
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanisms of Mg carbonates precipitation and implications for CO2 capture and utilization/storage
Hellen S.
Santos
 *a,
Hoang
Nguyen
a,
Fabricio
Venâncio
b,
Durgaprasad
Ramteke
a,
Ron
Zevenhoven
c and
Paivo
Kinnunen
a
*a,
Hoang
Nguyen
a,
Fabricio
Venâncio
b,
Durgaprasad
Ramteke
a,
Ron
Zevenhoven
c and
Paivo
Kinnunen
a
aFibre and Particle Engineering Research Unit, University of Oulu, Pentti Kaiteran katu 1, 90570 Oulu, Finland. E-mail: hellen.silvasantos@oulu.fi
bDevelopment Center of Real Time Chemical Processes and Analyses (NQTR), Institute of Chemistry, Federal University of Rio de Janeiro, Rua Hélio de Almeida, 40, Cidade Universitária da Universidade federal do Rio de Janeiro, 21941-614, Rio de Janeiro-RJ, Brazil
cProcess and Systems Engineering Laboratory, Åbo Akademi University, Henrikinkatu 2, 20500 Turku, Finland
First published on 2nd February 2023
Abstract
The mechanisms involved in the natural formations of dolomite (CaMg(CO3)2) and magnesite (MgCO3) have endured as challenging research questions over centuries, being yet a matter under investigation in multiple fields. From a geochemical perspective, it is still unknown why there are recent natural formations of dolomite and magnesite at ambient conditions, and yet most available synthetic routes for precipitating these minerals require high temperatures and/or pressures. The core scientific gap is that even though dolomite and magnesite are the most thermodynamically stable phases among the respective polymorphs/intermediates, their formation is controlled by slow kinetics and their syntheses at ambient conditions remain a challenge. Research findings lead to possible explanations based on the chemical and thermodynamical properties of the system: (i) the high energy barrier for dehydrating the Mg2+·6H2O cations hinders the carbonation of Mg precursors, inducing a preferential formation of the hydrated magnesium carbonates polymorphs, (ii) the intrinsic structural/spatial barrier of the CO32− groups in the rhombohedral arrangement of dolomite and magnesite shifts the system towards the formation of the respective polymorphs. However, further studies are still needed to enable a clearer understanding of the phenomenon. Recently, the research question at hand gained broader significance due to the relevance of Mg carbonates for routes of carbon capture and utilization/storage, which has been seen as one of the most promising solutions for such processes. The main socio-economic motivations behind such interest on these carbon mineralization methods are the high availability of Mg precursors (from natural sources to industrial waste-streams), the long-term geological storage of CO2 as magnesite, the possibility of utilizing the carbonate products in construction materials applications, and the relevance of the routes for climate mitigation actions. Therefore, understanding the mechanisms and kinetics of Mg carbonates precipitation is of fundamental importance for many fields, ranging from geology to necessary environmental actions. This review focuses on gathering the main information concerning the geochemical and chemical advances on the dynamics and mechanisms of Mg carbonates precipitation. It aims at providing a comprehensive summary of the developments from the fundamental sciences to the applications of Mg carbonates.
 Hellen S. Santos | Dr Hellen Santos holds a BSc degree in chemistry from the Federal University of Bahia (2012, Salvador-BA, Brazil), a M.S. in analytical chemistry from the Pontifical Catholic University of Rio de Janeiro (2014, Rio de Janeiro-RJ, Brazil), and a PhD in inorganic materials chemistry from the University of Turku (2017, Turku, Finland). Currently, she is a postdoctoral researcher at Magnesia-based materials and systems team, Fibre and Particle Engineering Research Unit of the University of Oulu (since 2020, Oulu, Finland), and has been dedicating her research efforts towards the recycling of inorganic industrial waste-streams for construction and energy applications. |
 Hoang Nguyen | Dr Hoang Nguyen is a postdoctoral researcher at Magnesia-based materials and systems team, Fibre and Particle Engineering Research Unit, University of Oulu (Finland). Hoang does research in construction and building materials with more than 10 years of training and experiences in the field. His research interests are low/no-carbon cements, cement chemistry, carbonatable materials, and thermodynamic modelling. |
 Fabricio Venâncio | Fabricio Venancio is PhD in chemistry specialized on crystallization process and control (PAT). His field of work is energy (oil & gas industry) and geochemistry. Fabricio has a research focus on crystallization kinetics, artificial rock maturation, catalysis and kinetics of chemical reactions. He is a former associate professor in Federal Center of Technology of Minas Gerais – IFRJ (Brasil) and former researcher in the Federal University of Rio de Janeiro – UFRJ (Brasil). Nowadays he is leader of the sustainability area in a multinational chemical company and a collaborator researcher in C3I Institute and UFRJ. |
 Durgaprasad Ramteke | Durgaprasad Ramteke is a PhD in physics specialized in condensed matter and material science. His research work focused on glasses, glass ceramics, ceramics, inorganic waste management, foam glass and geopolymers for direct applications. At present he is working as a postdoc in the University of Oulu as a researcher on the project CEMGlass (Cementitious Glass) aimed towards the developing a cementitious system with zero-to-negative CO2 emissions. Personal webpage: https://www.ddramteke.com/ |
 Ron Zevenhoven | Ron Zevenhoven has been professor in Engineering Thermodynamics and Modelling at Åbo Akademi University, Chemical and Process Engineering, in Turku Finland, since 2005. He holds an MSc (ChemEng) and PhD degrees (1988, 1992) from Delft University of Technology, the Netherlands. Current fields of research are CO2 capture and storage using mineral sequestration, material recycling and energy recovery, passive cooling based on thermal radiation, Stirling engine application and industrial heat pumps, thermal energy storage (TES), process scale-up and particle technology/multi-phase flow dynamics including nano-particulate material. Current h-index (Web of Science) 39. His personal webpage: https://users.abo.fi/rzevenho/. |
 Paivo Kinnunen | Dr Paivo Kinnunen holds a BSc degree in Physics from the University of Turku (2007, Turku, Finland), PhD degree in Applied Physics from the University of Michigan (2011, Ann Arbor, US). Currently he is an associate professor and principal investigator of Magnesia-based Materials and Systems research group at the Fibre and Particle Engineering Research Unit (University of Oulu, Oulu, Finland). His research is concentrating on carbon CO2 and utilization/storage as well as on developing novel cementitious binders. |
1. Introduction
Carbon dioxide mineralization is a promising pathway for enabling feasible and efficient routes of carbon capture and utilization/storage due to the large and globally widespread availability of suitable feedstocks.1 Among the large variety of precursors to carbon mineralization, alkaline earth-based materials, specially Ca and Mg-based materials, are considered as the most promising materials due to their large availability, high potential for carbonation, possible utilization in construction materials, and long term CO2 storage in geological time scale.2 Moreover, besides tailings and other waste streams from mining sites, several types of alkaline industrial waste-streams can be employed as precursors for carbon mineralization, for instance, only the resources of Ca and Mg bearing industrial waste streams (e.g. fly ash, cement kiln dust, steel slag and red mud) have the potential for capturing 200–300 Mt of CO2 annually.3 Routes of carbon mineralization from Mg silicate precursors have been greatly improved, leading to acceptable levels of conversion kinetics and energy requirements. However, the economic feasibility of these routes is still far from ideal, needing the creation of new business models and markets to direct the produced Mg carbonate products.4 Moreover, the profitability for carbon mineralization is directly dependent on the market size for the carbonated products, for which the construction market is considered the only sector large enough to enable profits. Yet, up to date reported LCA assessments have shown that the current routes of carbon capture and utilization/storage via carbon mineralization in Ca/Mg silicate minerals can reduce the CO2 emissions of the cement industry by 8–33%, with the profit forecast of up to 32 € per ton of cement.5 Recently developed thermal energy storage materials are considered also as an option for high-value applications, but it would require the creation of a disruptive business model aligned with legislative acceptance.6The economic feasibility of the current processes of carbon mineralization can be greatly improved if the mechanisms and kinetics of Mg carbonates are understood. Scientists such as Liebermann (1967), Berzelius (1820), and Bragg (1914) have investigated the geochemical aspects of carbonate minerals over centuries, providing to the scientific community relevant clues about the mechanisms and kinetics of most carbonate minerals precipitation.7 The Earth's surface is composed by over 277 types of carbonate-bearing minerals, which have been the scope of geochemical investigations since the 20th century.8 While considerable knowledge has been built on the chemistry orchestrating the formation of Ca carbonates, open questions remain on the chemistry of Mg carbonates formation. For example, a remaining geochemical problem is the comprehension of the dolomite precipitation mechanisms. Dolomite sediments represents 50% of the world's carbonate reservoirs, and ca. 80% of the oil and gas reservoirs in North America.9 Pre-halocene (Pre-Cenozoic Era) sediments display large amounts of dolomite formation in the carbonate platforms; contradictorily, the modern formation of dolomites is extremely rare even though contemporary sea water is thermodynamically oversaturated with relation to dolomite.10 Yet, the mechanisms of dolomitization in natural conditions of the Earth's surface are not well understood since synthetic dolomites have not been obtained under similar conditions. Thus far, the knowledge on dolomite nucleation and growth are based on high temperature (150–300 °C) experimental settings. These challenges on the synthesis of dolomite at ambient conditions, as well as the low amounts of modern dolomite in marine environments are the core of the so called “dolomite problem”.11
A closely related knowledge gap resides on the controversial dilemmas of magnesite (MgCO3) precipitation: magnesite is the most thermodynamically stable phase among its anhydrous and hydrated polymorphs, but its formation is hindered by kinetic barriers favoring the precipitation of its metastable hydrate phases.7 The stable phases in the equilibrium of solutions containing Mg ions and carbon dioxide are either brucite (pCO2 atmosphere <5 bar and low CO2 concentrations) or magnesite (most stable Mg carbonate at all conditions of pressure and temperature), based on the results obtained from the thermodynamic data fitted to the Pitzer model for the activity coefficients.12 Yet, the precipitation of magnesite requires moderate to high temperatures (60 to 200 °C) and/or high pressures (50 to 100 bar).12–14 Despite the simplicity of the stoichiometric reactions of Mg carbonates precipitation, they also involve complex chemical–morphological interactions that lead to challenging kinetics of carbonation nucleation and growth.3 Thus, although the thermodynamic variables of Mg carbonation are favourable at ambient conditions, the respective anhydrous carbonate minerals (magnesite – MgCO3 and dolomite – CaMg(CO3)2) have not yet been synthesized at ambient temperature and pressures using industrially feasible routes.11,15,16
As the dolomite and magnesite formation problems remain yet unsolved, several research efforts have been dedicated to increase the understanding on the mechanisms of Mg carbonates precipitation. While attempting to explain the mechanisms of dolomite/magnesite formation, the remaining dilemma is if the reaction is mainly hindered by (i) the high dehydration energy barrier of the Mg2+ cations16 or (ii) the intrinsic structural/spatial barrier of the CO32− groups in the magnesite/dolomite structure, avoiding the formation of long-range ordered structures.17 As the reasoning behind the dilemma is quite extensive, the current review aims to summarize all main findings on the subject, highlighting the remaining knowledge gaps and the main implications for carbon capture and utilization/storage routes with applications in construction materials.
2. Geochemistry of carbonate minerals
In nature, several factors may have contributed to the precipitation of carbonate minerals ranging from the geochemical impacts of large and small organisms to the influence on kinetics from diverse variables (fluctuating pH, gas-phase interactions, different subtracts and organic compounds).18 Most of the well-known carbonates are formed with divalent cations and carbonate ions (when there are no other joined anionic ligands).19 The alkaline-earth carbonates are usually the first to precipitate due to evaporation processes, being formed in dilute waters where the molar concentration of HCO3−(aq) is much higher than the summed concentrations of Mg2+(aq) and Ca2+(aq).18 Although there is a significant understanding on the geochemistry behind the natural formation of calcite (CaCO3) and other carbonates, it remains a challenge to explain the natural formation of Mg bearing carbonates, such as dolomite (MgCa(CO3)2) and magnesite (MgCO3).20A geochemical analysis of systematic mineralogy described by Railsback (2005) has associated the natural formation of simple carbonate minerals to the presence of hard cations (cations without outer shell electrons, such as Na+ and Mg2+) displaying low to medium polarizability. Following the Pearson's postulates on the interactions between hard and soft ions, Railsback observed that the hard anions of oxysalt radicals (such as carbonates) form minerals almost exclusively with hard cations. His study proposed that the incorporation of cations with higher ionic potential is enabled by neutral groups (H2O) promoting the initial shielding of the cations from each other, and the charge balance is maintained by the anionic groups (Fig. 1, top).21
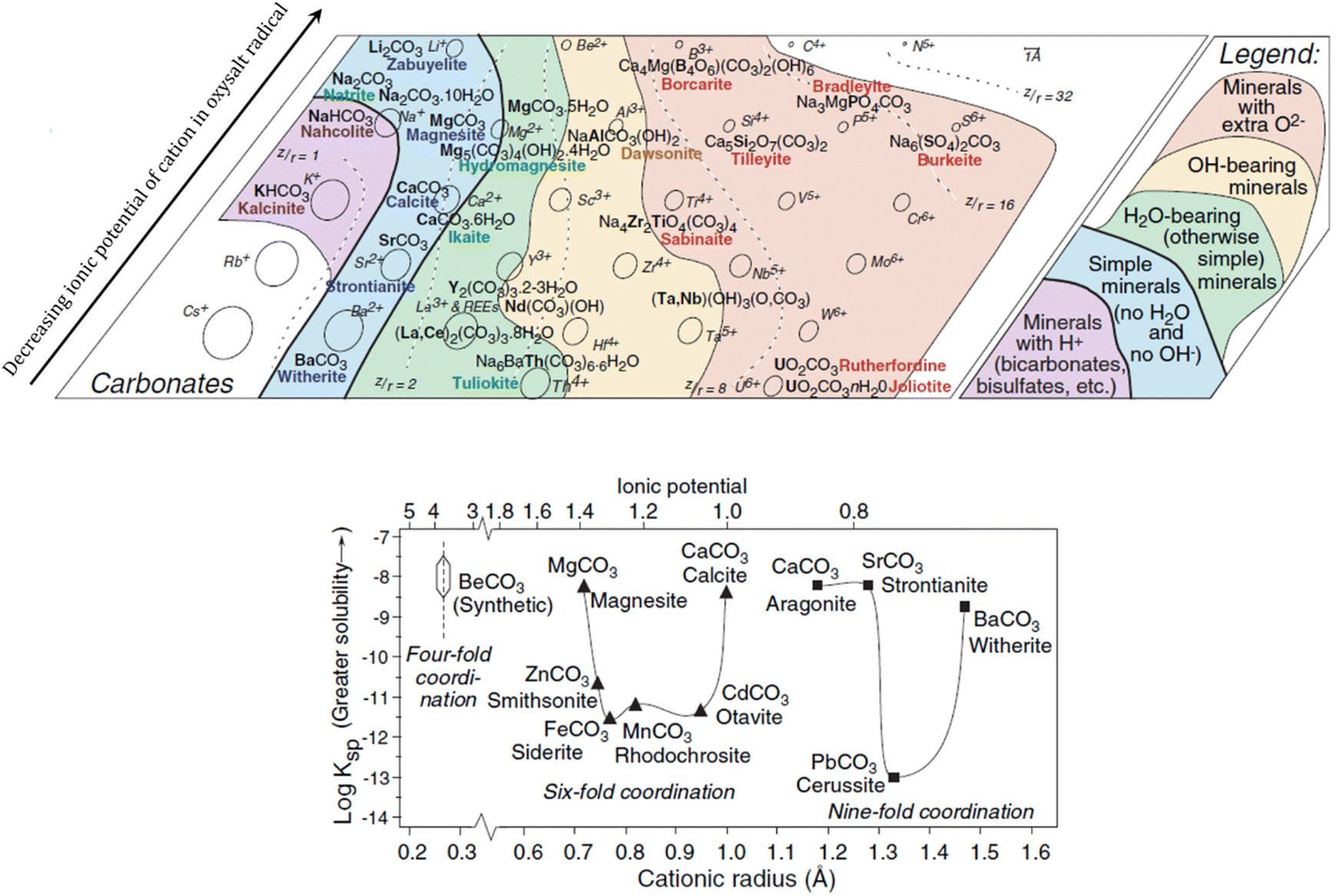 | ||
| Fig. 1 Carbonate minerals of hard cations (top) and solubility of alkaline-earth carbonates in function of ionic potential and radii, representing rhombohedral minerals with triangles and orthorhombic with squares (bottom). Adapted from Railsback (2005).21 Reproduced with the publisher's permission (© 2015 by Walter de Gruyter Berlin/Boston). | ||
The crystalline arrangement of simple carbonates of divalent cations is related to the ionic radii and coordination number of the cations, which can be classified in two structural groups:
(i) Rhombohedral carbonates, formed with smaller cations of sixfold coordination, and
(ii) Orthorhombic carbonates, formed with larger cations of ninefold coordination.21
Fig. 2 shows the unit cell structure of representatives from both structural classes, where the effect of the ionic radii on the distortions of the crystalline structures is noticeable: distortions on the ninefold coordination leads to shifts in the x, y, z axes, which determines whether the crystal structure of the carbonate has a triclinic, monoclinic, or orthorhombic space group. Likewise, distortions in the sixfold structure lead to carbonates belonging to either hexagonal/triclinic or rhombohedral/trigonal space groups.22
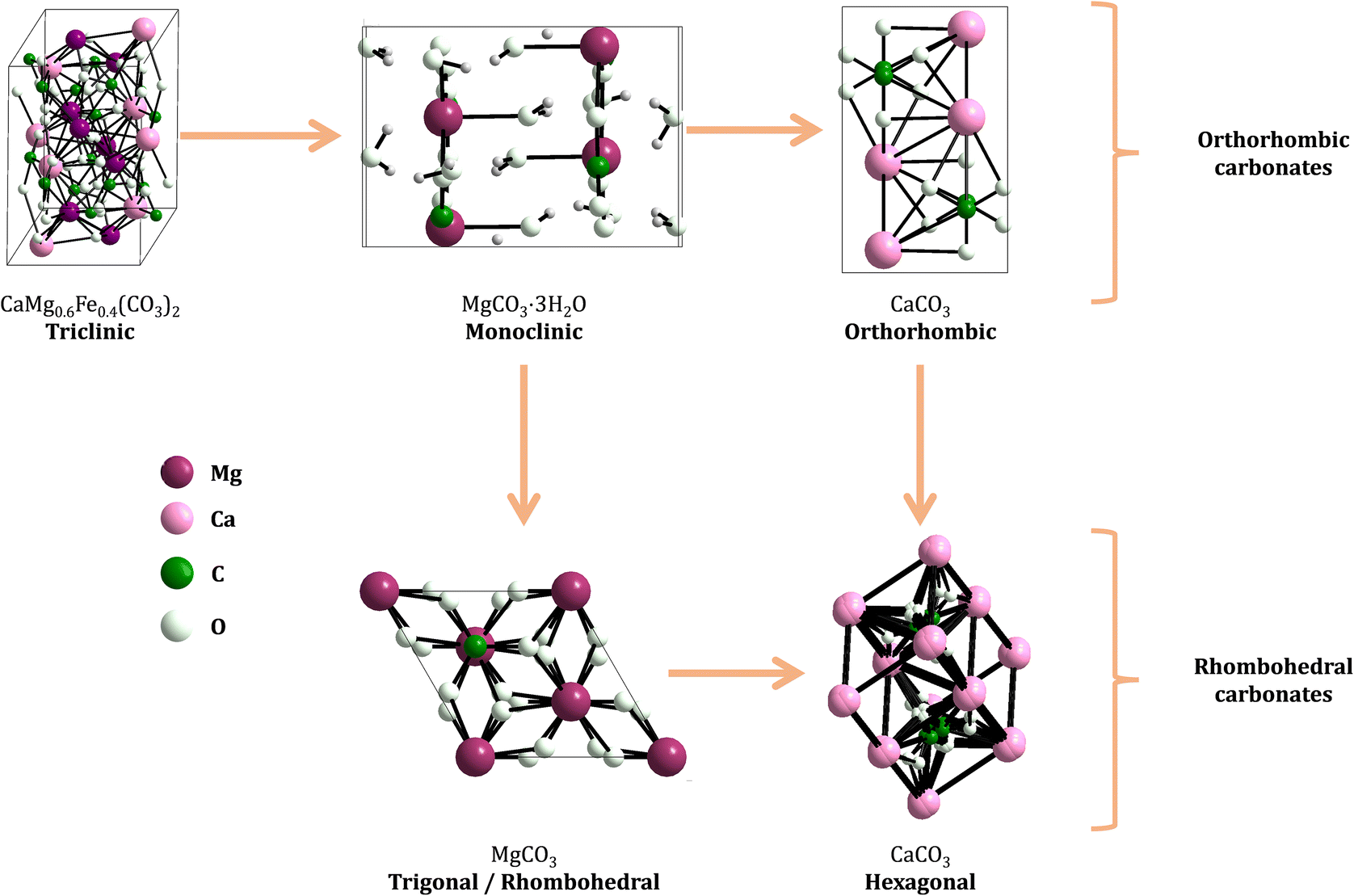 | ||
| Fig. 2 Structural groups of common Ca, Mg carbonate minerals, classified as orthorhombic or rhombohedral classes of carbonates. The frames around the structures represent the unit cell. Crystal structures were drawn with Diamond crystal impact software, version 4.6.4. The crystallographic information (.cif files) were obtained from the PDF4+ software (version 4.22.02, 2022). The utilized PDF cards were the following: triclinic calcium magnesium iron carbonate (04-019-1722), monoclinic nesquehonite (04-015-0663), orthorhombic aragonite (04-008-5421), hexagonal vaterite (00-060-0483), rhombohedral magnesite (04-010-3138). | ||
Fig. 1 (bottom) shows the solubility of minerals from both groups in function of ionic potential and ionic radii.21 Note the higher solubility for minerals in the extremes of the cation radii axis in each structural group, which can be connected to the variable distortion degrees of the octahedra units (Fig. 2). By instance, the rhombohedral carbonates formed with cations of intermediate radii (e.g. siderite, rhodochrosite) have much lower solubility than the ones formed with smaller cations (e.g. magnesite) or larger cations (e.g. calcite).19
While the formation of most simple carbonates can be explained via evaporative concentration mechanisms, the Ca/Mg carbonates have been proven to be formed via different geochemical routes. The precipitation of Ca and Mg carbonate minerals is accepted to be enabled by supersaturation conditions, being mainly explained by the ionic activities in solution when the precipitation occurs, and the kinetic effects inhibiting/favoring the precipitation. The ionic species influencing the precipitation of carbonate minerals are mainly Ca2+(aq), Mg2+(aq) and CO32−(aq) although the net charge-balance and nature of other ions also affect the precipitation mechanisms. The precipitation likelihood of a given mineral carbonate depends on the ionic activity of the solution which determines the respective solubility constant of the precipitate (eqn (1)).18
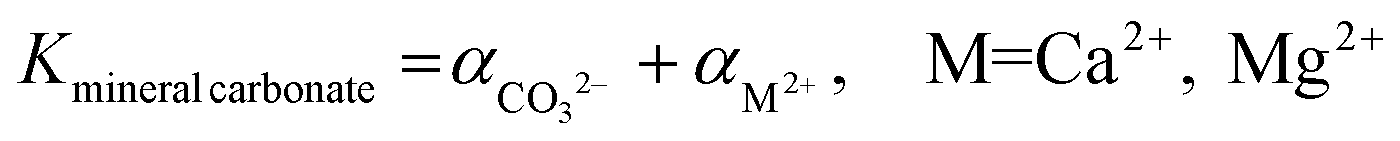 | (1) |
Calcite is the easier mineral carbonate to precipitate due to its lower solubility at 25 °C (Kcalcite = 10−8.48, Karagonite = 10−8.34), and dolomite the most difficult one, being nearly insoluble (Kdolomite = 10−17.2).18 However, the mechanism does not explain what inhibits magnesite formation despite the uncertainty about its solubility product constant (reported values range from 10−7.52 to 10−10.3).23 Therefore, the evaporative concentration mechanism cannot explain the precipitation of anhydrous Mg carbonates, and kinetic considerations are needed to justify the formation of these minerals.24,25
Dolomite is a special and intriguing case among carbonate minerals. It has been found in almost all sediments and rocks from the Archaean to the Holocene ages (Precambrian to Cenozoic Eras) and in different sedimentary conditions at Earth's surface, being inferred to precipitate during burial process in sedimentary basins. Dolomitizing fluids are speculated to occur in sea water (in the usual concentrations of seawater and in supersaturated conditions in regions where evaporative process occur), basinal brines and lacustrine waters.11 The natural formation of dolomite is unclear, as it is not well understood if it is formed as a primary product, hydrothermal, metamorphic phase or diagenetic replacement. However, it has been demonstrated that the dolomite formation occurs under sufficient supply of Mg and permeability of the fluid flow.9 The absence of dolomite is associated with the occurrence of sediments rich in calcite and Mg bearing clay minerals, which indicates that in such settings calcite does not undergo isomorphous replacement. Cases of diagenetic replacement have been observed in the replacement of aragonitic tufa by dolomite, explained by the dissolution of aragonite and consequent release of Ca, resulting in fast precipitation mechanisms assisted by microbial mediation, which allows dolomite supersaturation and precipitation at quasi-rhombohedral geometry, yielding protodolomite.18
The nucleation and growth of anhydrous carbonates are yet active research topics involving controversial dilemmas. Empirical investigations in aqueous systems have found carbonation routes via classical crystallization (spontaneous nucleation in supersaturated solutions), heterogeneous nucleation on pre-nucleation sites and subsequent crystal growth via continuous attachment of species on the nuclei, and non-classical crystallization pathways. The latter has been well identified in the formation of calcite, starting with the formation of pre-nucleation clusters from an ionic solution, which then aggregate to form an amorphous bulk phase, and subsequently, are converted to the crystalline bulk phase. However, it is still an open question if the amorphous carbonate nanoparticles do necessarily engage as precursors for the nucleation of anhydrous carbonates.26
It is broadly discussed in the literature that the high dehydration energy of Mg cations may be the main barrier to the dolomitization reactions. In aqueous systems, Mg2+ and Ca2+ are hydrated with different coordination number (6.0 for Mg2+ and ca. 7.3 for Ca2+), consequently Mg2+ has a stronger solvation shell and the removal of its coordinated water groups requires much more energy than Ca2+ (1926 versus 1579 kJ mol−1 for Mg2+ and Ca2+, respectively). Thus, calcium cations are more susceptible to carbonation and there is no formation of Ca/Mg carbonates without assisted dehydration of Mg2+. For instance, at the molar ratio Mg2+/Ca2+ = 4 the formation of Mg free aragonite is favored, and in modern seawater environments (Mg2+/Ca2+ ≅ 5.2) occurs a major precipitation of aragonite over calcite and Ca/Mg carbonates. Naturally, changes in the ionic strength and the presence of catalysts in the precipitating solution can destabilize the hydration shell of Mg2+ and facilitate the formation of Ca–Mg minerals. For example, it has been noticed that increasing salinity decreases the hydration energy of Mg2+, and the presence of negatively charged groups (such as the carboxyl moieties R-COO− of microorganisms) facilitate the growth of Ca–Mg carbonates.10
2.1. Crystallographic structure of the main Ca/Mg carbonate minerals
Calcite has a rhombohedral crystal structure, composed of sandwiched sheets of Ca2+ and CO32− oriented perpendicular to the c-axis, having the carbonate groups oriented opposed to the plane of each successive sheet. The cations are coordinated by six oxygen atoms, belonging to different carbonate groups. Each oxygen is shared between two cations of neighboring layers, originating a symmetric structure of space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. Other carbonate minerals (such as magnesite, siderite, rhodochrosite, etc.) also occur within the same space group (Table 1).11,27 Solid calcite precipitates at sufficient concentrations and activities of Ca2+(aq) and CO32−(aq) for supersaturating calcite. Due to its relatively low solubility, calcite shows easier precipitation than other carbonates, determined by the initial molar ratio [Ca2+]/[HCO3−] in dilute waters.18 When the solutions reach high Mg2+/Ca2+ ratios (>10), aragonite (rather than calcite or dolomite) begins to precipitate. This is attributed to the inhibitory effect of Mg2+ on the calcite precipitation: even if present at high concentrations, Mg2+ has a smaller ionic radii (consequently, wider hydration sphere) than Ca2+; thus, the dehydration of Mg2+ cations for allowing the fixation on the nucleation sites is not kinetically favorable and dolomite does not precipitate.18
c. Other carbonate minerals (such as magnesite, siderite, rhodochrosite, etc.) also occur within the same space group (Table 1).11,27 Solid calcite precipitates at sufficient concentrations and activities of Ca2+(aq) and CO32−(aq) for supersaturating calcite. Due to its relatively low solubility, calcite shows easier precipitation than other carbonates, determined by the initial molar ratio [Ca2+]/[HCO3−] in dilute waters.18 When the solutions reach high Mg2+/Ca2+ ratios (>10), aragonite (rather than calcite or dolomite) begins to precipitate. This is attributed to the inhibitory effect of Mg2+ on the calcite precipitation: even if present at high concentrations, Mg2+ has a smaller ionic radii (consequently, wider hydration sphere) than Ca2+; thus, the dehydration of Mg2+ cations for allowing the fixation on the nucleation sites is not kinetically favorable and dolomite does not precipitate.18
| Mineral | Chemical formula | Geometry/space group |
|---|---|---|
| a There is no crystal structure defined for dypingite up to date. | ||
| Calcite | CaCO3 | Rhombohedral/Rc (167)11 |
| Aragonite | CaCO3 | Orthorhombic/Pmcn (62)27 |
| Vaterite | CaCO3 | Hexagonal/P63/mmc (194)27 |
| Magnesite | MgCO3 | Rhombohedral/Rc (167)11 |
| Siderite | FeCO3 | Rhombohedral/Rc (167)11 |
| Rhodochrosite | MnCO3 | Rhombohedral/Rc (167)11 |
| Smithsonite | ZnCO3 | Rhombohedral/Rc (167)11 |
| Dolomite | CaMg(CO3)2 | Rhombohedral/R (148)11 |
| Huntite | CaMg3(CO3)4 | Rhombohedral/R (148)27 |
| Ankerite | Ca(Fe2+,Mg)(CO3)2 | Rhombohedral/R (148)27 |
| Calcium magnesium iron carbonate | CaMg0.6Fe0.4(CO3)2 | Triclinic/P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2)28 (2)28 |
| Kutnohorite | CaMn2+(CO3)2 | Rhombohedral/R (148)27 |
| Minrecordite | CaZn(CO3)2 | Rhombohedral R (148)27 |
| Norsethite | BaMg(CO3)2 | Trigonal/Rm (166)27 |
| Barringtonite | MgCO3·2H2O | Triclinic/P (2)27 |
| Nesquehonite | MgCO3·3H2O | Monoclinic/P21/c (14)27,29 |
| Lansfordite | MgCO3·5H2O | Monoclinic/P21/c (14)27,29 |
| Artinite | MgCO3·Mg(OH)2·3H2O | Monoclinic/C2/m (12)30 |
| Hydromagnesite | 4MgCO3·Mg(OH)2·4H2O | Monoclinic/P21/c (14)29 |
| Dypingite | 4MgCO3·Mg(OH)2·5H2O | Monoclinica![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 27 |
Dolomite also has a rhombohedral structure, deriving from calcite. Ideally, the dolomite crystal structure is formed by intercalated layers of Ca2+ and Mg2+, separated by CO32− sheets, having equal molar proportions of Mg and Ca, with the chemical formula CaMg(CO3)2. In ordered dolomites, Mg2+ and Ca2+ are segregated into different planes of the crystal structure. The crystal structure is similar to the calcite; however, the oxygen ligands shift towards the Mg2+ plane since the ionic radii of Ca2+ and Mg2+ differ greatly. Moreover, the bond lengths of Ca–O (2.38 Å) and Mg–O (2.08 Å) also differ significantly in the dolomite structure. This alternation of Ca and Mg cations results in the loss of the c-glide plane and diad axes of the calcite structure, and the crystals of ordered dolomite are formed with space group symmetry R (Fig. 3, middle). Consequently, the carbonate groups in the layers of ordered dolomite present a uniform rotation of 6.5° around the three-fold axis, when compared with their spatial arrangement in calcite.9,11,31 The majority of natural dolomites present iron impurities (<2 mol%) and trace amounts of Mn (up to several hundreds of ppm) as isomorphous replacement in the Mg sites. They are called natural ferroan dolomites and ankerites, presenting a composition range up to CaMg0.5Fe0.5(CO3)2; however the replacement of more than 50 mol% of Fe in the Mg sites has been obtained only experimentally.11
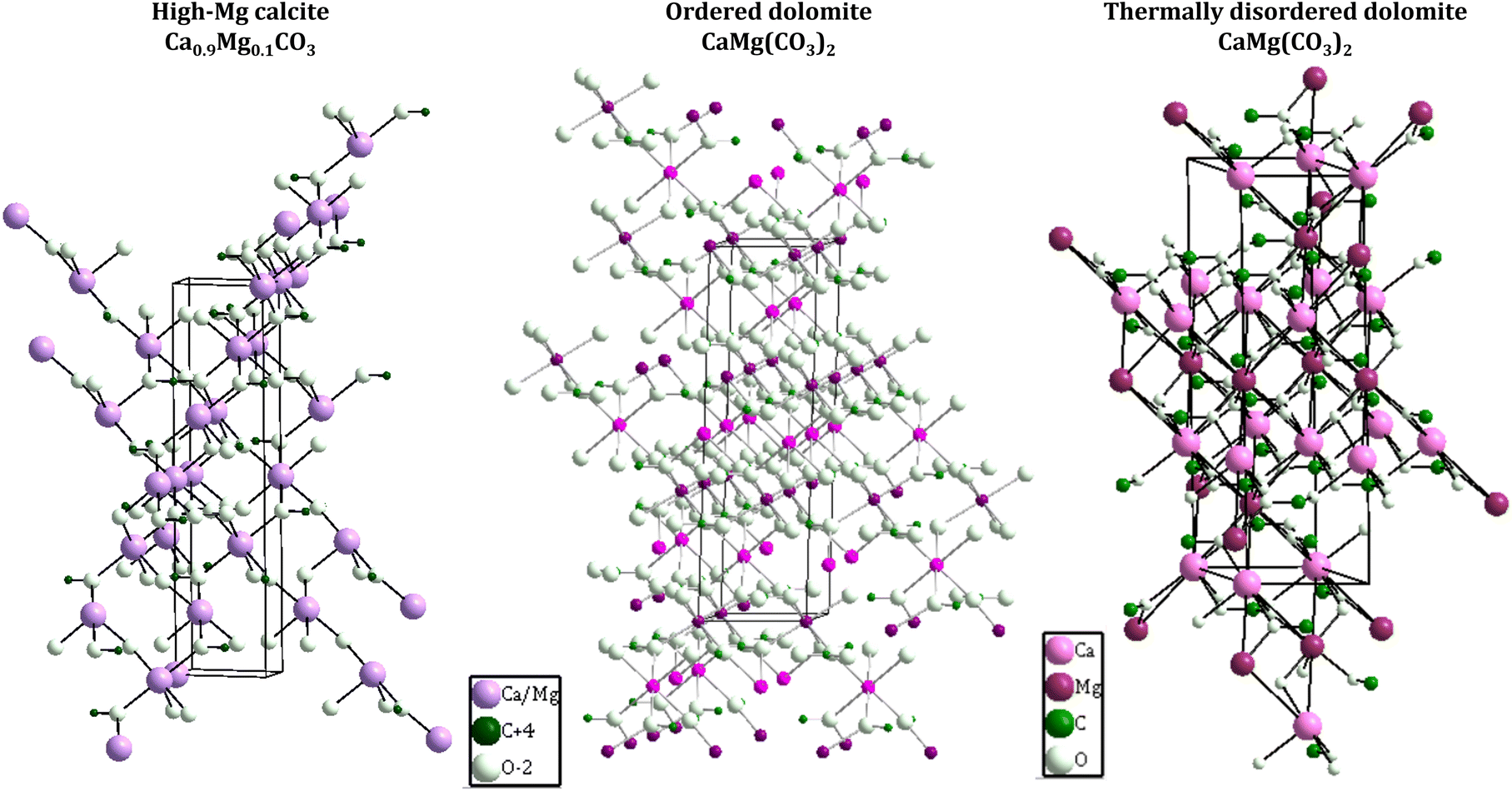 | ||
| Fig. 3 Crystal structure of Mg calcite33 (left), ordered dolomite34 (middle) and thermally disordered dolomite32 (right). Crystal structures were drawn with Diamond crystal impact software, version 4.6.4. The crystallographic information was obtained from the indicated references. The frames around the structures represent the unit cell. | ||
The crystallization of the ordered stoichiometric dolomite (Ca/Mg molar ratio 1:1) requires strict control of the kinetics of nucleation and crystallization, for this reason, synthetic ordered dolomites have been produced only at high temperature conditions via sequential mechanisms of dissolution–reprecipitation. In such experimental conditions, it has been observed that after the induction period, the dolomitization is conducted with relatively fast kinetics, forming first the metastable phases of high Mg calcite (Fig. 3, left) and calcian dolomite as intermediates of the reaction to obtain the stoichiometric ordered dolomite. It has been proposed that the energy barrier to the dolomite precipitation is surpassed by the high temperatures and the presence of seeds acting as nucleation sites, which is contradictory to the natural formation of dolomite that usually occurs at low temperatures (25–60 °C) under surface and shallow subsurface conditions. Sedimentary dolomites are found to be formed in types of sea/fresh waters with high Mg2+/Ca2+ ratios and elevated concentration of HCO3−(aq) and CO32−(aq), usually presenting weakly ordered and calcian structure.31
The poorly ordered phases of dolomites have much higher solubility than ordered dolomite, leading to a consequently higher reactivity. Such reactivity yields continuous structural and compositional changes over time; thus, the natural poorly ordered dolomites are replaced during diagenesis by more ordered and coarser-grained dolomites. For this reason, ancient sedimentary dolomites present composition closer to the ideal 50:50 ratio of stoichiometric dolomites, but still having an excess of Ca2+ up to 2–4 mol% of CaCO3. Since even ancient dolomites failed in building an ideal stoichiometric and ordered structure, dolomites have been classified as metastable mineral.31 Under high temperatures (>200 °C), dolomite becomes thermally disordered due to the expansion of the octahedra units (considerably greater for MgO than the CaO octahedra units), which is responsible for the thermal expansion of the unit-cell. The thermally disordered dolomite (Fig. 3, right) present the rigid-body libration of the carbonate groups with magnitude between that of calcite and magnesite; thus, neither rotation around the three-fold axis nor out-of-plane tilting is dominant.32
Temperature variations in experimental setups for dolomite precipitation led to the formation of different Mg carbonate minerals (Fig. 4). High Mg calcite (calcites containing more than 4 mol% of MgCO3) is formed as stable phase at high pressures (up to 103 bar) and temperatures above 1200 °C, but this is unstable at near-ambient conditions (when compared with dolomite and calcite). High Mg calcite is also formed at temperatures between 700 and 1200 °C, but this is then obtained mixed with dolomite and magnesite phases. The occurrence of ordered dolomite is observed with compositions higher than 43 mol% of MgCO3, at lower temperatures (<700 °C) and pressures (<50 bar). At about 1200 °C a solid solution of calcite and high Mg calcite with dolomite stoichiometry is formed. No solid solutions are observed at temperatures below than 1075 °C, when high Mg calcite exsolve into dolomite and calcite.11
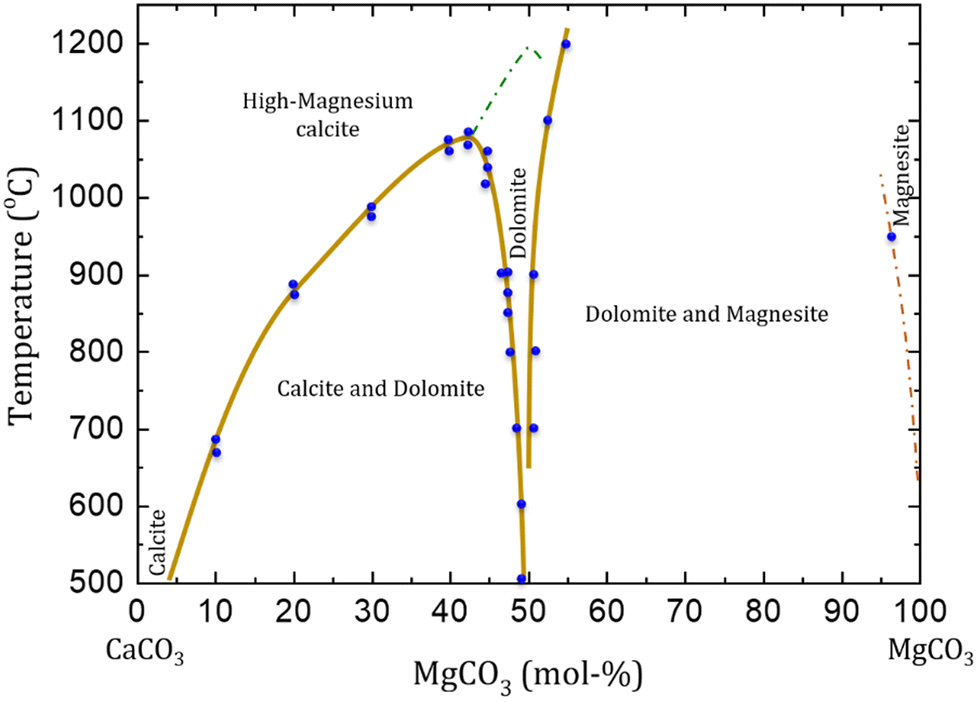 | ||
| Fig. 4 Phase diagram of CaCO3–MgCO3 system at high temperatures (above 500 °C) and pressures up to 10 kbar. The green dashed line indicates the approximate transition from calcite (Rc) to dolomite (R) symmetry. The diagram does not consider the transitions between calcite and aragonite, neither calcite I to calcite II that occur at high pressure. Variations in pressure is also not considered (reformulated from Gregg et al., 2015).11 | ||
When rhombohedral carbonates of Ca and Mg are formed at near-dolomite stoichiometry (ca. 40 to 50 mol%) with weakly or incompletely ordered structure, it is assumed that ordered dolomite would be precipitated by re-establishing the equilibrium (temperature, pressure, overhead gas phase). Therefore, even carbonate minerals with chemical composition very close to dolomite are not considered dolomite if there is no evidence of cation ordering. Those metastable minerals are referred as protodolomite, pseudo-dolomite or high/very high Mg calcite.11 Ordered dolomites that contain excess of Ca2+ are defined as calcian-dolomites, having a lower stability in marine environments than stoichiometric dolomites.11
High Mg calcite and very high Mg calcite have been successfully synthesized at ambient pressures and low temperatures (25 to 80 °C), being observed that the contents of Mg2+ (ranging from 22 to 58 mol% of MgCO3) can be controlled by varying the Mg2+/Ca2+ ratio and the salinity of the solutions. It has been speculated that organic compounds may facilitate the precipitation of very high magnesium calcite via the induced reduction of the dielectric constant of the solution, which decreases the hydration of Mg cations; thus, facilitating the Mg incorporation into the carbonate mineral. For instance, it has been demonstrated that the presence of dioxane and polysaccharides enhances the incorporation of Mg2+ into the precipitated carbonate mineral. The presence of sulphide species in the growth solution also showed a similar effect.11
Dolomites synthesized at low temperatures (below 100 °C) via isomorphous replacement in calcite or aragonite have extremely slow kinetics, and it is assumed to occur through the conversion of several intermediate disordered phases, resulting on the formation of calcite, high Mg calcite, very high Mg calcite and hydrated and hydroxylated Ca/Mg phases. Several experiments utilizing calcite and aragonite as reagents and temperatures above 150 °C have demonstrated the formation of intermediate metastable phases (disordered high or very high Mg calcite) before the formation of ordered dolomite. The reaction progresses until nearly total replacement of the calcium carbonate reagents, with continuous shift towards dolomite stoichiometry (50 mol% MgCO3) and increasing ordering of the crystal structure. It was also noticed that increasing the Mg2+/Ca2+ ratio favors the formation of phases with 1:1 stoichiometry close to dolomite but does not induce the needed cation ordering for precipitating ordered dolomite. It has been reported that very high Mg carbonates have preferential nucleation on aragonite rather than on calcite, if both phases are present.11
These structural differences indicate the current need of broadening the understanding on the chemical and physical properties of the Ca/Mg carbonates polymorphs. All metastable polymorphs have a possible role as intermediate estate in the formation of the more thermodynamically stable phase, and therefore, understanding well the properties of the metastable carbonate phases may be the key for proposing viable mechanisms of dolomite and magnesite precipitation.
3. Dolomite problem
Dolomite is a metastable mineral, and as such it undergoes different types of isomorphous replacement during the burial and metamorphism stages due to continuous dissolution and re-precipitation processes. Such processes of re-equilibrium without total pore cementation results in high inter-crystalline porosity in the dolomite crystals, which explains the efficient drainage and fluid storage capacity of dolomite reservoirs. However, there are yet many knowledge gaps about the mechanisms of dolomite formation, being not well understood the impacts of kinetics versus parity in dolomite precipitation.9,35,36 While explaining the evolution of dolomite sediments, the main challenging geochemical questions are: (i) Parity: is the amount of formed dolomite related to the composition of the seawater or does the precipitation/dissolution of dolomite affect the seawater composition? (ii) Why do the sediments of modern dolomite occur in much smaller volumes than ancient dolomites? (iii) How do the volume of dolomite sediments and sequential isomorphous replacements evolved over time? and (iv) What has been the role of bacterial mediation?9Dolomite often precipitates in lacustrine waters despite the small concentrations of Mg2+, and modern dolomite formation are found in lakes with high Mg2+/Ca2+ ratio (>10) and elevated carbonate alkalinity concentration (>5 × 10−3 g L−1). However, experimental observations supported by the geological timescales indicate that the dolomite is ideally precipitated in solutions with Mg2+/Ca2+ ratios ranging from 2 to 7.18 Several studies have been conducted attempting to explain the conditions of dolomite saturation at earth-surface conditions, but no synthetic dolomite has been obtained at such low temperatures and pressures. Some investigations have demonstrated that temperature control is essential for dolomite precipitation, which is achieved easier at high temperatures. Nevertheless, the degree of supersaturation also plays important role in the precipitation mechanism.11,18
Nowadays, it is broadly accepted that most of ancient dolomites have been formed in a process of diagenetic replacement of calcite following two possibilities of reactions as indicated in the eqn (2) and (3). This hypothesis is based on geological observations and on many synthetic experiments that demonstrate the difficulties of obtaining dolomite at Earth surface's temperatures, whereas synthesis at elevated temperatures conditions (>200 °C) leads to fast and easy formation of ordered dolomite. Moreover, is has been demonstrated that thermal dolomitization proceeds with the formation of intermediate metastable phases of calcite or aragonite (very high-Mg calcite), which converts to ordered dolomite at temperatures higher than 150 °C.10
| 2CaCO3 + Mg2+ → CaMg(CO3)2 + Ca2+ | (2) |
| CaCO3 + Mg2+ + CO32− → CaMg(CO3)2 | (3) |
Contradicting that hypothesis, modern dolomites are often found in evaporitic environments which has been demonstrated (via geological tracking) to have a history of low temperatures and saturated solution conditions; thus, leading to the assumption that dolomite had precipitated directly from those solutions as low-temperature primary dolomite (eqn (4)). Those dolomites are typically found in the upper sediments as very high Mg calcite or calcian dolomite, while more stoichiometric dolomites are the major phases in the deeper layers. That suggests that dolomite undergoes several steps of reprecipitation and recrystallization upon burial diagenesis. In such environments several factors (supersaturated conditions, high Mg/Ca ratio, high salinity, the effect of the biotic and abiotic environments) may have acted as catalysts, helping to overcome the kinetics barriers for dolomitization.10
| Ca2+ + Mg2+ + 2CO32− → CaMg(CO3)2 | (4) |
Other proposed models consider the effects of kinetic inhibitors of dolomitization. Experimental investigations have shown that sulphate ions retard the dolomite precipitation in modern sedimentary environments, implying that dolomite would precipitate only from solutions with sulphate concentrations much lower than in seawater. Further speculations suggest that sulphate-reducing bacteria have assisted the precipitation of dolomite by lowering the sulphate concentration in the saline brines and sea water. However, several modern dolomites have been found to form from brines at high sulphate concentrations, and microbial mediation cannot explain the geologic occurrences of many ancient sedimentary dolomite.31 Thus, these findings associate important broad questions on the dolomite problem: which catalyst existed in ancient ocean waters, which is no longer abundant in modern oceans? How did it promote such non-uniform stratigraphic distribution of dolomite? Why does modern dolomite not precipitate despite the supersaturation with relation to dolomite in seawater.25
3.1. Insights on the mechanisms of dolomite precipitation
The main discussions in the literature related to the “dolomite problem” is to determine whether natural dolomite crystallizes directly from aqueous solutions under certain conditions, or if the formation of crystalline and/or amorphous pre-nucleation clusters are involved in the mechanism.37 The precipitation of ordered dolomite is often described by Ostwald's step rule, which postulates that mineralogical reaction sequences have the intermediate products with Gibbs free energy closer to one of the precursors rather than the final stable phase. Therefore, the precipitation of the final product is facilitated by the formation of sequential intermediate phases, each phase having lower solubility than the precursor intermediate.11 This agrees with the experimental observations in dolomitization reactions where the solubility of the intermediate products decreases until the formation of ordered dolomite, as shown in Fig. 5.8 The role of cationic and anionic additives on the dolomite precipitation are still to be elucidated, but most proposed mechanisms consider a catalytic role for the ionic species. By instance, Vandeginste et al. (2019) have demonstrated that the presence of Zn2+ cations in solution have a catalytic effect on the dolomite precipitation. It was proposed that the higher dehydration energy of the Zn2+ cations facilitates the dehydration of the Mg2+ cations and the subsequent incorporation into the protodolomite structure.38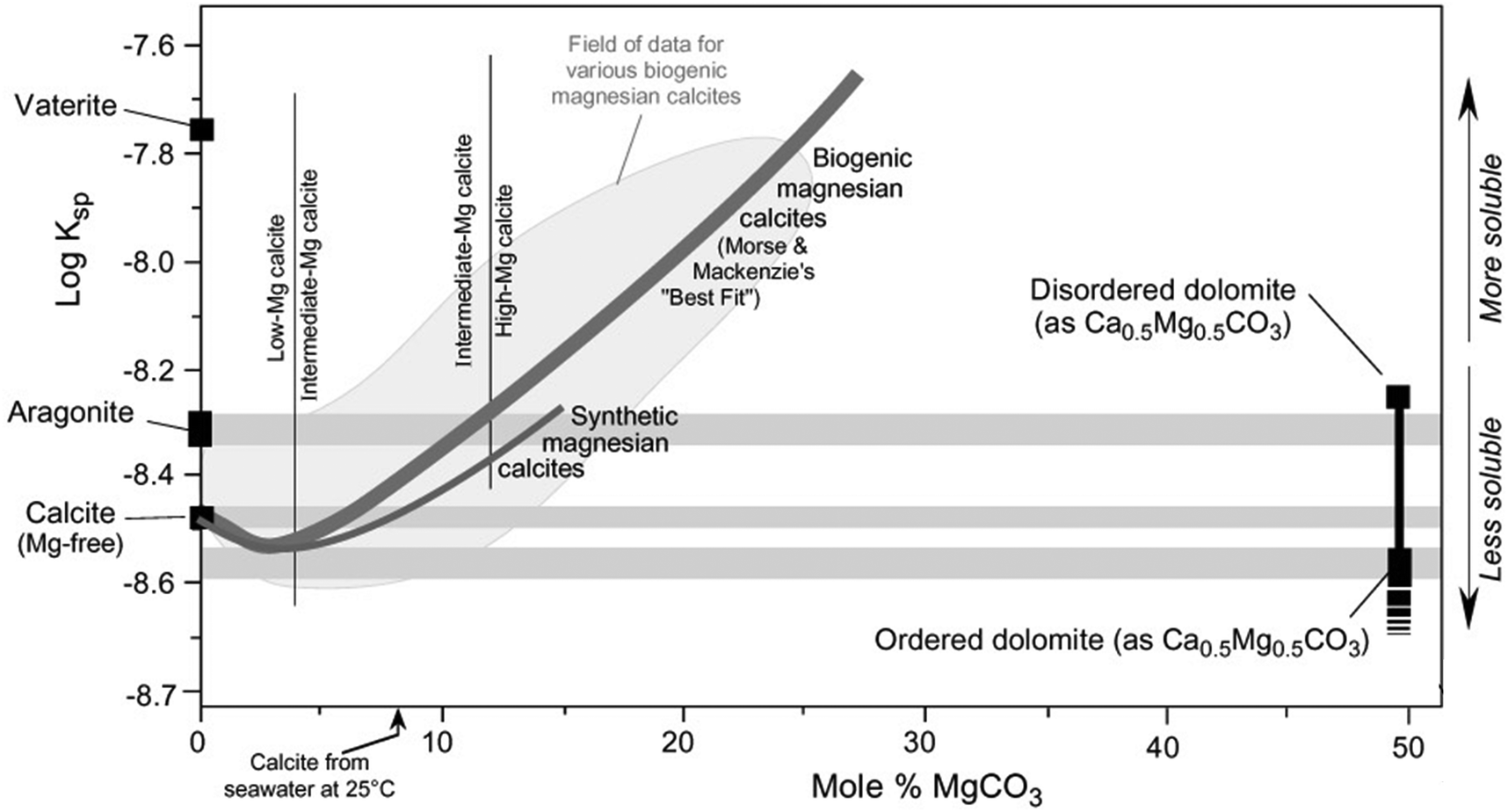 | ||
| Fig. 5 Solubility of Mg bearing carbonates as a function of the concentration of MgCO3, being the remaining CaCO3 (reproduced with the author's permission, © L. Bruce Railsback, Department of Geology, University of Georgia, Athens, Georgia 30602-2501 USA).8 | ||
Other proposed mechanisms are related to fluctuations of CO32−(aq) concentrations, which vary as function of pH (dependent on the dissociation of carbonic acid) and pCO2. Thus, the influence of biotic processes on the formation of Mg carbonates can be related to changes in CO2 levels due to the respiration and/or photosynthesis of aquatic organisms.18 The catalytic effects of biotic activity on the precipitation of carbonates are clearly seen in tufas, which are constituted of carbonates formed at low temperatures, and living organisms such as microbes, macrophytes and animals. The tufa precipitation is associated with the degassing of CO2, which elevates the pH and leads to supersaturation conditions. It has been demonstrated that the living organisms in tufas work as the alkaline engine for the precipitation conditions, acting also as nucleation sites due to the presence of extracellular polymeric substances (EPS).18
The microbial mediation for dolomitization has been speculated as successful route for synthesizing ordered dolomites at room temperature. The first work claiming this achievement was published in 1995 by Vasconcelos et al. when the authors reported the synthesis of ordered dolomite assisted by sulphate-reducing bacteria from the Desulfovibrio group,39 and following researches had utilized similar routes with variable means of microbial mediation (methanogenic archaea, fermenting bacteria and several aerobic halophiles). In those studies, it has been proposed that those microorganisms act as catalysts to dolomitization by increasing the dolomite saturation levels and acting as nucleation templates. However, it has been proven that none of those works had indeed succeed at obtaining synthetic ordered dolomite. A detailed criticism in that direction has been presented by Gregg et al. (2015): instead of dolomite, all the reports of such microbial mediation methods have shown XRD patterns compatible to very high Mg calcite.11 That is easily identified by comparing the intensity of the of the 100 and 015 X-ray reflections of dolomite: ordered, stoichiometric dolomite presents those reflections with nearly equal intensity in the XRD patterns. Since all the reports on microbial mediation methods failed in fulfilling this parameter in the presented XRD characterization, the synthesis of ordered dolomite at low temperatures remains as a research challenge up to date. Nevertheless, understanding the mechanisms of very high Mg calcite precipitation has brought valuable contributions to the knowledge on the mechanisms of dolomitization, since high Mg calcite is well accepted as intermediate for the formation of ordered dolomite.10,11
A comprehensive review on the investigations of microbially catalyzed dolomite formation has been provided by Petrash et al. (2017).25 The studies of such systems have shown that the initial building blocks of dolomite are clusters, being hypothesized that carboxyl groups present in the microorganisms act as template for the clusters formation. The proposed mechanism (Fig. 6) considers the direct effect of the carboxyl moieties (–COO−) found in the polysaccharide constituents of those microorganisms which lowers the energy requirements for dehydrating the Mg2+ cations while also serving as templates for the nucleation and growth of the Mg carbonates.25,40 The carboxyl groups play important role on overcoming the elevated enthalpy of the double hydration shell of [Mg(H2O)6]2+ cations (ΔH° = 1931 kJ mol−1)41 by dehydrating and binding of [Mg(H2O)6]2+ which has favourable enthalpy of formation ([Mg(H2O)6]2+ + R-CO2− → [Mg(H2O)5(R-CO2)]+ + H2O, ΔHf = −835.96 kJ mol−1). That is followed by the carbonation reaction through replacement of water molecules with CO32−(aq), creating MgCO3(H2O)4(R-CO2) as second intermediate complex which may act as template for clusters growth via ion attachment.25,42,43
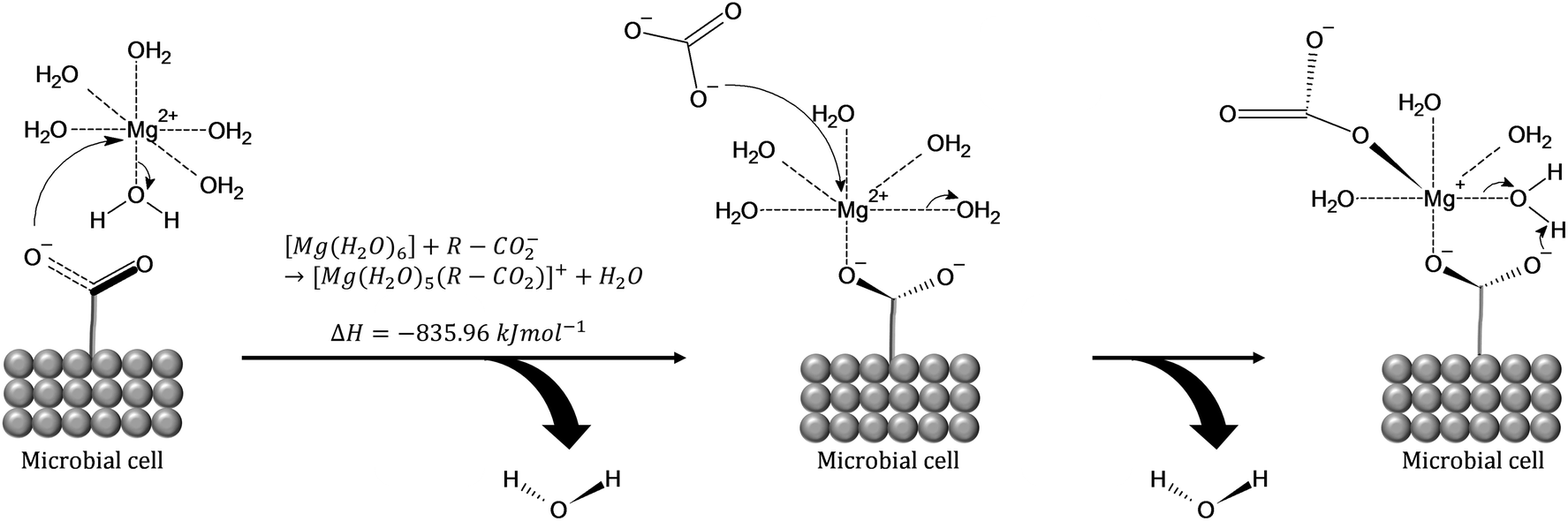 | ||
| Fig. 6 (1) [Mg(H2O)6]2+ attracted to carboxyl group on a microbial cell wall. (2) The complex attaches to the carboxyl group and ejects one hydration water in an energetically favorable reaction (inserted equation). The new [Mg (H2O)5(R-CO2)]+ complex carries a residual positive charge that is satisfied by the inclusion of aqueous carbonate (CO32−) or bicarbonate (HCO32−).42 Drawn with the ACD (ChemSketch (freeware) v. 2018.1.1). | ||
Petrographic and mineralogical studies have inferred that Mg bearing clay minerals have acted as abiotic catalysts for dolomite precipitation, proposing a direct relation between the clay minerals and the formation of abiotic dolomite (usually found in dolostones, cave speleothems and soils). It has been suggested that clay minerals play an important role as Mg source for dolomite precipitation and/or nucleation sites for the dolomite crystals. Nevertheless, such a relationship have not been observed in many sediments were both minerals are present.10 As a matter of fact, it has been observed that the dolomite authigenesis in many sedimentary settings indicates the growth of dolomite crystals in pore spaces around detrital clay particles.10,44
Publications dating from the 1960s have proposed that clay minerals may have influenced the natural formation of dolomites by attracting the population of cations to the surroundings of the clays via electrostatic interactions. This increases the concentration of Mg2+ in that region and facilitates the precipitation of high or very high Mg calcite. It was also speculated that the ion exchange properties of the clay minerals may have altered the concentration in the precipitating solution via absorption of Mg2+ and release of Ca2+, as predicted in the order of preferential cation exchange in clays: Na+ < K+ < Mg2+ < Ca2+. However, the influence of cation exchange would be expected to increase the rates of dolomitization towards the margins of the basin since the pore fluids in early stages of diagenesis have a usual movement in an upward direction. Therefore, the contribution of ion exchange processes in clay minerals for dolomite precipitation was disregarded as the predicted effects would oppose to the practical observations in dolomite sedimentary settings.44
Nevertheless, recent experimental findings propose that the negatively charged layers of the clay minerals act as nucleation templates for very high Mg calcite, similarly to the microbial mediation. Liu et al. (2019) have performed the synthesis of very high Mg calcite via carbonation reactions assisted by clay minerals, utilizing a molar ratio of Mg2+/Ca2+ = 8 in aqueous solution. Three different types of clay minerals, with large differences in surface charge and area (Table 2), have been evaluated in their study: illite, montmorillonite and kaolinite. Since illite and montmorillonite are T:O:T (tetrahedral:octahedral:tetrahedral) type of clays subjected to isomorphous replacement of cations, they present much higher surface charge and area than kaolite (T:O type), which is nearly charge neutral. The results showed that in the absence of clay minerals only aragonite is produced; however, when illite is introduced in the reacting system only dolomite-like material (very high Mg calcite) is formed containing 46.2 mol% of Mg. In presence of montmorillonite a very high Mg calcite with traces of aragonite was obtained whereas the kaolinite-assisted reactions produced monohydrocalcite. These results indicate that the surface charge of the clays is proportional to their catalytic effect on the precipitation of very high Mg calcite, which has been better facilitated at the highest charge density. The authors also noticed in the SEM images that the crystals of very high Mg calcite were precipitated only on the edges of the clay structure, which indicates that the hydroxyl groups present in the clay edges have been the catalytic sites for dolomitization rather than the basal surfaces. Also, a direct relation was noticed between the morphology of the precipitated phases and the negative surface charges of the clay employed as catalyst, which is similar to the morphological observation in microbially assisted experiments.10
| Clay mineral | BET surface area (m2 g−1) | Zeta potential (mV) | Products of the carbonation reactions |
|---|---|---|---|
| Illite | 20.5 | −41.99 | Very high Mg calcite |
| Montmorillonite | 22.7 | −34.51 | Very high Mg calcite and traces of aragonite |
| Kaolinite | 13.1 | −3.93 | Monohydrocalcite |
The dolomitization reactions in presence of microorganisms or negatively charges species have been explained based in two possible types of mechanisms (Fig. 7): metal-chelation or adsorption–displacement. The metal-chelation mechanism postulates that the negatively charged groups bind with the magnesium cations, decreasing its hydration sphere and generating Mg2+ complexes that requires much lower energy for effective carbonation. On the other hand, the adsorption–displacement mechanism proposes that the catalytic species are absorbed on the growing Ca–Mg carbonate, inducing the replacement of the water molecules coordinated to Mg2+, and promoting the diffusion of Mg2+ in the crystal structure. Although the adsorption–displacement mechanism may be feasible for microbially-assisted reactions, it cannot explain the catalytic effects of clay minerals, which are more compatible with the metal-chelation mechanism. Therefore, the presence of the clay minerals is speculated to promote the formation of Ca and Mg hydroxyl complexes on the clay's edges, which not only helps decreasing the energy barriers for shedding the hydration spheres of the cations, but also acts as nucleation centers for the carbonation and growth of the dolomite like structure.10
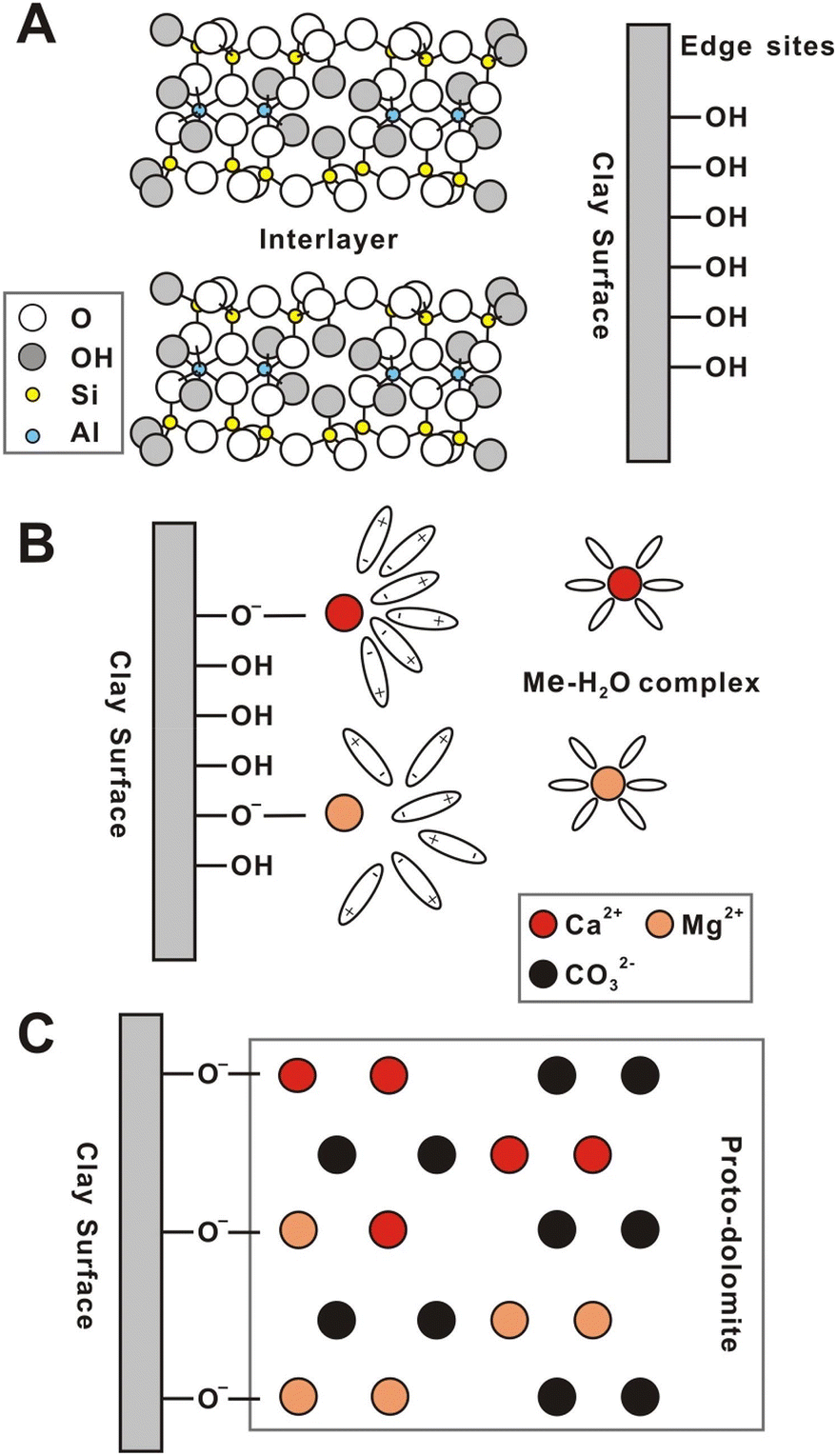 | ||
| Fig. 7 Schematic diagram of the proposed mechanism of the catalytic role of negatively charged clay minerals on dolomite precipitation. (A) Structure of a T:O:T type of clay mineral (e.g., illite and montmorillonite); (B) the adsorption and dehydration of Mg and Ca ions by hydroxyl groups; (C) the formation of Mg/Ca hydroxyl complexes favoring the carbonation reaction. Reproduced from Liu et al. (2019), with the publisher's permission.10 © 2018 Elsevier Ltd. All rights reserved. | ||
Some studies have also perceived a catalytic effect on the dolomite precipitation in low dielectric constant solvents. The room temperature precipitation of disordered dolomite and high Mg calcite has been achieved by partially replacing water with ethanol. Reacting systems with 75 vol% of ethanol, 50 mM of MgCl2·6H2O, 10 mM of CaCl2·H2O, and 50 mM of NaHCO3 yielded protodolomite with close stoichiometry (44 mol% of Mg) to the ordered dolomite at 25 °C. It was suggested that the lower dielectric constant of the medium (compared to pure water) assisted the dehydration of the Mg2+ cations, inducing a homogeneous classical nucleation in bulk solution (not on substrate surfaces), which do not exclude the formation of amorphous and intermediate crystalline phases. The results agreed with molecular dynamic calculations, which predicted a needed reduction of 2.9–4.6 kJ mol−1 in the energy barrier for dehydrating Mg2+ cation. Thus, it was argued that the 75 vol% ethanol solution decreased the solvation energy in 4.2–8.4 kJ mol−1, thus assisting the dolomitization reactions.24
The smaller growth rate of dolomite and magnesite when compared to calcite is often related to the slower water exchange rate and higher dehydration enthalpies for Mg cations compared with Ca cations (Kexchange = 5 × 105 and 6–9 × 108 s−1 and ΔH = 1922.1 and 1592.4 kJ mol−1 for Mg2+ and Ca2+ ions at room temperature, respectively).45 In evaporatively concentrated lakes, the precipitation of calcite leads to depletion of Ca2+, and consequent raise of the Mg2+/Ca2+ ratio followed by the precipitation of Mg minerals. In the presence of sulphate ions, the type of the Mg mineral to be formed will vary according to the kinetics of the calcite precipitation: the fast precipitation of calcite is followed by the precipitation of Mg carbonates; however, the slow precipitation of calcite extends the residence time of the Mg cations in solution favoring the nucleation of Mg sulphates minerals (Fig. 8). Therefore, the kinetics of calcite formation have direct effect on the type of Mg mineral to be formed in sediments, and the final molar ratios of [Mg2+(aq)]/[SO42−(aq)] and [SO42−(aq)]/[CO32−(aq)] in the residual fluid.18
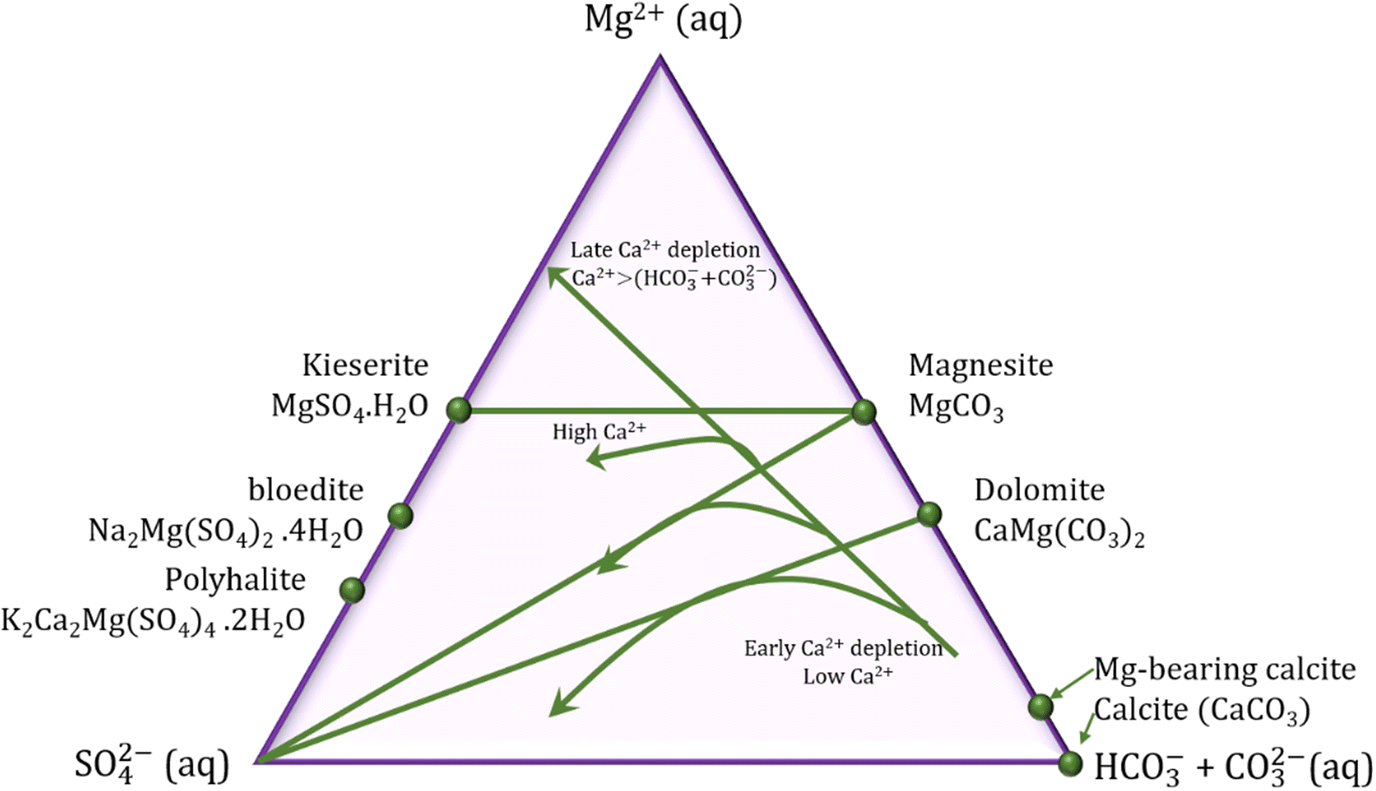 | ||
| Fig. 8 Ternary diagram in function of the molar ratios of solutes for precipitation of carbonate minerals. The calcite precipitation leads to Ca2+ depletion, which may favor the formation of dolomite or magnesium sulphate, depending on the kinetics of calcite precipitation (reformulated from Deocampo, 2010).18 | ||
Keller et al. have investigated the carbonation reaction of Ca(NO3)2 and MgSO4 with Na2CO3 at 81 °C for 1–23 days. The authors obtained a dolomite-like material with isomorphous replacement of carbonate groups, containing sulphate anions in the crystal structure (Ca1.056Mg0.944(SO4)0.035(CO3)1.965·0.26H2O) and, through microscope characterization techniques and XRD, they proposed a hydrated protodolomite phase as a precursor for dolomite formation via an Ostwald ripening model. It was suggested that the presence of sulphate increases the rate of nucleation but decreases the rate of crystal growth. The authors also observed that the precipitated materials displayed water losses (absorbed and coordinated) until 200 °C.46
Previous studies have also demonstrated that the growth of calcite crystals in presence of sulphate ions results in the formation of calcite polymorphs due to sulphate incorporation into the crystal structure, which disturbs the growth pattern of the crystal resulting in phases of lower solubility. As a matter of fact, molecular dynamic simulations have calculated stronger binding energy for calcite than for its polymorphs (aragonite and vaterite), indicating higher probability of selective preferential accumulation of sulphate in calcite rather than in the other polymorphs. Further experimental studies have shown that continuous increments in sulphate concentrations (from 500 to 4000 mg L−1) results in changes in the crystal habit of calcite, and in the stabilization of vaterite prior to aragonite phase. It also has been observed that mixed polymorph crystals of vaterite/calcite are formed at elevated sulphate concentrations (4000 mg L−1), but the sulphate incorporation is higher in the calcite crystals and can be magnified with increasing the salinity of the reacting system.47
3.2. Kinetics of dolomite precipitation
The unfavorable kinetics of dolomite precipitation at ambient temperatures and pressures has been associated to four main factors: (i) the high dehydration energy of Mg2+ cations, (ii) the energy barrier for long range ordering of the magnesium and carbonate ions in the crystalline structure, (iii) lattice stress build up correlated to the impurity incorporation model, and (iv) the self-limiting growth model.38 The challenges of synthesizing dolomite under temperatures and pressures close to (or at) ambient conditions are related to the kinetic barriers to the nucleation and growth, which may be associated to the ordered crystal structure of the mineral. Consequently, the thermodynamic data obtained for dolomite are mainly extracted from high-temperature (>100 °C) experimental settings, which are then extrapolated to lower temperatures. It also has been reported that the kinetics of dolomitization can be accelerated by increasing the surface area and solubility of the calcium carbonate precursors.11High-temperature synthesis of dolomite requires a long induction period (20 to >80% of the time needed for dolomitizing calcite or aragonite), followed by fast isomorphous replacement in the calcium carbonates and producing disordered phases of very high or high Mg calcite. The duration of the induction period has direct effect on the overall rate of dolomitization.11 In a work published by Malone et al. (1996)48 the maximum of protodolomite formation was obtained in 286 hours at 200 °C, while 336 days at 50 °C yielded the same results. These experiments were recrystallizations of Ca and Mg carbonates solutions conducted in conditions similar to seawater with 41.7 mol% of MgCO3. The authors also noticed the influence of surface area on the dolomitization. Thus, the crystallization was proposed to be influenced primarily by: (i) temperature, (ii) reacting mineralogy, (iii) solutions/environment chemistry and (iv) reacting surface area.48,49
The increasing of dolomitization rates have been correlated also with the pH of the medium (either high alkalinity or high acidity), high concentrations of Ca2+ and Mg2+, and high fluid to rock proportions. Some studies have shown that the kinetics of the dolomitization reaction can be accelerated by applying high Mg2+/Ca2+ molar ratios in the fluid, with a non-linear relation between the decrease of the induction time and the increment of the Mg2+/Ca2+ ratio. For example, solutions at Mg2+/Ca2+ ratios above 0.7 shows initial precipitation of very high Mg calcite within tens of hours, while below the 0.6 ratio the first precipitates are noted after hundreds of hours.11 The kinetics rates increase continuously until Mg2+/Ca2+ ratio of ca. 5, when the dolomitization rates starts to decrease. Some studies suggest that at this point the crystalline structure of dolomite is inhibited by the high magnesium content.50
The experimental observations reported by Higgins and Hu (2005) have underlined the kinetics of dolomite formation in function of a layered growth, investigated with in situ atomic force microscopy. The authors employed experimental settings with a broad range of Ca2+/Mg2+ activity ratios (from 0.01 to 4), employing solutions with approximately constant supersaturation conditions and ionic strength (0.1 M NaCl). It was noticed that the kinetics’ growth of the first single layer of dolomite is relative fast (0.1 ± 0.02 nm s−1) followed by a considerable slower growth of the second layer (ca. 50 times slower). The kinetics related to the growth of the first layer were found to be independent from the Ca2+/Mg2+ ratios, presenting similar velocity ratios for all molar ratios utilized by the authors. This may suggest that the formed initial layer is self-limiting and inhibits the further layer-by-layer growth. The structure and/or composition of the first layer varied according to the Ca2+/Mg2+ ratios, indicating that the dolomite (101) surface may act as nucleation template for a broad range of Ca–Mg carbonates.13
The kinetics constants of dolomite precipitation have been found to range from 10−22 to 10−16 mol cm2 s−1,51,52 and it was estimated that dolomite precipitation rate (at 25 °C) occurs within 3.98 × 10−12 cm per year (with a constant value of 10−22 mol cm2 s−1, i.e. slow kinetics) while it increases to 1.01 × 10−7 cm per year with the constant value of 10−16 mol cm2 s−1 (fast kinetics).51,53 Relevant insights regarding dolomite precipitation have been published by Gautelier et al. (2007). In this work the authors have found that the dolomite dissolution mechanism, could be described according with eqn (5).54
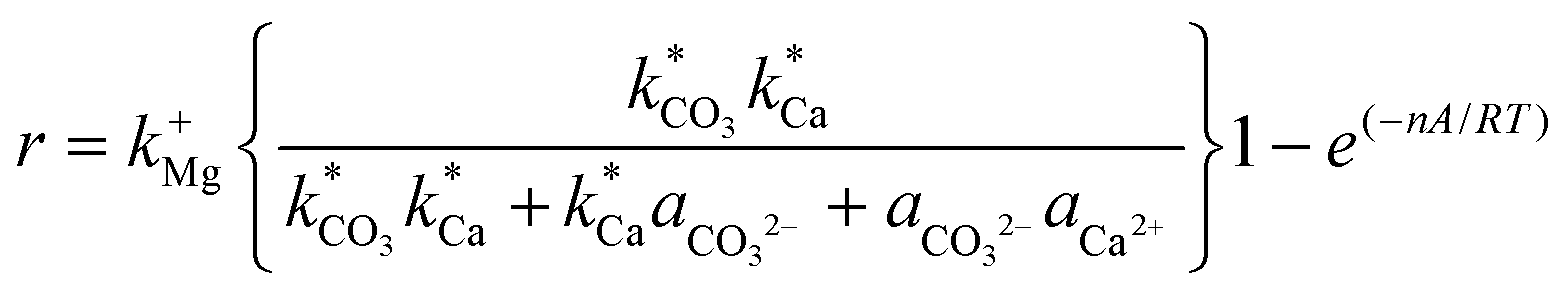 | (5) |
Where r represents the overall BET surface area considering a normalized dolomite dissolution rate at both near and far-from-equilibrium conditions, k+Mg has a rate constant equal to 4.0 × 10−12 mol cm−2 s−1, 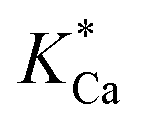 and
and 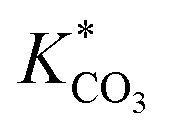 attribute equilibrium constants of 3.5 × 10−5 and 4.5 × 10−5, respectively, ai considers the activity of the subscripted aqueous species, A is the chemical affinity of the dissolving dolomite, R is the gas constant, T defines absolute temperature and n designates a stoichiometric coefficient of 1.9.54
attribute equilibrium constants of 3.5 × 10−5 and 4.5 × 10−5, respectively, ai considers the activity of the subscripted aqueous species, A is the chemical affinity of the dissolving dolomite, R is the gas constant, T defines absolute temperature and n designates a stoichiometric coefficient of 1.9.54
In an attempt to explain the kinetics rates of dolomite crystal growth, Arvidson and Mackenzie (1999) have performed a series of experiments in a circulating seeded reactor, employing temperatures ranging from 100 to 200 °C. The obtained results were applied to the parameters of a parabolic rate law following the eqn (6):36
| r = k(Ω − 1)n | (6) |
where, Ω represents the saturation index of ideal dolomite in the solution 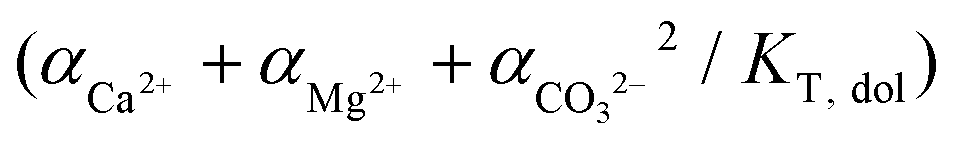 , n is the reaction order and rate constant applied followed the Arrhenius law
, n is the reaction order and rate constant applied followed the Arrhenius law 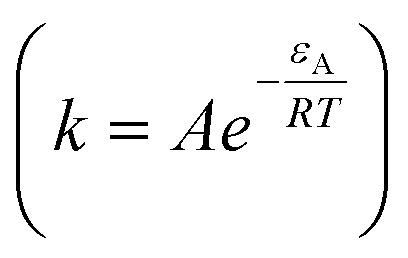 , where εA is the activation energy and A the pre-exponential term. Thus, the expansion of eqn (6) is shown in eqn (7):36
, where εA is the activation energy and A the pre-exponential term. Thus, the expansion of eqn (6) is shown in eqn (7):36
 | (7) |
The precipitation rate of dolomite at a given temperature and solution concentration were calculated via prior resolution of the free parameters in eqn (7) (n, εA, A). The values of the parameters A and n were extracted from previous reports were the authors used the data of existing experimental reports to build a kinetic model, and the selective fitting of rate data implied an overall reaction order (n) of 2.26, and a pre-exponential term (A) of 101.05. The value of activation energy was calculated to be 133.45 kJ mol−1, being a smaller value than the reported values in previous findings of other authors (176.15–205.02 kJ mol−1).36 This difference in the calculated activation energy were attributed to possible differences in the composition and cation ordering of dolomite phases obtained from different experiments. Considering the differences in the enthalpy of hydration of the cations, the authors estimated the activation energy related to cation ordering at approximately 41.84 kJ mol−1.36 The experiments employed temperatures between and 115 and 196 °C, and the utilized solutions had variable concentrations of CaCl2, MgCl2 and NaHCO3 under different pCO2 applied in the surrounding atmosphere. The retrieved dolomite seeds presented clear evidence of precipitation of newly formed material with composition close to dolomite. Additional co-precipitated phases included calcite, magnesian calcite and magnesite. The authors utilized their experimental findings to extrapolate the data and calculate the velocities rates of dolomite precipitation, evaluating that at least tens of years would be needed to precipitate dolomite at room temperature, even if high concentrations of MgCl2 (0.05 M) would be employed (Fig. 9).36
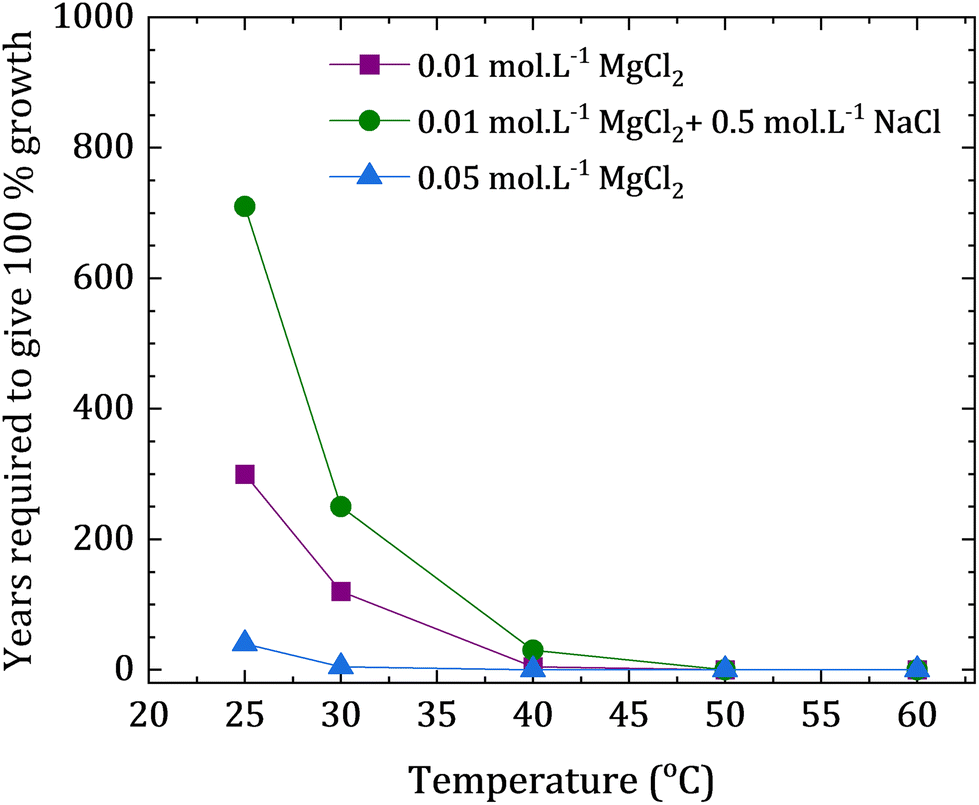 | ||
| Fig. 9 Extrapolated reaction times for dolomite growth at low temperatures, calculated by Arvidson and Mackenzie (1999) from their reported experimental data (graph plotted based on the reported data).36 | ||
The theoretical extrapolated results shown in Fig. 9 consider the calculated time needed for precipitating dolomite on a fixed substrate surface area, yielding a mass increase of 10 wt% (relative to the substrate). This kinetic evaluation suggests that the required time for the reaction at ca. 50 °C is still quite long, but yet reasonable. Further experimental observations of this same work led to the conclusion that the growth of calcite has an inhibitory effect on the growth of dolomite, describing a competitive mechanism between both phases. Thus, the authors did not find evidences that magnesian calcite could be the intermediate phase for dolomite formation. The authors have concluded that the clear close dependence between temperature and saturation state indicates an overall low rate of dolomite precipitation when compared with competing carbonate phases at low temperatures. Moreover, it was suggested that small increments of temperature could increase the dolomite precipitation rate relative to calcite (especially if the solution is undersaturated with relation to calcite) due to the close relationship between the activation energies of dolomite and calcite.36
Banerjee A. investigated the dolomite kinetic models via two different routes: (i) formation of primary dolomite from supersaturated solution (with relation to dolomite), and (ii) dolomitization via cation replacement in the calcite structure. The theoretical models have been built based on the kinetic constants of dolomite precipitation via these routes. The author utilized previous published work to calculate the kinetic parameters, considering the variation of reaction rate and transport expressions (rate constant, equilibrium constant, solution supersaturation, temperature, and Mg:Ca ratio), using the kinetic constant of 10−16.6 mol cm2 s−1 at 25 °C. It was then concluded that in ocean water conditions, dolomite only could be formed via microbially assisted mechanisms.51
Rodriguez-Blanco and co-workers (2015) proposed a new route for dolomite precipitation, where the dolomite formation was divided into three processes: (i) initially, an amorphous magnesium calcium carbonate phase precipitates almost instantaneously after mixing Na2CO3, CaCl2 and MgCl2 solutions (2:1:1 ratio) at ambient conditions, being the amorphous phase considered as precursor for the subsequent stages; (ii) the amorphous precursor leads to a proto-dolomite precipitation under thermal treatment at temperatures ranging from 25 to 220 °C and; (iii) the proto-dolomite is converted into the crystalline dolomite phase only at temperatures above 140 °C.55 The observations suggested dolomite precipitation via a non-classical crystallization model, where an “proto” nucleus is formed before the crystal nucleus.56 This transformation occurs with the spherulites via a growth front nucleation, in good agreement with the kinetic models previously published.46,57 The particle size growth suggested an Ostwald ripening dissolution–crystallization kinetics, transforming proto-dolomite into moderately to highly ordered dolomite.48 The authors also confirmed the influence of the temperature on the dolomite formation, measuring the kinetics of proto-dolomite formation, where the experimental temperature increase lead to an increasing in its constant formation.
The growth kinetics of dolomite compared with magnesite and calcite was performed by Davis et al. (2011), where they found the grain growth of dolomite to be much slower than the growth rates of magnesite or calcite. The experiments were performed at temperatures between 700 and 800 °C and the growth constant to dolomite was around 5 × 10−5 µm3 s−1, which represents a growth rate thirty times slower than magnesite (1–3 × 10−4 µm3 s−1) and three orders of magnitude slower than calcite (5–6 × 10−1 µm3 s−1) under similar conditions. The authors also observed that the precipitated phases differed greatly in porosity in the following order magnesite < dolomite ≪ calcite.58 Therefore, despite the large volume of studies on the kinetics of dolomite precipitation, all reported kinetic constants at ambient conditions are an extrapolation of the results obtained for the synthesis of dolomite at high temperatures and/or pressures. For this reason, there is not yet a consensual agreement on the reasons why dolomite does not precipitate at ambient conditions.
3.3. Thermodynamics of the dolomite family
There is a good agreement in the thermodynamic properties of dolomites among thermodynamic databases. Here we collected and evaluated the logK vs. temperature for dolomite among different databases including Minteq,59 PHREEQC,60 Lawrence Livermore National Laboratory (LLNL),61 Yucca Mountain Project (YMP),62 Thermoddem,63 and Thermochimie,64 which revealed a goodmatch among these databases (Fig. 10a). The logK value and the trend in relation with temperature are comparable among different dolomites. Notably, when one assesses thermodynamic datasets, the carbonate species should be considered for calculating the logK value, as differences may arise according to the charge of the carbonate anions, which can lead to a mismatch in the results among surveyed databases. Furthemore, there is a need to improve the quality of thermodynamic databases for other solid solutions of the CaCO3-MgCO3 system (Fig. 4) such as high and low magnesian calcites. Plus, as synthetic dolomites have not been obtained yet, the reported parameters of dolomite are an extrapolation of the characterized parameters at high temperatures which needs to be assessed and updated with new experimental data.
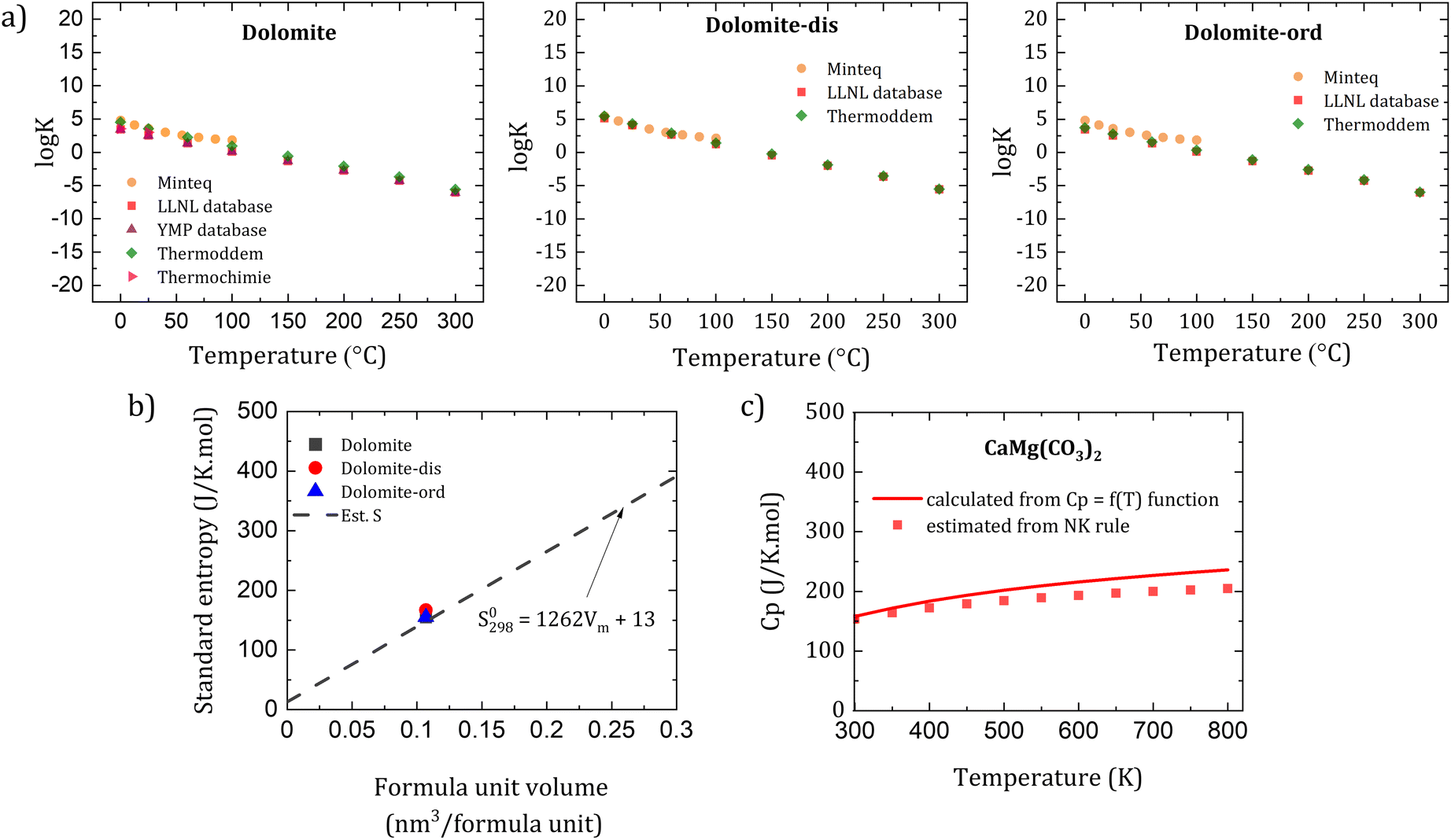 | ||
| Fig. 10 (a) Summary of logK with regard to temperature of dolomite, disordered dolomite (dolomite-dis) and ordered dolomite (dolomite-ord) among various thermodynamic databases, (b) comparison between data from thermodynamic databases and estimated values of standard molar entropy based on the formula unit volume proposed by Jenkins and Glasser,65 and (c) estimated Cp value of (a) dolomite and (b) estimated Cp values are estimated based on Newmann–Kopp rule66 compared with the Cp = f(T) function. | ||
The standard entropy of dolomite can be estimated using the correlation with formula unit volume as proposed by Jenkin and Glasser.65 The standard molar entropy (S°298, J mol−1 K−1) of anhydrous inorganic minerals (e.g., dolomites or any relevant solid solutions of alkali earth carbonates) can be correlated with the unit volume formula as follows:
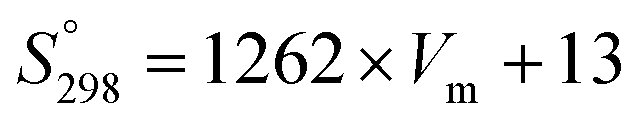 | (8) |
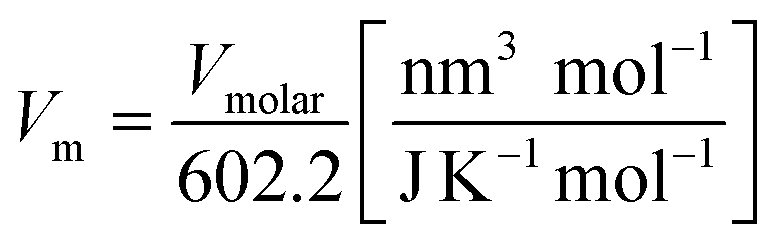 | (9) |
Fig. 10b shows the good agreement of the reported standard entropy of dolomites (from thermodynamic databases) in comparison to the estimated values. Therefore, one can estimate the standard entropy of dolomites or their solid solutions if the molar volume of the minerals is known.
To estimate the heat capacity of dolomites, we evaluate the possibility of using the empirical Neumann–Kopp rule66 for dolomites as a function of temperature. Fig. 10c exhibits a very good agreement between the Neumann–Kopp rule and the classic the Cp = f(T) function67 in predicting the heat capacity of dolomites. The rule, thus, can be used to estimate the heat capacity of dolomites or its solid solutions if there are known stoichiometric formulas of the carbonates. However, at a high temperature (above 600 K), there may be a need to adjust the discrepancy of estimated Cp considering the contribution of other factors at elevated temperatures such as the variation of thermal expansion and compressibility at high temperature beside the lattice vibrations and dilatation.
4. Thermodynamic enigma of the magnesite problem
Magnesite is widely employed as precursor in the pharmaceutical and cosmetic industry as well as flame retardant due to its high contents of MgO (47.8%) and high thermal stability (500–600 °C).68 Recently, research on the magnesite synthesis has gained added importance not only because of its shared issues with the enigmatic dolomite problem, but also driven by its possible technological applications. The durability of magnesite offers possibilities for long-term CO2 storage and applications as fillers in construction materials or flame retardants. While magnesia-based cements have been investigated over a century, the applications of Mg carbonates-based cements still need considerable developments. Since the hydrated Mg carbonates are thermodynamically metastable, they produce materials of low life span due to the fast interconversion between the hydrated phases.69,70 Therefore, the comprehension of the mechanisms and kinetics of Mg carbonates precipitation is to play a crucial role on the emerging solutions for the climate emergency.Considering that magnesite and dolomite belong to the same mineral class and space group of anhydrous carbonates, it is plausible to infer that the problem of synthesizing these minerals at ambient conditions share the same root.17 Likewise in the dolomite case, the main factors hindering the kinetics of magnesite precipitation at room temperature has been associated to: (i) the high energy barrier for [Mg2+·6H2O] dehydration (ΔhydG° = −1903.72 kJ mol−1),16 and/or (ii) to the intrinsic structural/spatial barrier of the CO32− groups17 in the magnesite symmetry group. Even though magnesite is the thermodynamically favored Mg carbonate (Table 3), the rate of magnesite precipitation at ambient conditions has been estimated as 23000 years for 10 g of magnesite to precipitate directly on a 1 m2 surface, or 200 years through hydromagnesite dehydration.71
| Phase | logK° |
ΔfG° [kJ mol−1] | ΔfH° [kJ mol−1] | S° [J (mol K)−1] | C 0p [J (mol K)−1] | V m [cm3 mol−1] |
|---|---|---|---|---|---|---|
| Brucite (BR), Mg(OH)2 | −11.16 | −832.2 | −923.3 | 63.1 | 77.3 | 24.6 |
| Magnesite (MS), MgCO3 | −8.29 | −1029.3 | −1112.9 | 65.7 | 75.9 | 28 |
| Nesquehonite (NQ), MgCO3·3H2O | −5.27 | −1724.0 | −1981.7 | 180.5 | 237.8 | 74.8 |
| Lansfordite (LF), MgCO3·5H2O | −5.24 | −2197.8 | −2574.3 | 249.5 | 317.9 | 103.2 |
| Hydromagnesite (HY), Mg5(CO3)4(OH)2·4H2O | −37.08 | −5856.8 | −6514.9 | 478.7 | 526.6 | 208.8 |
| Dypingite (DY), Mg5(CO3)4(OH)2·5H2O | −34.94 | −6081.7 | −6796.2 | 522.8 | 566.6 | 225.9 |
| Artinite (AT), Mg2CO3(OH)2·3H2O | −18.67 | −2568.6 | −2920.6 | 232.9 | 296.1 | 96.9 |
Table 3 shows the thermodynamic properties of most common minerals in MgO–CO2–H2O system collected and updated from current thermodynamic databases and literature. As seen in the thermodynamic parameters, the issue of magnesite precipitation at ambient conditions is rather a kinetic issue (see more details in section 4.2) than a thermodynamic limitation.72 This explains the prediction from thermodynamic calculations for the formation of magnesite as the main and sometimes only carbonate formed in the MgO–CO2–H2O system. In contrast, other hydrated Mg carbonates have more favorable kinetics to form at ambient conditions. Despite the relative fast precipitation of the hydrated phases, when carbon capture and utilization/storage applications are aimed these metastable minerals display losses of sequestrated CO2 and/or volumetric instability during the conversion among these carbonates.73
The MgO–CO2–H2O system has been extensively studied to elucidate the main research questions regarding the magnesite/dolomite problem. When analyzing the predicted solubilities for the expected Mg minerals to precipitate in such system without foreign ions (Fig. 11), magnesite is clearly expected to be the most stable phase compared to hydrated Mg carbonates under all conditions of temperature and pressure. Brucite solubility, however, varies with temperature, relative humidity and pressure, having higher stability only at high temperatures and low pressures.12 As the stability of the Mg carbonates is inversely proportional to temperature, the heat treatment of hydrated Mg carbonates lead to loss of coordinated water and carbonate groups until reaching the magnesite structure as follows:84
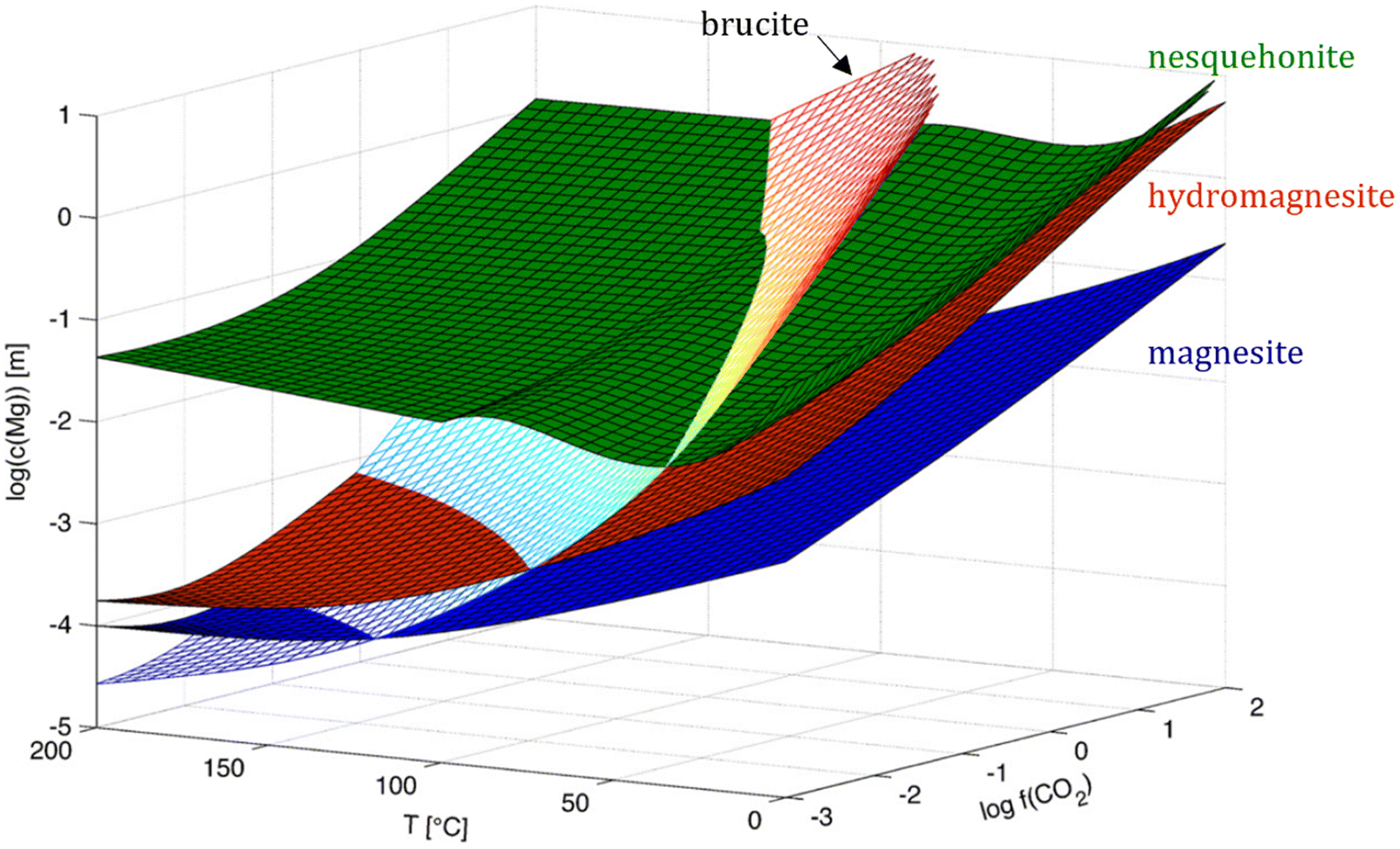 | ||
| Fig. 11 Solubilities of typical mineral to precipitate in the MgO–CO2–H2O system in absence of foreign ions. From top to bottom at bottom left corner: nesquehonite, hydromagnesite, magnesite and brucite. Adapted from Hänchen et al.,12 and reproduced with the publisher's permission (2012, Copyright © 2007 Elsevier Ltd.). | ||
Up to date the detailed crystallization pathway of magnesite is still a topic of active research. It is unclear whether crystallization occurs preferentially via a classical or non-classical model (Fig. 12). It is also important to note that one mechanism does not exclude the other.37 In the classical crystallization model, the nucleation is the initial step, followed by a monomer-by-monomer crystallization growth when the nuclei reach a critical size. Other studies have suggested the non-classical crystallization mechanism, which is initiated by the formation of pre-nucleation clusters, leading to particle attachment to form amorphous aggregates that can convert into the final crystalline phase. Thus, the non-classical crystallization can be seen as a preliminary step prior to the nucleation of the crystalline step, which may occur to overcome the kinetic barriers.37,56
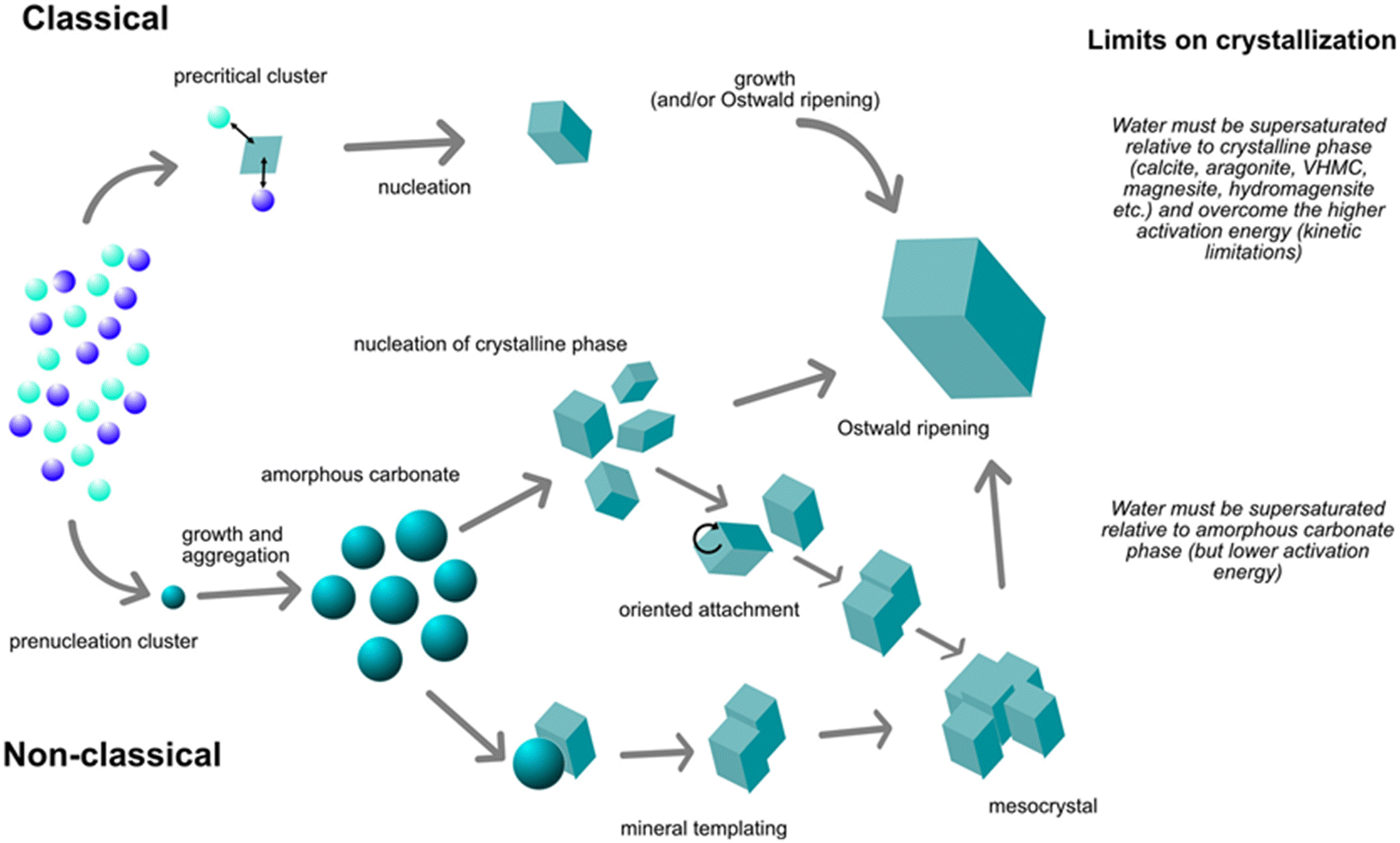 | ||
| Fig. 12 Schematic representation of the classical and non-classical crystallization mechanisms of Mg carbonate minerals. Reproduced with the publisher's permission (© 2022 Raudsepp et al. Sedimentology published by John Wiley & Sons Ltd on behalf of International Association of Sedimentologists).37 | ||
As a matter of fact, few works produced in the last decade have demonstrated the precipitation of amorphous magnesium carbonates employing variable precursors (MgCl2·6H2O,85 MgO,86,87 and anhydrous MgCl217,88). The majority of the works considers that the amorphous phase precipitates with a sphere of coordinated water (2 to 4 water molecules, resembling the composition of hydrated Mg carbonate phases),85,87,88 but anhydrous amorphous Mg carbonates have been formed in aprotic solvents.17 Nevertheless, the molecular structure and physico-chemical properties of amorphous Mg carbonates is still a topic under active discussion in the literature. Based on the assumptions of a cross-linked pathway between the non-classical and classical crystallization, it is expected that deepening the knowledge on the precipitation of amorphous Mg carbonates may lead to catalytic routes for obtaining magnesite.86
4.1. Molecular dynamics simulations
Computational characterization via combined classical molecular dynamics and enhanced sampling metadynamics have been used to simulate the catalytic and/or inhibitory role of the most common anionic species found in typical brine solutions. Thermodynamically, hydrated Mg2+ ions are stable only at the six-fold octahedral coordination state, which can transit to the metastable five-fold trigonal bipyramidal coordinate state assisted by out-of-shell solvent water molecules. However, the high free energy barrier between the six and five coordinate states at room temperature (65 kJ mol−1) makes the dehydration highly unfavorable since the available thermal energy at 300 K is 26 times lower (2.5 kJ mol−1) than the energy requirements. That results in a very slow water exchange in the first hydration shell of Mg2+(aq), making the carbonation steps of nucleation and growth highly susceptible to Mg (re)hydration, and thus leading to the formation of the hydrated polymorphs of magnesite.16,89The nucleation step of Mg carbonates is initiated by the creation of a vacant site at Mg2+, thus requiring the displacement of a water molecule from the first coordination shell of Mg2+(aq). Consequently, increasing the temperature facilitates overcoming the activation barrier between the six- and five-coordinated Mg2+ states and leads to magnesite precipitation, in agreement with former experimental evidence. Alternatively, the energy barrier for Mg2+ dehydration may be lowered by the formation of contact ion pairs (direct cation–anion contact) or solvent-shared ion pairs (cation–anion pairs intercalated with a water molecule), which may reduce the dehydration energy barrier of Mg2+. The calculated free energies of reaction and standard Gibbs activation energy for contact ion pairs commonly found in sea water is shown in Table 4. Since the Mg2+⋯SO42− pair is energetically more favorable than the Mg2+⋯CO32−, the presence of sulphate groups favors the nucleation of the Mg carbonate phases due to the non-competitive contact ion pairs formation. On the other hand, although the formation of the Mg2+⋯F− and Mg2+⋯HS− are thermodynamically favorable, the respective activation barrier is too high, thus, hindering the formation of these contact ion pairs. The values of the Gibbs free and activation energy for the acetate group shows that these do not inhibit the MgCO3 pairing. The study also shows that Cl− and NO3− ions form mainly solvent-shared ion pairs, thus the replacement of one water molecule with one Cl or O (from nitrate) occurs with high energy demand, without disturbing the inner shell of Mg2+.16
| Solvent-shared ion pairs | Anion | ΔG (kJ mol−1) | Δ‡G (kJ mol−1) |
|---|---|---|---|
| Mg2+⋯CO32− | Carbonate | −42.7 | 25.0 |
| Mg2+⋯SO42− | Sulphate | −29.0 | 44.7 |
| Mg2+⋯H2O | Water | −11.9 | 43.5 |
| Mg2+⋯F− | Fluoride | −62.8 | 47 |
| Mg2+⋯HS− | Bisulfide | −12.1 | 58.4 |
| Mg2+⋯HCO3− | Bicarbonate | −4.2 | 55.2 |
| Mg2+⋯CH3COO− | Acetate | −15.7 | 42.0 |
| Mg2+⋯PO42− | Phosphate | −111.0 | — |
| Mg2+⋯H2PO4− | Dihydrogen phosphate | −31.6 | 52.7 |
| Mg2+⋯C6H5O73− | Citrate | −43.6 | 41.2 |
| Mg2+⋯C6H4ONH2− | Aminophenolate | −41.2 | 47.5 |
| Mg2+⋯C2O4− | Oxalate | −23.5 | 30.8 |
| Mg2+⋯C4H4O52− | Malate | −16.7 | 38.5 |
Fig. 13 compares the free energy profiles of Mg2+ in water with those in presence of the investigated anions. Fig. 13 (top) shows the profiles of solutions with anions that tend to form solvent-shared ion pairs (not competing with the formation of Mg⋯CO3 contact ion pair), thus the simulations were done for the Mg2+⋯H2O⋯X ion pair (Table 4). The simulation profiles shows that nitrate ions do not interfere with the kinetics of Mg2+–H2O dehydration and chloride ions slightly increase the stabilization of the five coordinated Mg2+. Oppositely, bisulfide, fluoride, and acetate anions have a strong catalytic effect on the Mg2+ dehydration, promoting its five-fold coordinate state to similar level of stability of the six-fold one; thus, facilitating the creation of vacant coordinate state available for the nucleation and growth of the Mg carbonate species. The simulation profiles of contact ion pair (Mg2+⋯X) solutions (Fig. 13, bottom) shows that the counter anions can have even stronger catalytic effect, stabilizing also the three- and four-fold coordinate states of Mg2+.16
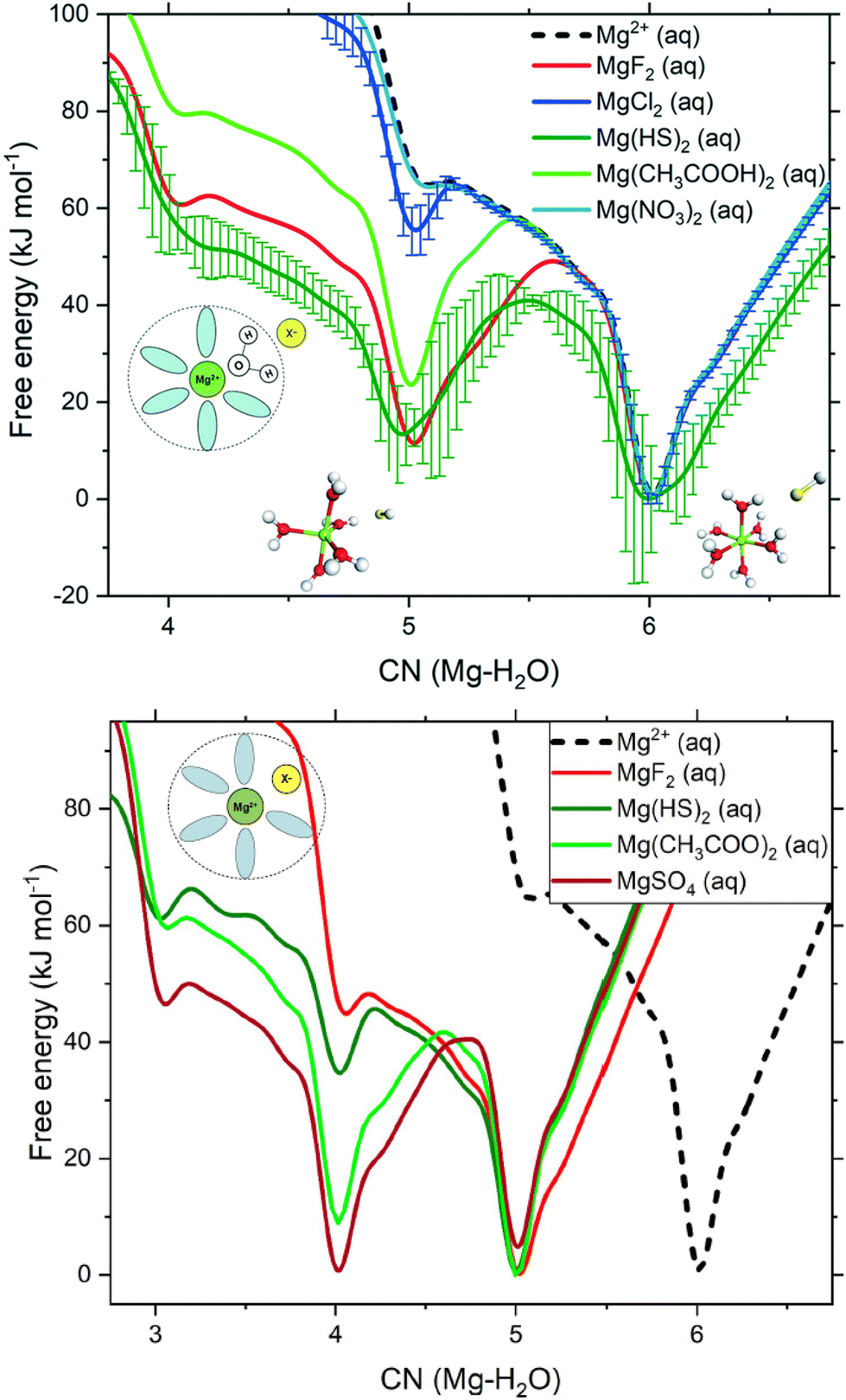 | ||
| Fig. 13 Top: Free energy as a function of the Mg2+–H2O coordination number, CN(Mg–H2O), for hydrated Mg2+ (single Mg2+, no counterions) and solvated Mg2+ with a counterion in the second hydration shell. The structures are the five- and six-coordinated states in Mg(HS)2(aq). Standard deviation computed from the average of the profiles of four independent MetaD simulations (300 K). Error bars are presented for the cases with the largest uncertainty, others have been removed for clarity. Bottom: Free energy as a function of the magnesium-water coordination number, CN(Mg–H2O), for a hydrated metal ion (single Mg2+, no counterions) and of Mg2+ with a counterion in the first hydration shell (©The Royal Society of Chemistry 2021).16 | ||
The most recent atomistic simulations published by Toroz et al. (2022) have evaluated three main criteria to probe the catalytic effect of a given additive:
1. The anionic additive can form solvent-shared ion pairs or contact ion pairs less stable than Mg2+⋯CO32−,
2. The anionic additive can stabilize the undercoordinated state of Mg(H2O)52+, and
3. The anionic additive can stabilize low hydration Mg(X)–(H2O)m2−m states via direct coordination to Mg2+.90
The study considered 30 species of anionic additives for calculating the thermodynamic stabilities of the solvent-shared ion pairs and contact ion pairs, and the potential of the anion to stabilize uncoordinated hydrated Mg2+ states without inhibiting the formation of the Mg2+⋯CO32− contact ion pair. It was found that only six species fulfilled the three criteria and were hypothesized as potential catalysts for Mg carbonate precipitation: citrate, aminophenolate, oxalate, sulphate, malate and dihydrogen phosphate (Table 5).90
| Anionic additive | formula | Criterion 1? | Criterion 2? | Criterion 3? | Promote carbonation? |
|---|---|---|---|---|---|
| Chloride | Cl− | N | N | N | N |
| Fluoride | F− | Y | Y | N | N |
| Iodide | I− | N | N | N | N |
| Nitrate | NO3− | N | N | N | N |
| Bicarbonate | HCO3− | Y | Y | N | N |
| Perchlorate | ClO4− | N | Y | N | N |
| Sulfate | SO 4 2− | Y | Y | Y | Y |
| Bisulfide | HS− | Y | Y | N | N |
| Formate | HCOO− | Y | Y | N | N |
| Acetate | CH3COO− | Y | Y | N | N |
| Phosphate | PO43− | N | N | Y | N |
| Hydrogen phosphate | HPO42− | N | Y | Y | N |
| Dihydrogen phosphate | H 2 PO 4 − | Y | Y | Y | Y |
| Metasilicate | SiO32− | N | Y | N | N |
| Taurate | C2H6NSO3− | Y | Y | N | N |
| Oxalate | C 2 O 4 2− | Y | Y | Y | Y |
| Salicylate | C7H5O3− | Y | Y | N | N |
| Citrate | C 6 H 5 O 7 3− | Y | Y | Y | Y |
| Aspartate | C4H6NO42− | Y | Y | N | N |
| Tartrate | C4H4O62− | N | N | Y | N |
| Malate | C 4 H 4 O 5 2− | Y | Y | Y | Y |
| Aminophenolate | C 6 H 4 ONH 2 − | Y | Y | Y | Y |
| Glycinate | C2H4NO2− | Y | Y | N | N |
| Glutamate | C5H8NO4− | Y | Y | N | N |
| Hydroxyl | OH− | N | Y | N | N |
| Phenolate | C6H5O− | N | Y | N | N |
| Isopropyl alcohol ionic | C3H7O2− | N | Y | N | N |
| Polyethylene glycol | C8O5H162− | N | Y | N | N |
| Hexafluorosilicate | SiF62− | N | Y | N | N |
4.2. Mechanisms and kinetics of the MgO–CO2–H2O system
As well detailed in the former sections, the precipitation of Mg carbonates is controlled by thermodynamically favorable reactions, and yet hindered by the kinetic constraints. In view of the highly hydrated character of Mg2+ ions, the formation of magnesite is often associated with an initial formation of hydrated Mg carbonates, other hydrate carbonates and even brucite. As dictated by Gibbs’ phase rule, the stable carbonate phase depends not only on temperature and pressure but on the humidity of the contacting gas phase as well. The formation of thermodynamically unstable phases as intermediary product towards a stable phase is an expected prediction of the Ostwald rule, which is aligned with the formation of hydrated Mg carbonates as intermediary products prior magnesite precipitation. It is speculated that the hydrated compounds convert into magnesite by two possible mechanisms: shrinkage (dehydration) and dissolution–precipitation solvent mediated transformation.12 Dissolved CO2 plays a key role on the precipitation of magnesite and hydrated Mg carbonates.91 It was showed that during controlled CO2 depressurization environment, the carbonation of brucite produces hydromagnesite through intermediates, including nesquehonite, dypingite and an unidentified structure with crystalline aspect.72 In a closed batch reactor system with brucite, temperature and pressure have been shown to play a key role on the magnesite formation.92The convergent features of the observed catalytic agents to anhydrous Mg carbonates precipitation are the negatively charged nature, and the partially filled π orbitals (seen in all compounds which have proven acceleratory effects on dolomite/magnesite precipitation). Interestingly, no direct relation between the ionic strength and kinetics of Mg carbonates precipitation can be found as a coherent observation in the existing studies. Thus, it is plausible to argue that the catalytic effect may be originating from the energy levels of the π* (antibonding) orbitals of the catalytic agents, which might modify the energy levels of the carbonate groups while bonding to it; thus, enabling the superposition alignment of the Mg orbitals with the necessary energy and symmetry for the crystalline growth of magnesite. If the catalytic mechanism involves the physisorption or chemisorption of CO2 into the aqueous medium, the phenomenon can spur various explanations. When CO2 is absorbed in water, it can be activated nucleophilically and/or electrophilically via the C or O atom, respectively, since the LUMO (lowest unoccupied molecular orbital) orbitals have predominant C-p character while the HOMO (highest occupied molecular orbital) have higher O-p character.93,94 Considering the linear and non-polar nature of the ground state of CO2, the electrophilic attack to the antibonding orbital 2πu results in the formation of radical anions, which are stabilized via geometric distortion in the angles of the C–O bonds.95 Interestingly, HCO2− and the CO2− anions have very similar bond length, angle and orbitals characters.93 Thus, the angular distortion in the C–O bonds has a direct effect on the energy levels of the HOMO and LUMO orbitals (Fig. 14),93–95 being plausible to assume that there should be an ideal symmetry in the carbonate groups to enable the precipitation of magnesite.
 | ||
| Fig. 14 Variations in total energy of the molecular orbitals of CO2 in function of (a) the C–O bond distances, (b) the OCO bond angles. (c) Walsh diagram and (d) graphical representation of the orbital energies of CO2 in function of the OCO bond angles.94 Reproduced with the publisher's permission (Copyright © 2014 Elsevier Ltd. All rights reserved). | ||
When the energy barrier for the dehydration of the Mg2+ ions is considered, the high required energies are associated with the high ratio between the electrical charge and the ionic radii of Mg2+. Thus, the reduction of water salinity (increasing the ionic strength) can help the Mg2+ dehydration by decreasing the thickness of the secondary hydration layer. Moreover, increasing the temperature and pCO2 often shows the formation of magnesite without discernible formation of the intermediary products.12
The saturation index (Ω) of magnesite is given by eqn (10), which considers the equilibrium of magnesite dissolution and precipitation, and accounts for the activity of the dissolved ions (ai) as well as the equilibrium constant (K) of magnesite, being also determined by the ratio between the reaction quotient (Q) and the equilibrium constant.23,96
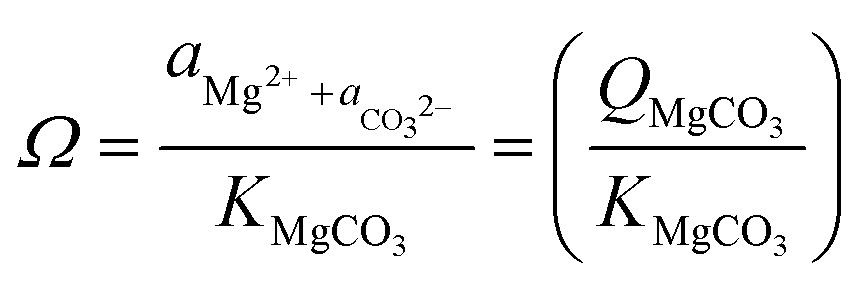 | (10) |
Thus, the thermodynamic parameters of magnesite formation could be determined from magnesium and carbonate ions. The Gibbs free energy of this reaction at 25 °C, and 1 bar is −1026.48 ± 2 kJ mol−1.23
As a conventional method for quantifying mineral growth, it has been demonstrated that mineral growth from solutions have its kinetics ruled by the growth mechanism, according to eqn (6). The coefficient n varies according to the growth mechanism: n = 1 when the growth is controlled by transport or adsorption, n = 2 for spiral growth, and exponential numbers are seen in process controlled by surface nucleation. Following these considerations, it was demonstrated that magnesite growth occurs via a spiral growth mechanism in artificial brine, at temperatures ranging from 80 to 120 °C, pressures between 1 to 2.5 bar, and saturation indices ranging from 20 to 100. By extrapolating the results, the rate constants was calculated for the obtuse and acute growth steps of magnesite are 2.4 × 10−13 and 2.0 × 10−14 cm s−1, respectively at Ω = 10 and 25 °C, meaning that following this kinetic model, it would take 1.5 years to precipitate 10 mm layer of magnesite at room temperature.96 However, the kinetic model of magnesite precipitation is still a matter of debate, other studies suggesting that magnesite grows via a kink site nucleation and propagation model.45
Despite the availability of few kinetic studies of magnesite precipitation (when compared to dolomite), the stability of magnesite has been investigated with dissolution studies. Acid leaching studies in natural magnesite using succinic acid,97 acetic acid,98 citric acid,99 and chlorine dissolution100 have estimated the activation energy of dissolution in 45.197 kJ mol−1, 34.60 kJ mol−1, 61.35 kJ mol−1 and 66.47 kJ mol−1, respectively (considering ambient conditions of temperature and pressure). A generic model shows the influence of pH under magnesite dissolution, where it was defined four distinct regions: an acid region (pH <2.5) in which rates are invariant with pH; between pH 3 and 5 in which rate is proportional to H3O+ activity; a second pH invariant region between pH 5 and 8; and an alkaline region (pH >8) in which rates declines with pH as well as bicarbonate and carbonate concentrations.101,102
Several routes have been proposed for magnesite precipitation, for instance, one usual pathway is the conversion of the intermediate hydrated magnesium carbonates species into magnesite: high alkaline conditions favors hydromagnesite precipitation, which yields magnesite upon slow dehydration.15 The following sub-sections discuss proposed mechanisms and routes of magnesite precipitation according to the main precursors investigated in the literature: MgCl2 and brine, Mg silicates, Mg sulphate, and Mg hydroxide (brucite).
4.3. Conventional systems of magnesite precipitation
The scientific relevance of the mechanisms involved in the carbonation of brine have generated many proposed routes based on the carbonation of MgCl2 with Na2CO3. However, these routes produce magnesite only at high temperatures and pressures. As observed for most reactions of Mg carbonation, the main products of the carbonation of MgCl2 are usually hydrated Mg carbonates (Table 6). It has been noted that the formation of magnesite is often associated with the formation of hydromagnesite as intermediary reaction product, being the hydromagnesite-to-magnesite conversion seen as thermodynamically spontaneous transformation of slow kinetics at room temperature and pressures. It has been observed that the increasing of temperature, salinity, pCO2 and the decreasing of Mg concentrations seems to accelerate the transformation. The utilization of high pCO2 has led to the direct precipitation of magnesite, without passing through the hydrated Mg carbonates metastable polymorphs as intermediary products.12
| [Mg2+]/[CO32−] | Carbonate precursor | Additional precursors? | Temp. (°C) | Pressure/stirring | Reaction time (min) | Main products |
|---|---|---|---|---|---|---|
| a Non specified rpm. b Magnesite was only observed with after several cycles of pH swing and high Mg concentrations. Lowering the Mg concentration shifts the product towards aragonite precipitation. | ||||||
| 0.4 to 25.4 | Na2CO3 | No | 25 | 1 bar/300 rpm | 300 to 1470 | NQ12 |
| 2.5 to 5.7 | Na2CO3 | No | 120 | 3 bar/300 rpm | 50 to 1140 | HY converted to MS after 15 h (ref. 12) |
| 8 to 129 | Na2CO3 | No | 120 | 100 bar/300 rpm | 50 to 1140 | HY converted to MS after 2 h (ref. 12) |
| 2.0 × 10−3 to 0.65 | NaHCO3 | 0.1 M NaCl (10−4 M HCl, 10−3 M NaOH in some runs) | 80 to 120 | 1 atm/mixed flow reactor | 420 to 4320 | MS96 |
| 2.0 × 10−4 to 0.42 | NaHCO3 | 0.1 to 1 M NaCl (10−3 M HCl, 10−3 M NaOH) | 100 to 200 | 1 atm/mixed flow reactor | 1020 to 4500 | MS103 |
| 0.7 to 1330 | NaHCO3 | 0.1 M NaCl | 80 and 90 | 1.7 atm/mixed flow reactor | 60 | MS45 |
| 3.23 (Liebermann exp. n. 57) | CaCO3 + daily additions of Na2CO3 | 2.83 M NaCl, 0.062 M KCl, pH swing: 1 h acidic (3.6 mmol HCl) and 23 h basic (2 mmol Na2CO3) | 40–43 | 1 atm/stirringa | 53280 (37 days) |
HY, AN and MS (16, 17 and 28%, respectively)108 |
| 3.23 (Liebermann exp. n. 57) | CaCO3 + daily additions of Na2CO3 | 2.83 M NaCl, 0.062 M KCl, 0.03 M ZnCl2, pH swing: 1 h acidic (3.6 mmol HCl) and 23 h basic (2 mmol Na2CO3) | 40–43 | 1 atm/stirringa | 53280 (37 days) |
97.3% MS and 2.7% AN108 |
| 17.99 (Liebermann exp. n. 57) | CaCO3 + CO2 bubbling in 12 × 60 h cycling | 2.78 M NaCl, 0.06 M KCl, pH swing: CO2 bubbling and ammonia titration | 40 | 1 atm | 60480 (42 days) |
MS and minor DY and AN20,109–111 |
| 0.9 to 25 (Liebermann exp. n. 57) | CaCO3 + CO2 bubbling | 2.78 M NaCl, 0.06 M KCl, 10 mM Na2SiO3, pH swing: CO2 bubbling and ammonia titration | 30, 40 and 50 | 1 atm | 240 and 720 | MS, CT and DLb71 |
| 1 | CaCl2, CsCO3 | Formamide used as solvent, CaCl2 | RT | 1 atm | Mg/Ca = 1:0, 5:1 and 2:1 – anhydrous amorphous Mg carbonate |
|
| 1:1 – disordered protodolomite |
||||||
| 1:2 and 1:5 – crystalline magnesian calcite17 |
||||||
| 1 | NaHCO3 | Low- and high-density polystyrene microshperes | RT | 1 atm | 103680 (72 days) |
MS with high-density polystyrene112 |
| 1 | NaHCO3 + Na2CO3 | — | RT | 1 atm/stirring in the initial 30 min | 4 h | NQ113 |
| 0.5 | urea | no | 140 | 1 bar | 6 h | MS single crystal14 |
| 1.5 to 0.5 | Urea, Na2CO3, NaHCO3, K2CO3, KHCO3, (NH4)2CO3 or NH4HCO3 | NaOH, KOH, H2SO4, HCl, HNO3, HAc, NaCl, Na2SO4, KCl or K2SO4 | 180 | 1 bar | 3 h | MS in the pH between 5.4 and 10.5114 |
Hänchen et al. (2008) have performed a systematic study of the carbonation of MgCl2 in different range of temperatures, pressures and [Mg2+]/[CO32−] molar ratios (Table 6), observing that in all experiments performed at 25 °C and 1 bar, nesquehonite precipitates as the main product, regardless of the employed [Mg2+]/[CO32−] molar ratios.12 These experiments have evidenced the preferential supersaturation with respect to nesquehonite at ambient conditions, thus governing the precipitation kinetics. It is important to highlight that the solutions were also saturated with respect to magnesite and hydromagnesite, but nesquehonite is the kinetically favored phase.12
Recent articles have reported the synthesis of magnesite at 40–43 °C based on the experiment number 57 of Liebermann (1967).107 The experiment performs the carbonation of an artificial brine-based system, which utilizes MgCl2 and MgSO4·7H2O as Mg source and CaCO3 for the carbonation. The system is submitted to a cyclic pH swing, leading to the precipitation of magnesite plus minor amounts of aragonite after 37–42 days.7,20,108–111 Two main approaches have been described for the pH swing step: (i) the pH is lowered by keeping 12 h of CO2 bubbling, raised again via titration with diluted ammonia solution to pH = 7, and kept under 40 °C in an open system for 60 h,7,20,109–111 or (ii) the pH is lowered with daily additions of 0.0036 mol of HCl, kept at pH 5.7–7.0 for 1 h and then the pH is raised again at 7.9–8.4 with 0.002 mol of Na2CO3 and kept for 23 h at 43 °C.108 The pH cycling method aims at the continuously dissolution of the metastable Mg carbonates polymorphs by lowering the pH, and then raising the pH to alkaline conditions again, inducing the precipitation of the more thermodynamically stable phases (magnesite and dolomite). The initial research on the pH swing method have proposed that the precipitation of magnesite occurred by breaking Ostwald's rule: by continuously precipitating the metastable phases, it was hypothesized that magnesite would crystalize without a metastable intermediate (at decreased potential energy), making the reaction irreversible.20,108,110 The hypothesis is supported by the fact that at 25 °C, the dissolution rate of magnesite is 100 times slower than dolomite and 1000 times slower than calcite.71 That would imply a continuous stepwise removal of the metastable phases (hydrated and anhydrous) by changing the reaction rates with the dissolution–precipitation cycles. That also could explain the contradictions in the thermodynamic data available for dolomite and magnesite.20,108,110
Hobbs et al. have proposed the magnesite precipitation in the pH swing method to occur via a mixed growth and incorporative magnesium mechanism: the acidic conditions break the Ca–CO3 bonds quicker than the Mg–CO3, thus inducing a net cation replacement. It was proposed that the first carbonated to precipitate depends on the solution chemistry, while the dissolution at low pH removes the metastable and Ca bearing phases. Subsequently, the pH raise will induce faster precipitation of magnesite or dolomite. Thus, it was proposed that the pH swing induces magnesite precipitation via a non-equilibrium cyclic growth and replacement mechanism.71 However, no further studies have corroborated with the grounds of this hypothesis.
The work published by Xu et al. in 201317 has evaluated the carbonation of MgCl2 in a nonaqueous solvent to probe the effect of the Mg2+ and Ca2+ cations solvation on the obtained products. The authors utilized formamide as solvent due to its similar properties to water (high dielectric constant, density, dipole moment, surface tension). It was observed that in systems with high Mg contents the main product was anhydrous amorphous Mg carbonate. The product of the reaction shifted towards protodolomite at Mg/Ca = 1, and in the systems with Mg/Ca < 1, the reaction shifted towards the precipitation of crystalline calcite. These findings demonstrated that high Mg contents hinders the long-range orders in the Mg–CO3 system, implying that there is an intrinsic difficulty in obtaining the rhombohedral arrangement of magnesite. The observations were associated to the disorder among the CO32− groups due to their limited degrees of freedom around the small Mg2+ cations (radii of Mg2+ is ca. 30% smaller than Ca2+), which may mean that the dolomite/magnesite problem is a stereochemical issue: the strong electrostatic attraction to the Mg2+ cations prevents the rotation and tilting of the carbonate groups, hindering the crystalline ordering in the precipitation step. Moreover, the tight cage arrangement in the RC geometry of the magnesite structure reduces the degrees of freedom of the carbonate groups, which also hampers the precipitation step.17,20
Inspired by the experimental findings on the catalytic role of the carboxyl functional groups on the precipitation of dolomite (the previously described studies on microbial mediation for dolomite precipitation), one interesting work investigated the use of carboxylated polystyrene microspheres as catalysts for magnesite precipitation at room temperature. The authors noticed that no precipitates were detected up to 60 days of reaction, and magnesite precipitated after 72 days in the system containing high-density polystyrene microspheres. Contradictorily, the carbonation with low density polystyrene did not yield magnesite. The reported results indicated that the site density and competitiveness of the ligands determines the success of their catalytic role on the Mg carbonates precipitation. Moreover, the authors also hypothesized direct precipitation of magnesite without an intermediate phase.112
It has been also demonstrated the utilization of deep eutectic solvents to enable the solvothermal synthesis of magnesite at ambient pressures and moderate temperatures.14,114 A single crystal of magnesite has been synthesized with 1 MgCl2·6H2O: 2 urea as deep eutectic solvent, keeping the system in autoclave for 6 h at 140 °C and pCO2 of 1 bar.14 More recent experiments have expanded the investigation on the hydrothermal synthesis of magnesite (3 h at 180 °C), utilizing deep eutectic solvents or molten salts (1 MgCl2·6H2O: 2 urea, MgCl2·6H2O, and MgSO4·7H2O) and variable sources of carbon and mineralizers (Table 6). It was observed that the precipitation of magnesite has a direct dependence with the pH of the system, noted to occur in the pH range between 5.4 to 10.5 (together with other Mg carbonates and impurity phases).114 Considering the speciation of carbonate ions according to the pH range in brines (Fig. 15), these findings indicate that the precipitation of magnesite might be more favorable in presence of HCO3− ions rather in systems rich in carbonate ions. Overall, the studies of the carbonation products of MgCl2 precursors indicate that the precipitation of magnesite requires high temperatures and moderate alkalinity, unless a catalytic agent (such as the high-density polystyrene microspheres) is added to assist the nucleation of magnesite in the system.
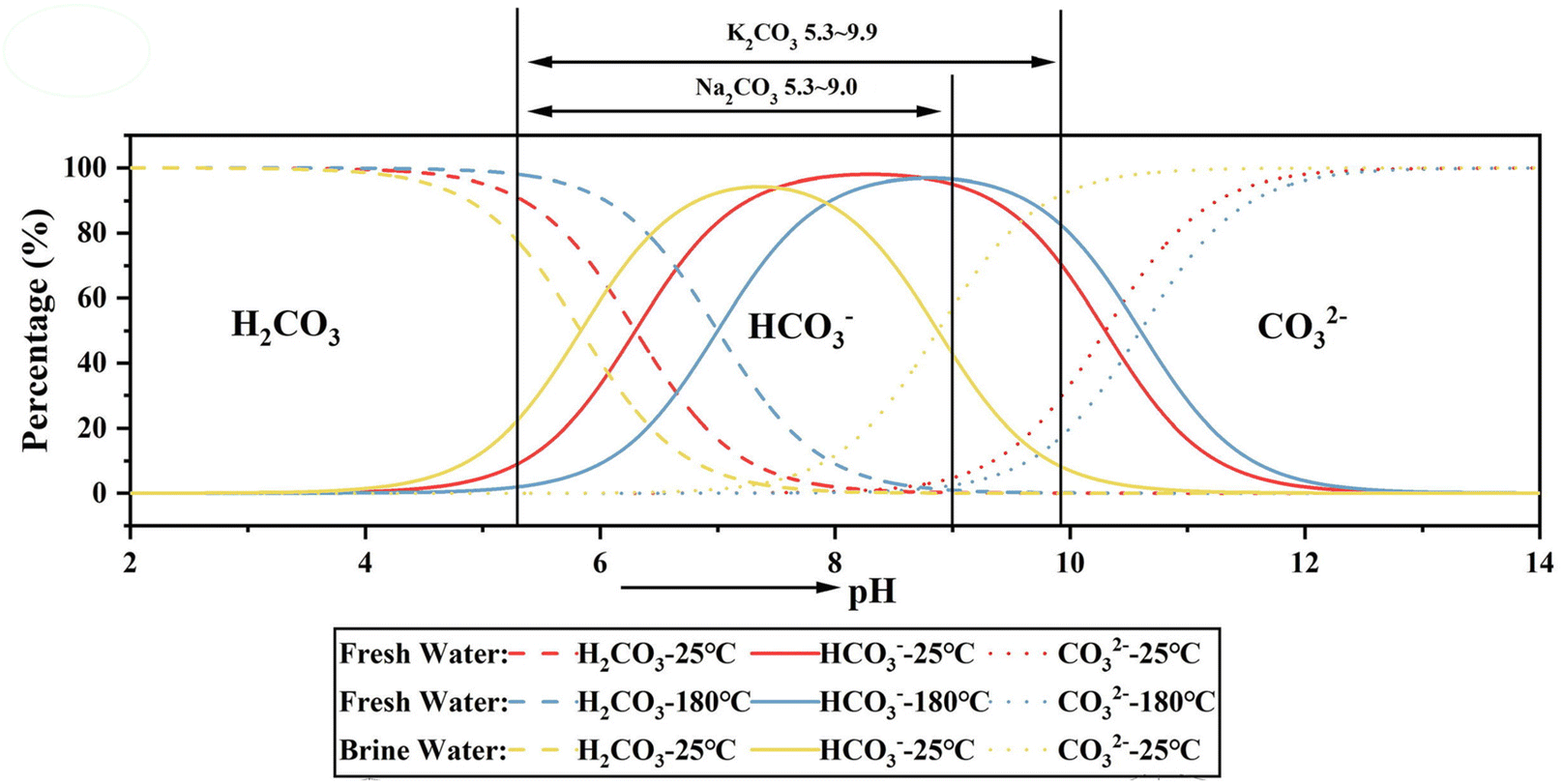 | ||
| Fig. 15 Carbonate ions speciation in brine and fresh water as a function of pH and temperature. Reproduced with the publisher's permission from Chai and Xu, 2022114 (© 2022 The Society of Powder Technology Japan. Published by Elsevier B.V. and The Society of Powder Technology Japan. All rights reserved). | ||
| Phenomena | Reactions | ΔH (kJ mol−1) | Eq. |
|---|---|---|---|
| Precipitation of Mg hydroxide | Mg2SiO4 + 2H2O → 2Mg(OH)2 + SiO2 | −99.7 | 11 |
| Mg recovery to solution | 2Mg(OH)2(l) ⇌ 2Mg2+(aq) + 2OH−(aq) | 12 | |
| 2Mg2+(aq) + 2OH−(aq) ⇌ 2MgO + 2H2O | 162.4 | 13 | |
| Dehydration of Mg2+ cations | Mg2+(H2O)7 → Mg2+(H2O)6 + H2O | 78.2 | 14117 |
| Mg2+(H2O)7 → Mg2+ + 7H2O | 1931 | 1541 |
|
| Global carbonation reaction of chrysotile | Mg3Si2O5(OH)4 + 3CO2 → 3MgCO3 + 2SiO2 + 2H2O | −64 | 16118 |
| Decomposition of AS and ABS | 2(NH4)2SO4(s) → (NH4)2S2O7(s) + 2NH3(g) + H2O(g) | — | 17 |
| (NH4)2SO4(s) → NH4HSO4(s) + NH3(g) | 110.5 | 18119 |
|
| 2NH4HSO4(s) ⇌ (NH4)2S2O7(s) + H2O(g) | 19 | ||
| NH4HSO4(s) → NH3(g) + H2O(g) + SO3(g) | 340.8 | 20119 |
|
| (NH4)2SO4(s) → 2NH3(g) + H2O(g) + SO3(g) | 451.4 | 21119 |
|
| ‘ÅA2’ carbonation route | (NH4)2SO4(aq) ⇌ NH4HSO4(aq) + NH3(g) | — | 22 |
| NH4HSO4(aq) ⇌ NH4+(aq) + HSO4−(aq) | — | 23 | |
| HSO4−(aq) ⇌ H+(aq) + SO42−(aq) | — | 24 | |
| NH4OH(l) + NH4HSO4(aq) ⇌ (NH4)2SO4(aq) + H2O(l) | — | 25 | |
| Mg3Si2O5(OH)4 + 3NH4HSO4 ⇌ 3MgSO4 + 2SiO2 + 3NH3 (g) + 5H2O | 188.3 | 26119 |
|
| Mg3Si2O5(OH)4 + 3(NH4)2SO4 ⇌ 3MgSO4 + 2 SiO2 + 6NH3 (g) + 5H2O (g) | 244.3 | 27119 |
|
| 5MgSO4(s) + 10H2O(l) + 10NH3(g) + 4CO2(g) ⇌ Mg5(OH)2(CO3)·4H2O(S) + 5(NH4)2SO4(aq) | — | 28 | |
These gas–liquid–solid reaction pathways are naturally occurring processes (within geological timescales of many years) and the challenges in mimicking the reaction network arise from the different effects promoted by changes in temperature and pH: high temperatures favour the kinetics of silicate dissolution and precipitation of carbonates, while also leading to lower CO2 solubility. On the other hand, low pHs (<4) results in higher solubility of the Mg2+ ions, whereas the carbonates precipitation requires alkaline pHs (>8). Consequently, there is a non-trivial dependence in the combined CO2 absorption–mineralization behaviour of Mg carbonates growth with temperature and pH. These contradictions have motivated the development of different methods for CO2 mineralization which can be broadly separated in three main approaches: the first based on pH-swing, the second based on the usage of elevated temperatures and CO2 pressures,105,115 and the third reacts Mg bearing silicates with CO and H2O via water–gas shift reaction to produce magnesium carbonates and H2 (g).115,120 However, the latest route results in a complex mixture of products that needs further separation, thus limiting their use to low-value rather than advanced technology applications.
The first two approaches have been extensively studied by Zevenhoven et al.,1,118,121–124 leading to two main carbonation routes, which are seen among the most economically feasible pathways for mineral carbonation of Mg silicates.5,125 They are referred as ÅA routes and separated into two main approaches: (ÅA1) semi-dry carbonation (at 500 °C and p(CO2) of 20 bar) conducted after thermal solid–solid Mg extraction,118 and (ÅA2) wet carbonation conducted after hydrothermal extraction.122 The ÅA1 route conducts the solid–solid extraction of Mg from serpentine utilizing a mixture of ammonium sulphate and/or bisulphate as flux agent under 400–440 °C for 30–60 min, leading to the precipitation of Mg and Fe sulphates (and double salts with NH4+ ions), which is then converted to Mg hydroxide through dissolution in water and addition of ammonia (raising the system pH to 10–12). The aqueous solvent is then recovered for a next extraction step; thus, allowing further semi-dry carbonation (at 500 °C and p(CO2) of 20 bar) for precipitating magnesite, obtaining a binding capacity of 240 kg CO2 per ton serpentinite.118 The ÅA2 route has lower energy requirements for Mg extraction but won't benefit from reaction heat released during a carbonation step at 500 °C. Alternatively, conducting the leaching of Mg2+ cations with solutions of ammonium bisulphate or sulphate produces MgSO4, and conducting subsequent carbonation at low temperatures (<60 °C) yields nesquehonite (MgCO3·3H2O). The usage of ammonium bisulphate or sulphate did not result in remarkable changes in the thermal extraction rates, but it was noticed that employing concentrations of ammonium sulphate slight above the stoichiometric amounts leads to better extraction of Mg.118 Dry/wet extraction of Mg and wet/dry carbonation of Mg salt can be combined in several ways, giving ÅA routes 3, 4 and 5 primarily depending on CO2 concentration in the gas processed, and the availability of waste heat.121 For the latest ÅA routes membrane electrodialysis is used to separate ions from aqueous streams, and for the recovery of the ammonium salts, processing it into sulphuric acid or ammonium bisulphate and aqueous ammonia, used for acid leaching of Mg from rock and raising pH for the carbonation of magnesium sulphate, respectively.126,127
In a third approach, the use of amines as catalysts have been demonstrated, indicating a promising pathway to avoid energy and/or chemical intensive processes.115 The amines act as nucleophilic agents to capture CO2, raise the alkalinity and facilitate the carbonate precipitation. The CO2 dissolution into the liquid phase can be achieved by employing amines or amino acid groups to bind CO2, or by increasing the CO2 hydration to generate HCO3−(aq) and CO32−(aq) via enzymatic catalysis of e.g. carbonic anhydrase. Depending on the composition of the solvent, CO2 capture via amino groups in aqueous environments proceeds through two main mechanisms: the formation of carbamates induced by primary and secondary amines, and the formation of bicarbonates promoted by sterically hindered or tertiary amines. Tertiary and cyclic amines are hypothesized to be more promising catalysts due to its higher chemical and thermal stability (thus, easier recovery), as well as the characteristic behaviour as Brønsted bases and the reaction stoichiometry of one molecule of amine to dissolve one CO2 molecule. However, the possibilities of generating harmful products makes most amines reagents undesirable, leading to the preferable selection of environmental-friendly alternatives such as amino acids salts. For example, glycine can induce the formation of the carbamic acid followed by the production of the respective carbamate and zwitterionic glycine.115,116 Other Mg chelating agents (such as citrate, acetate, oxalate, ethylenediaminetetraacetic acid, among other organic ligands) have been also proposed to increase the protonic concentration needed for dissolution. One limiting factor in the extraction of magnesium cations from silicates is the formation of a passivating layer of SiO2 on the silicates which hinders the extraction rate to the aqueous phase, having earlier speculated on the usage of Si chelating agents (e.g. catechol or metallic organic frameworks) to assist the cleavage of the Si–O bonds, thus producing Si oxyanions and facilitating the dissolution of silicate minerals.3,115
The phase equilibrium of the reaction system employed in the ÅA routes have been investigated through the solubility and reactivity of the magnesium and ammonium precursors.124 It was found that the solubility of Mg sulphate is highly affected by the concentrations of ammonium sulphate, which should be taken into consideration when evaluating the ideal Mg concentrations in carbonation reactions. It is expected that the saturation index of the carbonate minerals will be affected by the formation of intermediary double salts. This affects the degree of carbonation of the Mg precursors, and therefore, the overall feasibility and economic viability of the method. Moreover, it has direct effect on the favored type of carbonate to precipitate, and on the morphology of the carbonates. Fig. 16a shows a ternary phase diagram for the MgSO4–H2O–(NH4)2SO4 system, where the formation of mixed salts are noticed at 25 °C with MgSO4 <50 wt% and -(NH4)2SO4 <40 wt%, showing that the double salt MgSO4·(NH4)2SO4·6H2O had easy crystallization in the majority of the investigated SO42−:CO32− molar ratios.139
 | ||
| Fig. 16 (a) Ternary phase diagram of the ammonium sulphate, magnesium sulphate and water system at 25 °C. The letters in the diagram represents the coexisting solids in the supersaturated solution: A represents the equilibrium MgSO4–H2O, B shows the solubility of MgSO4, at C two main solids were identified MgSO4·xH2O and MgSO4·(NH4)2SO4·6H2O, D shows the precipitation of only MgSO4·(NH4)2SO4·6H2O, the E, F, and I indicates 100% or the represented phases in the triangle vertices, G shows the solubility of (NH4)2SO4, and H corresponds to (NH4)2SO4 and MgSO4·(NH4)2SO4·6H2O (Liu et al., Copyright© 2018, American Chemical Society).139 (b) Equilibrium phase diagram of the ternary diagram of MgCO3–MgSO4–H2O system at 25 °C. A and B represents pure MgSO4 and pure MgCO3, respectively, C and D shows the solubility of MgSO4·7H2O and MgSO4, respectively, and at point E, epsomite and magnesite co-precipitate, F is the solubility of MgCO3 (0.021%) (Li et al., Copyright © 2021, American Chemical Society).140 | ||
The phase diagram of the MgCO3–MgSO4–H2O system at 25 °C is shown in Fig. 16b, where the solubility of magnesite is seen at point F (0.021%). In this system no double salts were detected, but it was noticed preferential crystallization of epsomite MgSO4·7H2O and magnesite in the invariant curves DE and EF, respectively. The crystallization area of magnesite is seen in the area FEBF, while epsomite crystallizes in the CEDC area, and the eutectic region of both minerals is seen in the region CEBC.140 That indicates that for epsomite to precipitate, low contents of magnesite are needed.
Other authors have focused on the investigation of the carbonation mechanisms and the effect of variable reactional parameters. For all reported work on the carbonation of magnesium sulphate, the main products obtained are hydrated magnesium carbonates (mainly hydromagnesite and nesquehonite), double salts of Mg2+ and NH4+ or minerals of mixed cations. Table 8 summarizes the main reaction conditions applied and the obtained products.
| Precursors | Temperature (°C) | Pressure/stirring | Concentration | Reaction time (min) | Products |
|---|---|---|---|---|---|
| MgSO4·7H2O and (NH4)2CO3 | 20–50 | 1 atm/500 rpm | 0.2–0.5 M, equimolar | 480–72 | NQ2 |
| MgSO4 and CO2 after pH raise with NaOH | 20 | 36 atm/1000 rpm | 11700–13000 ppv Mg |
360 | HY, yield <0.1%134 |
| MgSO4 and (NH4)2CO3 | 80 | Up to 440 psi/800 | 2–3 M | 60 | HY, yield of 44–46%134 |
| MgSO4 and NH4HCO3 | 80 | Up to 758 psi/800 | 2–3 M | 60 | HY, yield of 18–22%138 |
| MgSO4·7H2O, (NH4)2CO3 and SDS | 20 | 1 atm/500 rpm | 0.2 M equimolar, SDS from 0 to 2.32 × 10−3 M | 210 | NQ141 |
| MgSO4·7H2O and (NH4)2CO3 | 20 to 60 | 1 atm/parallel, forward or reverse feeding | 0.25 M of MgSO4, and 0.5 M of (NH4)2CO3 | 60 | NQ (HY starts to form at 50 °C)142 |
| MgSO4·7H2O and (NH4)2CO3 | 70 to 80 | 1 atm/parallel feeding | 0.25 M of MgSO4, and 0.5 M of (NH4)2CO3 | 60 | HY142 |
| MgSO4·7H2O and (NH4)2CO3 | 40 | 1 atm/parallel feeding | 0.5 to 0.7 M of MgSO4, and 0.5 M of (NH4)2CO3 | 60 | NQ and ammonium magnesium carbonate → HY142 |
| MgSO4 and CO2 after pH raise with NH4OH | 25 | 1 atm/none (bubble action) | 0.06–0.36 | 60 | NQ, yield of 72% (Fe absent, removed during pH raise as FeOOH)122 |
Geng et al. (2019) have evaluated the kinetics of nesquehonite growth considering initial soluble systems of magnesium sulphate and ammonium carbonate with variable concentrations. It was assumed that the formation of nesquehonite passes through three main stages: (1) a nucleation induction period, (2) diffusion of dissolve species to crystal surface, (3) adsorption of solute on crystal surface, (4) construction of solute in crystal lattice, and (5) dissolution or dissociation of crystals in liquid phase. The authors verified that the reaction rate is controlled by the mass transfer rate of the step 2, while the steps 3 and 4 have stronger impact on the morphology of the final products. Thus, when the mass transfer rate is too high (due to e.g., high concentrations of reagents, elevated temperatures, or high stirring speed), the formation of the solid products will be faster than the crystal growth rate, which impacts the obtained morphology greatly. In practical terms, it was observed that the time required for the nucleation induction and equilibrium steps decreases with increasing temperature, concentration of the precursors, stirring speed and the [(NH4)2CO3]/[MgSO4] ratio, and that these parameters have a strong effect on the morphology of the precipitated nesquehonite.2
As a follow-up work, Geng et al. (2020) investigated the effect of sodium dodecyl sulphate on the carbonation of magnesium sulphate aiming precise tailoring of the morphology of the nesquehonite products. The authors noticed that when sodium dodecyl sulphate is ionized in solution, it has preferential combination with the active sites of Mg2+(H2O)5−x along the (100) crystallographic direction, this generates a repulsion of the liquid phase from the hydrophobic chain of sodium dodecyl sulphate, which in turn hinders the crystal growth in the (100) direction. It was also noticed that the presence of sodium dodecyl sulphate extended the nucleation induction and crystallization steps, also increasing the lattice spacing of the most significant (hkl) reflections of nesquehonite. The authors concluded that when the nesquehonite growth is hindered in the radial crystal plane, it shifts the growth towards the axial crystal plane, thus increasing the aspect ratio of the obtained nesquehonite's whiskers.141
Deng et al. (2019) have evaluated the pilot scale process of the carbonation of MgSO4 with (NH4)2CO3, studying the effect of varying the process’ parameters (feeding mode, pH, temperature, and reagents’ concentration) on the obtained products. It was observed that the highest carbonation ratio was obtained with the parallel feeding (83.3 wt% yield), when compared with the forward and reverse feeding (80.4 and 78.4 wt%, respectively). It was also reported that the raising of pH increased the carbonation ratio, favoring the precipitation of nesquehonite in a pH range of 7 to 9.5; above that the reaction shifts towards the formation of hydromagnesite. When the variations of temperature were considered, the authors noticed that nesquehonite was the main product from 20 to 40 °C, and after 50 °C the formation of hydromagnesite was induced and completely more favored at 70 °C. The effect of increasing the concentration of magnesium sulphate was also considered, noting that for molar ratios ([Mg2+]/[CO32−]) equal or greater than 1 nesquehonite starts to precipitate along with ammonium magnesium carbonate trihydrate. Thus, the increasing concentration of MgSO4 (above or equal equimolar concentrations) led to the formation of the double salt (NH4)2Mg(CO3)2·3H2O which was found to convert into hydromagnesite with time, decreasing the purity of the nesquehonite product. The authors have reported the molar ratio ([Mg2+]/[CO32−]) of 1.5 at 40 °C as the best performing conditions for higher yield and higher nesquehonite purity.142 Therefore, the reported investigations on the carbonation of Mg sulphate at low temperatures and ambient pressures indicate that nesquehonite in the major Mg carbonate phase expected to precipitate in such systems.
| Precursors | Temperature (°C) | Pressure (bar) | Additive/stirring | Reaction time | Products |
|---|---|---|---|---|---|
| 0.0025 M of Mg(OH)2 and NaHCO3 (1:1) |
20 | 1 | Mg acetate (0.01 to 1 M)/150–200 rpm in the initial 30 min | 1 day | • 0, 0.01 and 0.1 M of Mg acetate: BR, NQ, traces of Dy |
| • 1 M of Mg-acetate: BR and NQ151 | |||||
| 0.0025 M of Mg(OH)2 and NaHCO3 (1:1) |
20 | 1 | Mg acetate (0.01 to 1 M)/150–200 rpm in the initial 30 min | 28 days | • 0, 0.01 and 0.1 M of Mg acetate: DY and BR |
| • 1 M of Mg acetate: GI151 | |||||
| 0.34 M Mg(OH)2 | 150 | 70 | Potassium citrate (0–3 M)/800–1200 rpm | 6 to 12 h | • 0 M Kcit: HY after 6h, and MS after 12 h |
| • 1.5 M Kcit, 6 h: MS and HY | |||||
| • 2 M K cit, 6 h: MS68 | |||||
| Single crystal of Mg(OH)2, scCO2 | 50 | 90 | 0.14 wt% of dissolved water in the scCO2 | 3 and 5.5 h | Amorphous magnesium carbonate (3 h) and NQ (5.5)150 |
| Powdered Mg(OH)2 | 400 | 36 | 40 bar 10% H2O/PTGA | 2 h | MS (82% yield)146 |
| Mg(OH)2–H2O–CO2 slurry | 26 (plus 24 h heat aging step from 20 to 90 °C) | 50 | With and without NaOH (2 M) | 48 h | MS (with NaOH) and HY-MS-BR solid mixture (without)148 |
| Mg(OH)2–H2O–CO2 slurry | 90 (no heat aging step) | 50 | — | 12 days | HY148 |
| Mg(OH)2–H2O–CO2 slurry | 26 (no heat aging step) | 50 | 2 M NaOH | 24 h | DY148 |
| Mg(OH)2–H2O–CO2 slurry | 25 | 50 | NaOH | 1 h 45 min | NQ26 |
| Mg(OH)2–H2O–CO2 slurry | 25 | 1 | — | 40 min | NQ26 |
| Mg(OH)2–H2O–CO2 slurry | 90 | 50 | NaOH | 1 and 4 h | HY (1 h) and MS (4 h)26 |
| Mg(OH)2–H2O–CO2 slurry | 80–150 °C | 15.2 | Al2O3 or MS seeds | 2 h | • 150 °C, no seeds, Al2O3: HM |
| • 150 °C, MS seeds: MS149 | |||||
| Mg(OH)2–H2O and CO2 (g) | 50 | 91.2 | — | 1 to 56 days | • 1 to 10 days: HY, NQ and unreacted BR |
| • 14 days: 77% HY, 12% NQ, 6% BR, and 3% MS | |||||
| • 28 days: 60% HY and 40% MS | |||||
| • 56 days: 98% MS and 2% NQ15 | |||||
| Mg(OH)2–H2O and CO2 (g) | 35 | 91.2 | — | 1 to 135 days | • 63 days: 92% NQ and 8% HY |
| • 135 days: 92% NQ plus 2% HY and 6% MS15 | |||||
| 0.5 M Mg(OH)2 and CO2 (g) | 100 to 200 | 10 to 30 | α-Al2O3 particles or activated carbon (AC, 2000 m2 g−1) | 60 min | • 200 °C → magnesite and unreacted brucite |
| • 150 °C → MS is formed at 30 bar and all AC systems | |||||
| • 100 °C → HM145 | |||||
| Mg(OH)2–H2O and CO2 (g) | 35 | 111.5 | — | 1 to 140 days | • 63 days: 97% NQ and 3% HY |
| • 140 days: 84% MS and 16% NQ15 |
Some reported experiments have investigated the direct gas–solid carbonation of brucite at elevated temperature and pressures to convert it into magnesite, but the process parameters need to be tuned according to the kinetics of the system: if the rate of dehydroxylation is much higher than the carbonation, the obtained product will be MgO instead of MgCO3. It has been observed the complete conversion of Mg(OH)2 to magnesite under 500 °C, p(CO2) of 36 bar, and p(H2O) of 4 bar for ca. 7 h, being needed to apply some p(H2O) to prevent fast conversion in MgO. It was also noticed that intrinsic water (instead of steam) enhances the degree of carbonation of brucite, thus it was suggested that employing a fluidized bed reactor assists magnesite precipitation by avoiding the buildup of a passivation layer of carbonates on the active nucleation sites.146 The chain of reactions involved in the gas–liquid–solid carbonation processes are favored by the usage of moderate temperatures (greater than 90 °C) and p(CO2) higher than 50 atm. Such conditions help to overcome several rate limiting reactions such as CO2 hydration and carbonate precipitation but suffer from the buildup of passivating silicate-based layers that inhibit the chemical reactions.115,120
The Mg(OH)2–H2O–CO2 slurry systems can yield different hydrated/basic Mg carbonates polymorphs at ambient temperature and p(CO2) < 55 bar, but higher temperatures and longer reaction times are required to produce magnesite. In such slurry systems it has been observed that the precipitation of magnesite is preceded by the formation of hydromagnesite if temperature becomes ≫50 °C. The conversion of hydromagnesite to magnesite is the rate determining step for MgCO3 precipitation, which can be accelerated in conditions of high salinity, high temperatures, high p(CO2) and organic additives (e.g. monoethylene glycol).147 When high pressures (100–150 bar) and high temperatures (150–180 °C) are employed, the precipitation of magnesite is observed without any noticeable formation of intermediate precipitates. Montes-Hernandez et al. (2012) have observed that in conditions of ambient temperature and moderate pressures (50 bar), the precipitation of magnesite is facilitated by the presence of NaOH, which is assumed to have a catalytic role in the reaction. The authors propose that the alkalinity of the solution contributes to increase the CO32− concentration in the slurry, thus inducing the exclusive formation of magnesite in the heat aging step (20 to 90 °C).148 A follow-up work from this same research group have done in situ investigations on the nucleation and growth of magnesite in NaOH medium, high temperatures (90 °C and 50 bar), and moderate pressures, being observed that the initial precipitate in the system is hydromagnesite (60 min) followed by magnesite after 240 min.26
Other systems of wet carbonation evaluated the effect of surface wettability of heterogeneous nucleation sites (seeds) on the brucite carbonation. The authors utilized alumina particles as hydrophilic seeds and activated carbon powder145 or powdered magnesite149 as hydrophobic seeding sites. The seedless experiments at 200 °C yielded unreacted brucite and increasing magnesite amounts with pressure (20 to 35 bar), while at 100 and 150 °C mainly unreacted brucite and hydromagnesite were obtained (minor amounts of magnesite were observed at scCO2 of 30 bar). The authors observed that the addition of the nucleation seeds induced higher precipitation of magnesite only for the activated carbon systems at temperatures ≥150 °C. The use of alumina seeds did not promote remarkable effects on the carbonation products (Fig. 17).145 Thus, the role of the hydrophobic sites was evident in the work.
 | ||
| Fig. 17 Phase diagram with the reaction products of brucite carbonation at 100 to 200 °C and 10 to 30 bar, in presence or absence of alumina and activated carbon seeds (Atashin et al., ©2017 Elsevier Ltd. All rights reserved).145 | ||
The mechanism of nesquehonite precipitation from brucite at scCO2 (90 bar) and 50 °C have been investigated by Zhang et al. (2021) using in situ atomic force microscopy. The authors observed that the nucleation and growth of nesquehonite occurs through the initial formation of amorphous Mg carbonates as intermediate metastable precipitates. Over time, a consistent mass transfer between the amorphous and crystalline phases was observed, consistent with the metastability of the amorphous phase with respect to nesquehonite. The in situ observations led to identification of the following reaction steps involved in the carbonation of brucite at high temperatures and pressure: (i) formation of etch pits on the brucite via release of Mg2+ cations facilitated by the adsorbed water, (ii) precipitation of amorphous magnesium carbonates, (iii) nesquehonite nucleation and growth on the amorphous Mg carbonates, and (iv) surface passivation by the coverage of the nucleation template with the nesquehonite crystals.150
An earlier work on the carbonation of brucite at scCO2 (90 and 110 atm) and low to moderate temperatures (35 and 50 °C) have reported that in all utilized conditions, nesquehonite and hydromagnesite were the first phases to precipitate. However, a higher formation of magnesite was observed only after 56 days of reaction at 50 °C and 90 atm or after 140 days of reaction at 35 °C and 110 atm. Interestingly, it was noticed that moderate temperatures (50 °C) favors the formation of hydromagnesite and nesquehonite, which is slowly converted to nesquehonite as a major phase, being needed high pressures (110 bar) to finally form magnesite as sole product (Table 7).15 In that work, the authors do not mention the formation of amorphous Mg carbonates as intermediate, however the presented XRD patterns of the reaction products during the initial days clearly shows the broadening of the peaks and fluctuations on the XRD background, which are clear indications of the presence of amorphous contents. Indeed, the precipitation of amorphous magnesium carbonate phases as intermediate products prior the hydrate Mg carbonates precipitation has been confirmed with in situ atomic force microscopy, evaluating brucite carbonation in supercritical CO2.150
Organic additives can play a critical role in facilitating the carbonation kinetics and formation of different Mg carbonate polymorphs, yet the mechanisms of these reactions remain unclear. Dong et al. used potassium citrate to improve the kinetics of the brucite carbonation to form magnesite. The authors observed that potassium citrate shorten the synthesis duration for magnesite precipitation by 50% (total 6 h) under 0.7 MPa and 150 °C using 2 M solution of the ligand. The effects were attributed to the catalytic activity due to the increased ionic strength, which may have contributed to decreasing the activity and the thickness of the second hydration layer of the Mg(H2O)62+ cations.68 Nguyen et al.151 evaluated the effect of Mg acetate (from 0.01 to 1 M concentration) as an additive on the carbonation of brucite at ambient temperature and pressure. Acetate was found to enhance the reaction kinetics and degree of reaction in which brucite reached ca. 100% carbonation degree vs. only 50% in the plain sample. Nesquehonite was unstable and quickly converted to dypingite after 7 days of reaction. However, the presence of acetate delayed the conversion of nesquehonite, and once the conversion happened, giorgiosite [Mg5(CO3)4(OH)2-5·6H2O] was formed. The authors explained the phenomena by the strong catalytic effect of acetate in enhancing the carbonation of brucite and open a new pathway to form giorgiosite instead of dypingite as the normal conversion of nesquehonite. Given that giorgiosite is a relatively new hydrated Mg carbonate phase in the literature, more follow-up research is needed to understand its thermodynamic role in the Mg carbonates series.151 Note that for a large-scale (many tonnes per hour) CO2 mineralisation process, the use of chemical additives requires that these are almost completely recovered for re-use for economically viable operation.
Overall, the reported evaluation of the products obtained from the carbonation of brucite shows a continuous interconversion between the hydrated Mg carbonates over time, as a sign of the metastability of these phases. Moreover, the precipitation of magnesite is observed only at moderate to high temperatures (>80 °C) and/or moderate to high pressures (>15 bar).
5. Applications of Mg carbonates in construction materials
Mg based inorganic cements include a broad class of materials under development for over 150 years of research. A broad review on the types of Mg based cements and their most common applications has been published by Walling and Provis in 2016, which the reader is recommended to refer for broader understanding on the subject.69 These include Mg phosphate, Mg silicate hydrate, Mg oxysalt (both chloride and sulphate), and reactive MgO cements (RMC). Among them, only reactive MgO cements and Mg oxychloride cements have compositions including Mg carbonates, but in the later system the formation of carbonate phases is undesirable due to stability losses, offering low relevance for developing carbon capture and utilization routes within Mg oxychloride systems.69,70 As the scope of the current review is focused Mg carbonate materials, we offer a brief overview on the applications of Mg carbonate materials in the construction sector.As discussed above, even though Mg and Ca share generalized chemical properties comprising the alkali-earth metals group, they also possess substantial differences in reactivity arising from the distinctive features of electronegativity, ionic radii, and orbital energy levels. That leads to vast differences on the phase formation within the (MgO, CaO)–Al2O3–SiO2 system, and the attempts of mimicking the OPC systems with MgO does not lead to analogue key hydraulic phases in OPC such as Ca3SiO5, Ca3SiO5, and Ca3Al2O6.69 Alternatively, the CO2 capture within hydrated reactive MgO cements results on the precipitation of hydrated Mg carbonates (mainly nesquehonite, and hydromagnesite), which have the binding properties necessary to develop a dense microstructure network with the strength required in cementitious materials.152 As a matter of fact, the strength development of the reactive MgO cements based cements has been demonstrated to be higher than the ordinary Portland cement (OPC) counterparts with similar composition.153
The carbonation of MgO is usually associated with conditions of high pressures and temperatures, but the process conditions can be tuned according to the morphological properties of the MgO precursors.154 The reactivity of the reactive MgO cements precursor is affected by its particle size and surface area, which together with pressure, humidity, and temperature determines the type of hydrated Mg carbonate to precipitate. The hardening and mechanical properties as well as durability of the reactive MgO cements-based formulations are limited by the degree of hydration and carbonation of the reactive MgO cements. The hydration of the reactive MgO cements leads to brucite growth in the pore solution as well as the formation of a brucite passivation layer on the surface of unreacted MgO particles; but the later constitutes a different structure due to its connection with the MgO species, which facilitates its dissolution when compared with the precipitated brucite. Thus, the hydration of the unreacted MgO is determined by the solubility of the brucite passivation layer. For this reason, several studies have aimed at increasing the hydration degree of the reactive MgO cements via addition of hydration agents (e.g. HCl, MgCl2, Mg acetate, etc.), hydrated Mg carbonates blends or nucleation seeds, and/or high temperature curing.152 It has been found that a considerable increase in durability and strength of MgO cements can be achieved via addition of ferrous sulphate, which induces the formation of Mg oxysulphate phases during the curing procedure. Other approaches have probed cementitious materials based on MgO/OPC and blast furnace slag, resulting on the formation of magnesian calcite and nesquehonite, which led to lower porosity and higher microhardness than traditional OPC materials. However, the large-scale feasibility of the method needs further developments: reactive MgO cements have not yet demonstrated successful performance under ambient conditions although the demonstrated compressive strengths match the requirements for precast materials. The most optimistic forecast in the field considers that further developments to enhance stability are still to be met.69
Glasser et al. have investigated the production of hydrated Mg carbonates blocks by utilizing thermally-activated nesquehonite and following similar procedures for clay or cement-blocks preparation.113 The nesquehonite materials were obtained in a laboratory pilot-scale reactor, via carbonation (CO2 bubbled in alkaline solutions) of MgCl2. Two methods were proposed for preparing the nesquehonite blocks: (i) the nesquehonite powder undergo heat activation at 100–250 °C, then mixed with water, shaped, and cured at ambient conditions, and (ii) the nesquehonite is mixed with water, then shaped and cured at 60–80 °C. In the first method, the heat treatment partially removes the coordinated water of nesquehonite (forming an amorphous phase), which is then regenerated during the curing procedure upon considerable heat release from the hydration process. The authors observed that at room temperature, nesquehonite slowly decomposes into dypingite and finally hydromagnesite, but that would require several months to happen at ambient conditions. Employing higher temperatures (60–80 °C) decreases the curing period to several hours or few days. The obtained blocks showed compressive strength up to 8 MPa, thermal conductivity of 0.21–0.33 W K−1 m−1, and bulk densities of 600–900 kg m−3. Thus, the potential usage of nesquehonite blocks was suggested for producing panels for thermal insulation materials. However, the metastability of nesquehonite (depending strongly on the humidity of the gas (air) it is stored in, besides temperature and pressure) may hamper its commercial applications in such applications and further developments are still needed to bring the product to market.113
Indeed, the needle-like shape of nesquehonite is considered an important feature in making it the most promising hydrated Mg carbonate for producing alternative binders. The thermal activation of nesquehonite is also used for applications in plasterboard materials. The materials are also prepared via the traditional methods for cement/clay hardening from pastes (liquid to solid ratio of 0.8) into cubes subjected to seven days curing. It was observed that the rehydration of thermally activated nesquehonite not only reconstitutes its mineral phase, but also forms dypingite and hydromagnesite (leading to CO2 losses). The materials showed compressive strength between 4 to 15.6 MPa (larger particle sizes yielded higher compressive strength), which is higher than the observed for plasterboard materials made from gypsum (3–8 MPa). Despite the promising results, further studies are needed to evaluate the performance of nesquehonite plasterboard in terms of stability, thermal conductivity and fire suppression properties when compared to gypsum materials.84 Life cycle assessment (LCA) has envisaged promising prospects for the replacement of gypsum with nesquehonite in plasterboard materials. However, the market competitiveness of the product is dependent on the route for nesquehonite production, and the availability of renewable energy resources for the thermal activation (i.e. dehydration) of nesquehonite.155
Other investigations have produced composites of nesquehonite and silica gel (1:1 dry mass ratio) for applications as thermal energy storage (TES) materials. Both materials have potential for energy storage when dehydrated: silica gel dehydrates at 65–105 °C and has an energy storage capacity of 0.6 MJ kg−1, while nesquehonite at 25 °C has a theoretical energy storage of 1.0 MJ kg−1. That has been used to design a thermal energy storage rector based on the chemisorption properties of the materials, which is used to improve the efficiency of an exhaust air heat pump (standard Nordic) heating system. The nesquehonite composites undergo dehydration–hydration cycles, increasing the reactor temperature by 5–8 °C. Moreover, employing a three-reactor setup can increase the efficiency of the hybrid reactor by 49%. That is a promising application for the hydrated Mg carbonates materials, but further scale-up experiments are still needed before creating a disruptive business model based on the material.6,156
Given that dolomite and magnesite are the most stable phases among other Mg carbonates, they are the only Mg carbonates phases with current commercial applications in construction materials. Nowadays, supplementary cementitious materials containing both anhydrous minerals are used as fillers in cementitious composites produced with waste-streams rich in these phases e.g. fly ashes, steel slags and metakaolin.157–159 Thus, the direct utilization of anhydrous Mg carbonate phases offer the most promising prospects for direct commercial applications as filler agents in construction materials. However, lowering the energy requirements for magnesite/dolomite production is still needed for enabling the commercial feasibility within applications in the construction sector, depending largely on the market value of the carbonate and other (solid) products, besides the cost savings from CO2 emissions certificates or CO2 taxes.
6. Feasibility of carbon capture and utilization routes aiming for Mg carbonate production
Life cycle assessments (LCA) have been employed to evaluate the environmental feasibility of a given process. When a carbon capture and utilization route is analyzed, the feasibility is primarily dependent on the environmental impact of the product, energy requirements and transportation distance. Assessment of source, type of energy and transportation can impact significantly the feasibility of final product. The business opportunities for CO2 capture and utilization increased exponentially in the past decade, attracting new investors and governmental support. The estimated costs associated with different carbon capture and utilization routes reported in literature is discussed in this section. However, given that the level of the LCA estimations related to the technology readiness level (TRL) of the analysed technology, the level of the available LCA in the literature for such carbon capture and utilization routes ranges from the life cycle thinking to the prospective LCA.160 Capturing carbon for storage (CCS) or utilization (CCU) applications differ in their drawbacks and advantages. CCS applications suffer from low public acceptance, and uncertainty of the side effects of the storage.170 CCU on the other hand suffers from short product life cycles in the high volume applications, such as polymers, chemicals, and fuels.171Ex-situ Mg mineralization does not squarely fit in either CCU or CCS category, however it has the potential to achieve the best of both worlds: it has the near-permanent storage lifetime that is claimed in CCS and the high public acceptance associated with CCU.The CO2 capture step via integrated gasification combined cycle cost around $14.64 per ton for a plant with around 85% of capture efficiency. The feasibility of in situ CO2 sequestration mainly depend on the transportation and distance, besides the energy penalty for CO2 capture and purification (which for CO2 mineralization may be largely avoided). For example, the costs for storing CO2 in saline formations and in abandoned or depleted oil/gas sites would be around 0.49–7.81 € per ton CO2 with monitoring costs of 0.098 to 0.49 € per ton CO2. The ocean storage is also one of the most economically viable options for CO2 sequestration costing around 4.88 to 29.28 € per ton.161 In locations where suitable geological sites are not accessible, an ex situ process of CO2 sequestration is the most promising option, with the already two decades ago estimated costs (2003) of 48.79 to 292.76 € per ton CO2 without considering the potential revenue from carbon credit sales or utilization.162
The routes of CO2 mineralization currently available still faces hurdles of too high energy demands, challenging profitable business models unless supported by the economy of scale.5 The costs of mineral carbonation are still too high for large scale production (around 39.03–78.07 € per ton CO2) compared to geological sequestration. The estimated costs for CO2 sequestration within Mg silicates minerals such as olivine, wollastonite and serpentine are around 69.29 € per ton CO2, 79.04 € per ton CO2 and 109.3 € per ton CO2 respectively. If the energy requirements for the CO2 mineralization are subtracted, these costs decrease to 52.7 € per ton CO2, 62.45 € per ton CO2 and 76.12 € per ton CO2, respectively. However, the costs associated with the pre-treatment of the minerals and chemicals demand would also affect the cost of the mineralization process.163,164 Gálvez-Martos et al. have evaluated the cost to produce 1 ton of nesquehonite as ca. 380 €, where 80% of the reported cost was due to the alkali reagents utilized for converting the MgO precursor into nesquehonite.165 By utilizing waste-streams precursor, the costs of the carbonation process can be greatly improved. Moreover, more optimistic forecast can be achieved by deducting the price of CO2 emission allowances (ca. 97 € per metric ton of CO2 in the European Union emission trading system166).
The ÅA routes developed in Finland have achieved relevant progresses in lowering the energy demands and reagent consumption in the process of Mg extraction and carbonates, being demonstrated technology readiness level (TRL) 5–6 for the wet carbonation route.1,167 Even though the steps of pre-treatment for Mg extraction and post treatment of the carbonate products have already reached TRL 5–9, reactors for the carbonation process are still at TRL 6 (pilot trials).168 Few start-up companies have initiated field trials in the European Union, Australia and in North America,5,125 but the profitability of the process can only be enabled via high-volume applications of the carbonate products. In this sense, the only market with high enough volume to support the economy of scale of Mg carbonate production would be the construction sector. In this scenario, the state-of-art technologies have the forecast of being able to reduce the current global CO2e emissions by 8–33% per year while generating a profit up to 32 € per tonne of cement.5
The most available sources for CO2 uptake within Mg feedstocks are Mg silicate minerals which are highly abundant in the European Union and offers a total potential uptake ranging from 63 to 38% as described in Table 10.125 That offers a promising solution for addressing the current supply risks in Mg raw materials. For example in the European Union 89% of the primary supply of Mg is imported;169 however, sources of Mg silicates in natural resources (ultramafic rocks) and in inorganic waste-streams (steel slags, mining tailings, fly ash, etc.) are abundant globally. Thus, the main issue in establishing local production of Mg raw materials is related to the feasibility and economic viability of the extraction route.105
| Mineral | Formula | Reaction | Max. CO2 uptake (wt%) |
|---|---|---|---|
| Olivine (fosterite) | Mg2SiO4 | Mg2SiO4 + 2H2CO3 → 2MgCO3 + H4SiO4 | 63 |
| Serpentine polytype | Mg3Si2O5(OH)4 | Mg3Si2O5(OH)4 + 3H2CO3 → 3MgCO3 + 2H4SiO4 + H2O | 48 |
| Akermanite | Ca2MgSi2O7 | Ca2MgSi2O7 + 3H2CO3 + H2O → 2CaCO3 + MgCO3 + 2H4SiO4 | 48 |
| Pyroxene (diopside) | CaMgSi2O6 | CaMgSi2O6 + 2H2CO3 + 2H2O → CaCO3 + MgCO3 + 2H4SiO4 | 41 |
| Tremolite | Ca2Mg5Si8O22(OH)2 | Ca2Mg5Si8O22(OH)2 + 7H2CO3 + 8H2O → 2CaCO3 + 5MgCO3 + 8H4SiO4 | 38 |
| Enstatite | MgSiO3 | MgSiO3 + H2CO3 + H2O → MgCO3 + H4SiO4 | 44 |
7. Final remarks and conclusions
Mg carbonates are a class of materials to play a fundamental role on climate change mitigation actions, as they offer possibilities for carbon capture and utilization in construction materials, while having the long-term storing expected from carbon capture and storage applications, being also of high relevance for addressing the current criticality of Mg supply in, for example, the European Union. It seems plausible that the existing knowledge gaps on the mechanisms and kinetics of anhydrous Mg carbonate precipitation are the main reasons for the lack of economically feasible methods for producing these materials. Thus, novel methods for catalyzing the carbonation of Mg precursors are essential to open the market space for such routes, even when only applications for carbon capture and utilization/storage are aimed at. In turn, follow-up research in this direction can address the climate change emergency while also addressing the enigmatic geochemical problems of the natural dolomite/magnesite formations.Moreover, a deeper understanding on the surface chemistry of hydrated Mg carbonates directly relates to the possible applications of these materials. Currently, these minerals have shown potential for applications as thermal insulation panels, plasterboard structures and thermal energy storage (TES) materials. Still, considerable developments are needed to address the stability of these materials and verify their suitability for commercial applications. Doubtless, this class of materials offer broad perspectives for establishing disruptive business models within the carbon capture and utilization/storage and the construction sectors. However, the market deployment will also require a shift in legislation requirements aligned with the research developments in the field. The authors hope that the current review will motivate research in the field, fomenting new ideas and broadening the directions to be exploited in the future.
Author contributions
HSS has conceptualized the review paper, done the research investigation and its formal analysis as well as written the original draft and revised versions. HN has assisted the conceptualization, written the thermodynamic considerations in the original draft, as well as done the review and editing in the following versions. FV has co-written the kinetics considerations in the original draft and contributed with the review and editing in the following versions. DR has co-written the life cycle assessment discussion in the original draft. RZ has assisted the writing of the CCU/S considerations, and the “Mg silicate precursors” and “Mg sulphate precursors” sections in the original draft and contributed with the review and editing in the following versions. PK has assisted the conceptualization of the review paper, supervised the research investigation and analyses, acquired, and administrated the resources for performing the research, as well as actively revised all versions of the manuscript.Conflicts of interest
The work does not present any conflicts of interest.Acknowledgements
This work has been executed as part of the Magnex project (2022–2025), for which H. S. S., R. Z. and P. K. acknowledge the financial support from the Academy of Finland (funding decision 347183). P. K. and H. N. acknowledges financial support from Academy of Finland (grant 329477), as well as from the University of Oulu & The Academy of Finland Profi5 (326291). D. R. and P. K. acknowledges the funding of the Academy of Finland (Cemglass project, 322085). The authors thank Professor Bruce Railsback for providing the Fig. 4 for the paper, and Dr. Ellina Bernard (Empa, Switzerland) for valuable discussion to improve our thermodynamic data assessment.References
- R. Zevenhoven, M. Slotte, E. Koivisto and R. Erlund, Serpentinite Carbonation Process Routes Using Ammonium Sulfate and Integration in Industry, Energy Technol., 2017, 5(6), 945–954, DOI:10.1002/ente.201600702.
- X. Geng, L. Lv, C. Li, T. Zhang, B. Liang, Y. Chen and S. Tang, The Kinetics of CO2 Indirect Mineralization of MgSO4 to Produce MgCO3·3H2O, J. CO2 Util., 2019, 33, 64–71, DOI:10.1016/J.JCOU.2019.04.017.
- G. Gadikota, Carbon Mineralization Pathways for Carbon Capture, Storage and Utilization, Commun. Chem., 2021, 4(1), 1–5, DOI:10.1038/s42004-021-00461-x.
- H. Geerlings and R. Zevenhoven, CO2 Mineralization—Bridge Between Storage and Utilization of CO2, Annu. Rev. Chem. Biomol. Eng., 2013, 4, 103–117, DOI:10.1146/ANNUREV-CHEMBIOENG-062011-080951.
- T. Strunge, P. Renforth and M. Van der Spek, Towards a Business Case for CO2 Mineralisation in the Cement Industry, Commun. Earth Environ., 2022, 3, 59, DOI:10.1038/s43247-022-00390-0.
- R. Erlund and R. Zevenhoven, Thermal Energy Storage (TES) Capacity of a Lab Scale Magnesium Hydro Carbonates/Silica Gel System, J. Energy Storage, 2019, 25, 100907, DOI:10.1016/j.est.2019.100907.
- J. C. Deelman, Low-Temperature Formation of Dolomite and Magnesite, 2.2, Compact Disc Publications, Eindhoven, The Netherlands, 2011 Search PubMed.
- L. B. Railsback, Some Fundamentals of Mineralogy and Geochemistry: Carbonates, 2006http://railsback.org/FundamentalsIndex.html (accessed Jul 15, 2021) Search PubMed.
- J. Warren, Dolomite: Occurrence, Evolution and Economically Important Associations, Earth-Sci. Rev., 2000, 52(1–3), 1–81, DOI:10.1016/S0012-8252(00)00022-2.
- D. Liu, Y. Xu, D. Papineau, N. Yu, Q. Fan, X. Qiu and H. Wang, Experimental Evidence for Abiotic Formation of Low-Temperature Proto-Dolomite Facilitated by Clay Minerals, Geochim. Cosmochim. Acta, 2019, 247, 83–95, DOI:10.1016/j.gca.2018.12.036.
- J. M. Gregg, D. L. Bish, S. E. Kaczmarek and H. G. Machel, Mineralogy, Nucleation and Growth of Dolomite in the Laboratory and Sedimentary Environment: A Review, Sedimentology, 2015, 62(6), 1749–1769, DOI:10.1111/SED.12202.
- M. Hänchen, V. Prigiobbe, R. Baciocchi and M. Mazzotti, Precipitation in the Mg-Carbonate System—Effects of Temperature and CO2 Pressure, Chem. Eng. Sci., 2008, 63(4), 1012–1028, DOI:10.1016/J.CES.2007.09.052.
- S. R. Higgins and X. Hu, Self-Limiting Growth on Dolomite: Experimental Observations with in Situ Atomic Force Microscopy, Geochim. Cosmochim. Acta, 2005, 69(8), 2085–2094, DOI:10.1016/J.GCA.2004.10.010.
- N. Li, Z. Chang, Y. Zhan, H. Dang, N. A. Khan, W. Li, H. Zhou and C. Sun, Growth of Cubic Anhydrous Magnesium Carbonate Single Crystal in Deep Eutectic Solvent, J. Solid State Chem., 2020, 292, 121684, DOI:10.1016/J.JSSC.2020.121684.
- O. Qafoku, D. A. Dixon, K. M. Rosso, H. T. Schaef, M. E. Bowden, B. W. Arey and A. R. Felmy, Dynamics of Magnesite Formation at Low Temperature and High PCO2 in Aqueous Solution, Environ. Sci. Technol., 2015, 49(17), 10736–10744, DOI:10.1021/ACS.EST.5B02588.
- D. Toroz, F. Song, G. A. Chass and D. Di Tommaso, New, Insights into the Role of Solution Additive Anions in Mg2+ Dehydration: Implications for Mineral Carbonation, CrystEngComm, 2021, 23, 4896–4900, 10.1039/D1CE00052G.
- J. Xu, C. Yan, F. Zhang, H. Konishi, H. Xu and H. H. Teng, Testing the Cation-Hydration Effect on the Crystallization of Ca–Mg–CO3 Systems, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(44), 17750–17755, DOI:10.1073/PNAS.1307612110.
- D. M. Deocampo, The Geochemistry of Continental Carbonates, in Carbonates in Continental Settings: Geochemistry, Diagenesis and Applications, ed. A. M. Alonso-Zarza and L. H. Tanner, Elsevier, 2010, vol. 62, pp. 1–59, DOI:10.1016/S0070-4571(09)06201-3.
- L. B. Railsback, Patterns in the Compositions, Properties, and Geochemistry of Carbonate Minerals, Carbonates Evaporites, 1999, 14(1), 1–20, DOI:10.1007/BF03176144.
- J. C. Deelman, Magnesite, Dolomite and Carbonate Groups, HAL open Science ([Research Report] (formerly) Technische Universiteit Eindhoven), 2021, hal-03412979v2, https://hal.science/hal-03412979/document Search PubMed.
- L. B. Railsback, A Synthesis of Systematic Mineralogy, Am. Mineral., 2005, 90(7), 1033–1041, DOI:10.2138/AM.2005.1842.
- D. F. Shriver and P. W. Atkins, Inorganic Chemistry, Oxford University Press, Oxford, UK, 3rd edn, 1999 Search PubMed.
- P. Bénézeth, G. D. Saldi, J. L. Dandurand and J. Schott, Experimental Determination of the Solubility Product of Magnesite at 50 to 200 °C, Chem. Geol., 2011, 286(1–2), 21–31, DOI:10.1016/J.CHEMGEO.2011.04.016.
- Y. Fang, F. Zhang, G. A. Farfan and H. Xu, Low-Temperature Synthesis of Disordered Dolomite and High-Magnesium Calcite in Ethanol–Water Solutions: The Solvation Effect and Implications, ACS Omega, 2022, 7(1), 281–292, DOI:10.1021/acsomega.1c04624.
- D. A. Petrash, O. M. Bialik, T. R. R. Bontognali, C. Vasconcelos, J. A. Roberts, J. A. McKenzie and K. O. Konhauser, Microbially Catalyzed Dolomite Formation: From near-Surface to Burial, Earth-Sci. Rev., 2017, 171, 558–582, DOI:10.1016/J.EARSCIREV.2017.06.015.
- G. Montes-Hernandez and F. Renard, Time-Resolved in Situ Raman Spectroscopy of the Nucleation and Growth of Siderite, Magnesite, and Calcite and Their Precursors, Cryst. Growth Des., 2016, 16(12), 7218–7230, DOI:10.1021/ACS.CGD.6B01406.
- mindat.org. Mineral database https://www.mindat.org/ (accessed Sep 12, 2022).
- M. Merlini, W. A. Crichton, M. Hanfland, M. Gemmi, H. Müller, I. Kupenko and L. Dubrovinsky, Structures of Dolomite at Ultrahigh Pressure and Their Influence on the Deep Carbon Cycle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(34), 13509–13514, DOI:10.1073/pnas.1201336109.
- M. Akao and S. Iwai, The Hydrogen Bonding of Hydromagnesite, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33(4), 1273–1275, DOI:10.1107/S0567740877005834.
- H. Jagodzinski, Kristallstruktur Und Fehlordnung Des Artinits Mg2[CO3(OH)2]3 H2O, Tschermaks Mineral. Petrogr. Mitt., 1965, 10(1–4), 297–330, DOI:10.1007/BF01128636.
- S. Frisia, Dolomite and Dolomitization, in Encyclopedia of Geochemistry, ed. C. P. Marshall and R. W. Fairbridge, Kluwer Academic Publisher, Dordrecht, NL, 1999, pp. 140–142, DOI:10.1007/1-4020-4496-8_79.
- R. J. Reeder and S. A. Markgraf, High-Temperature Crystal Chemistry of Dolomite, Am. Mineral., 1986, 71(5–6), 795–804 CAS.
- P. L. Althoff, Structural Refinements of Dolomite and a Magnesian Calcite and Implications for Dolomite Formation in the Marine Environment, Am. Mineral., 1977, 62(7–8), 772–783 CAS.
- J. A. Wasastjerna, The Crystal Structure of Dolomite, in Commentationes physico-mathematicae II, Societas Scientiarum Fennica, Helsingfors, 1924, pp. 1–14 Search PubMed.
- J. A. Roberts, P. C. Bennett, L. A. González, G. L. Macpherson and K. L. Milliken, Microbial Precipitation of Dolomite in Methanogenic Groundwater, Geology, 2004, 32(4), 277–280, DOI:10.1130/G20246.2.
- R. S. Arvidson and F. T. Mackenzie, The Dolomite Problem; Control of Precipitation Kinetics by Temperature and Saturation State, Am. J. Sci., 1999, 299(4), 257–288, DOI:10.2475/AJS.299.4.257.
- M. J. Raudsepp, S. A. Wilson, B. Morgan, A. Patel, S. G. Johnston, E. J. Gagen and S. J. Fallon, Non–classical Crystallization of Very High Magnesium Calcite and Magnesite in the Coorong Lakes, Australia, Sedimentology, 2022, 69(5), 2246–2266, DOI:10.1111/sed.12991.
- V. Vandeginste, O. Snell, M. R. Hall, E. Steer and A. Vandeginste, Acceleration of Dolomitization by Zinc in Saline Waters, Nat. Commun., 2019, 10(1), 1851, DOI:10.1038/s41467-019-09870-y.
- C. Vasconcelos, J. A. McKenzie, S. Bernasconi, D. Grujic and A. J. Tiens, Microbial Mediation as a Possible Mechanism for Natural Dolomite Formation at Low Temperatures, Nature, 1995, 377(6546), 220–222, DOI:10.1038/377220a0.
- F. Zhang, H. Xu, H. Konishi, E. S. Shelobolina and E. Roden, Polysaccharide-Catalyzed Nucleation and Growth of Disordered Dolomite: A Potential Precursor of Sedimentary Dolomite, Am. Mineral., 2012, 97(4), 556–567, DOI:10.2138/AM.2012.3979.
- N. Ichieda, M. Kasuno, K. Banu, S. Kihara and H. Nakamatsu, Evaluation of Hydration Enthalpies of Monatomic Cations by Considering Both Long-Range and Short-Range Interactions, J. Phys. Chem. A, 2003, 107(38), 7597–7603, DOI:10.1021/JP0348171.
- P. A. Kenward, D. A. Fowle, R. H. Goldstein, M. Ueshima, L. A. González and J. A. Roberts, Ordered Low-Temperature Dolomite Mediated by Carboxyl-Group Density of Microbial Cell WallsLow-Temperature Dolomite Mediated by Microbial Cell Wall Character, Am. Assoc. Pet. Geol. Bull., 2013, 97(11), 2113–2125, DOI:10.1306/05171312168.
- P. A. Kenward, R. H. Goldstein, L. A. González and J. A. Roberts, Precipitation of Low-Temperature Dolomite from an Anaerobic Microbial Consortium: The Role of Methanogenic Archaea, Geobiology, 2009, 7(5), 556–565, DOI:10.1111/j.1472-4669.2009.00210.x.
- C. F. Kahle, Possible Roles of Clay Minerals in the Formation of Dolomite, J. Sediment. Res., 1965, 35(2), 448–453, DOI:10.1306/74d712a3-2b21-11d7-8648000102c1865d.
- J. N. Bracco, A. G. Stack and S. R. Higgins, Magnesite Step Growth Rates as a Function of the Aqueous Magnesium:Carbonate Ratio, Cryst. Growth Des., 2014, 14(11), 6033–6040, DOI:10.1021/CG501203G/SUPPL_FILE/CG501203G_SI_001.PDF.
- I. J. Kelleher and S. A. T. Redfern, Hydrous Calcium Magnesium Carbonate, a Possible Precursor to the Formation of Sedimentary Dolomite, Mol. Simul., 2002, 28(6–7), 557–572, DOI:10.1080/08927020290030134.
- V. Kartnaller, E. M. Ribeiro, F. Venancio, F. F. Rosario and J. Cajaiba, Preferential Incorporation of Sulfate into Calcite Polymorphs during Calcium Carbonate Precipitation: An Experimental Approach, CrystEngComm, 2018, 20(16), 2241–2244, 10.1039/C8CE00172C.
- M. J. Malone, P. A. Baker and S. J. Burns, Recrystallization of Dolomite: An Experimental Study From, Geochim. Cosmochim. Acta, 1996, 60(12), 2189–2207, DOI:10.1016/0016-7037(96)00062-2.
- A. Katz and A. Matthews, The Dolomitization of CaCO3: An Experimental Study at 252–295 °C, Geochim. Cosmochim. Acta, 1977, 41(2), 297–308, DOI:10.1016/0016-7037(77)90238-1.
- A. M. Gaines, Dolomitization Kinetics: Recent Experimental Studies, in Concepts and Models of Dolomitization, SEPM (Society for Sedimentary Geology), 1980, pp. 81–86, DOI:10.2110/pec.80.28.0081.
- A. Banerjee, Estimation of Dolomite Formation: Dolomite Precipitation and Dolomitization, J. Geol. Soc. India, 2016, 87(5), 561–572, DOI:10.1007/s12594-016-0430-9.
- J. Haas, T. Budai, O. Győri and S. Kele, Multiphase Partial and Selective Dolomitization of Carnian Reef Limestone (Transdanubian Range, Hungary), Sedimentology, 2014, 61(3), 836–859, DOI:10.1111/sed.12088.
- F. L. Sayles and W. S. Fyfe, The Crystallization of Magnesite from Aqueous Solution, Geochim. Cosmochim. Acta, 1973, 37(1), 87–99, DOI:10.1016/0016-7037(73)90246-9.
- M. Gautelier, J. Schott and E. H. Oelkers, An Experimental Study of Dolomite Dissolution Rates at 80 °C as a Function of Chemical Affinity and Solution Composition, Chem. Geol., 2007, 242(3–4), 509–517, DOI:10.1016/j.chemgeo.2007.05.008.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, A Route for the Direct Crystallization of Dolomite, Am. Mineral., 2015, 100(5–6), 1172–1181, DOI:10.2138/AM-2015-4963.
- D. Gebauer and H. Cölfen, Prenucleation Clusters and Non-Classical Nucleation, Nano Today, 2011, 6(6), 564–584, DOI:10.1016/J.NANTOD.2011.10.005.
- L. Gránásy, T. Pusztai, G. Tegze, J. A. Warren and J. F. Douglas, Growth and Form of Spherulites, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72(1), 011605, DOI:10.1103/PhysRevE.72.011605.
- N. E. Davis, J. Newman, P. B. Wheelock and A. K. Kronenberg, Grain Growth Kinetics of Dolomite, Magnesite and Calcite: A Comparative Study, Phys. Chem. Miner., 2011, 38(2), 123–138, DOI:10.1007/s00269-010-0389-9.
- minteq.v4.dat.
- PHREEQC Version 3 https://www.usgs.gov/software/phreeqc-version-3 (accessed Oct 15, 2022).
- J. Johnson, F. Anderson and D. Parkhurst, Database Thermo.Com. Lawrence Livermore National Laboratory: Livermore, California. 2000.
- C. E. A. Palmer, R. J. Silva and J. J. Bucher, Thermodynamic Data Base Needs for Modeling Studies of the Yucca Mountain Project, 1996 Search PubMed.
- P. Blanc, A. Lassin, P. Piantone, M. Azaroual, N. Jacquemet, A. Fabbri and E. C. Gaucher, Thermoddem: A Geochemical Database Focused on Low Temperature Water/Rock Interactions and Waste Materials, Appl. Geochem., 2012, 27(10), 2107–2116, DOI:10.1016/j.apgeochem.2012.06.002.
- E. Giffaut, M. Grivé, P. Blanc, P. Vieillard, E. Colàs, H. Gailhanou, S. Gaboreau, N. Marty, B. Madé and L. Duro, Andra Thermodynamic Database for Performance Assessment: ThermoChimie, Appl. Geochem., 2014, 49, 225–236, DOI:10.1016/j.apgeochem.2014.05.007.
- H. D. B. Jenkins and L. Glasser, Standard Absolute Entropy, Values from Volume or Density. 1. Inorganic Materials, Inorg. Chem., 2003, 42(26), 8702–8708, DOI:10.1021/ic030219p.
- J. Leitner, P. Voňka, D. Sedmidubský and P. Svoboda, Application of Neumann–Kopp Rule for the Estimation of Heat Capacity of Mixed Oxides, Thermochim. Acta, 2010, 497(1–2), 7–13, DOI:10.1016/j.tca.2009.08.002.
- R. A. Robie and B. S. Hemingway, The Heat Capacities at Low-Temperatures and Entropies at 298.15 K of Nesquehonite, MgCO3–3H2O, and Hydromagnesite, Am. Mineral., 1972, 57(11–12), 1768–1781 CAS.
- W. Dong, R. Liu, H. Zhao and X. Han, Preparation and Characterization of Anhydrous Magnesium Carbonate under the Present of Potassium Ions Solution, Powder Technol., 2020, 360, 741–746, DOI:10.1016/J.POWTEC.2019.09.018.
- S. A. Walling and J. L. Provis, Magnesia-Based Cements: A Journey of 150 Years, and Cements for the Future?, Chem. Rev., 2016, 4170–4204, DOI:10.1021/acs.chemrev.5b00463 . American Chemical Society.
- M. A. Shand, J. Qian, F. Jin, A. Al-Tabbaa and L. Mo, Magnesia Cements, Elsevier, 2020, DOI:10.1016/C2010-0-68998-X.
- F. W. C. Hobbs and H. Xu, Magnesite Formation through Temperature and PH Cycling as a Proxy for Lagoon and Playa Paleoenvironments, Geochim. Cosmochim. Acta, 2020, 269, 101–116, DOI:10.1016/J.GCA.2019.10.014.
- L. Hopkinson, P. Kristova, K. Rutt and G. Cressey, Phase Transitions in the System MgO-CO2-H2O during CO2 Degassing of Mg-Bearing Solutions, Geochim. Cosmochim. Acta, 2012, 76, 1–13, DOI:10.1016/j.gca.2011.10.023.
- A. M. Chaka and A. R. Felmy, Ab Initio Thermodynamic Model for Magnesium Carbonates and Hydrates, J. Phys. Chem. A, 2014, 118(35), 7469–7488, DOI:10.1021/jp500271n.
- W. Hummel, U. Berner, E. Curti, F. J. Pearson and T. Thoenen, Nagra/PSI Chemical Thermodynamic Data Base 01/01, Radiochim. Acta, 2002, 90(9–11), 805–813, DOI:10.1524/ract.2002.90.9-11_2002.805.
- H. C. Helgeson, J. M. Delany, H. W. Nesbitt and D. K. Bird, Summary and Critique of the Thermodynamic Properties of Rock-Forming Minerals, Am. J. Sci., 1978, 278-A, 1–201 Search PubMed.
- R. A. Robie and B. S. Hemingway, Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 Bar (105 Pascals) Pressure and at Higher Temperatures, U.S. Geological Survey, Denver, CO, USA, 1st edn, 1995 Search PubMed.
- A. L. Harrison, V. Mavromatis, E. H. Oelkers and P. Bénézeth, Solubility of the Hydrated Mg-Carbonates Nesquehonite and Dypingite from 5 to 35 °C: Implications for CO2 Storage and the Relative Stability of Mg-Carbonates, Chem. Geol., 2019, 504, 123–135, DOI:10.1016/j.chemgeo.2018.11.003.
- R. J. Hill, J. H. Canterford and F. J. Moyle, New Data for Lansfordite, Mineral. Mag., 1982, 46(341), 453–457, DOI:10.1180/minmag.1982.046.341.08.
- Q. Gautier, P. Bénézeth, V. Mavromatis and J. Schott, Hydromagnesite Solubility Product and Growth Kinetics in Aqueous Solution from 25 to 75 °C, Geochim. Cosmochim. Acta, 2014, 138, 1–20, DOI:10.1016/j.gca.2014.03.044.
- G. Raade, Dypingite, a New Hydrous Basic Carbonate of Magnesium, from Norway, Am. Mineral., 1970, 55(9–10), 1457–1465 CAS.
- E. Bernard, B. Lothenbach, D. Rentsch, A. German and F. Winnefeld, Effect of Carbonates on the Formation of Magnesium Silicate Hydrates, Mater. Struct., 2022, 55(7), 183, DOI:10.1617/s11527-022-02018-3.
- E. Königsberger, L.-C. Königsberger and H. Gamsjäger, Low-Temperature Thermodynamic Model for the System Na2CO3−MgCO3−CaCO3−H2O, Geochim. Cosmochim. Acta, 1999, 63(19–20), 3105–3119, DOI:10.1016/S0016-7037(99)00238-0.
- J. W. Johnson, E. H. Oelkers and H. C. Helgeson, SUPCRT92: A Software Package for Calculating the Standard Molal Thermodynamic Properties of Minerals, Gases, Aqueous Species, and Reactions from 1 to 5000 Bar and 0 to 1000 °C, Comput. Geosci., 1992, 18(7), 899–947, DOI:10.1016/0098-3004(92)90029-Q.
- R. Chaliulina, J.-L. Galvez-Martos, A. Hakki, A. Elhoweris, J. Mwanda and Y. Al-Horr, Eco Materials from CO2 Capture: Compressive Strengths of a Plasterboard Alternative, Constr. Build. Mater., 2021, 312, 125276, DOI:10.1016/j.conbuildmat.2021.125276.
- A. V. Radha, A. Fernandez-Martinez, Y. Hu, Y.-S. Jun, G. A. Waychunas and A. Navrotsky, Energetic and Structural Studies of Amorphous Ca1−xMgxCO3·nH2O (0≤x≤1), Geochim. Cosmochim. Acta, 2012, 90, 83–95, DOI:10.1016/j.gca.2012.04.056.
- J. Yang, Y. Han, J. Luo, K. Leifer, M. Strømme and K. Welch, Synthesis and Characterization of Amorphous Magnesium Carbonate Nanoparticles, Mater. Chem. Phys., 2019, 224, 301–307, DOI:10.1016/j.matchemphys.2018.12.037.
- O. Cheung, P. Zhang, S. Frykstrand, H. Zheng, T. Yang, M. Sommariva, X. Zou and M. Strømme, Nanostructure and Pore Size Control of Template-Free Synthesised Mesoporous Magnesium Carbonate, RSC Adv., 2016, 6(78), 74241–74249, 10.1039/C6RA14171D.
- C. E. White, N. J. Henson, L. L. Daemen, M. Hartl and K. Page, Uncovering the True Atomic Structure of Disordered Materials: The Structure of a Hydrated Amorphous Magnesium Carbonate (MgCO3 ·3D2O), Chem. Mater., 2014, 26(8), 2693–2702, DOI:10.1021/cm500470g.
- D. Toroz, F. Song, A. Uddin, G. A. Chass and D. Di Tommaso, Solution Additives Promoting the Onset of MgCO3 Nucleation, ChemRxiv. Cambridge: Cambridge Open Engage, 2021 DOI:10.26434/chemrxiv-2021-kl62l , Cambridge Univ. Press.
- D. Toroz, F. Song, A. Uddin, G. A. Chass and D. Di Tommaso, A Database of Solution Additives Promoting Mg2+ Dehydration and the Onset of MgCO3 Nucleation, Cryst. Growth Des., 2022, 22(5), 3080–3089, DOI:10.1021/ACS.CGD.1C01525.
- O. S. Pokrovsky, S. V. Golubev and J. Schott, Dissolution Kinetics of Calcite, Dolomite and Magnesite at 25 °C and 0 to 50 Atm PCO2, Chem. Geol., 2005, 217(3–4), 239–255, DOI:10.1016/j.chemgeo.2004.12.012.
- D. E. Ellis and P. J. Wyllie, A Model of Phase Relations in the System MgO-SiO2-H2O-CO2 and Prediction of the Compositions of Liquids Coexisting with Forsterite and Enstatite, in Kimberlites, Diatremes, and Diamonds: Their Geology, Petrology, and Geochemistry, American Geophysical Union, Washington, D. C., 1979, pp. 313–318, DOI:10.1029/SP015p0313.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, CO2 Activation over Catalytic Surfaces, ChemPhysChem, 2017, 18(22), 3135–3141, DOI:10.1002/cphc.201700782.
- B. Mondal, J. Song, F. Neese and S. Ye, Bio-Inspired Mechanistic Insights into CO2 Reduction, Curr. Opin. Chem. Biol., 2015, 25, 103–109, DOI:10.1016/j.cbpa.2014.12.022.
- U. J. Etim, C. Zhang and Z. Zhong, Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces, Nanomaterials, 2021, 11(12), 3265, DOI:10.3390/nano11123265.
- G. D. Saldi, G. Jordan, J. Schott and E. H. Oelkers, Magnesite Growth Rates as a Function of Temperature and Saturation State, Geochim. Cosmochim. Acta, 2009, 73(19), 5646–5657, DOI:10.1016/J.GCA.2009.06.035.
- N. Raza, Z. I. Zafar, M. Najam-ul-Haq and R. V. Kumar, Leaching of Natural Magnesite Ore in Succinic Acid Solutions, Int. J. Miner. Process., 2015, 139, 25–30, DOI:10.1016/j.minpro.2015.04.008.
- B. Dönmez, F. Demir and O. Laçin, Leaching Kinetics of Calcined Magnesite in Acetic Acid Solutions, J. Ind. Eng. Chem., 2009, 15(6), 865–869, DOI:10.1016/j.jiec.2009.09.014.
- F. Demir, O. Laçin and B. Dönmez, Leaching Kinetics of Calcined Magnesite in Citric Acid Solutions, Ind. Eng. Chem. Res., 2006, 45(4), 1307–1311, DOI:10.1021/ie0507629.
- H. Özbek, Y. Abali, S. Çolak, I. Ceyhun and Z. Karagölge, Dissolution Kinetics of Magnesite Mineral in Water Saturated by Chlorine Gas, Hydrometallurgy, 1999, 51(2), 173–185, DOI:10.1016/S0304-386X(98)00069-3.
- J. W. Morse and R. S. Arvidson, The Dissolution Kinetics of Major Sedimentary Carbonate Minerals, Earth-Sci. Rev., 2002, 58(1–2), 51–84, DOI:10.1016/S0012-8252(01)00083-6.
- O. S. Pokrovsky and J. Schott, Processes at the Magnesium-Bearing Carbonates/Solution Interface. II. Kinetics and Mechanism of Magnesite Dissolution, Geochim. Cosmochim. Acta, 1999, 63(6), 881–897, DOI:10.1016/S0016-7037(99)00013-7.
- G. D. Saldi, J. Schott, O. S. Pokrovsky, Q. Gautier and E. H. Oelkers, An Experimental Study of Magnesite Precipitation Rates at Neutral to Alkaline Conditions and 100–200 °C as a Function of pH, Aqueous Solution Composition and Chemical Affinity, Geochim. Cosmochim. Acta, 2012, 83, 93–109, DOI:10.1016/J.GCA.2011.12.005.
- Y. Sun, Q. Wang, Y. Wang, R. Yun and X. Xiang, Recent Advances in Magnesium/Lithium Separation and Lithium Extraction Technologies from Salt Lake Brine, Sep. Purif. Technol., 2021, 256, 117807, DOI:10.1016/j.seppur.2020.117807.
- C. D. Hills, N. Tripathi and P. J. Carey, Managed Pathways for CO2 Mineralisation: Analogy with Nature and Potential Contribution to CCUS-Led Reduction Targets, Faraday Discuss., 2021, 230, 152–171, 10.1039/D0FD00142B.
- X. Cao, S. Zhang, H. Zhao, Y. Zhong, R. Zhang and R. Liu, Technoeconomic Analysis of a Brine Purification Process–Combined Carbon Dioxide Mineralization and Hydromagnesite Recovery, Ind. Eng. Chem. Res., 2022, 61(20), 6802–6812, DOI:10.1021/acs.iecr.2c00353.
- O. Liebermann, Synthesis of Dolomite, Nature, 1967, 213(5073), 241–245, DOI:10.1038/213241a0.
- V. Vandeginste, Effect of PH Cycling and Zinc Ions on Calcium and Magnesium Carbonate Formation in Saline Fluids at Low Temperature, Minerals, 2021, 11(7), 723, DOI:10.3390/MIN11070723.
- J. C. Deelman , Note on Magnesite Formation (Studies on Irreversible Geochemical Reactions n 9). Carnets Géologie/Notebooks Geol. 2003, Letter 200, CG2003_L03_JCD.
- J. C. Deelman, Low-Temperature Nucleation of Magnesite and Dolomite, Neues Jahrb. Mineral., 1999, 7(Monatshefte), 289–302 Search PubMed.
- D. Anjos, A. P. A. Sifeddine, A. Sanders, C. J. Patchineelam and S. R, Synthesis of Magnesite at Low Temperature, Carbonates Evaporites, 2011, 26(3), 213–215, DOI:10.1007/S13146-011-0063-4/FIGURES/1.
- I. M. Power, P. A. Kenward, G. M. Dipple and M. Raudsepp, Room Temperature Magnesite Precipitation, Cryst. Growth Des., 2017, 17(11), 5652–5659, DOI:10.1021/acs.cgd.7b00311.
- F. P. Glasser, G. Jauffret, J. Morrison, J.-L. Galvez-Martos, N. Patterson and M. S.-E. Imbabi, Sequestering CO2 by Mineralization into Useful Nesquehonite-Based Products, Front. Energy Res., 2016, 4, 3, DOI:10.3389/fenrg.2016.00003.
- K. Chai and S. Xu, Synthesis and Mechanism of a New Environment-Friendly Flame Retardant (Anhydrous Magnesium Carbonate) by Hydrothermal Method, Adv. Powder Technol., 2022, 33(11), 103776, DOI:10.1016/j.apt.2022.103776.
- G. Gadikota, Multiphase Carbon Mineralization for the Reactive Separation of CO2 and Directed Synthesis of H2, Nat. Rev. Chem., 2020, 4(2), 78–89, DOI:10.1038/s41570-019-0158-3.
- M. Liu, A. Hohenshil and G. Gadikota, Integrated CO2 Capture and Removal via Carbon Mineralization with Inherent Regeneration of Aqueous Solvents, Energy Fuels, 2021, 35(9), 8051–8068, DOI:10.1021/ACS.ENERGYFUELS.0C04346.
- S. E. Rodriguez-Cruz, R. A. Jockusch and E. R. Williams, Hydration Energies and Structures of Alkaline Earth Metal Ions, M2+·(H2O)n, n = 5–7, M = Mg, Ca, Sr, and Ba, J. Am. Chem. Soc., 1999, 121(38), 8898–8906, DOI:10.1021/JA9911871.
- E. Koivisto, R. Erlund, M. Fagerholm and R. Zevenhoven, Extraction of Magnesium from Four Finnish Magnesium Silicate Rocks for CO2 Mineralisation - Part 1: Thermal Solid/Solid Extraction, Hydrometallurgy, 2016, 166, 222–228, DOI:10.1016/J.HYDROMET.2016.07.005.
- I. S. Romão, L. M. Gando-Ferreira, M. M. V. G. Da Silva and R. Zevenhoven, CO2 Sequestration with Serpentinite and Metaperidotite from Northeast Portugal, Miner. Eng., 2016, 94, 104–114, DOI:10.1016/J.MINENG.2016.05.009.
- M. Liu, H. Asgar, S. Seifert and G. Gadikota, Novel Aqueous Amine Looping Approach for the Direct Capture, Conversion and Storage of CO2 to Produce Magnesium Carbonate, Sustainable Energy Fuels, 2020, 4(3), 1265–1275, 10.1039/C9SE00316A.
- R. Zevenhoven and N. Häggqvist, Several Process Routes for Stepwise Carbonation of Serpentinite – When Use Which Route?, SSRN Electron. J., 2022, 1–16, DOI:10.2139/SSRN.4141031.
- R. Zevenhoven, M. Slotte, J. Åbacka and J. Highfield, A Comparison of CO2 Mineral Sequestration Processes Involving a Dry or Wet Carbonation Step, Energy, 2016, 117, 604–611, DOI:10.1016/J.ENERGY.2016.05.066.
- E. Koivisto and R. Zevenhoven, Methods for Recovery and Re-Use of Additive Chemicals during CO2 Mineralisation, J. Water Process Eng., 2017, 20, 61–70, DOI:10.1016/J.JWPE.2017.09.003.
- J. Highfield, H. Lim, J. Fagerlund and R. Zevenhoven, Activation of Serpentine for CO2 Mineralization by Flux Extraction of Soluble Magnesium Salts Using Ammonium Sulfate, RSC Adv., 2012, 2(16), 6535, 10.1039/c2ra01347a.
- C. D. Hills, N. Tripathi and P. J. Carey, Mineralization Technology for Carbon Capture, Utilization, and Storage, Front. Energy Res., 2020, 8, 142, DOI:10.3389/FENRG.2020.00142.
- E. Koivisto and R. Zevenhoven, Energy Use of Flux Salt Recovery Using Bipolar Membrane Electrodialysis for a CO2 Mineralisation Process, Entropy, 2019, 21(4), 395, DOI:10.3390/e21040395.
- E. Koivisto and R. Zevenhoven, Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation, Geosciences, 2018, 8(4), 123, DOI:10.3390/geosciences8040123.
- G. Pasupathi and P. Philominathan, Investigation on Growth and Characterization of a New Inorganic NLO Material: Zinc Sulphate (ZnSO4:7H2O) Doped with Magnesium Sulphate (MgSO4:7H2O), Mater. Lett., 2008, 62(28), 4386–4388, DOI:10.1016/j.matlet.2008.07.023.
- I.-M. Chou and R. R. Seal, Determination of Epsomite-Hexahydrite Equilibria by the Humidity-Buffer Technique at 0.1 MPa with Implications for Phase Equilibria in the System MgSO4 -H2O, Astrobiology, 2003, 3(3), 619–630, DOI:10.1089/153110703322610708.
- L. Palmer and B. D. Newby, Development of a Simplified Protocol for Administration of 20% Magnesium Sulphate for Prophylaxis and Treatment of Eclampsia, Can. J. Hosp. Pharm., 2009, 62(6), 490–495, DOI:10.4212/cjhp.v62i6.847.
- K. Eddy, J. Vogel, R. Zahroh and M. Bohren, Factors Affecting Use of Magnesium Sulphate for Pre–eclampsia or Eclampsia: A Qualitative Evidence Synthesis, BJOG, 2022, 129(3), 379–391, DOI:10.1111/1471-0528.16913.
- G. H. Neilsen and D. Neilsen, Consequences of Potassium, Magnesium Sulphate Fertilization of High Density Fuji Apple Orchards, Can. J. Soil Sci., 2011, 91(6), 1013–1027, DOI:10.4141/cjss2011-023.
- Statista, Leading Magnesium Sulfates Exporting Countries Worldwide in 2020, Based on Value, 2023 Search PubMed.
- M. M. Maroto-Valer, D. J. Fauth, M. E. Kuchta, Y. Zhang and J. M. Andrésen, Activation of Magnesium Rich Minerals as Carbonation Feedstock Materials for CO2 Sequestration, Fuel Process. Technol., 2005, 86(14–15), 1627–1645, DOI:10.1016/j.fuproc.2005.01.017.
- G. Alexander, M. M. Maroto-Valer and P. Gafarova-Aksoy, Evaluation of Reaction Variables in the Dissolution of Serpentine for Mineral Carbonation, Fuel, 2007, 86(1–2), 273–281, DOI:10.1016/j.fuel.2006.04.034.
- S. Teir, H. Revitzer, S. Eloneva, C.-J. Fogelholm and R. Zevenhoven, Dissolution of Natural Serpentinite in Mineral and Organic Acids, Int. J. Miner. Process., 2007, 83(1–2), 36–46, DOI:10.1016/j.minpro.2007.04.001.
- X. Wang and M. M. Maroto-Valer, Dissolution of Serpentine Using Recyclable Ammonium Salts for CO2 Mineral Carbonation, Fuel, 2011, 90(3), 1229–1237, DOI:10.1016/J.FUEL.2010.10.040.
- X. Wang and M. M. Maroto-Valer, Optimization of Carbon Dioxide Capture and Storage with Mineralisation Using Recyclable Ammonium Salts, Energy, 2013, 51, 431–438, DOI:10.1016/j.energy.2013.01.021.
- W. Liu, G. Chu, H. Yue, B. Liang, D. Luo and C. Li, Phase Equilibrium of the MgSO4-(NH4)2SO4-H2O Ternary System: Effects of Sulfuric Acid and Iron Sulfate and Its Application in Mineral Carbonation of Serpentine, J. Chem. Eng. Data, 2018, 63(5), 1603–1612, DOI:10.1021/acs.jced.7b01113.
- Z. W. Li, H. P. Zheng, J. H. Tang, D. B. Xie, D. X. Qin and L. Y. Mao, Stable Solid–Liquid Equilibrium of the Quaternary System Na+, Mg2+//CO32−, SO42−–H2O at 298.15 K, J. Chem. Eng. Data, 2021, 66(5), 2012–2021, DOI:10.1021/ACS.JCED.0C01056.
- X. Geng, L. Lv, T. Zhang and S. Tang, The Regulating Mechanism of MgCO3·3H2O Whisker Growth Orientation with the Presence of SDS, J. CO2 Util., 2020, 42, 101307, DOI:10.1016/J.JCOU.2020.101307.
- C. Deng, W. Liu, G. Chu, D. Luo, G. Zhang, L. Wang, H. Yue, B. Liang and C. Li, Aqueous Carbonation of MgSO4 with (NH4)2CO3 for CO2 Sequestration, Greenhouse Gases: Sci. Technol., 2019, 9(2), 209–225, DOI:10.1002/GHG.1840.
- G. J. Simandl, S. Paradis and M. Irvine, Brucite – Industrial Mineral with a Future, Geosci. Canada, 2007, 34(2), 57–64 Search PubMed.
- F. Shahbaz, I. Singh, P. Krishnan and K. Celik, Life Cycle Assessment of Brucite and Synthetic MgO Produced from Reject Brine Using Different Alkalis, J. Cleaner Prod., 2022, 380, 135071, DOI:10.1016/j.jclepro.2022.135071.
- S. Atashin, R. A. Varin and J. Z. Wen, Directed Precipitation of Anhydrous Magnesite for Improved Performance of Mineral Carbonation of CO2, J. Environ. Chem. Eng., 2017, 5(4), 3362–3372, DOI:10.1016/J.JECE.2017.06.048.
- J. Fagerlund, J. Highfield and R. Zevenhoven, Kinetics Studies on Wet and Dry Gas–Solid Carbonation of MgO and Mg(OH)2 for CO2 Sequestration, RSC Adv., 2012, 2(27), 10380–10393, 10.1039/C2RA21428H.
- X. Zhang, Y. Zeng and Z. Li, Accelerated Phase Transition and Synthesis of Magnesite from Hydromagnesite in Aqueous Mono-Ethylene Glycol Solution, J. Cryst. Growth, 2021, 563, 126105, DOI:10.1016/J.JCRYSGRO.2021.126105.
- G. Montes-Hernandez, F. Renard, R. Chiriac, N. Findling and F. Toche, Rapid Precipitation of Magnesite Microcrystals from Mg(OH)2-H2O-CO2 Slurry Enhanced by NaOH and a Heat-Aging Step (from ∼20 to 90 °C), Cryst. Growth Des., 2012, 12(11), 5233–5240, DOI:10.1021/CG300652S.
- E. J. Swanson, K. J. Fricker, M. Sun and A. H. A. Park, Directed Precipitation of Hydrated and Anhydrous Magnesium Carbonates for Carbon Storage, Phys. Chem. Chem. Phys., 2014, 16(42), 23440–23450, 10.1039/C4CP03491K.
- X. Zhang, A. S. Lea, A. M. Chaka, J. S. Loring, S. T. Mergelsberg, E. Nakouzi, O. Qafoku, J. J. De Yoreo, H. T. Schaef and K. M. Rosso, In Situ Imaging of Amorphous Intermediates during Brucite Carbonation in Supercritical CO2, Nat. Mater., 2021, 21(3), 345–351, DOI:10.1038/s41563-021-01154-5.
- H. Nguyen, H. S. Santos, H. Sreenivasan, W. Kunther, V. Carvelli, M. Illikainen and P. Kinnunen, On the Carbonation of Brucite: Effects of Mg-Acetate on the Precipitation of Hydrated Magnesium Carbonates in Aqueous Environment, Cem. Concr. Res., 2022, 153, 106696, DOI:10.1016/j.cemconres.2021.106696.
- N. T. Dung and C. Unluer, Advances in the Hydration of Reactive MgO Cement Blends Incorporating Different Magnesium Carbonates, Constr. Build. Mater., 2021, 294, 123573, DOI:10.1016/j.conbuildmat.2021.123573.
- N. T. Dung and C. Unluer, Influence of Accelerated Hydration and Carbonation on the Performance of Reactive Magnesium Oxide Concrete, Adv. Cem. Res., 2020, 32(2), 78–90, DOI:10.1680/JADCR.17.00186.
- S. Hamdi, L. Vieille, K. Nahdi and L. Favergeon, Synthesis, Characterization and Low-Temperature Carbonation of Mesoporous Magnesium Oxide, J. Therm. Anal. Calorim., 2019, 138(3), 1923–1933, DOI:10.1007/S10973-019-08431-1/FIGURES/14.
- J.-L. Gálvez-Martos, R. Chaliulina, E. Medina-Martos, A. Elhoweris, A. Hakki, J. Mwanda and Y. Al-Horr, Eco-Efficiency of a Novel Construction Material Produced by Carbon Capture and Utilization, J. CO2 Util., 2021, 49, 101545, DOI:10.1016/j.jcou.2021.101545.
- R. Erlund and R. Zevenhoven, Simulations on Design and System Performance of Building Heating Boosted by Thermal Energy Storage (TES) with Magnesium Hydro Carbonates/Silica Gel, Energies, 2020, 13(17), 4520, DOI:10.3390/en13174520.
- R. Snellings, G. Mertens and J. Elsen, Supplementary Cementitious Materials, Rev. Mineral. Geochem., 2012, 74(1), 211–278, DOI:10.2138/rmg.2012.74.6.
- M. Zajac, P. Durdzinski, C. Stabler, J. Skocek, D. Nied and M. B. Haha, Influence of Calcium and Magnesium Carbonates on Hydration Kinetics, Hydrate Assemblage and Microstructural Development of Metakaolin Containing Composite Cements, Cem. Concr. Res., 2018, 106, 91–102, DOI:10.1016/j.cemconres.2018.01.008.
- Á. Fernández, J. L. G. Calvo and M. C. Alonso, Ordinary Portland Cement Composition for the Optimization of the Synergies of Supplementary Cementitious Materials of Ternary Binders in Hydration Processes, Cem. Concr. Compos., 2018, 89, 238–250, DOI:10.1016/j.cemconcomp.2017.12.016.
- N. M. Holden, A Readiness Level Framework for Sustainable Circular Bioeconomy, EFB Bioeconomy J., 2022, 2, 100031, DOI:10.1016/j.bioeco.2022.100031.
- Carbon Dioxide Capture and Storage, ed. B. Metz, O. Davidson, H. Coninck, M. de Loos and L. Meyer, IPCC, Cambridge, UK, 2005 Search PubMed.
- K. S. Lackner, A Guide to CO2 Sequestration, Science, 2003, 300(5626), 1677–1678, DOI:10.1126/SCIENCE.1079033.
- S. J. Gerdemann, W. K. O'Connor, D. C. Dahlin, L. R. Penner and H. Rush, Ex Situ Aqueous Mineral Carbonation, Environ. Sci. Technol., 2007, 41(7), 2587–2593, DOI:10.1021/es0619253.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, S. J. Gerdemann, L. R. Penner and D. N. Nilsen, Aqueous Mineral Carbonation, DOE, US, 2005 Search PubMed.
- J.-L. Gálvez-Martos, A. Elhoweris, A. Hakki and Y. Al-horr, Techno-Economic Assessment of a Carbon Capture and Utilization Process for the Production of Plaster-like Construction Materials, J. CO2 Util., 2020, 38, 59–67, DOI:10.1016/j.jcou.2019.12.017.
- Trading Economics, EU Carbon Permits 2023, https://tradingeconomics.com/commodity/carbon, accessed on 04/02/23.
- M. Slotte, Two Process Case Studies on Energy Efficiency, Life Cycle Assessment and Process Scale-Up, PhD thesis, Åbo Akademi University, 2017 Search PubMed.
- G. Faber, A. Ruttinger, T. Strunge, T. Langhorst, A. Zimmermann, M. van der Hulst, F. Bensebaa, S. Moni and L. Tao, Adapting Technology Learning Curves for Prospective Techno-Economic and Life Cycle Assessments of Emerging Carbon Capture and Utilization Pathways, Front. Clim., 2022, 4, 820261, DOI:10.3389/fclim.2022.820261.
- G. A. Blengini, C. E. L. Latunussa, U. Eynard, C. T. de Matos, D. Wittmer, K. Georgitzikis, C. Pavel, S. Carrara, L. Mancini, M. Unguru, D. Blagoeva, F. Mathieux and D. Pennington, Study on the EU's List of Critical Raw Materials (2020). Final Report, European Comission, Luxembourg, 2020, DOI:10.2873/11619.
- D. Daval, Carbon dioxide sequestration through silicate degradation and carbon mineralisation: promises and uncertainties, npj Mater. Degrad., 2018, 2, 11, DOI:10.1038/s41529-018-0035-4.
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Transformation technologies for CO2 utilisation: Current status, challenges and future prospects, Chem. Eng. J., 2021, 409, 128138, DOI:10.1016/j.cej.2020.128138.
| This journal is © the Partner Organisations 2023 |