
Dual ligand-assisted assembly of metal–organic frameworks on upconversion nanoparticles for NIR photodynamic therapy against hypoxic tumors†
Xinyue
Zhang‡
a,
Jiasen
Cui‡
 b,
Jinhui
Liu
a,
Xi
Chen
b,
Mingli
Chen
*a and
Jianhua
Wang
*a
b,
Jinhui
Liu
a,
Xi
Chen
b,
Mingli
Chen
*a and
Jianhua
Wang
*a
aDepartment of Chemistry, Northeastern University, Box 332, Shenyang 110819, China. E-mail: chenml@mail.neu.edu.cn; jianhuajrz@mail.neu.edu.cn; Fax: +86 24 83676698; Tel: +86 24 83688944
bSchool and Hospital of Stomatology, Department of Oral Pathology, China Medical University, Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, 110001, China
First published on 22nd September 2023
Abstract
The hypoxic nature of tumor microenvironments significantly impedes the effectiveness of photodynamic therapy (PDT). To address this challenge, we constructed a pioneering nanohybrid by integrating upconversion nanoparticles (UCNPs) and metal–organic frameworks (MOFs) through a dual-ligand-assisted assembly approach. We functionalized UCNPs with polyvinyl pyrrolidone (PVP) and branched polyethylenimine (PEI), enabling the in situ growth of MOFs on multiple UCNP-conjugates. This nanohybrid, termed UCM, possesses a unique heterogeneous structure that facilitates effective energy transfer from UCNPs to MOFs, enhancing NIR-activated PDT. A distinguishing feature of UCMs is biocatalytically active MOFs, which provide them with a peroxidase-like capability. This characteristic allows UCMs to utilize the excess H2O2 in the tumor microenvironment, ensuring continuous oxygen production essential for type II PDT. Our research indicates that UCMs not only amplify the efficacy of PDT but also address the therapeutic challenges in hypoxic tumor microenvironments by supplying in situ oxygen.
1. Introduction
Malignant tumors pose a significant threat to human lives, with an increasing incidence.1,2 Photodynamic therapy (PDT) is a precise cellular-level treatment that relies on the photoactivation of photosensitizers (PS) to generate reactive oxygen species (ROS), leading to therapeutic effects and cancer cell death. ROS disruption of tumor metastasis homeostasis induces severe apoptosis in tumor cells, effectively halting tumor progression.3–5 Metal–organic frameworks (MOFs) have gained extensive applications in biomedical fields due to their tunable functionality and favorable biocompatibility.6–11 In particular, MOFs can integrate photosensitizers into periodic arrays and self-assemble porphyrins and metal ions through coordination, enabling high photosensitizer loading. The unique structure of MOFs prevents photosensitizer aggregation and enhances photodynamic therapy efficiency.12–14 The type II PDT mechanism typically involves an energy transfer process, where the photosensitizer in the triplet state transfers energy directly to the surrounding oxygen, generating cytotoxic singlet oxygen (1O2). However, PDT exacerbates tumor hypoxia, thereby diminishing its efficacy.15 Intriguingly, certain characteristics of the tumor microenvironment present opportunities to augment therapeutic outcomes. For instance, the tumor milieu is often replete with hydrogen peroxide (H2O2). Some researchers have harnessed the metal-mediated Fenton reaction to decompose H2O2, generating toxic hydroxyl radicals, thereby synergistically enhancing antitumor effects.16 Alternatively, leveraging peroxidase-mimetic activity to decompose H2O2, leading to in situ oxygen generation, offers a promising avenue to bolster the efficacy of photodynamic therapy.17–19 Conventional porphyrin MOFs lack central metals with redox activity and exhibit no biocatalytic properties. To address this, Fe(III)–porphyrin can be employed as an organic ligand, enabling Fe3+ to serve as an active metal center. The resulting iron–porphyrin MOFs can act as biocatalysts, catalyzing H2O2 (50–100 μM) within the tumor microenvironment to alleviate hypoxia.20,21MOFs as photosensitizers require photoirradiation to initiate photodynamic therapy. However, in living organisms, the weak penetration capability of visible light to tissues significantly limits the therapeutic depth of MOFs and weakens the photodynamic treatment effect.22–25 Near-infrared light (NIR) provides deeper penetration capability in tissues, and thus it is highly desired to develop a NIR activated photodynamic therapy platform. Photostimulated luminescence involves the release of stored energy from a previously excited state when exposed to light, leading to the emission of luminescence. In contrast, upconversion refers to the process of absorbing two or more photons to emit light at a shorter wavelength. This process typically converts near-infrared or infrared light to visible or ultraviolet light, exhibiting potential intra-biological photomodulation and photoconversion.26–28 In this respect, the integration of UCNPs with MOFs provides a promising avenue for improving the efficiency of PDT. UCNPs receive near-infrared photons to enable energy transfer to MOFs via FRET and initiate NIR activated PDT. Recently, a number of UCNP-MOF composites with a heterogeneous structure have emerged and found wide applications in photodynamic therapy, targeted drug delivery, bioassay and bioimaging.29–32 The fundamental studies have demonstrated that a key issue in the synthesis of MOF crystals is to regulate the deprotonation of the organic ligands in order to control the reaction rate with metal ions.33 In this respect, the synthesis of MOFs in a homogeneous medium is used to control the nucleation and crystal growth, which is a competitive process with the assembly of UCNPs-MOFs in a heterogeneous medium. Therefore, it is critical to functionalize UCNPs with various functional moieties for endowing them with specific surface properties to initiate the growth of MOFs on the UCNP surface.
In this study, we constructed novel UCNP-MOF nanohybrids (UCMs) with asymmetric hetero zstructures by employing the so-called dual-ligand-assisted self-assembly strategy. The two ligands, i.e., polyvinyl pyrrolidone (PVP) and branched polyethylenimine (PEI), promote heterogeneous nucleation and enable in situ growth of iron porphyrin-based MOFs [PCN-224(Fe)] on multiple UCNPs. Porphyrin-based MOFs exhibit great potential as photosensitizers for PDT. The integration of UCNPs with PCN-224(Fe) ensures deeper tissue treatment. In addition, PCN-224(Fe) possesses peroxidase-like activity and catalyzes the breakdown of endogenous H2O2 in tumors to produce oxygen and alleviate hypoxia in the tumor microenvironment. By taking advantage of the absorption of NIR by UCNPs and the biocatalytic activity of PCN-224(Fe), the obtained UCMs nanohybrids serve as a multifunctional platform for synergistic NIR-activated PDT to show excellent anti-tumor capability for both in vitro and in vivo treatments.
2. Experimental
2.1. Preparation of UCNPs, PEI/PVP-UCNPs and UCMs
In a typical procedure, 1.0 mmol of LnCl3·6H2O (Ln = Y, Yb, Tm, Nd) was mixed with 6 mL of oleic acid and 15 mL of octadecene. The mixture solution was heated to 160 °C with stirring under argon and kept at 160 °C for 60 min to remove oxygen and water. The solution was cooled to room temperature. Then a 10 mL methanol solution of NaOH (2.5 mmol) and NH4F (4.0 mmol) was added dropwise and stirred for another 60 min. The solution mixture was heated to 120 °C, kept at this temperature for another 60 min to remove methanol and then heated to 300 °C and maintained for 60 min under an argon atmosphere. The products were centrifuged and washed three times with cyclohexane solution. The final products were stored in cyclohexane. Basic surface modification of the as-synthesized NaYF4:25% Yb3+, 0.9% Tm3+, 0.6% Nd3+ nanocrystals with PEI, was performed by the widely accepted solvent evaporation method described elsewhere.34 In brief, the branched PEI (300 mg) was added to the chloroform dispersion of OA-UCNPs (10 mg mL−1, 6 mL). The mixture was sonicated in an ultrasonic bath for 10 min and then stirred for 4 h at room temperature. After evaporation of the solvent, the resulting residue was added to ethanol. After further sonication, the resulting dispersion was stirred vigorously at room temperature for 1 h. Then, PVP (600 mg) was added to the obtained solution, subjected to ultrasonic treatment for 10 minutes, and the mixed solution was stirred for 24 h. Centrifuge the reacted solution and wash it three times with ethanol.Add Fe(III)–TCPP (4 mg) and ZrCl4 (4 mg) into a mixture solution of dmf/ethanol/methanol (2 mL/1 mL/1 mL), then add PEI/PVP-UCNPs (8 mg) and sonicate the mixed solution for 10 minutes. Further, add glacial acetic acid (400 μL) to the mixed solution. Continue stirring the mixed solution at room temperature for 4 h. Afterwards, benzoic acid (35 mg) was added, stirring continued, and the mixture was heated to 90 °C to conduct the reaction for another 5 h. Finally, the product was washed and obtained by centrifugation.
2.2. Cellular uptake of UCMs
HepG2 cells were cultured overnight in confocal dishes, the original medium was removed. DMEM medium solution containing UCMs (100 μg mL−1) was added, and the cells continued to incubate for 15, 30, 60 and 120 min. The medium was then removed, and DMEM medium containing DAPI was added. After incubation at 37 °C for various times, the medium was removed, the cells were washed three times with PBS, and the cells were observed under a confocal laser scanning microscope. UCMs were excited by a 488 nm laser and the red emission from the porphyrin-MOF was collected using a band-pass filter within the range 600–700 nm.2.3. Cell photodynamic therapy experiments
CCK-8 assay, dead and live cell staining assay and flow cytometry were used to determine the capability of UCMs to inhibit the growth of tumor cells. For CCK-8 experiments, to simulate the tumor hypoxic environment, HepG2 cells were incubated in oxygen-depleted bags. HepG2 cells were seeded into 96-well plates and cultured for 24 h. Thereafter the original medium was removed, and the cells were treated with UCMs (with or without 100 μM H2O2). The cells were then divided into two groups, i.e., treated with or without laser at 980 nm at 1.5 W cm−2. The cells were then incubated for 4 h to ensure that UCMs was internalized and phagocytosed by the cells. The cells were irradiated with laser for 10 min. After 24 h of further incubation, 10 μL of CCK-8 solution was added, and the relative cell viability was determined using a microplate reader.For the dead and live cell staining assay, the HepG2 cells were incubated for 24 h, and then the original medium was replaced with DMEM containing different components (H2O2, UCMs or UCMs + H2O2) and incubated for another 4 h. The treated cells were divided into two groups. The irradiation group was irradiated using a 980 nm laser (1.5 W cm−2) for 10 min. Then the cells were washed with PBS and stained with FDA (10 μM) and PI (20 μM) simultaneously. After staining for 20 min, the medium was removed and washed with PBS. Finally, fluorescence imaging of cells was performed by CLSM.
For flow cytometry analysis of apoptosis, HepG2 cells were treated by using the above FDA/PI assays. Then, the treated cells were dissociated for 2 min, and were collected by centrifugation. Afterwards, 500 μL of binding buffer was added to the re-disperse the cells, and then the PBS solution containing Annexin V (5 μL) and PI (5 μL) were utilized to dye the live and dead cells for 30 min. Finally, the apoptosis ratio of tumor cells after different treatments were analyzed by flow cytometry.
2.4. In vivo anticancer effect
The HepG2 tumor model was constructed by a subcutaneous injection of HepG2 cells into the dorsal flank of mice. Tumor-bearing mice were collected until the tumor volume reached about 200–400 mm3, and then used for in vivo antitumor therapy. Tumor volume = (tumor length) × (tumor width)2/2. The relative tumor volume was calculated as Vt/V0. (Vt and V0 represent the tumor volume at the end and the beginning of the treatment, respectively).HepG2 tumor-bearing mice were randomly divided into four groups (3 mice in each group), and treated with PBS, PBS with laser, UCMs, UCMs with laser via a tail-vein injection. UCMs were injected intravenously at a concentration of 4 mg mL−1 (100 μL), and the 980 nm laser (1.5 W cm−2, 10 min, 2 min break after 2 min irradiation) was used for the irradiation of tumors at 8 h post-injection. After each two days, another injection and treatment were applied using the same parameters. During antitumor treatment in mice, the tumor size and the body weights were measured and recorded every other day by using Vernier calipers and an electronic balance. After 14 days of treatment, the mice were euthanized, and their tumor tissues were harvested and weighed. The collected major organs (lungs, liver, spleen, and kidneys) were sectioned and stained with hematoxylin and eosin (H&E) for histological analysis. In addition, tumor tissue was sectioned and stained with Ki67 and ALDH1 for histological analysis.
Furthermore, hypoxia in vivo was tested using HIF-1α. At first, the extracted tumors of mice were divided into two groups, including tail-vein injection of saline as a blank group, and tail-vein injection of UCMs (4 mg mL−1, 100 μL). Next, the frozen sections were dyed using HIF-1α and DAPI. Finally, the tumor slices of each group were observed by CLSM.
3. Results and discussion
3.1. Preparation and characterizations of UCNPs and UCMs
UCNPs (NaYF4:25% Yb3+, 0.9% Tm3+, 0.6% Nd3+) were first prepared in a mixed solution containing oleic acid and octadecene with thermal decomposition.35 UCNPs were then sequentially modified with dual-ligands, i.e., PEI and PVP. Thereafter, the controlled assembly of PCN-224(Fe) on multiple UCNPs was conducted for in situ growth and thermal crystallization to obtain UCMs. The process for the preparation of UCMs and the potential mechanisms of type II photodynamic therapy treatment in hypoxic microenvironment are illustrated in Scheme 1.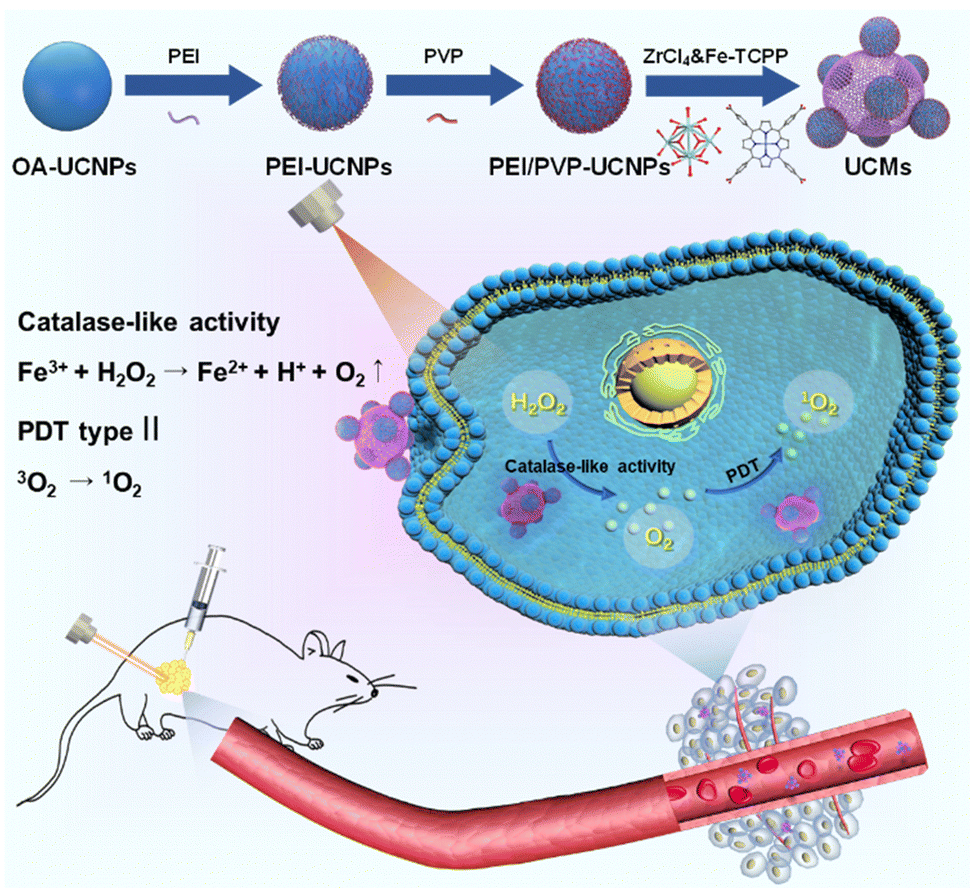 | ||
| Scheme 1 Illustration for the dual ligand functionalization of UCNPs and MOF assembly on multiple UCNPs to obtain the nanohybrid UCMs, and the potential mechanisms by employing UCMs in type II PDT treatment against cancer cells in a hypoxic microenvironment. | ||
Fig. 1a illustrates that the obtained UCNPs were well dispersed with favorable homogeneity and size distribution at ∼32.7 nm. Afterwards, the hydrophobic UCNPs were modified with PEI to convert into hydrophilic and disperse in PVP solution, wherein the PVP moiety can continuously attach to UCNPs. The hexagonal nanostructure of UCNPs was almost unchanged when two layers of ligands were covered on UCNPs with an average size of ∼37.8 nm (Fig. 1b). Ligand exchange led to no obvious aggregation of UCNPs which were well dispersed in aqueous medium. The synthesis process for the heterogeneous structured UCMs was observed by TEM images in Fig. S1 (ESI†). PEI/PVP-modified UCNPs were mixed with PCN-224(Fe) precursors, i.e., ZrCl4 and Fe(III)–TCPP, in a specific ratio of dmf/ethanol/methanol. The double-ligand modification of UCNPs drove the onset of heterogeneous nucleation process under the catalysis of acetic acid. After 5 min of mixing, the presence of many clusters of free MOFs in the solution may be clearly observed (Fig. S1b, ESI†), and a layer of MOFs could be identified on the surface of UCNPs. After 4 h of mixing, larger heterogeneous clusters of irregular morphology were formed on the surface of UCNPs (Fig. S1c, ESI†). Then the heterogeneous clusters on the surface of UCNPs were further thermally crystallized under thermodynamic drive (Fig. S1(d)–(f), ESI†), wherein it indicated that the growth size of PCN-224(Fe) may be well regulated by controlling the thermal crystallization time. For the construction of heterostructures, it is important to manipulate the surface properties of the heterogeneous nucleation interface to create the required conditions for triggering heterogeneous nucleation.36 On the one hand, PEI possesses –NH2 functional groups to facilitate the enrichment of Fe(III)–TCPP to the nucleation surface through an amide reaction.37 On the other hand, PVP is capable for chelating metal ions,29 and thus the co-modification of PVP and PEI create heterogeneous nucleation sites on UCNPs. In addition, both PEI and PVP ligands can drive multiple UCNP aggregates to further expand heterogeneous nucleation sites. When only PEI modification is available, multiple MOFs are generated on the surface of UCNPs to initiate irreversible aggregation (Fig. S2a, ESI†). When only PVP modification was performed, multiple MOFs grew on the surface of UCNPs simultaneously (Fig. S2e, ESI†). In contrast, moderate growth of single MOFs on the surface of multiple UCNPs allows for better energy transfer between heterogeneities.38 By changing the addition ratio of PEI and PVP (Fig. S2(b)–(d), ESI†), the dual-ligand co-modification further expanded the heterogeneous nucleation sites and provided a suitable growth environment for the regular growth of MOFs, resulting in heterogeneous UCMs with good dispersion and uniform morphology. Thermogravimetric analysis was performed on different UCNPs to analyze the quantity of modified ligands on the surface of UCNPs. The results illustrated sequential increment on the loss of the sample weight, which indicated the continuous attachment of the dual ligands on the surface of the UCNPs. The modified amount of ligands on the UCNPs was derived to be ca. 4.8% and ca. 4.4% for PEI and PVP, respectively (Fig. S4, ESI†). The addition of methanol is necessary for the self-assembly of UCMs.39 The solvent likewise affects the heterogeneous growth environment, and the PCN-224(Fe) growth morphology is transformed from longitudinal rods to ellipsoidal crystals of appropriate size by continuously changing the methanol concentration (Fig. S3a and b, ESI†). Irregular Zr-TCPP products were generated on the surface of UCNPs when no DMF was introduced into the reaction environment. (Fig. S3c, ESI†). In addition, it was verified that the heterostructure was not affected by the electrostatic force effect. The upconversion luminescence on the UCMs did not recover in the presence of different concentrations of NaCl. This observation well indicated the favorable structural stability of UCMs, which encountered no heterostructure separation at the ionic strength of the solution (Fig. S5, ESI†).
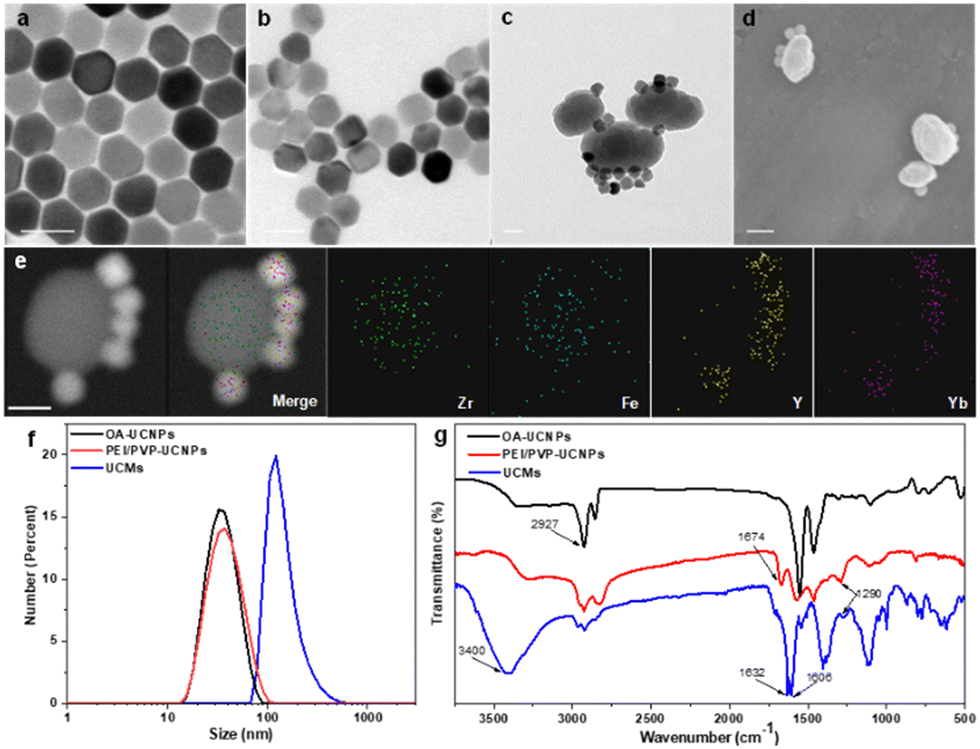 | ||
Fig. 1 TEM images of (a) UCNPs (NaYF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25% Yb3+, 0.9% Tm3+, 0.6% Nd3+), (b) dual-ligand PEI/PVP modified UCNPs, and (c) UCMs. (d) SEM images of UCMs. (e) Dark-field TEM images and the corresponding area-elemental mapping of UCMs, including Zr, Fe, Y and Yb. (f) The statistical data showing the size distributions of OA-UCNPs, PEI/PVP-UCNPs and UCMs. (g) FT-IR spectra for OA-UCNPs, PEI/PVP-UCNPs and UCMs. The scale bars indicate 50 nm in all the tested cases. :25% Yb3+, 0.9% Tm3+, 0.6% Nd3+), (b) dual-ligand PEI/PVP modified UCNPs, and (c) UCMs. (d) SEM images of UCMs. (e) Dark-field TEM images and the corresponding area-elemental mapping of UCMs, including Zr, Fe, Y and Yb. (f) The statistical data showing the size distributions of OA-UCNPs, PEI/PVP-UCNPs and UCMs. (g) FT-IR spectra for OA-UCNPs, PEI/PVP-UCNPs and UCMs. The scale bars indicate 50 nm in all the tested cases. | ||
UCMs with asymmetric structures were clearly identified from the TEM images as illustrated in Fig. 1c. Meanwhile, the morphology of PCN-224(Fe) was found to remain unchanged (Fig. S6, ESI†). This well indicated the stabilization effect of dual-ligand by in situ growth on the surface of UCNPs (Fig. 1d). TEM images of the UCMs and elemental mapping of their corresponding regions confirmed the asymmetric nanostructure of the UCMs (Fig. 1e). The size of the UCMs was derived to be ca. 122.1 nm (Fig. 1f). The elemental mapping images in Fig. 1e further indicated that the representative elements Y and Yb of UCNPs were distributed only in the area on the MOF side, while the elements Zr and Fe of MOFs were uniformly distributed throughout the NPs. This observation indicated the tight in situ growth of MOFs on the surface of UCNPs. The standard cards of OA-UCNPs and PEI/PVP-UCNPs with β-NaYF4 were well indexed according to the X-ray diffraction (XRD) patterns of different samples (Fig. S7, ESI†) (JCPDS: 16-0334). When the asymmetric UCNPs-MOFs nanostructures were formed, the XRD patterns of UCMs showed two new peaks at 7.1° and 9.9°, respectively. These peaks well matched the simulated curve of PCN-224(Fe), which indicated the successful synthesis of UCMs. In addition, zeta potential values in different steps were recorded to evaluate the formation of nanoparticles with different components. It revealed the potential variation throughout the reaction, and this provided further evidence for the synthesis of UCMs (Fig. S8, ESI†). Furthermore, we analyzed the surface chemistry of UCMs by X-ray photoelectron spectroscopy (XPS), and the characteristic elements Y, Zr and Fe of UCNPs and PCN-224(Fe) could be well found from the XPS full spectrum (Fig. S9a, ESI†). Fig. S9(b)–(e) (ESI†) illustrates that the C 1s spectrum was deconvoluted into three peaks at 284.8, 286.5, and 288.4 eV, which can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C/C–H, and CO/CN, respectively. The deconvolution of N 1s and Fe 2p spectra derived the characteristic peaks of Fe–N,40 and C–N attributed to porphyrin iron located at 712.3 eV, 725.1 eV and 399.2 eV. In the O 1s spectrum, the CO peak located at 533.05 eV is obviously stronger than the C–O peak at 534.7 eV, and the characteristic OC–N peak was identified at 401.3 eV. This observation indicated the successful reaction of porphyrin with the imine group. FT-IR spectra in Fig. 1g indicate that the modified dual-ligand UCNPs showed a typical CO stretching peak at 1673.9 cm−1 and a C–N characteristic stretching vibration peak at 1290.1 cm−1 in comparison with OA-UCNPs. In addition, the modification of dual-ligands significantly weakened the characteristic –CH3 peak, which further demonstrated the assembly of the dual ligands on OA-UCNPs. In the FT-IR spectra of UCMs, the sharp characteristic bands (CO) at 1631.7 cm−1 and 1606 cm−1, the enhancement of the characteristic band (N–H) at 3400 cm−1 and the weakening of the characteristic band (C–N) at 1290 cm−1 clearly indicated that the porphyrin monomer reacted with the amine group on the surface of UCNPs by condensation. These observations well demonstrated in situ growth of the MOFs on the surface of UCNPs.
C, C–C/C–H, and CO/CN, respectively. The deconvolution of N 1s and Fe 2p spectra derived the characteristic peaks of Fe–N,40 and C–N attributed to porphyrin iron located at 712.3 eV, 725.1 eV and 399.2 eV. In the O 1s spectrum, the CO peak located at 533.05 eV is obviously stronger than the C–O peak at 534.7 eV, and the characteristic OC–N peak was identified at 401.3 eV. This observation indicated the successful reaction of porphyrin with the imine group. FT-IR spectra in Fig. 1g indicate that the modified dual-ligand UCNPs showed a typical CO stretching peak at 1673.9 cm−1 and a C–N characteristic stretching vibration peak at 1290.1 cm−1 in comparison with OA-UCNPs. In addition, the modification of dual-ligands significantly weakened the characteristic –CH3 peak, which further demonstrated the assembly of the dual ligands on OA-UCNPs. In the FT-IR spectra of UCMs, the sharp characteristic bands (CO) at 1631.7 cm−1 and 1606 cm−1, the enhancement of the characteristic band (N–H) at 3400 cm−1 and the weakening of the characteristic band (C–N) at 1290 cm−1 clearly indicated that the porphyrin monomer reacted with the amine group on the surface of UCNPs by condensation. These observations well demonstrated in situ growth of the MOFs on the surface of UCNPs.
3.2. Photodynamic properties of UCMs
The upconversion luminescence properties of UCNPs were investigated. Under 980 nm laser irradiation, the upconversion luminescence spectra are shown in Fig. 2b, emitting mainly short-wavelength light centered at 450 nm (1D2 → 3F4), 475 nm (1G4 → 3H6) and 645 nm (1G4 → 3F4) and 700 nm (3F3 → 3H6) (as shown in Fig. S10, ESI†). It is obvious that the upconversion luminescence intensity of UCNPs after modification with dual-ligands is not significantly weakened with respect to that of OA-UCNPs (Fig. 2b). Fig. 2a indicates that UCMs present a typical broad UV-vis absorption band for PCN-224(Fe) within the range of 300-650 nm. The overlap of absorption spectra and upconversion spectra may indicate the possibility of energy resonance transfer between PCN-224(Fe) and UCNPs. The intensity of the upconversion luminescence spectra of UCMs was significantly decreased compared to that of PEI/PVP-UCNPs (Fig. 2b). This observation provided an indication for the energy transfer process in heterogeneous structures of UCMs. To further verify the occurrence of energy transfer, time-resolved photoluminescence lifetimes were investigated for PEI/PVP-UCNPs and UCMs. As shown in Fig. S11 (ESI†), the decay curves show a significant decrease in the lifetime of Tm3+ emission from 443.9 μs to 366.6 μs at 475 nm and from 457.1 μs to 362.6 μs at 645 nm. Thus, UCNPs can harvest low-energy photons in the NIR light region to produce emission at short wavelengths, which can be resonated by fluorescence resonance energy transfer (FRET) absorbed by PCN-224(Fe). Due to the presence of the porphyrin ligand, PCN-224(Fe) can generate 1O2 under 660 nm light irradiation.12 Therefore, in the UCM system, the activated porphyrin ligand can effectively absorb energy-delivering light and generate toxic 1O2 to facilitate photodynamic type II treatment.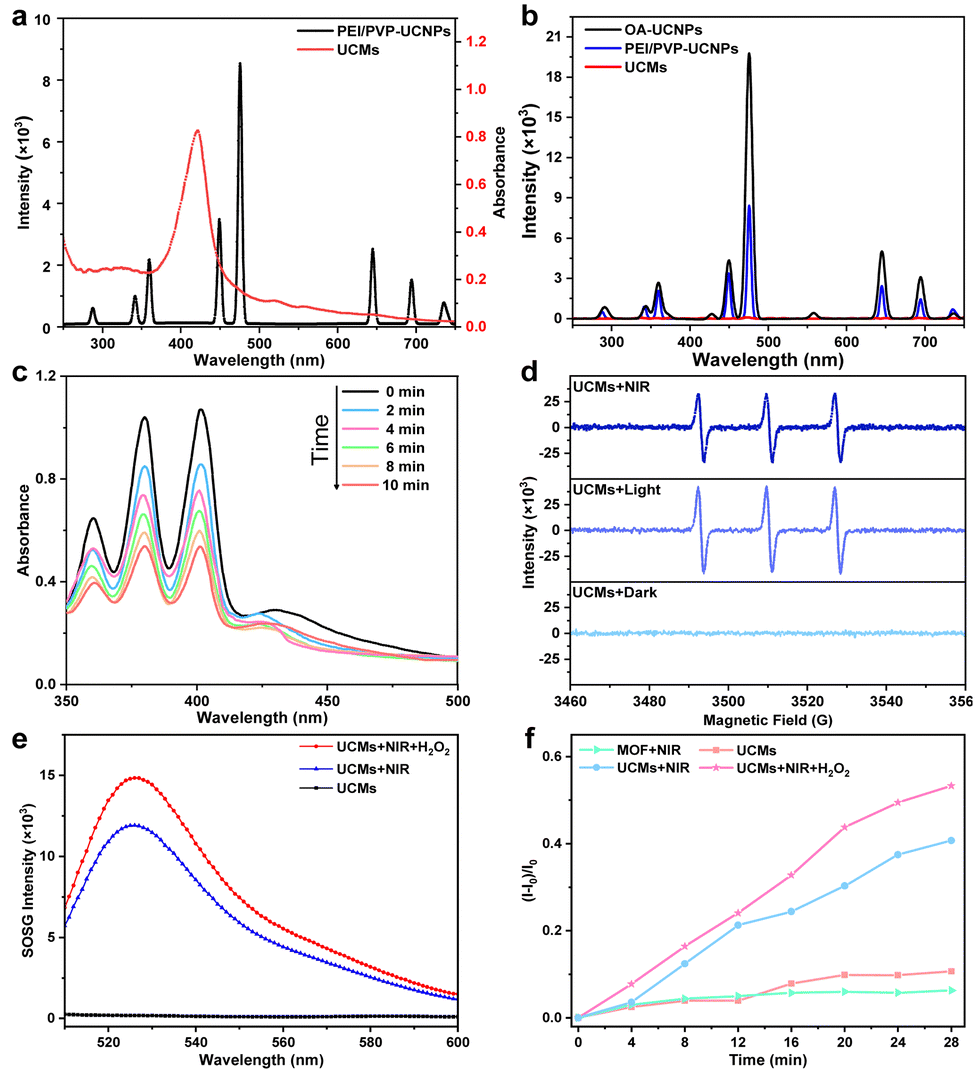 | ||
| Fig. 2 (a) Upconversion luminescence spectra of the PEI/PVP-UCNPs and the UV-vis absorption spectra of UCMs. (b) Upconversion luminescence spectra of OA-UCNPs (1 mg mL−1), PEI/PVP-UCNPs (1 mg mL−1) and UCMs (1 mg mL−1). (c) 1O2 generation under normoxic conditions upon 980 nm laser irradiation (1.5 W cm−2). The concentration of UCMs was 200 μg mL−1. (d) ESR spectra of UCMs with different conditions (dark, light: 660 nm laser, NIR: 980 nm laser) upon the addition of TEMP (trapping agent of 1O2). (e) 1O2 generation under different conditions tested by SOSG. (f) Time-dependent generation of 1O2 treated with different conditions tested by SOSG. The concentration of H2O2 was 100 μM. | ||
For investigating the capability of UCMs for the production of 1O2 with excitation by NIR irradiation, 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) was used for the characteristic detection of singlet oxygen. ABDA reacts with 1O2 to produce the corresponding peroxide and leads to a decrease in its characteristic absorption. Therefore, the variation of 1O2 production can be detected by monitoring the change of ABDA absorbance. Fig. 2c illustrates a sharp decrease in the absorbance of ABDA within 10 min of the reaction, which indicated that the mixed system produced a large amount of 1O2 under 980 nm light excitation. In addition to the ABDA test, 2,2,6,6-tetramethylpiperidine (TEMP) was further employed as a spin trapping agent, and electron spin resonance (ESR) spectroscopy was used to determine the 1O2 production. Fig. 2d indicates 1O2 characteristic triple peaks 1:1:1 under 980 nm irradiation. This further confirmed the production of a large amount of 1O2 by energy transfer process in UCMs under NIR laser irradiation. In addition, the quantum yield (Δφ) of 1O2 production by UCMs was evaluated. The Δφ value by methylene blue (MB) was derived to be 0.42 with the 1O2 quantum yield calculation formula (Fig. S12, ESI†).
3.3. Peroxidase-like activity of UCMs
The tumor microenvironment is characterized by hypoxia and a high concentration of H2O2. Therefore, the tumor microenvironment may pose limitations for the photodynamic therapy due to the fact that photodynamic type II therapy occurs in the presence of oxygen. The previous studies have demonstrated that Fe(III)–porphyrins exhibit high biocatalytic activity, the chelation with Fe3+ can generate oxygen with H2O2.20,21 In this respect, UCMs may be able to alleviate hypoxic environments and facilitate photodynamic therapy. The capability of UCMs to decompose H2O2 and produce oxygen was first investigated, and the amount of oxygen produced by UCMs under different conditions was tested using a dissolved oxygen meter. Fig. S13 (ESI†) illustrates that UCMs can continuously produce oxygen in the presence of H2O2 compared to the cases with H2O2-only and UCMs-only. Furthermore, the capability of UCMs to produce 1O2 under different conditions was examined by using the singlet oxygen sensor green (SOSG) characterization kit. Fig. 2e and f demonstrates that UCMs could produce more singlet oxygen in the presence of H2O2 under NIR light irradiation. By considering the O2-dependent characteristics of photodynamic type II therapy, UCMs can consume more H2O2 to generate O2 and ameliorate tumor hypoxia and thus to enhance 1O2 production. That is to say, the biocatalytic nature of UCMs shows promising potential in improving the efficacy of photodynamic therapy. As nanomaterials for biological applications, it is important to study the stability of UCMs. UCMs in different concentrations were dispersed in PBS, DMEM medium and fetal bovine serum (FBS). Notably, no aggregation of UCMs was observed after two days in PBS, DMEM medium, or FBS (Fig. S14, ESI†). Furthermore, the size of UCMs remained largely consistent across the different solutions (Fig. S15, ESI†). Based on these observations, it can be concluded that UCMs exhibit commendable stability under different solvent media studied.3.4. In vitro cellular uptake and cell toxicity
The photodynamic treatment capability of UCMs to enhance the therapeutic effect was investigated at the cellular level. First, the uptake of UCMs by HepG2 cells was evaluated by using laser confocal microscopy (CLSM) after culturing the cells with UCMs for different times. For subsequent observation, the cells were localized by staining the nuclei with DAPI, and in addition, UCMs could produce red fluorescent signals under 488 nm excitation.12 According to the CLSM analysis (Fig. S16, ESI†), the fluorescence of UCMs in the cell interior was barely observed after 15 min of incubation, and a faint fluorescence could be identified within the cells after 30 min of incubation, which was afterwards significantly enhanced at 60 min. It was seen that the intracellular fluorescence intensity and distribution were almost the same at 60 min and 120 min of incubation, indicating that the cells could engulf UCMs well within 60 min. In addition, the DAPI nuclear localization illustrated that UCMs were taken up by the cells to the cytoplasmic site. After incubating HepG2 cells with different concentrations of UCMs for 24 h, the cell viability was kept at >85% at 200 μg mL−1 and >80% at 1 mg mL−1 (Fig. 3a). In addition, different concentrations of UCMs were incubated with normal liver cells LX-2 for 24 h. Fig. S17 (ESI†) shows that even at the highest concentration of 200 μg mL−1, the viability of LX-2 cells reached 95%. These indicated that UCMs exhibit favorable biocompatibility which may facilitate the subsequent photodynamic therapy of tumors.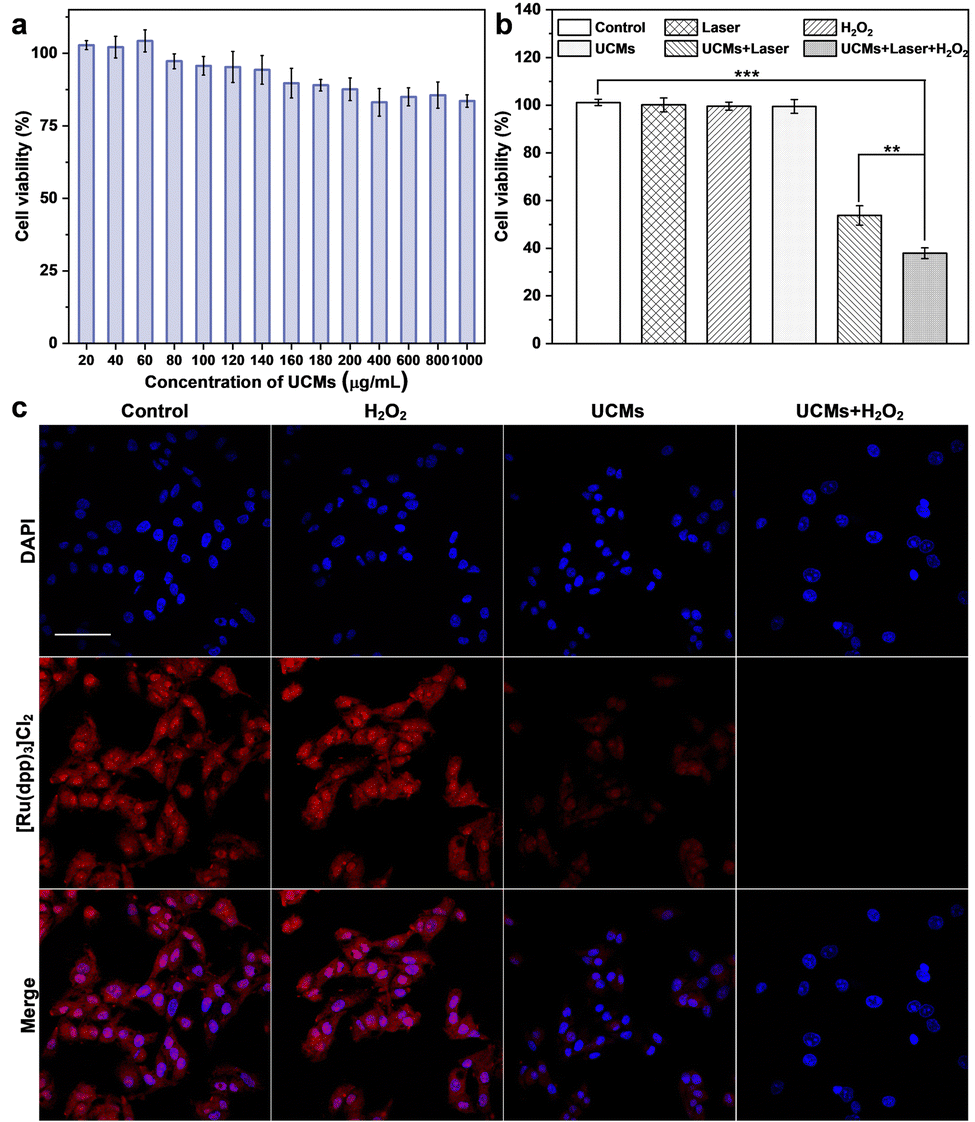 | ||
| Fig. 3 (a) HepG2 cell viability treated with different concentrations of UCMs for 24 h. (b) HepG2 cell viability after incubating with different treatments for 24 h. Data are shown as means ± SD. n = 3. **p < 0.01, ***p < 0.001. (c) CLSM images of the produced intracellular O2. After various treatments (control, H2O2, UCMs, UCMs + H2O2), HepG2 cells were stained with cell nuclear dye DAPI, and [Ru(dpp)3]Cl2 were used as the dissolved O2 probe. The concentration of H2O2 was 100 μM. The power density of 980 nm laser was 1.5 W cm−2 for 10 min. The scale bar indicates 50 μm. | ||
3.5. Evaluation of photodynamic therapy in living cells
The effect of photodynamic treatment of UCMs was evaluated (Fig. S18, ESI†), with the evaluation of cell viability as analyzed by using CCK-8 assay. With the increase of UCM concentration for cell incubation, the viability of cells after NIR irradiation was gradually decreased. At a concentration level of 100 μg mL−1 for UCMs, the cell viability was remarkably decreased to 63.2%. This indicated that UCMs exhibit a significant PDT effect and obvious value-added inhibition on tumor cells. Further studies have found that UCMs may enhance the effects of photodynamic therapy. For examining the capability of UCMs to decompose H2O2 in cell interior and generate O2, [Ru(dpp)3]Cl2 reagent was chose as an oxygen indicator, and the fluorescence of [Ru(dpp)3]Cl2 would be quenched by oxygen. The cells were first pretreated with anoxia and incubated in anoxic bags for 12 h. The anoxic cells were then incubated with UCMs. Fig. 3c clearly shows the fluorescence quenching of [Ru(dpp)3]Cl2 in the cells treated with UCMs, indicating the intracellular decomposition of H2O2 to produce O2. In addition, the cells pre-incubated with H2O2 showed complete dye quenching after the addition of UCMs, illustrating the production of more oxygen by the H2O2-treated cells with respect to those without incubation with H2O2. This observation further demonstrated the capability of H2O2 consumption by UCMs to produce O2.The ability of UCMs to enhance photodynamic therapy was investigated by culturing HepG2 cells under hypoxic conditions and by introducing additional H2O2-treated cells. First, the biocompatibility of H2O2 with cells was studied. The results of Fig. S19 (ESI†) showed that cells were still active after 1 mM H2O2 treatment. CCK-8 was used to detect the relative viability of hypoxic cells under different treatments, and the results showed (Fig. 3b) that the relative viability of cells was significantly reduced under near-infrared light irradiation at UCMs concentration of 100 μg mL−1. When the cells were treated with H2O2, the relative viability rate of the cells was further significantly reduced under near-infrared irradiation, indicating that the addition of H2O2 promoted the photodynamic treatment effect of UCMs. It is indirectly proved that UCMs can overcome the hypoxic environment and play a better photodynamic therapeutic effect in the tumor cell microenvironment. To explore the amount of intracellular production of reactive oxygen species (ROS), a common intracellular ROS fluorescent probe, DCFH-DA, was used to detect the production of intracellular ROS in HepG2 cells after different treatments. As shown in Fig. S20 (ESI†), UCMs were capable of producing ROS under NIR light irradiation, while in H2O2-treated cells, UCMs could produce even more ROS under laser irradiation. This further confirmed the aforementioned results for the enhancement of photodynamic therapy.
In addition, further assessment of apoptosis effect by different treatments was performed under CLSM by staining the cells with FDA/PI (Fig. S21, ESI†); it was found that the control group and the group treated only with 980 nm laser, UCMs as well as H2O2, showed almost no cell death (green fluorescence). In contrast, cells treated with UCMs irradiated with NIR showed mostly dead results (red fluorescence). By NIR irradiation of the cells pretreated with H2O2, virtually only dead cells were observed in the field of view. Meanwhile, the results of flow cytometry in Fig. S22 (ESI†) showed that NIR and H2O2-treated cells were almost killed, which well demonstrated the observations in live-dead cell staining experiments. Collectively, the above studies confirmed that UCMs have the capability to facilitate PDT while overcoming the lack of oxygen in the tumor microenvironment and they are able to utilize the excess H2O2 therein to enhance the PDT treatment effect further.
3.6. Evaluation of in vivo treatment effect
UCMs have been demonstrated to exhibit excellent photodynamic treatment effect in vitro. Herein, their anti-tumor effect in vivo was further exploited. The anti-tumor capacity of UCMs was evaluated in detail in BALB/C mice by tail intravenous injection. To further investigate the safety of UCMs on the body, the biodistribution of UCMs was studied with detection by inductively coupled plasma mass spectrometry (ICP-MS) and the levels of yttrium (Y) in the brain, liver, kidneys, spleen, lungs and tumors of the mice were measured at 6 h, 24 h, and 48 h after the injection of UCMs. The experiments indicated that at 6 h after injection, UCMs were found to distribute in all these organs, with the majority identified in kidneys, spleen, liver and tumors. After 48 h, UCMs were almost completely metabolized in all organs except tumor tissues (Fig. S23, ESI†).Furthermore, to investigate the effect of UCM treatment, the tumor-bearing mice were divided into four groups to receive different treatments. The mice were treated every two days for two weeks, and the variations of tumors as well as body weight were recorded during the treatment period. The tumor volume growth curve analysis showed that the growth of tumors was not inhibited during the 14 days of treatment with PBS and laser. However, tumor growth was significantly inhibited in the group that received UCMs and UCMs + laser treatment (Fig. 4a). Fig. S24 (ESI†) demonstrates significant difference in tumor size after 14 days. In addition, the body weight of mice was relatively stable during the treatment period, further indicating negligible biological toxicity and side effects of UCMs (Fig. 4b). Immunohistochemical experiments were performed on each organ after treatment, and the results of H&E staining illustrated that there were no significant changes in the morphology of each tissue in all groups after treatment (Fig. S25, ESI†). In addition, the blood routine was performed on the tumor-bearing mice injected intravenously with UCMs, and those hematological indicators presented no discernible differences from the control group (Fig. S26, ESI†). Those data indicate the excellent biocompatibility of UCMs in the therapy conditions. Immunohistochemical staining of tumor tissues was performed for Ki67 and ALDH1, with Ki67 as the cell proliferation marker and ALDH1 as a tumor stem cell marker. Fig. 4c indicates that the value-added of tumor cells was inhibited and apoptosis of tumor cells was found after treatment with UCMs and laser. To confirm the capability of UCMs to ameliorate hypoxia in solid tumors, HIF-1α was selected for immunofluorescence assays of tumors, with the results illustrated in Fig. S27 (ESI†). After treatment, the tumors in the PBS group showed a significant HIF-1α fluorescence signal (red fluorescence) due to hypoxia, while the characteristic immunofluorescence of HIF-1α in tumors in the UCMs group decreased dramatically. This was mainly due to the catalytic activity of UCMs, which broke down intracellular H2O2 and produced O2 in the tumor microenvironment. This further promoted the enhancement efficacy of oxygen-dependent PDT type II in vivo. It demonstrated the promising potential antitumor therapeutic effect of UCMs under laser irradiation in the hypoxic tumor environment. The above experimental results clearly confirmed that UCMs have favorable biosafety and anti-tumor therapeutic effects in vivo, and furthermore, they may have great prospects for the application in tumor therapy.
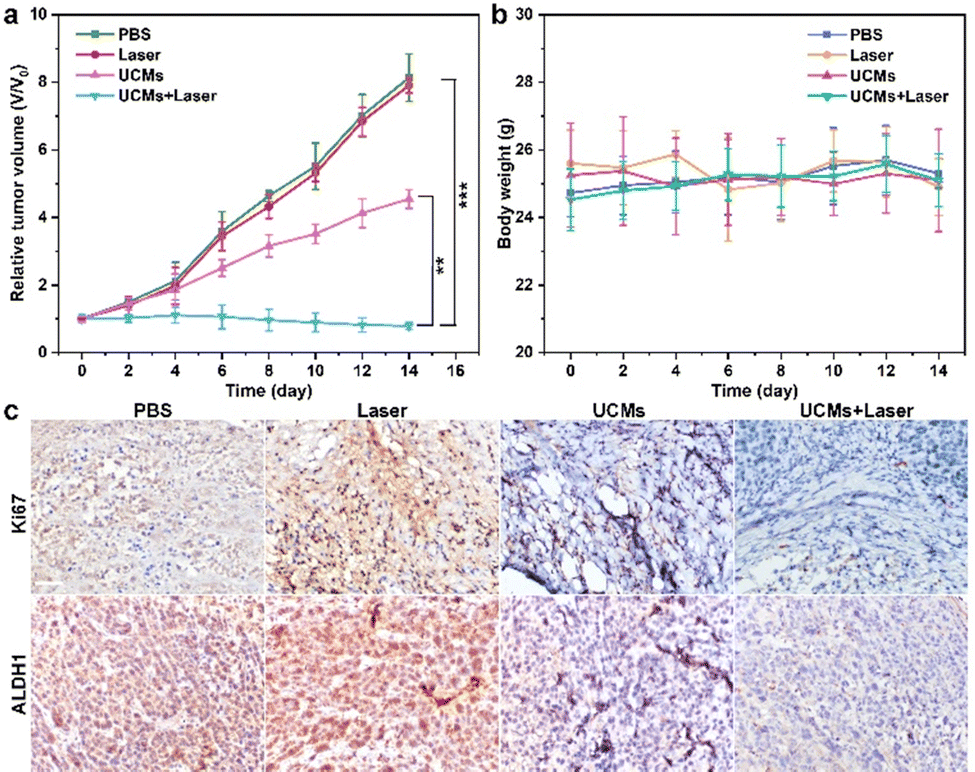 | ||
| Fig. 4 (a) The relative tumor volume and (b) the body weight of mice bearing HepG2 tumors after various treatments during a 14 day treatment. Data are shown as means ± SD; n = 3 mice/group. **P < 0.01, ***P < 0.001. (c) The Ki67 staining and ALDH1 staining in the tumor site with different treatments. The concentration of UCMs was 4 mg mL−1 (100 μL). The power density of 980 nm laser was 1.5 W cm−2. The scale bar is 50 μm. | ||
4. Conclusions
In summary, UCNPs-MOFs (UCMs) nanocomposites have been prepared utilizing a dual-ligand-assisted self-assembly approach. This method generates asymmetric heterostructures composed of UCNPs and Fe(III)–porphyrin MOFs. The nucleation and growth kinetics of PCN-224(Fe) can be meticulously controlled by the dual ligands, i.e., PEI and PVP, that are present on the surface of the functional nanoparticles. The integrated UCMs nanosystem showcases promising optical properties, as the UCNPs undergo an energy transfer with the MOFs. This characteristic endows the UCMs with the ability to activate photodynamically when exposed to near-infrared light. Additionally, UCMs possess the ability to decompose endogenous H2O2 in the tumor microenvironment. The inherent ability of UCMs to self-oxygenate helps overcome the limitations imposed by the tumor microenvironment. This feature promotes the subsequent activation of photodynamic therapy using near-infrared light, significantly enhancing the capability of photodynamic treatment. In vitro and in vivo studies have demonstrated that UCMs display significant biocatalytic activity and a remarkable PDT effect when driven by near-infrared irradiation. The current work leverages a unique dual-ligand-assisted self-assembly protocol to highlight the immense potential of MOF-based heterostructures in biomedicine. This innovative strategy could pave the way for the integration and construction of more heterostructured nanoplatforms for potential future applications in the field of biomedicine.Author contributions
Xinyue Zhang: methodology, investigation, software, writing – original draft, resources. Jiasen Cui: responsible for animal experiment. writing – review & editing. Jinhui Liu: investigation, validation. Xi Chen: responsible for animal experiment. Ming-Li Chen: conceptualization, methodology, writing – review & editing, supervision, funding acquisition. Jianhua Wang: conceptualization, methodology, revising & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support of the National Natural Science Foundation of China (22274017 and 22074011). Special thanks are due to the Analytical and Testing Center, Northeastern University, for the assistance in instrumental analysis.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- A. S. Thakor and S. S. Gambhir, Ca-Cancer J. Clin., 2013, 63, 395–418 CrossRef PubMed.
- C. J. Allegra, J. Natl. Cancer Inst., 2012, 104, 961–962 CrossRef PubMed.
- G. Yang, X. Sun, J. Liu, L. Feng and Z. Liu, Adv. Funct. Mater., 2016, 26, 4722–4732 CrossRef CAS.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS PubMed.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2015, 137, 7600–7603 CrossRef CAS PubMed.
- G. Li, S. Zhao, Y. Zhang and Z. Tang, Adv. Mater., 2018, 30, 1800702 CrossRef PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed.
- F. S. Liao, W. S. Lo, Y. S. Hsu, C. C. Wu, S. C. Wang, F. K. Shieh, J. V. Morabito, L. Y. Chou, K. C. Wu and C. K. Tsung, J. Am. Chem. Soc., 2017, 139, 6530–6533 CrossRef CAS PubMed.
- D. Wang, J. Zhou, R. Shi, H. Wu, R. Chen, B. Duan, G. Xia, P. Xu, H. Wang, S. Zhou, C. Wang, H. Wang, Z. Guo and Q. Chen, Theranostics, 2017, 7, 4605–4617 CrossRef CAS PubMed.
- J. Park, Q. Jiang, D. Feng, L. Mao and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS PubMed.
- Y. Wang, W. Wu, J. Liu, P. N. Manghnani, F. Hu, D. Ma, C. Teh, B. Wang and B. Liu, ACS Nano, 2019, 13, 6879–6890 CrossRef CAS PubMed.
- J. Liu, T. Liu, P. Du, L. Zhang and J. Lei, Angew. Chem., Int. Ed., 2019, 58, 7808–7812 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- L. Chudal, N. K. Pandey, J. Phan, O. Johnson, L. Lin, H. Yu, Y. Shu, Z. Huang, M. Xing, J. P. Liu, M. L. Chen and W. Chen, ACS Appl. Bio Mater., 2020, 3, 1804–1814 CrossRef CAS PubMed.
- F. Muhammad, A. Wang, L. Miao, P. Wang, Q. Li, J. Liu, J. Du and G. Zhu, Langmuir, 2015, 31, 514–521 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS PubMed.
- L. Chudal, N. K. Pandey, J. Phan, O. Johnson, X. Li and W. Chen, Mater. Sci. Eng., C, 2019, 104, 109979 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- S. Dong, Y. Dong, T. Jia, S. Liu, J. Liu., D. Yang, F. He, S. Gai, P. Yang and J. Lin, Adv. Mater., 2020, 32, 2002439 CrossRef CAS PubMed.
- N. Kheshtchin, S. Arab, M. Ajami, R. Mirzaei, M. Ashourpour, N. Mousavi, N. Khosravianfar, F. Jadidi-Niaragh, A. Namdar, F. Noorbakhsh and J. Hadjati, Cancer Immunol. Immunother., 2016, 65, 1159–1167 CrossRef CAS PubMed.
- Z. Cai, F. Xin, Z. Wei, M. Wu, X. Lin, X. Du, G. Chen, D. Zhang, Z. Zhang, X. Liu and C. Yao, Adv. Healthcare. Mater., 2020, 9, 1900996 CrossRef CAS PubMed.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 12502–12510 CrossRef CAS PubMed.
- G. Lan, K. Ni, Z. Xu, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2018, 140, 5670–5673 CrossRef CAS PubMed.
- L. D. Sun, H. Dong, P. Z. Zhang and C. H. Yan, Annu. Rev. Phys. Chem., 2015, 66, 619–642 CrossRef CAS PubMed.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- Y. Li, Z. Di, J. Gao, P. Cheng, C. Di, G. Zhang, B. Liu, X. Shi, L. D. Sun, L. Li and C. H. Yan, J. Am. Chem. Soc., 2017, 139, 13804–13810 CrossRef CAS PubMed.
- Y. Shao, B. Liu, Z. Di, G. Zhang, L. D. Sun, L. Li and C. H. Yan, J. Am. Chem. Soc., 2020, 142, 3939–3946 CrossRef CAS PubMed.
- Y. Zhang, F. Wang, C. Liu, Z. Wang, L. Kang, Y. Huang, K. Dong, J. Ren and X. Qu, ACS Nano, 2018, 12, 651–661 CrossRef CAS PubMed.
- H. J. Cai, T. T. Shen, J. Zhang, C. F. Shan, J. G. Jia, X. Li, W. S. Liu and Y. Tang, J. Mater. Chem. B, 2017, 5, 2390–2394 RSC.
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed.
- A. E. Guller, A. Nadort, A. N. Generalova, E. V. Khaydukov, A. V. Nechaev, I. A. Kornienko, E. V. Petersen, L. Liang, A. B. Shekhter, Y. Qian, E. M. Goldys and A. V. Zvyagin, ACS Biomater. Sci. Eng., 2018, 4, 3143–3153 CrossRef CAS PubMed.
- L. Zhang, D. Jin and M. H. Stenzel, Biomacromolecules, 2021, 22, 3168–3201 CrossRef CAS PubMed.
- Y. Zhang, X. Zhu and Y. Zhang, ACS Nano, 2021, 15, 3709–3735 CrossRef CAS PubMed.
- T. Wan, W. Li and Z. Chen, J. Pharm. Biomed. Anal., 2021, 199, 114049 CrossRef CAS PubMed.
- L. He, M. Brasino, C. Mao, S. Cho, W. Park, A. P. Goodwin and J. N. Cha, Small, 2017, 13, 1700504 CrossRef PubMed.
- L. He, Q. Ni, J. Mu, W. Fan, L. Liu, Z. Wang, L. Li, W. Tang, Y. Liu, Y. Cheng, L. Tang, Z. Yang, Y. Liu, J. Zou, W. Yang, O. Jacobson, F. Zhang, P. Huang and X. Chen, J. Am. Chem. Soc., 2020, 142, 6822–6832 CrossRef CAS PubMed.
- H. Ang and L. Hong, ACS Appl. Mater. Interfaces, 2017, 9, 28079–28088 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01398g |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2023 |