
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface and optical properties of phase-pure silver iodobismuthate nanocrystals†
Anastasia
Matuhina
 a,
G. Krishnamurthy
Grandhi
a,
Ashanti
Bergonzoni
b,
Laurent
Pedesseau
b,
Roberto
Grisorio
c,
Shambhavee
Annurakshita
d,
Harri
Ali-Löytty
e,
Riya
Varghese
d,
Kimmo
Lahtonen
f,
George
Volonakis
g,
Vincenzo
Pecunia
h,
Godofredo
Bautista
d,
Jacky
Even
b and
Paola
Vivo
*a
a,
G. Krishnamurthy
Grandhi
a,
Ashanti
Bergonzoni
b,
Laurent
Pedesseau
b,
Roberto
Grisorio
c,
Shambhavee
Annurakshita
d,
Harri
Ali-Löytty
e,
Riya
Varghese
d,
Kimmo
Lahtonen
f,
George
Volonakis
g,
Vincenzo
Pecunia
h,
Godofredo
Bautista
d,
Jacky
Even
b and
Paola
Vivo
*a
aHybrid Solar Cells, Faculty of Engineering and Natural Sciences, Tampere University, P.O. Box 541, FI-33014 Tampere, Finland. E-mail: paola.vivo@tuni.fi
bUniv Rennes, INSA Rennes, CNRS, Institut FOTON – UMR 6082, F-35000 Rennes, France
cDipartimento di Ingegneria Civile, Ambientale, del Territorio, Edile e di Chimica (DICATECh), Politecnico di Bari, Via Orabona 4, 70125 Bari, Italy
dPhotonics Laboratory, Physics Unit, Faculty of Engineering and Natural Sciences, Tampere University, FI-33014 Tampere, Finland
eSurface Science Group, Photonics Laboratory, Tampere University, P.O. Box 692, FI-33014 Tampere, Finland
fFaculty of Engineering and Natural Sciences, Tampere University, P.O. Box 692, Tampere FI-33014, Finland
gUniv Rennes, ENSCR, INSA Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, 35700 Rennes, France
hSchool of Sustainable Energy Engineering, Simon Fraser University, 5118–10285 University Drive, Surrey, British Columbia V3T 0N1, Canada
First published on 22nd August 2023
Abstract
The study of surface defects is one of the forefronts of halide perovskite research. In the nanoscale regime, where the surface-to-volume ratio is high, the surface plays a key role in determining the electronic properties of perovskites. Perovskite-inspired silver iodobismuthates are promising photovoltaic absorbers. Herein, we demonstrate the colloidal synthesis of phase pure and highly crystalline AgBiI4 nanocrystals (NCs). Surface-sensitive spectroscopic techniques reveal the rich surface features of the NCs that enable their impressive long-term environmental and thermal stabilities. Notably, the surface termination and its passivation effects on the electronic properties of AgBiI4 are investigated. Our atomistic simulations suggest that a bismuth iodide-rich surface, as in the case of AgBiI4 NCs, does not introduce surface trap states within the band gap region of AgBiI4, unlike a silver iodide-rich surface. These findings may encourage the investigation of surfaces of other lead-free perovskite-inspired materials.
Introduction
The colloidal synthesis of halide perovskites is an effective tool to flexibly control size, shape, and optoelectronic properties of the resulting nanocrystals (NCs)1,2 through the fine-tuning of parameters like temperature,3,4 ligand nature and concentration,3,5 additives,6 and quenching time. When compared to thin film fabrication, the versatility of NC synthesis leads to several advantages like tuning the crystallization phase, eliminating the impurities, and/or enhancing stability.7,8 Furthermore, the colloidal synthesis at the nanoscale enables the formation of some lead-free perovskite compositions that do not exist in the bulk form but can be only synthesized at the nanoscale.9 The vast majority of the reported studies on lead-free perovskite-inspired NCs focus on halide double perovskites, such as Cs2AgMX6 (M = Bi, Sb; X = Cl, Br, or I).10 On the other hand, silver iodobismuthates (some embodiments of which are known as rudorffites) were previously studied for their high ionic conduction properties.11,12 However, they recently proved to be appealing candidates for photovoltaics, photodetectors, memristors, and high-energy radiation detector applications although they have been mostly investigated in thin films rather than in NCs.13–16 Premkumar et al. reported the ligand-assisted reprecipitation synthesis of Ag–Bi–I quantum dots (QDs) of various compositions with an optical band gap significantly higher than that of the bulk counterparts.17 For example, AgBiI4 QDs exhibited a band gap of 2.9 eV (absorption maximum ∼415 nm), whereas the reported bulk band gap values lie between 1.63 and 1.93 eV. The blue shift in the band gap was attributed to the quantum confinement (size) effect.17 However, the quantum confinement effect was not discussed in the relationship with the Bohr radii to explain the optical properties of these QDs. Moreover, Ag–Bi–I QDs have low crystallinity (based on the reported X-ray diffraction (XRD) patterns), hinting at poor phase purity, which is a common drawback of bulk films as well. Indeed, impurities, such as AgI (which produces an absorption peak at ∼415–425 nm) and BiI3, often co-exist with the various compositions of Ag–Bi–I and may influence or contribute to the observed optical band gap, in turn causing impairment of the optoelectronic device performance.13,15,18,19 Moreover, recent studies show that synthesis conditions also influence the stability of halide perovskite NCs.20,21 Therefore, there is a need to develop alternative synthesis protocols with systematically tuned reaction conditions (e.g., type and ratio of precursor materials, temperature, and post-synthesis purification22) to obtain highly crystalline and stable Ag–Bi–I NCs with phase and/or compositional purity.Surface defects represent one of the most limiting factors in the performance and stability of Ag–Bi–I-based photovoltaic cells and photodetectors. Despite the various strategies to suppress these surface defects or imperfections,13,14,23,24 their influence on the electronic properties of Ag–Bi–I remains largely unexplored. On the other hand, the surface atomic structure of lead-based NCs also influences their electronic structure as well as the structural and other material properties.25–27 For example, theoretical calculations suggest that PbX2 surface termination results in the loss of defect tolerance for CsPbX3 NCs due to the appearance of mid-gap states,28 and band gap reduction in MAPbX3.25 The chemical nature of the surface termination can also influence the stability and performance of solar cells,26,28 ultimately suggesting and stressing even more the importance of surface studies. Thus, it is paramount to investigate the influence of the surface atomic structure on the electronic band structure of Ag–Bi–I and other lead-free perovskite-inspired materials (PIMs), which in turn significantly impacts their optoelectronic device performance.
In this work, we optimize the colloidal synthesis of AgBiI4 NCs using a hot-injection approach under iodide-rich conditions. We also study their surface properties combining experimental characterizations with atomistic simulations. A thorough tuning of the synthesis parameters (temperature, choice of the precursors, and ratio between them) reveals that it is possible to obtain phase-pure and highly crystalline AgBiI4 NCs, conversely to what has been shown earlier for Ag–Bi–I QDs. Our NCs can withstand the degradation in polar solvents, thus significantly simplifying their post-synthesis purification and film formation. We found that the synthesized AgBiI4 NCs display non-linear optical (NLO) responses that can be detected unambiguously and visualized using multiphoton absorption (MPA) microscopy. Given the increasing interest in finding low-toxicity NLO-active alternatives for various photonic and optoelectronic applications, our findings may promote the growing need to develop NLO microscopy techniques to study the non-linear optical responses and other characteristics of lead-free PIMs.29–32
The synthesized NCs can preserve their structural integrity under ambient storage for months as well as when exposed to elevated temperatures. The combination of X-ray photoelectron spectroscopy (XPS) and nuclear magnetic resonance (NMR) techniques reveal the complex surface features of these NCs. Our density functional theory (DFT) calculations suggest that the AgBiI4 NCs possess an electronically favorable surface that does not negatively influence their band structure. This is the first-ever demonstration of the influence of surface termination and its passivation on the electronic properties of AgBiI4, which, in general, is rarely studied for lead-free PIMs.
Experimental section
Materials
Bismuth(III) acetate (Bi(Ac)3, ≥99.99%), silver acetate, (Ag(Ac), ≥99.99%), octadecene (ODE, 90%), oleic acid (OA, 90%), oleylamine (OlAm, technical grade, 70%), hexane (for spectroscopy, ≥99%), and ethyl acetate (EA, ≥99.5%), iodotrimethylsilane (TMS-I, 97%), were purchased from Sigma-Aldrich. All chemicals were used without further purification.Synthesis details
0.401 mmol each of Bi(Ac)3 and Ag(Ac), along with 5 mL ODE, 1.25 mL of OA, and 0.2 mL of OlAm were loaded into a 25 mL three-neck flask. The scaling and handling were carried out in ambient conditions. The precursor solution was degassed in vacuum for 1 h at 120 °C until the metal salts were fully dissolved, and the reaction mixture turned into a dark reddish color (Fig. S1a†). Then the temperature was increased to 190 °C and 0.3 mL of TMS-I was quickly injected into it followed by an instant quenching of the reaction using an ice-water bath. After cooling down to room temperature, the reaction solution attained a non-transparent dark-brown color (Fig. S1b†). The crude NC solution was purified by centrifugation at 4500 rpm for 5 min. The supernatant was discarded, and the precipitate was left in a vacuum desiccator for 30 min to remove the excess solvent and then redispersed in hexane (Fig. S1c†). For all characterizations, the NCs were washed with EA by the following method. The NC dispersion was initially drop-casted on the substrate, and then the substrate was quickly dipped into EA (for 3–5 s). The resultant film was dried under Ar flow. After evaporating EA, a black NC film was formed (Fig. S1d†).Characterization techniques
XRD patterns were recorded by Malvern Panalytical Empyrean Alpha 1 in a powder diffraction mode using Cu Kα radiation (λ = 1.5406 Å) and a cathode voltage and current of 45 kV and 40 mA, respectively.Transmission electron microscope (TEM) images were taken using JEM-F200 (200 kV). 20 μl of the NC dispersion in hexane was drop casted on the carbon-coated Cu grids following by dipping of the grid into EA.
Scanning electron microscope (SEM) images were taken using field emission SEM Zeiss Ultraplus (at 3–15 kV). The Energy-dispersive X-ray spectroscopy (EDS) was performed using the Oxford Instruments X-MaxN 80 (EDS) combined with field emission SEM Zeiss Ultraplus at 15 kV.
Ultraviolet–visible (UV-vis) reflectance spectra of thick AgBiI4 NC films were recorded using an integrating sphere attached to a Shimadzu UV-3600 UV-vis-NIR spectrophotometer.
Inductively coupled plasma mass spectroscopy (ICP-MS) measurements were conducted with Thermo Scientific iCAP™ RQ equipment. After purification, NC precipitate samples were dissolved in concentrated 1HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4HCl (v/v) followed by dilution and measurement in 2% 1HNO3:4HCl (v/v) matrix. Ionic standard solutions with a concentration range of 0.001–1000 μg l−1 for Bi and Ag were prepared in 2% 1:4 HNO3:HCl using super-pure chemicals (Romil-SpA™) and applied to measure calibration curves. Ultrapure H2O (18.2 MΩ cm, Merck Milli-Q®) was used for sample dilutions.
:4HCl (v/v) followed by dilution and measurement in 2% 1HNO3:4HCl (v/v) matrix. Ionic standard solutions with a concentration range of 0.001–1000 μg l−1 for Bi and Ag were prepared in 2% 1:4 HNO3:HCl using super-pure chemicals (Romil-SpA™) and applied to measure calibration curves. Ultrapure H2O (18.2 MΩ cm, Merck Milli-Q®) was used for sample dilutions.
The XPS measurements were performed in an ultrahigh vacuum (UHV) system. The AgBiI4 film samples were drop casted on Si wafer and let dry in a vacuum desiccator overnight before loading into the UHV system. XPS spectra were measured using non-monochromatized Al Kα X-rays (hν = 1486.6 eV) generated by a twin anode X-ray source (8025 Twin anode X-Ray source, V. G. Microtech) operated at 300 W X-ray power and a hemispherical electron spectrometer (CLAM4 MCD LNo5, V. G. Microtech). To monitor changes in surface chemical composition under X-rays, five consecutive cycles of spectra (Bi 4f → I 3d → Ag 3d → O 1s → C 1 s → N 1s, duration of a cycle ∼10 min) were recorded and analyzed separately. Survey spectrum was measured at the end. The chemical states of the elements were determined from the XPS spectra by least-squares fitting of asymmetric Gaussian–Lorentzian lineshapes after background subtraction. The analysis was made in CasaXPS software version 2.3.17PR1.1.33 The binding energy scale was calibrated by setting C 1s (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C) to 284.8 eV.
–C) to 284.8 eV.
1H solution NMR spectra were recorded on Agilent 500/54 Premium shielded spectrometer. 1H chemical shifts were referenced using the internal residual peak of the solvent (CDCl3, δ 7.27 ppm).
Thermogravimetric analysis (TGA) was carried out on a PerkinElmer Pyris 6 TGA instrument under a nitrogen flow (40 mL min−1) at a heating rate of 10 °C min−1.
For the NLO measurements, a drop casted AgBiI4 NC sample on a pre-cleaned glass substrate was prepared and instantly probed using a customized multiphoton absorption (MPA) microscope. A femtosecond laser source (Coherent) with wavelength of 1060 nm, pulse length of 140 fs, and repetition rate of 80 MHz was employed as the multiphoton excitation source. The linearly polarized output of the laser was spatially cleaned, collimated, and then directed to the multiphoton microscopy setup that is equipped with a microscope objective with numerical aperture of 0.8 (Nikon), and two cooled photomultiplier tubes (Becker & Hickl). The nonlinear signals from the sample were excited at normal incidence using the microscope objective and then collected using the same objective. The nonlinear signals were discriminated from the fundamental ones using appropriate dichroic and short pass filters, restricting the detection wavelengths to 300 to 400 nm, and 400 to 650 nm in separate detection channels. To create a scanning map, the outputs of the detectors were synchronized to the movement of the sample that is fixed onto a 3D piezo-scanner (Mad City Labs). The measurements were carried out at room temperature with a pixel dwell time of 50 ms, a pixel resolution of 500 nm and an average power of 0.5 mW as measured before the sample. Although the experimental imaging parameters are not fully optimized, these parameters were found earlier to be suitable in acquiring nonlinear luminescence without overexposing this particular sample to femtosecond laser radiation.
Different theoretical and computational approaches are employed in this work. For the details, see the ESI.†
Results and discussion
Synthesis of AgBiI4 NCs
TMS-I was injected into a metal acetate precursor solution containing Ag(Ac), Bi(Ac)3, and capping ligands (OA and OlAm) at 190 °C, which instantaneously caused the nucleation and growth of the AgBiI4 NCs (see Fig. S1 and the Synthesis section of the ESI,† for details). This approach has a few advantages compared to the traditional hot-injection method involving metal oleate injection.34 For instance, metal acetates typically demonstrate a higher solubility than the metal salts (like AgI in the present case) in the presence of acid- and amine-based capping ligands. Importantly, the trimethylsilyl halide (TMS-X) injection route involves halide-rich synthesis conditions, which ensure the formation of NCs with enhanced stability and optoelectronic properties.7,9,34–37 Moreover, the TMS-X injection method allows the tuning of the amounts of halide and cation precursors independently, which permits the injection of excess halide while maintaining the targeted cation ratio in contrast to the conventional metal oleate injection route.7,9 Nonetheless, it guarantees neither the formation nor the phase purity of the NCs; thus, optimizing the synthesis conditions is still necessary. To obtain the desired AgBiI4 NCs, various reaction parameters, i.e., injection temperature, metal acetate to halide ratio, precursor concentration, and the ratio between the capping ligands, were screened, as shown in the scheme of Fig. 1. We first varied the injection temperature between 120 and 190 °C and characterized the as-obtained NCs after purification through XRD analysis (see Fig. 2 and Fig. S2 in ESI†). The desired NCs were achieved when the injection temperature was ≥190 °C, below which AgI was predominantly formed (Fig. S2a in ESI†). However, the AgBiI4 NCs obtained at ≥190 °C demonstrated only a moderate colloidal stability in non-polar solvents like hexane, toluene, and chloroform. Therefore, we aimed to alter the synthesis conditions further to improve the colloidal stability, while at the same time tuning the shape and size of the NCs. We realized that excess OA is crucial to obtain AgBiI4 as the exclusive product. For instance, reducing the OA to OlAm ratio from 1:0.16 to 0.125:0.075, i.e., by nearly half, led to the formation of BiI3 as the only phase (Fig. S2b in ESI†), probably due to the strong affinity between the amine ligand molecules and Bi3+. This is similar to the case of CsPbBr3 NCs, wherein more OA than OlAm is required to obtain the NCs in a desired form.38 Furthermore, syntheses involving lower precursor concentration or excess TMS-I (which was thought to be beneficial for obtaining NCs with reduced surface traps and modified size and shape) resulted in the formation of unidentified products (Fig. S2c and d in ESI†). In other words, the desired AgBiI4 NCs were not obtained under these two conditions. These observations suggest that the synthesis of the ternary Ag–Bi–I NCs through the TMS-I injection route demands careful balancing of the individual components involved, such as precursor stoichiometries, capping ligand concentrations, and injection temperature, to prevent the formation of undesired products or NCs with poor phase purity. The AgBiI4 NCs were successfully obtained in the present work by injecting TMS-I into a mixture of metal acetates and excess OA (OA:OlAm = 1:0.16) at 190 °C. Further synthesis tuning with shorter chain ligands or ligands with other functional groups may improve morphologies, colloidal stability, and size tunability of the NCs.39 The synthesis of the AgBiBr4 NCs by employing the reaction parameters optimized for AgBiI4 NCs led to the formation of AgBr (see Fig. S2e in ESI†). This may have occurred due to the unfavorable formation of AgBiBr4, which is also supported by the lack of reports on this material in the literature.
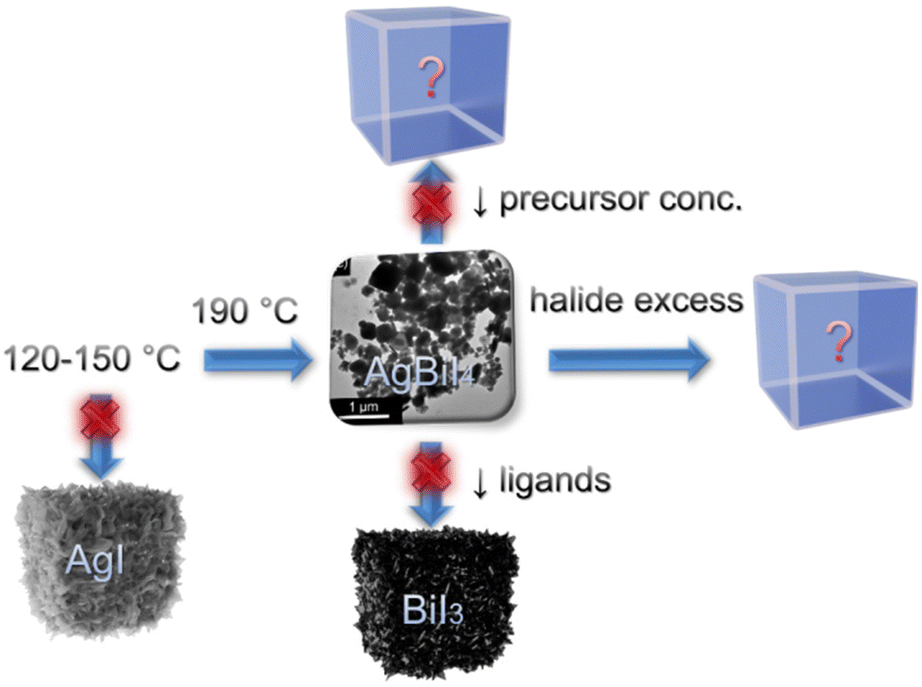 | ||
| Fig. 1 Schematic representation of the synthesis optimization routes to obtain phase-pure AgBiI4 NCs. | ||
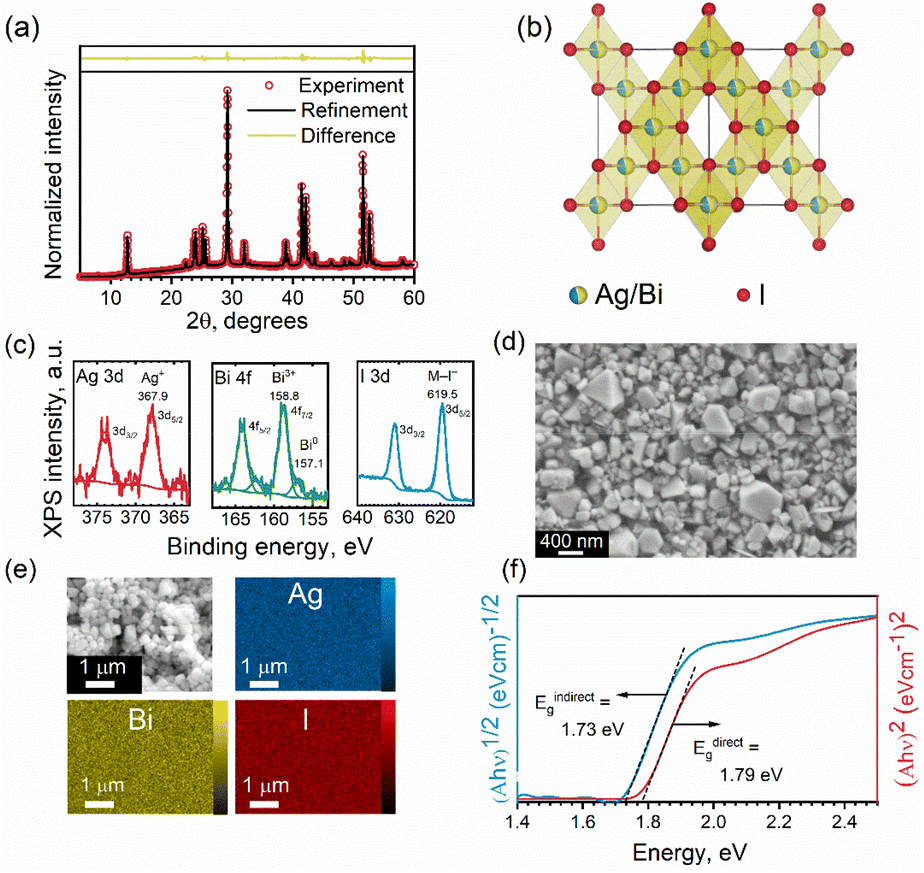 | ||
| Fig. 2 (a) Experimental (open circles) and refined (solid line) profiles and their difference (yellow line) obtained after the full-pattern Rietveld refinement of the XRD patterns of AgBiI4 NC film. (b) The refined crystal structure of AgBiI4. (c) XPS core-level spectra of Ag 3d, Bi 4f, and I 3d (d) high magnification SEM image of the AgBiI4 NCs. (e) SEM-EDS color maps showing the distribution of Ag, Bi, and I in the AgBiI4 NC film. (f) Tauc plots of AgBiI4 NC film for direct and in-direct band gap assumptions. | ||
Structure and morphology of the NCs
The XRD patterns of the as-obtained NCs closely match those of various Ag–Bi–I compositions, making the assignment of the correct composition challenging. Thus, we performed Rietveld fitting on the XRD patterns (Fig. 2a) to identify the phase purity of the NCs. The refinement outcomes (summarized in Table S1 in ESI†) revealed that the experimental XRD pattern of the NCs fully converges to the cubic model of AgBiI4, with a corresponding space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and no co-existing secondary (impurity) phases were observed. Moreover, the presence of only one diffraction peak at 2θ ≈ 42° (Fig. S3 in ESI†) confirms the crystallization of AgBiI4 NCs in the Fdm space group.15,40 Both Ag and Bi occupy the same edge-sharing octahedral sites of 16c with a cation deficiency (see Table S1 in ESI†), consistently with the bulk AgBiI4.40 On the other hand, the tetrahedral sites of the structure remain vacant. The refined crystal structure of the NCs is shown in Fig. 2b. The elemental analysis by ICP-MS showed a Ag:Bi atomic ratio of 1.08, which is close to the nominal value of 1, further confirming the atomic composition of the AgBiI4 NCs. XPS measurements were conducted to identify the valence X-ray effect states of Ag, Bi, and I (see Fig. S4 in ESI† for the XPS survey spectrum for AgBiI4 NCs). One chemical state was determined for Ag (Ag 3d5/2 at 367.9 eV), Bi (Bi 4f7/2 at 158.8 eV), and I (I 3d5/2 at 619.5 eV), corresponding to the oxidation states of 1+ (Ag+), 3+ (Bi3+), and 1− (I−), respectively (see Fig. 2c and Table 1). Further, some metallic Bi (Bi0 4f7/2 at 157.1 eV) evolved at the surface during the XPS measurement, which was assigned to an X-ray effect.
m, and no co-existing secondary (impurity) phases were observed. Moreover, the presence of only one diffraction peak at 2θ ≈ 42° (Fig. S3 in ESI†) confirms the crystallization of AgBiI4 NCs in the Fdm space group.15,40 Both Ag and Bi occupy the same edge-sharing octahedral sites of 16c with a cation deficiency (see Table S1 in ESI†), consistently with the bulk AgBiI4.40 On the other hand, the tetrahedral sites of the structure remain vacant. The refined crystal structure of the NCs is shown in Fig. 2b. The elemental analysis by ICP-MS showed a Ag:Bi atomic ratio of 1.08, which is close to the nominal value of 1, further confirming the atomic composition of the AgBiI4 NCs. XPS measurements were conducted to identify the valence X-ray effect states of Ag, Bi, and I (see Fig. S4 in ESI† for the XPS survey spectrum for AgBiI4 NCs). One chemical state was determined for Ag (Ag 3d5/2 at 367.9 eV), Bi (Bi 4f7/2 at 158.8 eV), and I (I 3d5/2 at 619.5 eV), corresponding to the oxidation states of 1+ (Ag+), 3+ (Bi3+), and 1− (I−), respectively (see Fig. 2c and Table 1). Further, some metallic Bi (Bi0 4f7/2 at 157.1 eV) evolved at the surface during the XPS measurement, which was assigned to an X-ray effect.
| Element | Ag | Bi | I | O | N | C | |||
| Transition | Ag 3d5/2 | Bi 4f7/2 | I 3d5/2 | O 1s | N 1s | C 1s | |||
| E b (eV) | 367.9 | 158.8 | 157.1 | 619.5 | 532.5 | 534.0 | 402.3 | 284.8 | 286.5 |
| Possible chemical state | Ag+ | Bi3+ | Bi0 * | I− | ![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) –C, –C, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
–C |
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) H3+ H3+ |
–C |
–O/N |
| Atomic ratio (I = 4) | |||||||||
| Cycle 1 | 0.20 | 0.91 | 0.00 | 4.00 | 4.44 | 3.74 | 3.39 | 80.57 | 11.27 |
| Cycles 2–5 | 0.16 | 0.72 | 0.13 | 4.00 | 1.46 | 0.55 | 1.09 | 32.64 | 5.03 |
Fig. S5 in ESI† shows the TEM images of the NCs. However, the thickness of the NCs is higher than the penetration depth of the electron beam, which limits the information that can be extracted from TEM in this case. Therefore, we studied the NCs through SEM. The NC films were prepared via an antisolvent-based “dipping method”. Briefly, the NCs dispersed in hexane were drop-casted (or spin-coated) on the substrate. The as-obtained NC film was dipped into EA for 2–3 s and immediately dried under Ar flow. This resulted in the formation of dry AgBiI4 NC films. The XRD patterns of the NC films before and after dipping into EA remained the same, suggesting that the EA treatment did not disintegrate the NC structure but removed the excess ligands from the NC film (Fig. S2f in ESI†). The SEM image of the NC film shows scattered individual NCs (Fig. S6†), indicating that the NCs had not coalesced despite their modest colloidal stability. The NCs are a few hundred nanometers thick (see the SEM images in Fig. 2d and Fig. S6 in ESI†), as also noticed from the TEM images (Fig. S5 in ESI†), with an average size of 296 ± 70 nm (Fig. 2d). In addition to the halide-rich synthesis environment and high injection temperature, the large particle size of ∼300 nm of the AgBiI4 NCs can be attributed to their high crystallinity.41,42 The Energy Dispersive Spectroscopy (EDS) maps in Fig. 2e indicate a uniform distribution of Ag, Bi, and I in AgBiI4 NCs.
Optical properties and bulk electronic band structure of the NCs
We estimated the optical band gap of AgBiI4 NCs from Tauc plots (Fig. 2f). The direct and indirect band gap assumptions resulted in band gap values of 1.79 eV and 1.73 eV, respectively. The direct band gap of 1.79 eV falls within the broad range of the values (1.63–1.93 eV) reported for the bulk AgBiI4 (see Table S2 for the summary in ESI†). The co-existence of other Ag–Bi–I compositions might have contributed to the variations in the reported band gap values of AgBiI4. Also, no quantum confinement effects are anticipated for the NCs due to their size of few hundred nanometers. This size is indeed much larger than the exciton Bohr radius of approximately 6 Å (0.6 nm) for AgBiI4, estimated using the 3D Wannier exciton model assuming a relative dielectric constant of 3.36,24 and an effective average reduced mass of 0.33m0.43 Therefore, no specific enhancement of the emission is expected from excitonic effects. Moreover, a vanishing quantum confinement effect on the electronic band structure is also predicted for such big sizes (Fig. S10 in ESI†), in turn explaining the comparable band gap of the NCs with that of bulk AgBiI4. A slightly indirect band gap nature of AgBiI4 NCs indicated by a lower value of Eindirectg than Edirectg as shown in Fig. 2f, aligns with previously reported findings.40,43 This observation is further supported by our own DFT electronic band structure calculations, which demonstrate a lower Eindirectg value from Γ to R, though very close to that of Edirectg at Γ (Fig. S8 and S9 in ESI†). The orbital contributions to the conduction band minimum (CBM) and valence band maximum (VBM) were analyzed by computing the spatial localization of the density of states (LDOS) (Fig. 3). Their analysis revealed, in agreement with the literature,43,44 that the CBM results from hybridization between Bi 6p and I 5p orbitals, while the VBM is a mix of I 5p and Ag 4d orbitals. Most importantly, the LDOS highlighted the existence of two set of I atoms with only apical ones contributing to the VBM (Fig. 3). This specific localization of the VBM gives rise to unique physical behavior within this material family, setting it apart from similar compounds such as lead perovskites.45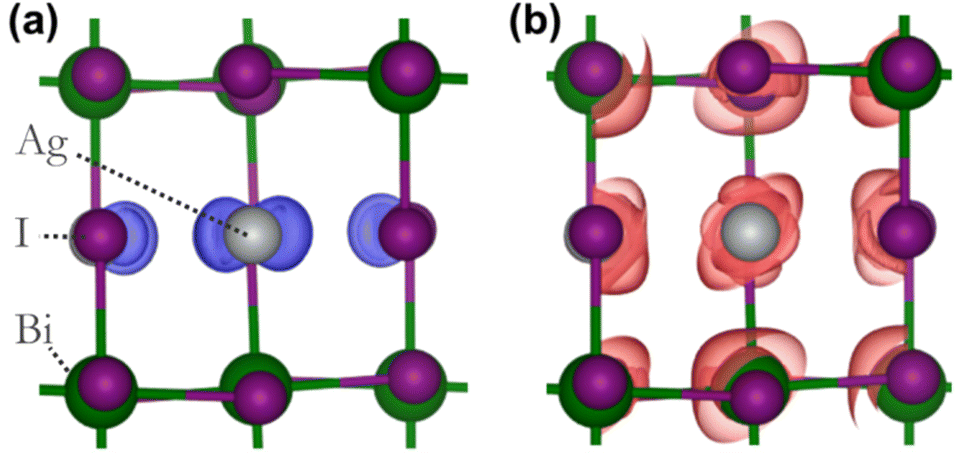 | ||
| Fig. 3 Localized density of states at the (a) VBM and (b) CBM computed for AgBiI4 with a Imma model crystallographic structure. The grey, green, and purple spheres indicate Ag, Bi, and I atoms respectively. | ||
Despite no luminescence was detected from the NCs, similar as with bulk AgBiI4 films, the analysis of the CBM and VBM symmetries suggests that band-to-band transitions both at Γ and R might be optically active (Table S3 in ESI†). Therefore, the absence of luminescence could potentially be rather attributed to the weakness of the oscillator strengths as well as the existence of nonradiative processes arising from a high concentration of bulk defects with a high capture cross-section on the order of 10−19 cm2 present in the band gap region quenching the emission at room temperature within the material.46
Multiphoton absorption
Although the AgBiI4 NCs are non-emissive under linear absorption, when exciting a microscopic region of the AgBiI4 NC film with femtosecond pulses centered at 1060 nm, the sample was found to emit nonlinear signals that are distinct and much stronger than the background, as depicted in the scanning maps (Fig. 4). Importantly, the nonlinear signals were apparent at two spectral regions (300 to 400 nm and 400 nm to 650 nm), corresponding to the two separate detection channels of our multiphoton microscope. Furthermore, the results from these two detection channels exhibited the same qualitative features (i.e., highly colocalized or overlapped in space). Here, multiple, i.e., two (or three), monochromatic photons, are simultaneously absorbed by the sample to generate a conventional or single photon luminescence at wavelengths that are greater than half (or a third) of the excitation wavelength. Altogether, the results imply that the nonlinear signals are strongly indicative of MPA-induced luminescence from AgBiI4 NCs and are not dominated by high-order scattering associated with bulk and relative differences in the third-order nonlinear susceptibility of material interfaces47 or molecular symmetry-breaking,48 as earlier reported in the context of halide perovskites. The intensity variations in the nonlinear maps were likely due to large-scale sample nonuniformities.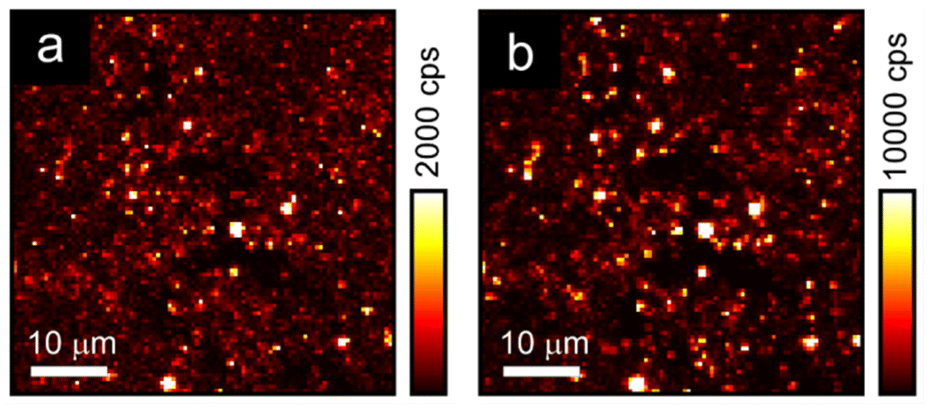 | ||
| Fig. 4 Multiphoton absorption-based luminescence maps of the drop-casted AgBiI4 NCs at spectral regions corresponding to (a) 300 to 400 nm, and (b) 400 nm to 650 nm. The input polarization is along the vertical. The collected signals are not polarization-analyzed. The maps are plotted at different scales (in terms of signal counts per second or cps) for emphasis. The resolution of the maps is 100 × 100 pixels. | ||
Ambient and thermal stabilities of AgBiI4 NCs
Lead-free and tin-free PIMs typically show higher stability than lead perovskites under air exposure, humidity, and high temperatures.49–52 Sansom et al. reported that the XRD pattern of bulk AgBiI4 sample remains unchanged up to 90 °C, above which a small quantity of Ag2BiI5 started appearing as an impurity while the crystal structure of the film was still intact.53 We investigated the structural and morphological stabilities of the AgBiI4 NC films under ambient air exposure and heat treatment through XRD and SEM. The NC films displayed a nearly identical XRD pattern before and after 18 weeks (∼4 months) of storage in the ambient conditions (dark, relative humidity (RH) ≈30%, T = 25 °C), suggesting their long-term stability in the air (Fig. 5a). The aged NC film (see the SEM image in Fig. 5b) also does not demonstrate any significant changes (no apparent fusion of the particles noticed) compared to the fresh film (Fig. 2d and Fig. S6 in ESI†). The thermal stability of the films was monitored for 10 minutes in the temperature range of 25–160 °C in the air. No obvious changes in the corresponding XRD patterns were detected (Fig. S7 in ESI†), thus suggesting no structural degradation occurring even at 160 °C, in contrast with what is known for the bulk AgBiI4 films.53 The TGA data of the NCs (Fig. 6a) show only a negligible weight loss of 2% at 160 °C. We ascribe the superior ambient and thermal stabilities of AgBiI4 NCs to their surface characteristics (strongly bound ligand shell and surface atomic composition).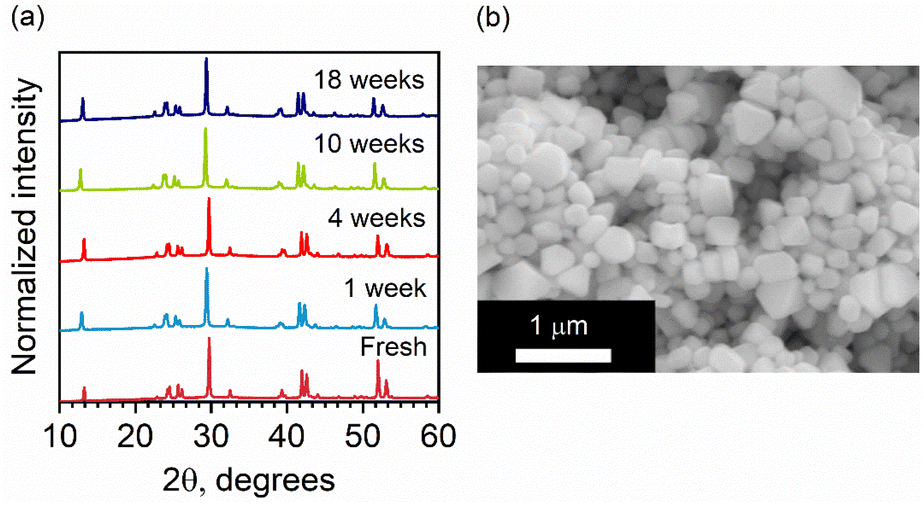 | ||
| Fig. 5 (a) Evolution of the XRD patterns of a AgBiI4 NC film stored in the ambient conditions in dark (RH ∼ 30% T = 25 °C). The patterns were recorded under identical measurement settings. (b) SEM image of an aged NC film (two months old) stored in the ambient conditions in dark. | ||
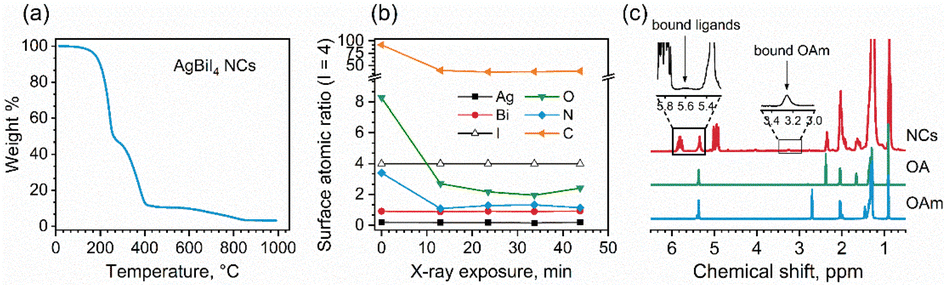 | ||
| Fig. 6 (a) TGA data for a AgBiI4 NC sample. (b) Changes in surface composition (atomic ratios normalized to I = 4) of AgBiI4 film sample during XPS measurement of five consecutive cycles of spectra (Bi 4f → I 3d → Ag 3d → O 1s → C 1 s → N 1s, duration of a cycle ∼10 min). (c) 1H NMR spectra of AgBiI4 NCs, pure OA, and pure OlAm. The insets highlight the proton resonances related to bound ligands. | ||
Surface chemistry of the NCs
To investigate the binding nature of the capping ligands (OA and OlAm) on the NC surface, we analyzed the XPS data related to the O 1s and N 1s peaks of the NCs. The O 1s core-level signal can be fit to two peaks with the binding energies of 532.5 eV and 534.0 eV, which can be attributed to monodentate (O–C and OC) carboxylate and bidentate (O–CO) carboxylate or carboxylic acid bonds, respectively, present on the NC surface.54 This might indicate the presence of OA in both oleate and acid forms on the NC surface. The N 1s core-level signal demonstrated a single peak at ∼402 eV related to the nitrogen atom of oleylammonium ions (OlAm+).55,56 It is possible that the OlAm+ act as a capping ligand through the interaction with the surface I− or by substituting some of the Ag+ ions, being the NC surface silver-deficient, as experimentally supported by XPS measurements (Table 1). The binding of OlAm+ to the AgBiI4 surface might resemble the case of CsPbBr3 NCs.38,57 We further monitored the changes in the surface composition of the AgBiI4 NC film during XPS measurement of five consecutive cycles (Fig. 6b). While Ag, Bi, and I concentrations remain largely unchanged during the X-ray exposure for ∼45 min in an ultra-high vacuum environment, those of C, N, and O decrease markedly during the first 10 min. This corresponds to the desorption of a portion of the capping molecules from the NC surface under a high vacuum atmosphere. The relative decrease is the strongest for the O 1s peak at 534.0 eV, which leads us to speculate that the OA molecules that are weakly bound on the NC surface desorb easily compared to the bound OA and OlAm+. After the initial fast change, the O 1s to N 1s ratio stabilizes to ∼2:1, which corresponds to OA:OlAm ratio of ∼1:1 (since each oleic acid molecule contains two ‘O’ atoms), best represents the ligand shell composition that is strongly bound to the NC surface.
Fig. 6c shows the 1H NMR spectra of the AgBiI4 NCs, pure OA, and pure OlAm in CDCl3. The peak at 2.4 ppm is exclusive for OA (i.e., CH2–COOH), indeed being absent in the pure OlAm. Therefore, the 1H NMR spectrum of the NCs is dominated by the peaks related to OA, suggesting an apparent excess of OA compared to OlAm. The slightly broadened OA peaks of the NC sample might be due to the equilibria established in solution between bound and unbound OA molecules,58 portion of which was also detected by XPS. On the other hand, the broad NMR resonances at ∼3.3 and 5.6 ppm are typical of the methylene protons attached to the nitrogen atom (i.e., CH2–N) of OlAm+ and of the vinylene protons that can arise from tightly bound ligands.59 Overall, the NMR data suggest that the organic shell of the AgBiI4 NCs is composed of both weakly bound OA and strongly bound OA and OlAm+. The existence of the weakly and strongly bound organic species attached to the NCs was further confirmed by TGA (Fig. 6a), which shows two different thermal events (T < 400 °C) ascribable to the weight loss/decomposition of the organic ligands. The first thermal event (T ≈ 200 °C) responsible for the main weight loss (∼52%) is due to the detachment of weakly bound or unbound species (including OA and ODE traces). The second one (T ≈ 350 °C) concerns the removal of tightly bound organic species with a lower weight variation (∼35%) of the sample.
The atomic composition of the NCs from XPS (probing depth of ∼10 nm) was found to be Ag0.2Bi1.0I4.7, which is indicative of their surface atomic composition. An earlier study on the bulk AgBiI4 also exhibited a surface composition of Ag0.2Bi1.0I3.1.53 In the case of lead-based perovskite NCs, halide rich surfaces have been shown to improve both phase and thermal stabilities.34,60 We hypothesize that the excess I− on the surface (I/Bi = 4.7) of the AgBiI4 NCs contributed to their structural integrity in the air and at elevated temperatures. In addition, the composition is different from their bulk counterpart with a large Ag deficiency (0.2 ≪ 1), thus possessing a bismuth iodide-rich surface.53
To assess the potential interest, in terms of electronic properties, brought by a Bi-rich surface compared to an Ag-rich surface, we designed a BiI and an AgI terminated nanoplatelets (Fig. 7a and b). Further, to capture the influence of the OA rich environment, the surfaces of the modeled slabs were coated with OA molecules facing the undercoordinated metal atoms. We chose only OA here instead of both OA and OlAm for the simplicity of the calculations. While the atomic positions of surface atoms for all slabs were relaxed, those of the central regions of the slabs were kept frozen in order to mimic the bulk material (Fig. 7a and b). The band structures of the coated and uncoated systems were computed at the PBE + SOC level of theory (Fig. 7c and d). In the case of the BiI terminated slab, the resulting band structures (Fig. 7c) did not reveal the presence of surface states at the vicinity of the band gap even in the uncoated system, and the bulk electronic structure was fairly well-recovered with such slab calculations. Likewise, apart from slightly decreasing the charge carriers’ effective masses, the passivation with OA did not influence much the electronic band structure. Employing computationally demanding hybrid functionals calculations on a smaller slab confirms that there is a minimum effect on the electronic structure when OA is present (Fig. S11 in ESI†). On the other hand, the AgI-terminated surface exhibits greater sensitivity to surface defects with the emergence of shallow surface states (Fig. 7d). These states can be attributed to the presence of 4 apical I atoms on this surface influenced by surface distortions, thus leading to a degeneracy splitting of the corresponding states and narrowing the band gap (Table 2). The OA coating appeared to be ineffective in passivating these surface defects, even closing further the band gap.
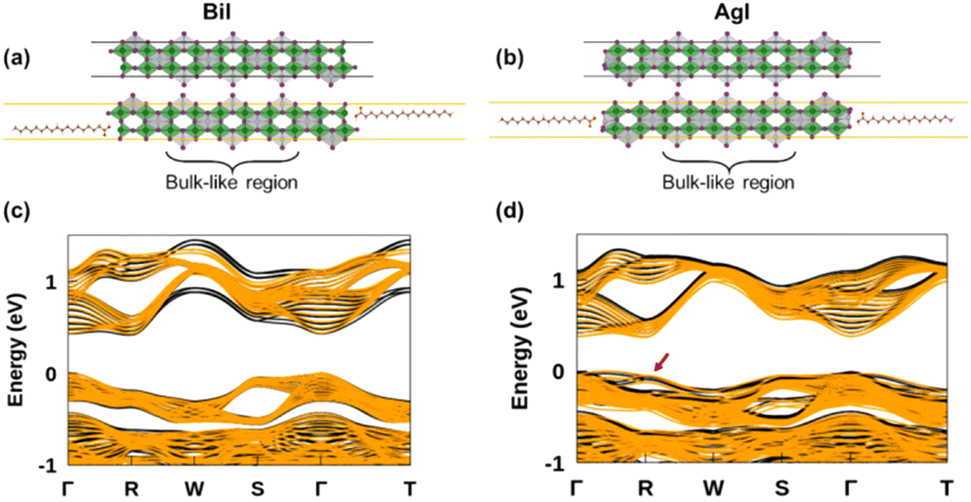 | ||
| Fig. 7 Illustration of influence of surfaces defects on the electronic properties. Models of the (a) BiI terminated and (b) AgI terminated slabs with the corresponding PBE + SOC band structures represented below (c and d). The band structures of the coated surfaces are represented in yellow on top of that of the pristine surfaces (black) for better comparison. | ||
| Slabs’ surface | E g (eV) | |
|---|---|---|
| Pristine surfaces | BiI | 0.41 |
| AgI | 0.40 | |
| Coated surfaces | BiI + OA | 0.42 |
| AgI + OA | 0.37 |
Conclusions
We have demonstrated the first colloidal synthesis of highly crystalline and phase-pure AgBiI4 NCs upon the thorough tuning of the synthesis protocol. The NCs displayed impressive long-term and thermal stabilities, which were attributed to the iodide-rich surface and surface encapsulation by the capping ligands. This study outlined an unambiguous surface-electronic structure relationship for Ag–Bi–I NCs. The electronic band structures of the AgI and BiI terminated surfaces revealed that no additional trap states are introduced in the latter case that closely resembles the experimentally observed surface atomic composition of the AgBiI4 NCs. Our work represents a relevant starting point for the investigation of the surface properties of other lead-free PIMs. The MPA-based NLO response from Ag–Bi–I NCs encourages further thorough studies on various silver iodobismuthate and perovskite-inspired compositions in this promising yet unexplored direction.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. A. M.: conceptualization, data curation, methodology, formal analysis, writing – original draft; G. K. G.: conceptualization, data curation, formal analysis, writing – review & editing; A. B.: methodology, data curation, formal analysis, investigation, writing – review & editing; L. P.: methodology, data curation, formal analysis, investigation, writing – review & editing; R. G.: methodology, data curation, formal analysis, investigation, writing – review & editing; S. A.: methodology, data curation, formal analysis, investigation, writing – review & editing; H. A.-L.: data curation, formal analysis, writing – review & editing; R. V.: methodology, data curation, writing – review & editing; K. L.: data curation, formal analysis, writing – review & editing; G. V. – methodology, writing – review & editing; V. P. formal analysis, writing – review & editing; G. B.: supervision, formal analysis, writing – review & editing; J. E.: supervision, formal analysis, writing – review & editing; P. V.: supervision, conceptualization, funding acquisition, project administration, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Academy of Finland Flagship Programme, Photonics Research and Innovation (PREIN), decision No 320165. A. M. and P. V. thank the financial support of Tampere University, Faculty of Engineering and Natural Sciences. P. V. also thanks Jane and Aatos Erkko foundation for the financial support (SOL-TECH project). G. K. G. acknowledges Tampere Institute for Advanced Study for postdoctoral funding. S. A. thanks Jenny and Antti Wihuri Foundation, R. V. thanks Finnish Foundation for Technology Promotion, G. V. acknowledges support from the Agence Nationale pour la Recherche through the CPJ program. G. B. thanks PREIN Flagship support No. 31001491. J. E. acknowledges the financial support from the Institut Universitaire de France. This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement no 862656 (project DROP-IT). DFT calculations were performed at FOTON Institute, and the work was granted access to the HPC resources of TGCC/CINES under the allocation A0140911434 made by GENCI. Tampere Microscopy Center is gratefully acknowledged for providing the access to the characterization facilities.References
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- A. Matuhina, G. K. Grandhi, M. Liu, J. H. Smått, N. S. M. Viswanath, H. Ali-Löytty, K. Lahtonen and P. Vivo, Nanoscale, 2021, 13, 14186–14196 RSC.
- C. Chen, L. Zhang, T. Shi, G. Liao and Z. Tang, Nanomaterials, 2019, 9, 1751 CrossRef CAS PubMed.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed.
- R. Grisorio, F. Fasulo, A. B. Muñoz-García, M. Pavone, D. Conelli, E. Fanizza, M. Striccoli, I. Allegretta, R. Terzano, N. Margiotta, P. Vivo and G. P. Suranna, Nano Lett., 2022, 22, 4437–4444 CrossRef CAS PubMed.
- M. Liu, S. K. Matta, H. Ali-Löytty, A. Matuhina, G. K. Grandhi, K. Lahtonen, S. P. Russo and P. Vivo, Nano Lett., 2021, 22, 311–318 CrossRef PubMed.
- F. Locardi, M. Cirignano, D. Baranov, Z. Dang, M. Prato, F. Drago, M. Ferretti, V. Pinchetti, M. Fanciulli, S. Brovelli, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 12989–12995 CrossRef CAS PubMed.
- J. Yang, M. K. Choi, D.-H. Kim and T. Hyeon, Adv. Mater., 2016, 28, 1176–1207 CrossRef CAS PubMed.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- S. Ghosh and B. Pradhan, ChemNanoMat, 2019, 5, 300–312 CrossRef CAS.
- T. Oldag, T. Aussieker, H.-L. Keller, C. Preitschaft and A. Pfitzner, Z. Anorg. Allg. Chem., 2005, 631, 677–682 CrossRef CAS.
- A. Kulkarni, F. Ünlü, N. Pant, J. Kaur, C. Bohr, A. K. Jena, S. Öz, M. Yanagida, Y. Shirai, M. Ikegami, K. Miyano, Y. Tachibana, S. Chakraborty, S. Mathur and T. Miyasaka, Sol. RRL, 2021, 5, 2100077 CrossRef CAS.
- N. Pai, J. Lu, T. R. Gengenbach, A. Seeber, A. S. R. Chesman, L. Jiang, D. C. Senevirathna, P. C. Andrews, U. Bach, Y. Cheng and A. N. Simonov, Adv. Energy Mater., 2019, 1803396, 1–11 Search PubMed.
- V. Pecunia, Y. Yuan, J. Zhao, K. Xia, Y. Wang, S. Duhm, L. Portilla and F. Li, Nano-Micro Lett., 2020, 12, 1–12 CrossRef PubMed.
- A. Chakraborty, N. Pai, J. Zhao, B. R. Tuttle, A. N. Simonov and V. Pecunia, Adv. Funct. Mater., 2022, 32, 2203300 CrossRef CAS.
- E. V. Ushakova, A. I. Matuhina, A. V. Sokolova, S. A. Cherevkov, K. V. Bogdanov, A. Dubavik, M. A. Baranov, A. P. Litvin, K. Takai, A. V. Fedorov and A. V. Baranov, Opt. Spectrosc., 2019, 127, 1110–1116 CrossRef CAS.
- S. Premkumar, D. Liu, Y. Zhang, D. Nataraj, S. Ramya, Z. Jin, B. B. Mamba, A. T. Kuvarega and J. Gui, ACS Appl. Nano Mater., 2020, 3, 9141–9150 CrossRef CAS.
- F. Yu, L. Wang, K. Ren, S. Yang, Z. Xu, Q. Han and T. Ma, ACS Sustainable Chem. Eng., 2020, 8, 9980–9987 CrossRef CAS.
- A. Z. Chen and J. J. Choi, J. Vac. Sci. Technol., 2020, 38, 1 Search PubMed.
- S. Akhil, V. G. V. Dutt and N. Mishra, ChemNanoMat, 2021, 7, 342–353 CrossRef CAS.
- V. G. V. Dutt, S. Akhil, R. Singh, M. Palabathuni and N. Mishra, ACS Appl. Nano Mater., 2022, 5, 5972–5982 CrossRef CAS.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410–5423 CrossRef CAS.
- Q. Zhang, C. Wu, X. Qi, F. Lv, Z. Zhang, Y. Liu, S. Wang, B. Qu, Z. Chen and L. Xiao, ACS Appl. Energy Mater., 2019, 2, 3651–3656 CrossRef CAS.
- M.-C. Wu, Q.-H. Wang, K.-C. Hsiao, S.-H. Chen, C.-M. Ho, M.-H. Jao, Y.-H. Chang and W.-F. Su, Chem. Eng. J. Adv., 2022, 10, 100275 CrossRef CAS.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S.-H. Jang, J. Zhang, X. Lu, D. Lei, C.-S. Lee, Z. Zhu and A. K.-Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed.
- R. Grisorio, C. Dibenedetto, A. Matuhina, G. K. Grandhi, P. Vivo, E. Fanizza, M. Striccoli and G. P. Suranna, ACS Appl. Nano Mater., 2023, 6, 8082–8092 CrossRef CAS.
- M. I. Bodnarchuk, S. C. Boehme, S. ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef CAS PubMed.
- G. Walters, B. R. Sutherland, S. Hoogland, D. Shi, R. Comin, D. P. Sellan, O. M. Bakr and E. H. Sargent, ACS Nano, 2015, 9, 9340–9346 CrossRef CAS PubMed.
- B. Yang, J. Chen, Q. Shi, Z. Wang, M. Gerhard, A. Dobrovolsky, I. G. Scheblykin, K. J. Karki, K. Han and T. Pullerits, J. Phys. Chem. Lett., 2018, 9, 5017–5022 CrossRef CAS PubMed.
- C. Stavrakas, A. A. Zhumekenov, R. Brenes, M. Abdi-Jalebi, V. Bulović, O. M. Bakr, E. S. Barnard and S. D. Stranks, Energy Environ. Sci., 2018, 11, 2846–2852 RSC.
- B. Li, H. Li, Y. Sun, M. G. Humphrey, C. Zhang and Z. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 10858–10867 CrossRef CAS PubMed.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- Y. Cai, H. Wang, Y. Li, L. Wang, Y. Lv, X. Yang and R.-J. Xie, Chem. Mater., 2019, 31, 881–889 CrossRef CAS.
- S. Akhil, V. G. V. Dutt and N. Mishra, Nanoscale, 2021, 13, 13142–13151 RSC.
- P. Liu, W. Chen, W. Wang, B. Xu, D. Wu, J. Hao, W. Cao, F. Fang, Y. Li, Y. Zeng, R. Pan, S. Chen, W. Cao, X. W. Sun and K. Wang, Chem. Mater., 2017, 29, 5168–5173 CrossRef CAS.
- S. Akhil, M. Palabathuni, S. Biswas, R. Singh and N. Mishra, ACS Appl. Nano Mater., 2022, 5, 13561–13572 CrossRef CAS.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS PubMed.
- N. Fiuza-Maneiro, K. Sun, I. López-Fernández, S. Gómez-Graña, P. Müller-Buschbaum and L. Polavarapu, ACS Energy Lett., 2023, 8, 1152–1191 CrossRef CAS.
- A. Koedtruad, M. Goto, M. A. Patino, Z. Tan, H. Guo, T. Nakamura, T. Handa, W. T. Chen, Y. C. Chuang, H. S. Sheu, T. Saito, D. Kan, Y. Kanemitsu, A. Wakamiya and Y. Shimakawa, J. Mater. Chem. A, 2019, 7, 5583–5588 RSC.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, Angew. Chem., Int. Ed., 2018, 57, 9083–9087 CrossRef CAS PubMed.
- F. Liu, J. Jiang, T. Toyoda, M. A. Kamarudin, S. Hayase, R. Wang, S. Tao and Q. Shen, ACS Appl. Nano Mater., 2021, 4, 3958–3968 CrossRef CAS.
- B. Cucco, L. Pedesseau, C. Katan, J. Even, M. Kepenekian and G. Volonakis, Sol. RRL, 2022, 6, 2200718 CrossRef CAS.
- V. T. Barone, B. R. Tuttle and S. V. Khare, J. Appl. Phys., 2022, 131, 245701 CrossRef CAS.
- J. Even, L. Pedesseau, M.-A. Dupertuis, J.-M. Jancu and C. Katan, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205301 CrossRef.
- V. Pecunia, J. Zhao, C. Kim, B. R. Tuttle, J. Mei, F. Li, Y. Peng, T. N. Huq, R. L. Z. Hoye, N. D. Kelly, S. E. Dutton, K. Xia, J. L. MacManus-Driscoll and H. Sirringhaus, Adv. Energy Mater., 2021, 11, 2003968 CrossRef CAS.
- G. K. Grandhi, A. Matuhina, M. Liu, S. Annurakshita, H. Ali-Löytty, G. Bautista and P. Vivo, Nanomaterials, 2021, 11, 1458 CrossRef CAS PubMed.
- J. I. Jang, J. B. Ketterson, C. C. Stoumpos, S. H. Rhim, Y. S. Kim, M. G. Kanatzidis, A. J. Freeman, D. J. Clark and L. Frazer, J. Am. Chem. Soc., 2015, 137, 6804–6819 CrossRef PubMed.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- M. Liu, A. Matuhina, H. Zhang and P. Vivo, Materials, 2019, 12, 3733 CrossRef CAS PubMed.
- A. Matuhina, G. K. Grandhi, F. Pan, M. Liu, H. Ali-Löytty, H. M. Ayedh, A. Tukiainen, J.-H. Smått, V. Vähänissi, H. Savin, J. Li, P. Rinke and P. Vivo, ACS Appl. Nano Mater., 2023, 6, 953–965 CrossRef CAS.
- E. V. Ushakova, A. I. Matuhina, A. V. Sokolova, S. A. Cherevkov, A. Dubavik, O. S. Medvedev, A. P. Litvin, D. A. Kurdyukov, V. G. Golubev and A. V. Baranov, Nanotechnology, 2019, 30, 405206 CrossRef CAS PubMed.
- H. C. Sansom, G. F. S. Whitehead, M. S. Dyer, M. Zanella, T. D. Manning, M. J. Pitcher, T. J. Whittles, V. R. Dhanak, J. Alaria, J. B. Claridge and M. J. Rosseinsky, Chem. Mater., 2017, 29, 1538–1549 CrossRef CAS.
- D. Wilson and M. A. Langell, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- S. Gonzalez-Carrero, R. E. Galian and J. Pérez-Prieto, J. Mater. Chem. A, 2015, 3, 9187–9193 RSC.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- R. Grisorio, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, V. Vergaro, G. Ciccarella, N. Margiotta and G. P. Suranna, Nanoscale, 2020, 12, 623–637 RSC.
- R. Grisorio, M. E. D. Clemente, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, M. Irimia-Vladu and G. P. Suranna, Nanoscale, 2019, 11, 986–999 RSC.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02742b |
| This journal is © The Royal Society of Chemistry 2023 |