. Li, H. Li, X. Zhou, X. Liu, Y. Duan, P. Lin, P. Duann, X. Xie, J. Ma, Z. Liu and J. Guan, Biomacromolecules, 2017, 18, 2820–2829 CrossRef CAS PubMed.
- M. Neri, I. Riezzo, N. Pascale, C. Pomara and E. Turillazzi, Mediators inflammation, 2017, 2017 Search PubMed.
- H. N. Sabbah, M. Wang, R. C. Gupta, S. Rastogi, I. Ilsar, M. S. Sabbah, S. Kohli, S. Helgerson and R. J. Lee, JACC Heart Fail., 2013, 1, 252–258 CrossRef PubMed.
- Y. Efraim, H. Sarig, N. C. Anavy, U. Sarig, E. de Berardinis, S. Chaw, M. Krishnamoorthi, J. Kalifa, H. Bogireddi, T. V. Duc, T. Kofidis, L. Baruch, F. Y. C. Boey, S. S. Venkatraman and M. Machluf, Acta Biomater., 2017, 50, 220–233 CrossRef CAS PubMed.
- R. J. Lee, A. Hinson, R. Bauernschmitt, K. Matschke, Q. Fang, D. L. Mann, R. Dowling, N. Schiller and H. N. Sabbah, Int. J. Cardiol., 2015, 199, 18–24 CrossRef PubMed.
- S. V. Rao, U. Zeymer, P. S. Douglas, H. Al-Khalidi, J. A. White, J. Liu, H. Levy, V. Guetta, C. M. Gibson, J. F. Tanguay and P. Vermeersch, J. Am. Coll. Cardiol., 2016, 68, 715–723 CrossRef PubMed.
- D. M. Clifford, S. A. Fisher, S. J. Brunskill, C. Doree, A. Mathur and S. Watt., Cochrane Database Syst. Rev., 2012, 2, CD006536 Search PubMed.
- H. Yang, Nat. Biotechnol., 2011, 29, 857–858 CrossRef CAS.
- Z. Chen, L. Chen, C. Zeng and W. Wang, Stem Cells Int., 2018, 2018, 7045245 Search PubMed.
- G. Vassalli and T. Moccetti, Swiss Med. Wkly., 2011, 141, 1–6 Search PubMed.
- A. Attar, A. Monabati, M. Montaseri, M. Vosough, S. Hosseini, J. Kojouri, A. Ardekani, P. Izadpanah, N. Azarpira, G. Pouladfar and M. Ramzi, Trials, 2022, 23, 1–7 CrossRef.
- P. van der Meer, E. Lipsic, R. H. Henning, K. Boddeus, J. van der Velden, A. A. Voors, D. J. van Veldhuisen, W. H. van Gilst and R. G. Schoemaker, J. Am. Coll. Cardiol., 2005, 46, 125–133 CrossRef CAS PubMed.
- K. U. Hong, Y. Guo, Q. H. Li, P. Cao, T. A. Maqtari, B. N. Vajravelu, J. Du, M. J. Book, X. Zhu, Y. Nong, A. Bhatnagar and R. Bolli, PLoS One, 2014, 9, e96725 CrossRef.
- Y. Imanishi, A. Saito, H. Komoda, S. Kitagawa-Sakakida, S. Miyagawa, H. Kondoh, H. Ichikawa and Y. Sawa, J. Mol. Cell. Cardiol., 2008, 44, 662–671 CrossRef PubMed.
- F. Van Den Akker, J. C. Deddens, P. A. Doevendans and J. P. G. Sluijter, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 2449–2458 CrossRef CAS PubMed.
- Z. Wu, G. Chen, J. Zhang, Y. Hua, J. Li, B. Liu, A. Huang, H. Li, M. Chen and C. Ou, Sci. Rep., 2017, 7, 15826 CrossRef PubMed.
- M. Song, H. Jang, J. Lee, J. Kim, S. Kim, K. Sun and Y. Park, Biomaterials, 2014, 35, 2436–2445 CrossRef CAS PubMed.
- L. J. F. Peters, E. A. L. Biessen, M. Hohl, C. Weber, E. P. C. van der Vorst and D. Santovito, Front. Physiol., 2020, 11, 793 CrossRef PubMed.
- G. Tong, Y. Liang, M. Xue, X. Chen, J. Wang, N. An, N. Wang, Y. Chen, Y. Wang, L. Jin and W. Cong, Biochem. Biophys. Res. Commun., 2020, 527, 15–21 CrossRef CAS PubMed.
- Z. Li, X. Guo, A. F. Palmer, H. Das and J. Guan, Acta Biomater., 2012, 8, 3586–3595 CrossRef CAS PubMed.
- M. Hirata and T. Yamaoka, Acta Biomater., 2018, 65, 44–52 CrossRef CAS PubMed.
- Z. Li, J. Guo, Q. Chang and A. Zhang, Biol. Pharm. Bull., 2009, 32, 1343–1346 CrossRef CAS.
- J. Rezaie, R. Rahbarghazi, M. Pezeshki, M. Mazhar, F. Yekani, M. Khaksar, E. Shokrollahi, H. Amini, S. Hashemzadeh, S. E. Sokullu and M. Tokac, J. Cell. Physiol., 2019, 234, 21732–21745 CrossRef CAS.
- S. Sahoo and D. W. Losordo, Circ. Res., 2014, 114, 333–344 CrossRef CAS.
- L. L. Zhang, Y. Y. Xiong and Y. J. Yang, Stem Cells Dev., 2021, 30, 561–577 CrossRef CAS PubMed.
- Z. Wei, Z. Chen, Y. Zhao, F. Fan, W. Xiong, S. Song, Y. Yin, J. Hu, K. Yang, L. Yang, B. Xu and J. Ge, Biomaterials, 2021, 275, 121000 CrossRef CAS PubMed.
- S. Bheri and M. E. Davis, ACS Nano, 2019, 13, 9702–9706 CrossRef CAS PubMed.
- S. Zhu, C. Yu, N. Liu, M. Zhao, Z. Chen, J. Liu, G. Li, H. Huang, H. Guo, T. Sun, J. Chen, J. Zhuang and P. Zhu, Bioact. Mater., 2022, 13, 119–134 CrossRef CAS PubMed.
- Y. Zou, L. Li, Y. Li, S. Chen, X. Xie, X. Jin, X. Wang, C. Ma, G. Fan and W. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 56892–56908 CrossRef CAS PubMed.
- Q. Huang, Y. Zou, M. C. Arno, S. Chen, T. Wang, J. Gao, A. P. Dove and J. Du, Chem. Soc. Rev., 2017, 46, 6255–6275 RSC.
- P. Zhou, L. Qin, Z. Ge, B. Xie, H. Huang, F. He, S. Ma, L. Ren, J. Shi, S. Pei, G. Dong, Y. Qi and F. Lan, J. Biomed. Mater. Res., Part B, 2022, 110, 1968–1990 CrossRef CAS.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- Y. Xu, K. Liu, Y. Yang, M. Kim, C. Lee, R. Zhang, T. Xu, S. Choi and C. Si, Front. Bioeng. Biotechnol., 2023, 10, 1–18 Search PubMed.
- Q. Huang, Y. Zou, M. C. Arno, S. Chen, T. Wang, J. Gao, A. P. Dove and J. Du, Chem. Soc. Rev., 2017, 46, 6255–6275 RSC.
- M. Liu, C. Chen, J. Yu, H. Zhang, L. Liang, B. Guo, Y. Qiu, F. Yao, H. Zhang and J. Li, Mater. Today Bio, 2022, 17, 100477 CrossRef CAS PubMed.
- Y. Kambe and T. Yamaoka, Biomater. Sci., 2019, 7, 4153–4165 RSC.
- X. Chen, L. Zhu, X. Wang and J. Xiao, Mol. Pharmaceutics, 2022, 20, 57–81 CrossRef PubMed.
- T. D. Johnson, S. Y. Lin and K. L. Christman, Nanotechnology, 2011, 22, 494015 CrossRef PubMed.
- M. Rigol, N. Solanes, J. Farré, S. Roura, M. Roqué, A. Berruezo, N. Bellera, L. Novensà, D. Tamborero, C. Prat-Vidal, M. Huzman, M. Batlle, M. Hoefsloot, M. Sitges, J. Ramírez, A. Dantas, A. Merino, G. Sanz, J. Brugada, A. Bayés-Genís and M. Heras, J. Card. Failure, 2010, 16, 357–366 CrossRef CAS PubMed.
- S. Dimmeler and A. M. Zeiher, Cardiology, 2009, 113, 155–160 CrossRef PubMed.
- M. Gyöngyösi, P. M. Haller, D. J. Blake and E. Rendon, Circ. Res., 2018, 123, 301–308 CrossRef PubMed.
- N. Yamamoto, H. Akamatsu, S. Hasegawa, T. Yamada, S. Nakata, M. Ohkuma, E. Miyachi, T. Runouchi and K. Matsunaga, J. Dermatol. Sci., 2007, 48, 43–52 CrossRef CAS PubMed.
- M. Al-Nbaheen, R. Vishnubalaji, D. Ali, A. Bouslimi, F. Al-Jassir, M. Megges, A. Prigione, J. Adjaye, M. Kassem and A. Aldahmash, Stem Cell Rev. Rep., 2013, 9, 32–43 CrossRef CAS.
- N. Takehara, Y. Tsutsumi, K. Tateishi, T. Ogata, H. Tanaka, T. Ueyama, T. Takahashi, T. Takamatsu, M. Fukushima, M. Komeda, M. Yamagishi, H. Yaku, Y. Tabata, H. Matsubara and H. Oh, J. Am. Coll. Cardiol., 2008, 52, 1858–1865 CrossRef CAS.
- A. Kędziora, J. Konstanty-Kalandyk, R. Litwinowicz, P. Mazur, B. Kapelak and J. Piątek, Adv. Int. Cardiol., 2022, 18, 360–365 Search PubMed.
- T. Wang, X. J. Jiang, Q. Z. Tang, X. Y. Li, T. Lin, D. Q. Wu, X. Z. Zhang and E. Okello, Acta Biomater., 2009, 5, 2939–2944 CrossRef CAS PubMed.
- T. H. Qazi, L. Tytgat, P. Dubruel, G. N. Duda, S. Van Vlierberghe and S. Geissler, ACS Biomater. Sci. Eng., 2019, 5, 5348–5358 CrossRef CAS PubMed.
- E. Y. Shin, L. Wang, M. Zemskova, J. Deppen, K. Xu, F. Strobel, A. J. García, R. Tirouvanziam and R. D. Levit, J. Am. Heart Assoc., 2018, 7, e006949 CrossRef.
- Y. Chen, C. Li, C. Li, J. Chen, Y. Li, H. Xie, C. Lin, M. Fan, Y. Guo, E. Gao, W. Yan and L. Tao, J. Am. Heart Assoc., 2020, 9, e013784 CrossRef CAS PubMed.
- E. Koivunotko, J. Snirvi, A. Merivaara, R. Harjumäki, S. Rautiainen, M. Kelloniemi, K. Kuismanen, S. Miettinen, M. Liperttula and R. Koivuniemi, Biomedicines, 2022, 10, 2584 CrossRef CAS PubMed.
- Z. Wu, W. Li, S. Cheng, J. Liu and S. Wang, Nanomedicine, 2023, 47, 102616 CrossRef CAS PubMed.
- H. Ding, J. Ding, Q. Liu, J. Lin, M. He, X. Wu, X. Chen, C. Xiao, T. Ren, Y. Zhu, C. Gao, X. Hu and J. Wang, Chem. Eng. J., 2022, 433, 133511 CrossRef CAS.
- K. Zhu, D. Jiang, K. Wang, D. Zheng, Z. Zhu, F. Shao, R. Qian, X. Lan and C. Qin, J. Nanobiotechnol., 2022, 20, 1–16 CrossRef PubMed.
- X. Li, J. Zhou, Z. Liu, J. Chen, S. Lü, H. Sun, J. Li, Q. Lin, B. Yang, C. Duan, M. Xing and C. Wang, Biomaterials, 2014, 35, 5679–5688 CrossRef CAS PubMed.
- I. M. Williams and J. C. Wu, Arterioscler., Thromb., Vasc. Biol., 2019, 39, 1317–1329 CrossRef CAS PubMed.
- J. Li, Y. Ma, X. Miao, J. Guo and D. Li, Eeurological Sci., 2021, 42, 3585–3593 CrossRef PubMed.
- G. Krenning, M. J. A. van Luyn and M. C. Harmsen, Trends Mol. Med., 2009, 15, 180–189 CrossRef CAS PubMed.
- B. Vrtovec, G. Poglajen, L. Lezaic, M. Sever, D. Domanovic, P. Cernelc, A. Socan, S. Schrepfer, G. Torre-Amione, F. Haddad and J. C. Wu, Circ. Res., 2013, 112, 65–173 CrossRef PubMed.
- P. Tluri, J. S. Miller, R. J. Emery, G. Hung, A. Trubelja, J. E. Cohen, K. Lloyd, J. Han, A. C. Gaffey, J. W. MacArthur, C. S. Chen and Y. J. Woo, J. Thorac. Cardiovasc. Surg., 2014, 148, 1090–1098 CrossRef PubMed.
- A. C. Gaffey, M. H. Chen, C. M. Venkataraman, A. Trubelja, C. B. Rodell, P. V. Dinh, G. Hung, J. W. MacArthur, R. V. Soopan, J. A. Burdick and P. Atluri, J. Thorac. Cardiovasc. Surg., 2015, 150, 1268–1277 CrossRef PubMed.
- K. T. Campbell, R. S. Stilhano and E. A. Silva, Biomaterials, 2018, 179, 109–121 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, T. Mei, H. Cao, Y. Hu, W. Jia, J. Wang, Z. Zhang, Z. Wang, W. Le and Z. Liu, Adv. Sci., 2022, 9, 2104299 CrossRef PubMed.
- H. Zhang, Y. Wang, Y. Tan, H. Wang, P. Tao and P. Zhou, Basic Res. Cardiol., 2019, 114, 1–17 CrossRef CAS PubMed.
- A. C. Gaffey, M. H. Chen, A. Trubelja, C. M. Venkataraman, C. W. Chen, J. J. Chung, S. Schultz, C. M. Sehgal, J. A. Burdick and P. Atluri, J. Thorac. Cardiovasc. Surg., 2019, 157, 79–1490 CrossRef PubMed.
- B. J. Bain, Medicine, 2017, 45, 187–193 CrossRef.
- S. Menezes, D. Melandri, G. Anselmi, T. Perchet, J. Loschko, J. Dubrot, R. Patel, E. L. Gautier, S. Hugues, M. P. Longhi, J. Y. Henry, S. A. Quezada, G. Lauvau, A. Lennon-Duménil, E. G. Martínez, A. Bessis, E. G. Perdiguero, C. E. Jacome-Galarza, H. Garner, F. Geissmann, R. Golub, M. C. Nussenzweig and P. Guermonprez, Immunity, 2016, 45, 1205–1218 CrossRef CAS PubMed.
- C. Peet, A. Ivetic, D. I. Bromage and A. M. Shah, Cardiovasc. Res., 2020, 116, 1101–1112 CrossRef CAS PubMed.
- S. Smith and R. Ascione, Pharmacol. Ther., 2023, 108397 CrossRef CAS PubMed.
- C. H. Chen, M. Y. Chang, S. S. Wang and P. C. Hsieh, Am. J. Physiol.: Heart Circ. Physiol., 2014, 306, H1078–H1086 CrossRef CAS PubMed.
- P. J. Psaltis, N. Schwarz, D. Toledo-Flores and S. S. J. Nicholls, Curr. Cardiol. Rev., 2016, 12, 195–215 CrossRef PubMed.
- T. D. Henry, J. H. Traverse, B. L. Hammon, C. A. East, B. Bruckner, A. E. Remmers, D. Recker, D. A. Bull and A. N. Patel, Circ. Res., 2014, 115, 730–737 CrossRef CAS PubMed.
- X. Y. Li, T. Wang, X. J. Jiang, T. Lin, D. Q. Wu, X. Z. Zhang, E. Okello, H. X. Xu and M. J. Yuan, Cardiology, 2010, 115, 194–199 CrossRef CAS PubMed.
- Y. L. Tang, Y. J. Wang, L. J. Chen, Y. H. Pan, L. Zhang and N. L. Weintraub, Exp. Biol. Med., 2013, 238, 294–300 CrossRef CAS PubMed.
- T. Hosoda, Am. J. Cardiovasc. Dis., 2012, 2, 58 CAS.
- L. Barile, E. Messina, A. Giacomello and E. Marbán, Prog. Cardiovasc. Dis., 2007, 50, 31–48 CrossRef CAS PubMed.
- M. Kreke, R. R. Smith, L. Marbán and E. Marbán, Expert Rev. Cardiovasc. Ther., 2012, 10, 1185–1194 CrossRef CAS PubMed.
- M. Valente, D. S. Nascimento, A. Cumano and P. Pinto-do-Ó, Stem Cells Dev., 2014, 23, 2263–2273 CrossRef CAS PubMed.
- A. Marui, Y. Tabata, S. Kojima, M. Yamamoto, K. Tambara, T. Nishina, Y. Saji, K. Inui, T. Hashida, S. okoyama, R. Onodera, T. Ikeda, M. Fukushima and M. Komeda, Circ. J., 2007, 71, 1181–1186 CrossRef CAS PubMed.
- N. Takehara, Y. Tsutsumi, K. Tateishi, T. Ogata, H. Tanaka, T. Ueyama, T. Takahashi, T. Takamatsu, M. Fukushima, M. Komeda, M. Yamagishi, H. Yaku, Y. Tabata, H. Matsubara and H. Oh, J. Am. Coll. Cardiol., 2008, 52, 1858–1865 CrossRef CAS PubMed.
- C. Prat-Vidal, V. Crisóstomo, I. Moscoso, C. Báez-Díaz, V. lanco-Blázquez, G. mez-Mauricio, G. Bericio, S. Uilar, M. Fernández-Santo, F. Fernández-Avilés, F. M. Sánchez-Margallo, A. Bayes-Genis and A. Bernad, Cells, 2021, 10, 2571 CrossRef CAS PubMed.
- P. Menasché, V. Vanneaux, A. Hagège, A. Bel, B. Cholley, A. Parouchev, I. Cacciapuoti, R. Al-Daccak, N. Benhamouda, H. Blons, O. Agbulut, L. Tosca, J. Trouvin, J. Fabreguettes, V. Bellamy, D. Charron, E. Tartour, G. Tachdjian, M. Desnos and J. Larghero, J. Am. Coll. Cardiol., 2018, 71, 429–438 CrossRef PubMed.
- M. Kanda, T. Nagai, N. Kondo, K. Matsuura, H. Akazawa, I. Komuro and Y. Kobayashi, Cell Transp., 2023, 32, 09636897231174078 Search PubMed.
- C. Y. Chang, A. T. Chan, P. A. Armstrong, H. C. Luo, T. Higuchi, I. A. Strehin, S. Vakrou, X. Lin, S. N. Brown, B. O. Rourke, T. P. Abraham, R. L. Wahl, C. J. Steenbergen, J. H. Elisseeff and M. R. Abraham, Biomaterials, 2012, 33, 8026–8033 CrossRef CAS PubMed.
- J. Morgan and F. Muntoni, Nat. Cell Biol., 2007, 9, 249–251 CrossRef CAS PubMed.
- H. K. Haider, Y. Lei and M. Ashraf, Curr. Opin. Mol. Ther., 2008, 10, 611 CrossRef CAS PubMed.
- H. K. Haider, L. Ye and M. Ashraf, Recent Pat. Cardiovasc. Drug Discovery, 2007, 2, 205–213 CrossRef CAS PubMed.
- K. Malliaras, M. Kreke and E. Marban, Clin. Pharm. Ther., 2011, 90, 532–541 CrossRef CAS PubMed.
- G. Xu, A. Fatima, M. Breitbach, A. Kuzmenkin, C. J. Fügemann, D. Ivanyuk, K. Kim, O. Cantz, K. Pfannkuche, H. Schöler, B. K. Fleischmann, J. Hescheler and T. Šarić, Int. J. Mol. Sci., 2023, 24, 6546 CrossRef CAS PubMed.
- Q. Bai, R. Desprat, B. Klein, J. Lemaître and J. De Vos, Curr. Gene Ther., 2013, 13, 93–98 CrossRef CAS PubMed.
- O. Lindvall, R. A. Barker, O. Brüstle, O. Isacson and C. N. Svendsen, Cell Stem Cell, 2012, 10, 151–155 CrossRef CAS PubMed.
- M. Csöbönyeiová, N. Beerová, M. Klein, M. Debreová-Čeháková and Ľ. Danišovič, Int. J. Mol. Sci., 2022, 23, 10314 CrossRef PubMed.
- E. I. Parrotta, V. Lucchino, L. Scaramuzzino, S. Scalise and G. Cuda, Int. J. Mol. Sci., 2020, 21, 54 Search PubMed.
- H. Wang, Z. Liu, D. Li, X. Guo, F. Kurtis Kasper, C. Duan, J. Zhou, A. G. Mikos and C. Wang, J. Cell. Mol. Med., 2012, 16, 1310–1320 CrossRef CAS PubMed.
- K. Nakajima, J. Fujita, M. Matsui, S. Tohyama, N. Tamura, H. Kanazawa, T. Seki, Y. Kishino, A. Hirano, M. Okada, R. Tabei, M. Sano, S. Goto, Y. Tabata and K. Fukuda, PLoS One, 2015, 10, e0133308 CrossRef PubMed.
- A. Chow, D. J. Stuckey, E. Kidher, M. Rocco, R. J. Jabbour, C. A. Mansfield, A. Darzi, S. E. Harding, M. M. Stevens and T. Athanasiou, Stem Cell Rep., 2017, 9, 1415–1422 CrossRef CAS PubMed.
- S. Khazaei, M. Soleimani, S. H. A. Tafti, R. M. Aghdam and Z. Hojati, Cell Trans., 2021, 30, 09636897211048786 Search PubMed.
- K. Wu, Y. Wang, H. Yang, Y. Chen, K. Lu, Y. Wu, C. Liu, H. Zhang, H. Meng, Q. Yu, Y. Zhang and Z. Shen, ACS Appl. Mater. Interfaces, 2023, 15, 2578–2589 CrossRef CAS PubMed.
- Y. Lu, Q. Hu, C. Jiang and Z. Gu, Curr. Opin. Biotechnol, 2019, 58, 81–91 CrossRef CAS PubMed.
- D. Shen, Z. Li, S. Hu, K. Huang, T. Su, H. Liang, F. Liu and K. Cheng, Nano Lett., 2019, 19, 1883–1891 CrossRef CAS PubMed.
- J. Tang, T. Su, K. Huang, P. Dinh, Z. Wang, A. Vandergriff, M. T. Hensley, J. Cores, T. Allen, T. Li, E. Sproul, E. Mihalko, L. J. Lobo, L. Ruterbories, A. Lynch, A. Brown, T. G. Caranasos, D. Shen, G. A. Stouffer, Z. Gu, J. Zhang and K. Cheng., Nat. Biomed. Eng., 2018, 2, 17–26 CrossRef CAS PubMed.
- Y. Liu, D. Zhong, Y. He, J. Jiang, W. Xie, Z. Tang, J. Qiu, J. Luo and X. Wang, Adv. Sci., 2022, 9, 2202920 CrossRef CAS PubMed.
- A. Turchinovich, O. Drapkina and A. Tonevitsky, Front. Immunol., 2019, 10, 202 CrossRef CAS PubMed.
- J. C. Akers, D. Gonda, R. Kim, B. S. Carter and C. C. Chen., J. Neuro-Oncol., 2013, 113, 1–11 CrossRef PubMed.
- R. Crescitelli, C. Lässer, T. G. Szabo, A. Kittel, M. Eldh, I. Dianzani, E. I. Buzás and J. Lötvall, J. Extracell. Vesicles, 2013, 2, 20677 CrossRef PubMed.
- P. Hauser, S. Wang and V. V. Didenko, Signal Trans. Immunohistochem.: Methods and Protocols, 2017, 193–200 CAS.
- S. N. Hurwitz, J. M. Olcese and D. G. Meckes Jr., JoVE, 2019, e59143 CAS.
- F. Jansen, G. Nickenig and N. Werner, Circ. Res., 2017, 120, 1649–1657 CrossRef CAS PubMed.
- G. Van Niel, G. d'Angelo and G. Raposo, Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228 CrossRef CAS PubMed.
- C. M. Boulanger, X. Loyer, P. E. Rautou and N. Amabile, Nat. Rev. Cardiol., 2017, 14, 259–272 CrossRef CAS PubMed.
- X. Loyer, I. Zlatanova, C. Devue, M. Yin, K. Y. Howangyin, P. Klaihmon, C. L. Guerin, M. Kheloufi, J. Vilar, K. Zannis, B. K. Fleischmann, D. Hwang, J. Park, H. Lee, P. Menasché, J. S. Silvestre and C. M. Boulanger, Circ. Res., 2018, 123, 100–106 CrossRef CAS PubMed.
- N. Bie, T. Yong, Z. Wei, L. Gan and X. Yang, Adv. Drug Delivery Rev., 2022, 114450 CrossRef CAS PubMed.
- H. Song, X. Chen, Y. Hao, J. Wang, Q. Xie and X. Wang, J. Nanobiotechnol., 2022, 20, 1–23 CrossRef PubMed.
- Z. Zhang, H. Fu, Z. Li, J. Huang, Z. Xu, Y. Lai, X. Qian and S. Zhang, Chem. Eng. J., 2022, 135756 CrossRef CAS.
- H. Xu, Y. Q. Ni and Y. S. Liu, Front. Cardiovasc. Med., 2021, 8, 733985 CrossRef PubMed.
- ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). March 17, 2023, Identifier NCT00287391, Treatment of Non-ischemic Cardiomyopathies by Intravenous Extracellular Vesicles of Cardiovascular Progenitor Cells (SECRET-HF), Available from: https://clinicaltrials.gov/ct2/show/NCT05774509.
- R. Waters, P. Alam, S. Pacelli, A. R. Chakravarti, R. P. H. Ahmed and A. Paul, Acta Biomater., 2018, 69, 95–106 CrossRef CAS PubMed.
- S. Firoozi, S. Pahlavan, M. H. Ghanian, S. Rabbani, M. Barekat, A. Nazari, M. Pakzad, F. Shekari, S. N. Hassani, F. Moslem, F. N. Lahrood, M. Soleimani and H. Baharvand, Biochem. Biophys. Res. Commun., 2020, 524, 903–909 CrossRef CAS PubMed.
- P. Gil-Cabrerizo, L. Saludas, F. Prósper, G. Abizanda, M. E. de Anleo, A. Ruiz-Villalba, E. Garbayo and M. J. Blanco-Prieto, Int. J. Pharm., 2022, 629, 122356 CrossRef CAS PubMed.
- E. A. Mol, Z. Lei, M. T. Roefs, M. H. Bakker, M. J. Goumans, P. A. Doevendans, P. Y. W. Dankers, P. Vader and J. P. G. Sluijter, Adv. Healthcare Mater., 2019, 8, 1900847 CrossRef CAS PubMed.
- M. Monguió-Tortajada, C. Prat-Vidal, D. Martínez-Falguera, A. Teis, C. Soler-Botija, Y. Courageux, M. Munizaga-Larroudé, M. Moron-Font, A. Bayes-Genis, F. E. Borràs, S. Roura and C. Gálvez-Montón1, Theranostics, 2022, 12, 4656 CrossRef PubMed.
- Q. Li, Z. Huang, Q. Wang, J. Gao, J. Chen, H. Tan, S. Li, Z. Wang, X. Weng, H. Yang, Z. Pang, Y. Song, J. Qian and J. Ge, Biomaterials, 2022, 284, 121529 CrossRef CAS PubMed.
- S. Hu, X. Wang, Z. Li, D. Zhu, J. Cores, Z. Wang, J. Li, X. Mei, X. Cheng, T. Su and K. Cheng, Nano Today, 2021, 39, 101210 CrossRef CAS PubMed.
- L. Sun, Y. Ji, B. Chi, T. Xiao, C. Li, X. Yan, X. Xiong, L. Mao, D. Cai, A. Zou, Y. Wang, L. Zhang, L. Tang and Q. Wang, Biomed. Pharmacother., 2023, 161, 114557 CrossRef CAS PubMed.
- X. Hu, X. Ning, Q. Zhao, Z. Zhang, C. Zhang, M. Xie, W. Huang, Y. Cai, Q. Xiang and C. Ou, ACS Appl. Mater. Interfaces, 2022, 14, 289–36303 Search PubMed.
- B. Yu, H. Li, Z. Zhang, P. Chen, L. Wang, X. Fan, X. Ning, Y. Pan, F. Zhou, X. Hu, J. Chang and C. Ou, Nat. Commun., 2023, 14, 2094 CrossRef CAS PubMed.
- D. Kretzschmar, S. Betge, A. Windisch, R. Pistulli, I. Rohm, M. Fritzenwanger, C. Jung, K. Schubert, B. Theis, I. Petersen, S. Drobnik, G. Mall, H. R. Figulla and A. Yilmaz, Clin. Sci., 2012, 123, 387–398 CrossRef CAS PubMed.
- T. L. Kennedy, A. J. Russell and P. Riley, Trends Cardiovasc. Med., 2021, 31, 405–415 CAS.
- Y. Zhang, Z. Cai, Y. Shen, Q. Lu, W. Gao, X. Zhong, K. Yao, J. Yuan and H. Liu, J. Nanobiotechnol., 2021, 19, 1–16 CrossRef PubMed.
- T. T. Tang, J. Yuan, Z. F. Zhu, W. C. Zhang, H. Xiao, N. Xia, X. X. Yan, S. F. Nie, J. Liu, S. F. Zhou, J. J. Li, R. Yao, M. Y. Liao, X. Tu, Y. H. Liao and X. Cheng, Basic Res. Cardiol., 2012, 107, 1–17 CrossRef PubMed.
- P. Cheng, L. Cheng, H. Han, J. Li, C. Ma, H. Huang, J. Zhou, J. Feng, Y. Huang, Y. Lv, H. Huang, Y. Wang, L. Hou, Y. Chen and G. Li, Adv. Healthcare Mater., 2022, 11, 2200971 CrossRef CAS PubMed.
- E. J. Woods, A. Bagchi, W. S. Goebel and V. D. Vilivalam, Regener. Med., 2010, 5, 659–667 CrossRef CAS.
- J. Wu, G. Li, T. Ye, G. Lu, R. Li, L. Deng, L. Wang, M. Cai and W. Cui, Chem. Eng. J., 2020, 393, 124715 CrossRef CAS.
- R. P. Harrison, N. Medcalf and Q. A. Rafiq, Regener. Med., 2018, 13, 159–173 CrossRef CAS.
- P. P. Zwetsloot, A. M. D. Végh, S. J. J. of Lorkeers, G. P. J. van Hout, G. L. Currie, E. S. Sena, H. Gremmels, J. W. Buikema, M. Goumans, M. R. Macleod, P. A. Doevendans, S. A. J. Chamuleau and J. P. G. Sluijter, Circ. Res., 2016, 118, 1223–1232 CrossRef CAS PubMed.
- P. Cheng, X. Wang, Q. Liu, T. Yang, H. Qu and H. Zhou, Front. Pharmacol., 2023, 14, 420 Search PubMed.
- M. Zhang, S. Cheng, Y. Jin, N. Zhang and Y. Wang, Clin. Transl. Med., 2021, 11, e292 CrossRef PubMed.
- S. He, Z. Zhang, R. Luo, Q. Jiang, L. Yang and Y. Wang, Adv. Healthcare Mater., 2023, 2300029 CrossRef CAS PubMed.
- X. Liu, X. Zhong and C. Li, Chin. Chem. Lett., 2021, 32, 2347–2358 CrossRef CAS.
- J. Dai, Z. Chen, S. Wang, F. Xia and X. Lou, Mater. Today Bio, 2022, 15, 100279 CrossRef CAS PubMed.
- D. Han, F. Wang, Z. Qiao, B. Wang, Y. Zhang, Q. Jiang, M. Liu, Y. Zhuang, Q. An, Y. Bai, J. Shangguan, J. Zhang and G. Liang, Bioact. Mater., 2023, 23, 369–382 CrossRef PubMed.
- W. Song, P. Jia, T. Zhang, K. Dou, L. Liu, Y. Ren, F. Liu, J. Xue, M. Hasanin, H. Qi and Q. Zhou, J. Nanobiotechnol., 2022, 20, 1–29 CrossRef PubMed.
- E. S. Yastrebova, A. V. Gisich, V. M. Nekrasov, K. V. Gilev, D. I. Strokotov, A. V. Chernyshev, A. A. Karpenko and V. P. Maltsev, Cytometry, Part A, 2023, 103, 39–53 CrossRef CAS PubMed.
- J. Tang, D. Shen, T. G. Caranasos, Z. Wang, A. C. Vandergriff, T. A. Allen, M. T. Hensley, P. U. Dinh, J. Cores, T. Li, J. Zhang, Q. Kan and K. Cheng, Nat. Commun., 2017, 8, 13724 CrossRef CAS PubMed.
- Y. Zhai, J. Su, W. Ran, P. Zhang, Q. Yin, Z. Zhang, H. Yu and Y. Li, Theranostics, 2017, 7, 2575 CrossRef CAS PubMed.
- M. Wang, Y. Xin, H. Cao, W. Li, Y. Hua, T. J. Webster, C. Zhang, W. Tang and Z. Liu, Biomater. Sci., 2021, 9, 1088–1103 RSC.
- H. E. Bøtker, H. A. Cabrera-Fuentes, M. Ruiz-Meana, G. Heusch and M. Ovize, J. Cell. Mol. Med., 2020, 24, 2717–2729 CrossRef PubMed.
- M. Meloni, M. Marchetti, K. Garner, B. Littlejohns, G. Sala-Newby, N. Xenophontos, I. Floris, M. Suleiman, P. Madeddu, A. Caporali and C. Emanueli, Mol. Ther., 2013, 21, 1390–1402 CrossRef CAS PubMed.
- S. Li, S. Liu, M. Sun, J. Wang, C. Wang and Y. Sun, Front. Pharmacol., 2020, 11, 24 CrossRef CAS PubMed.
- S. Zhang, Q. Fu, Y. Zhang, J. Pan, L. Zhang, Z. Zhang and Z. Liu, Acta Pharmacol. Sin., 2021, 42, 1040–1054 CrossRef CAS PubMed.
- P. Chowdhury, U. Ghosh, K. Samanta, M. Jaggi, S. C. Chauhan and M. M. Yallapu, Bioact. Mater., 2021, 6, 3269–3287 CrossRef CAS PubMed.
- X. Zhang, Y. Zhang, R. Zhang, X. Jiang, A. C. Midgley, Q. Liu, H. Kang, J. Wu, A. Khalique, M. Qian, D. An, J. Huang, L. Ou, Q. Zhao, J. Zhuang, X. Yan, D. Kong and X. Huang, Adv. Mater., 2022, 34, 2110352 CrossRef CAS PubMed.
- K. Huang, S. Wen, W. Wang, J. Zhou, J. Huang, F. Wang, L. Pang, Y. Wang and X. Sun, Nanomedicine, 2020, 16, 465–480 CrossRef PubMed.
- X. Li, G. Hong, G. Zhao, H. Pei, J. Qu, C. Chun, Z. Huang and Z. Lu, Front. Pharmacol., 2022, 13, 1274 Search PubMed.
- J. Lu, J. Zhang, W. Yan, C. Ge, Y. Zhou, R. Zhu, S. Duan, L. Yin and Y. Chen, Nano Res., 2023 Search PubMed.
- M. Ziegler, X. Wang and K. Peter, Cardiovasc. Res., 2019, 115, 1178–1188 CrossRef CAS PubMed.
- T. Gremmel, R. Koppensteiner, A. Kaider, B. Eichelberger, C. Mannhalter and S. Panzer, Thromb. Haemostasis, 2015, 113, 806–812 CrossRef PubMed.
- Z. Zhang, Z. Chen, L. Yang, J. Zhang, Y. Li, C. Li, R. Wang, X. Wang, S. Huang, Y. Hu, J. Shi and W. Xiao, J. Funct. Biomater., 2022, 13, 181 CrossRef CAS PubMed.
- T. Zhou, X. Yang, T. Wang, M. Xu, Z. Huang, R. Yu, Y. Jiang, Y. Zhou and J. Shi, Membranes, 2022, 12, 605 CrossRef CAS PubMed.
- V. Triaca, V. Carito, E. Fico, P. Rosso, M. Fiore, M. Ralli, A. Lambiase, A. Greco and P. Tirassa, Aging, 2019, 11, 11770 CrossRef CAS PubMed.
- P. Kanda, E. I. Alarcon, T. Yeuchyk, S. Parent, R. A. de Kemp, F. Variola, D. Courtman, D. J. Stewart and D. R. Davis, ACS Nano, 2018, 12, 4338–4350 CrossRef CAS PubMed.
- C. Yao, W. Wu, H. Tang, X. Jia, J. Tang, X. Ruan, F. Li, D. T. Leong, D. Luo and D. Yang, Biomaterials, 2020, 257, 120256 CrossRef CAS PubMed.
- S. Frantz, U. Hofmann, D. Fraccarollo, A. Schäfer, S. Kranepuhl, I. Hagedorn, B. Nieswandt, M. Nahrendorf, H. Wagner, B. Bayer, C. Pachel, M. P. Schön, S. Kneitz, T. Bobinger, F. Weidemann, G. Ertl and J. Bauersachs, FASEB J., 2013, 27, 871–881 CrossRef CAS PubMed.
- Y. Cavusoglu, B. Timuralp, T. Us, Y. Akgün, G. Kudaiberdieva, B. Gorenek, A. Unalir, O. Goktekin and N. Ata, Angiology, 2004, 55, 397–402 CrossRef PubMed.
- Y. Wei, M. Zhu, S. Li, T. Hong, X. Guo, Y. Li, Y. Liu, X. Hou and B. He, ACS Appl. Mater. Interfaces, 2021, 13, 33756–33766 CrossRef CAS PubMed.
- Y. Zhou, Q. Liang, X. Wu, S. Duan, C. Ge, H. Ye, J. Lu, R. Zhu, Y. Chen, F. Meng and L. Yin, Adv. Mater., 2023, 2210691 CrossRef CAS PubMed.
- V. Frodermann and M. Nahrendorf, Eur. Heart J., 2017, 38, 198–200 CAS.
- L. Bao, G. Dou, R. Tian, Y. Lv, F. Ding, S. Liu, R. Zhao, L. Zhao, J. Zhou, L. Weng, Y. Dong, B. Li, S. Liu, X. Chen and Y. Jin, Bioact. Mater., 2022, 9, 183–197 CrossRef CAS PubMed.
- M. A. Gavin, J. P. Rasmussen, J. D. Fontenot, V. Vasta, V. C. Manganiello, J. A. Beavo and A. Y. Rudensky, Nature, 2007, 445, 771–775 CrossRef CAS PubMed.
- A. Saxena, M. Dobaczewski, V. Rai, Z. Haque, W. Chen, N. Li and N. G. Frangogiannis, Am. J. Physiol.: Heart Circ. Physiol., 2014, 307, H1233–H1242 CrossRef CAS PubMed.
- K. Matsumoto, M. Ogawa, J. Suzuki, Y. Hirata, R. Nagai and M. Isobe, Int. Heart J., 2011, 52, 382–387 CrossRef CAS PubMed.
- F. Li, D. Liu, M. Liu, Q. Ji, B. Zhang, Q. Mei, Y. Cheng and S. Zhou, J. Nanobiotechnol., 2022, 20, 251 CrossRef CAS PubMed.
- P. Chen, L. Wang, X. Fan, X. Ning, B. Yu, C. Ou and M. Chen, Theranostics, 2021, 11, 2263 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |