
An efficient, green, and residual oxidant-free wastewater treatment technique enabled by coupling a dual-cathode heterogeneous electro-Fenton process and UV radiation in tandem†
Lele
Cui
 abc,
Mingming
Sun
abc and
Zhenghua
Zhang
*abc
abc,
Mingming
Sun
abc and
Zhenghua
Zhang
*abc
aMembrane & Nanotechnology-Enabled Water Treatment Center, Institute of Environment and Ecology, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, Guangdong, China. E-mail: zhenghua.zhang@sz.tsinghua.edu.cn
bGuangdong Provincial Engineering Research Centre for Urban Water Recycling and Environmental Safety, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, Guangdong, China
cSchool of Environment, Tsinghua University, Beijing 100084, China
First published on 13th July 2023
Abstract
Efficient implementation of a catalyst-integrated cathode-based heterogeneous electro-Fenton (HEF) process usually suffers from the conflict between the optimal potentials of the two targeted reduction reactions (O2 to H2O2 and metal(n+1)+ to metaln+). In addition, the residual H2O2 in the HEF effluent poses a potential environmental risk to aquatic ecosystems and would further make the HEF technology impractical. Herein, a tandem system of dual-cathode HEF and UV radiation is proposed to bypass these issues. In the dual-cathode HEF unit, the air-diffusion cathode (ADC) without aeration and the FeOCl-functionalized graphite felt (FeOCl/GF) perform their respective functions at different operating current densities (100 mA cm−2 for ADC and 5 mA cm−2 for FeOCl/GF) to achieve efficient H2O2 production, ˙OH formation and Fe(III) electroreduction simultaneously. Subsequently, the underutilized H2O2 in the HEF effluent will be reactivated in the UV module to eliminate the risk of oxidant residues while further improving the decontamination efficiency. Importantly, interesting antagonistic and synergistic phenomena governed by the cathodic current density of the ADC in this tandem system were also highlighted by constructing different standalone/coupled processes. The HEF/UV tandem strategy proposed in the present work offers a promising scheme for distributed wastewater treatment due to the demonstrated preeminent process efficiency, as well as the reagent- and residual-oxidant-free green chemistry concept.
1. Introduction
Decentralized water treatment systems with small-scale characteristics offer a promising means of accessing clean water, as they theoretically substantially reduce the energy input while enabling the fulfillment of various end-user-specific treatment needs.1,2 Unfortunately, the challenge of anthropogenically exacerbated accumulation of persistent organic pollutants (POPs) in aquatic ecosystems makes distributed schemes impractical, due to the rather inefficient decontamination performance of conventional wastewater treatment facilities for POPs.2,3 Electrochemical advanced oxidation processes (EAOPs), capable of degrading most hazardous organic compounds, are appealing module options for such decentralized systems.4 Among the EAOP family, those based on the Fenton reaction chemistry, such as electro-Fenton (EF), have proven to be the most robust techniques.5,6 The efficiency of the EF process lies in the generation of strongly oxidizing hydroxyl radicals (˙OH) via the Fenton reaction between the cathode-produced hydrogen peroxide (H2O2) and the externally added Fe2+ catalyst.4,7 Nevertheless, the conventional homogeneous EF process suffers from fundamental drawbacks, such as a narrow working pH window (2.8–3.5) and the formation of iron sludge.8 In this context, heterogeneous EF (HEF) with solid-phase iron catalyst-functionalized carbonaceous cathode supports has recently been extensively developed and validated to effectively overcome these bottlenecks while bypassing the time-consuming and costly dispersion/separation procedures of catalyst particles in typical suspended heterogeneous processes.4,6Over the past few years, substantial efforts have focused on the rational design of advanced catalytic cathodes to maximize the production of ˙OH, such as heteroatom-incorporated spinel-grafted carbon fibers,9 transition metal-encapsulated carbon aerogels,10 and even atomically dispersed iron-anchored porous carbons.11 Despite these encouraging achievements, the reaction kinetics of HEF processes are generally significantly inferior (at least by one order of magnitude) to their homogeneous counterparts at equivalent catalyst loadings.12 In addition to the inherent limited atom utilization of solid catalysts, this phenomenon may also be closely related to the difficulty in executing the expected dual functions (i.e., generation of H2O2 and ˙OH in a stepwise manner) of such composite cathodes in real working scenarios. For one thing, most foreign metal species were identified as being thermodynamically more favorable for the 4e− pathway (O2 + 4H+ + 4e− → 2H2O) than the 2e− pathway (O2 + 2H+ + 2e− → H2O2) during the oxygen reduction reaction (ORR), which would cause deterioration of H2O2 production in the system.6,10,13 Although previous reports have attributed the observed drop in H2O2 yield entirely to an on-site activation mechanism,14,15 this has not been subjected to rigorous molecular-level justification due to the hitherto elusive interaction between the catalyst and the carbon matrix in the ORR. For another, such integrated cathode-catalyzed HEF processes generally suffer from the working current density problem, namely the mismatch of the required potentials for H2O2 generation (E° = 0.695 V per SHE) and high-valent metal electroreduction (E° = 0.77 V per SHE for Fe(III)).16,17 As a result, it is difficult to achieve optimal H2O2 production and active metal regeneration as well as ˙OH formation simultaneously in conventional operation with a single cathode. Although there are several recent studies of homogeneous EF designed dual-cathode systems to cope with this bottleneck, they were not green and sustainable due to the necessity of either complex iron salt dosing or costly oxygen/air aeration procedures.12,16,18
Another important but underappreciated issue is that most of the current works exclusively pursue high decontamination efficiency without concern for the residual H2O2 in the HEF effluent. Indeed, excessive accumulation of H2O2 at the end of EF treatment was frequently detected, either for homogeneous or heterogeneous modes,4,14,19 which not only causes an undesired wastage of electrical energy but also poses a potential risk to aquatic organisms and phytoplankton if the effluent is discharged directly.20 A recent endeavor couples a HEF system with a Fe3O4–carbon filter to quench residual H2O2 and thus makes the effluent safe for discharge into the environment.21 However, this process relies on the catalytic disproportionation of H2O2 to O2 by the Fe3O4–carbon filter, resulting in a decrease in H2O2 utilization and potential secondary pollution by Fe leaching. UV/H2O2 is a well-established technique in potable water facilities, which can effectively remove trace organic contaminants by ˙OH generated from the photolytic homolysis of H2O2.3,19 More strikingly, benefiting from its excellent activation efficiency, even a low concentration of H2O2 (e.g., 3 mg L−1) can still be rapidly converted to ˙OH under UV irradiation.2 Therefore, it would be highly desirable if a low-power UV module was integrated into HEF systems. This combined strategy is expected to yield H2O2-free treatment solution while accelerating the mineralization of target pollutants through the generation of additional ˙OH and the direct photolysis of photolabile intermediates.
Herein, a tandem system combining dual-cathode-based HEF and UV irradiation was developed for the first time, in an attempt to address the aforementioned challenges encountered in distributed EAOP treatment systems. For the dual-cathode HEF module, an air-diffusion cathode (ADC) with self-sustained aeration is employed for in situ electrosynthesis of H2O2, and a graphite felt catalytic cathode with high iron oxychloride loading (FeOCl/GF) is used for rapid conversion of H2O2 into active species. These two cathodes perform their respective functions at different optimal working potentials to simultaneously achieve efficient H2O2 production, Fe(III) electroreduction, and ˙OH formation. Of note is that the underutilized H2O2 remaining in the HEF effluent will be continuously activated by the subsequently deployed UV irradiation module to eliminate the risk of oxidant residues and further improve the degradation efficiency. In addition, by constructing different independent/coupled systems (UV, H2O2, UV/H2O2, HEF, and HEF/UV), the reaction kinetics and H2O2 concentration evolution corresponding to each isolated unit can be well discriminated. Thus, this study also enables the decoupling of the exact contribution of each subprocess and the elucidation of interesting antagonistic and synergistic phenomena that occurred in the dual-cathode HEF and UV tandem system.
2. Materials and methods
2.1. Electrode fabrication
A detailed description of the chemicals used is provided in Text S1.† ADC for in situ H2O2 generation was prepared by a modified coating method.22 Specifically, the pristine graphite felt (GF) was sequentially cleaned with ethanol and deionized water in an ultrasonic bath for 30 min, and then annealed at 400 °C for 4 h to remove impurities. The cleaned GF was soaked in the PTFE suspension (5 wt%) for 30 min and then taken out and dried at room temperature before use. Carbon black (CB) particles were ultrasonically dispersed in anhydrous ethanol for 10 min, followed by the dropwise addition of PTFE (at a CB-to-PTFE mass ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and vigorous stirring at 70 °C until a wet paste was formed. The resulting paste was uniformly coated on one side of GF with a CB loading of 12.44 mg cm−2, and calcined at 350 °C for 60 min.
:3) and vigorous stirring at 70 °C until a wet paste was formed. The resulting paste was uniformly coated on one side of GF with a CB loading of 12.44 mg cm−2, and calcined at 350 °C for 60 min.
Considering that the catalytic cathode here is only responsible for the activation of ADC-produced H2O2 but not for H2O2 electrosynthesis, a FeOCl/GF composite with high FeOCl loading was fabricated in this study by improving our previously reported thermal-induced strategy.15 In brief, the above pretreated GF was immersed in 5 mL FeCl3·6H2O ethanol solution with a concentration of 1.5 g mL−1 for 3 h, and then evaporated and dried at 45 °C for 12 h to obtain the FeCl3/GF precursor. Afterwards, the precursor was transferred to a covered crucible and calcined at 220 °C for 1 h under atmospheric conditions with a temperature increase rate of 10 °C min−1, and the resulting product was washed several times with deionized water to remove unreacted soluble iron salts. Note that in order to alleviate the shedding and leaching of active metals from the catalytic cathode during the degradation process, the FeOCl/GF composite was additionally subjected to a PTFE post-treatment process (i.e., soaking in 1 wt% PTFE suspension followed by calcination at 180 °C). This not only improves the adhesion between FeOCl and the GF matrix, but also slows down the corrosive dissolution of the solid metal by the electrolyte due to the slight hydrophobic evolution at the solid–liquid interface.9 A detailed description of the characterization methods used for different cathodes is presented in Text S2.†
2.2. Electrolyzer and degradation system
The electrolysis experiments involved in this work were all carried out in a custom-designed undivided reactor with a cylindrical cavity (Fig. 1). The ADC was embedded on the side wall of the reactor near the water inlet allowing one side of the cathode to be exposed to the air, while the FeOCl/GF catalytic cathode was placed inside the cavity for sufficient interaction with the upstream H2O2 solution. Two Ti/Pt mesh electrodes were used as anodes and placed in parallel with the corresponding cathodes with an inter-electrode gap of 1.5 cm. All electrodes have an active area of 7.1 cm2 (3 cm in diameter) unless otherwise specified. Note that each electrode pair was independently provided with the optimal current (galvanostatic mode) by different DC power supplies.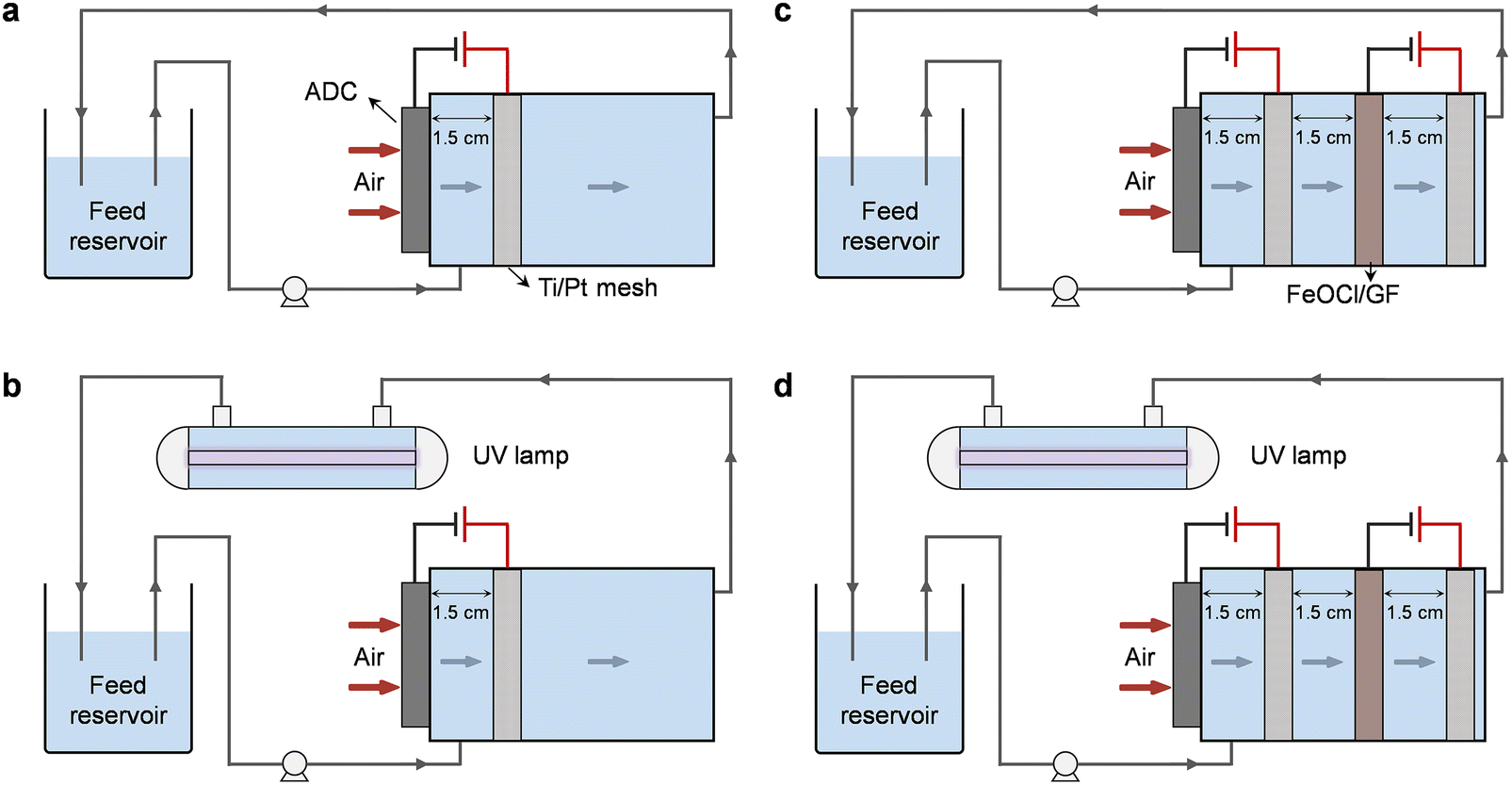 | ||
| Fig. 1 Schematic illustration of different standalone or coupled degradation systems: (a) H2O2, (b) UV/H2O2, (c) HEF, and (d) HEF/UV. | ||
Degradation experiments were carried out with oxytetracycline (OTC) as the model pollutant in a tandem system of a dual-cathode electrolyzer and a pipeline type sterilizer equipped with a 6 W UV lamp (emission at 254 nm). A total volume of 300 mL of OTC aqueous solution (20 mg L−1) containing 50 mM Na2SO4 was used as the simulated wastewater and supporting electrolyte in the feed reservoir, with its initial pH adjusted using 1 M of H2SO4 or NaOH. Typical water treatment processes were performed in a continuous recirculation mode. Driven by the peristaltic pump (Longer), the sewage containing OTC first enters the electrolysis unit and the rapid accumulation of H2O2 occurs when it comes in contact with the ADC. The solution will then be forced to flow through the FeOCl/GF catalytic cathode during which considerable reactive radicals will be generated due to the activation of H2O2 by the iron active center. Finally, the effluent from the electrolysis unit will enter the UV lamp module to completely consume the residual H2O2 and further mineralize pollutants and intermediate molecules. In order to decouple the respective contributions of different subprocesses in the system, five different standalone or coupled systems, including UV (only UV irradiation unit, Fig. S1†), H2O2 (electrolytic unit with ADC only, Fig. 1a), UV/H2O2 (ADC + UV irradiation, Fig. 1b), HEF (electrolysis unit with dual cathodes, Fig. 1c), and HEF/UV (dual cathode + UV irradiation, Fig. 1d), were purposely constructed. H2O2 accumulation over time was also performed in these settings, in the absence of pollutants.
2.3. Analytical methods
The concentrations of H2O2 and total Fe in the solution were measured with a UV-vis spectrophotometer (UV-1100, Shanghai Mapada Instruments Co. Ltd, China) by the potassium titanium(IV) oxalate method at 400 nm and the 1,10-phenanthroline method at 510 nm, respectively. The current efficiency (CE) and electric energy consumption (EEC, kW h kg−1) for H2O2 electrosynthesis were calculated by the following two equations: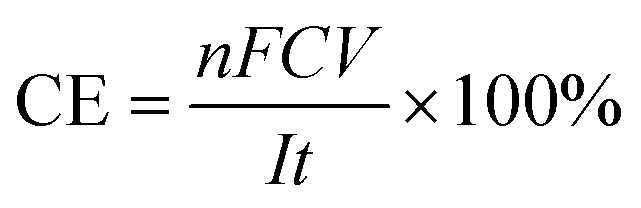 | (1) |
486 C mol−1), C is the concentration of H2O2 (mol L−1), V is the volume of the electrolyte (L), I is the current (A), and t is the electrolysis time (s).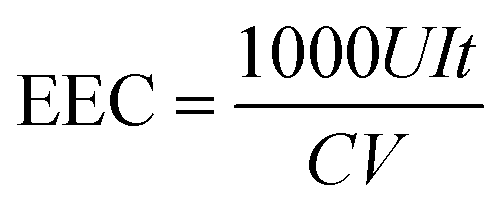 | (2) |
The evolution of the OTC concentration was monitored using the same spectrophotometer at 357 nm. Prior to performing the analyses, the collected samples were filtered using a 0.22 μm filter. The OTC degradation kinetics for different systems were determined using a pseudo-first-order model as described in eqn (3):
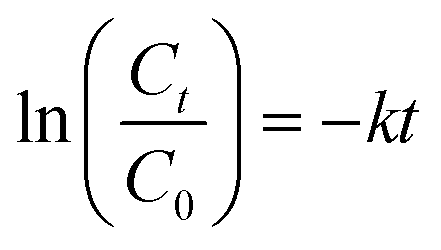 | (3) |
The concept of synergistic factor (sf) was introduced herein to characterize the antagonistic and synergistic effects occurring in the dual-cathode HEF-coupled UV radiation system, and was calculated as follows:
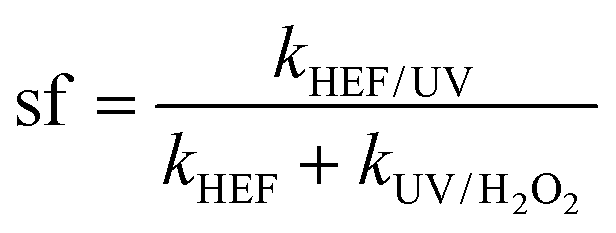 | (4) |
The OTC mineralization rate was monitored by a total organic carbon (TOC) analyzer (TOC-L, Shimadzu, Japan). The oxidation by-products of OTC were identified by ultrahigh performance liquid chromatography coupled with a mass spectrometry (LC-MS-8050, Shimadzu, Japan) system equipped with a C18 column (2.1 mm × 100 mm, 1.7 μm), and the detailed measurement method is given in our previous work.15 Ecological Structure–Activity Relationship (ECOSAR) software was utilized to predict the acute and chronic toxicity of OTC and its degradation intermediates. The active species generation in the system was identified by electron paramagnetic resonance (EPR) spectroscopy (Micro-EPR, Bruker). DMPO was applied as the spin-tapping agents for ˙OH, ˙O2− (methanol system), and SO4˙−. TEMP was used to detect 1O2.
3. Results and discussion
3.1. Characterization and electrocatalytic performance of different cathodes
Scanning electron microscopy (SEM) images reveal that the prepared ADC is characterized by a double-layer structure with PTFE-treated GF as the diffusion layer and CB as the catalytic layer (Fig. S2†). The high macroporosity of the GF layer allows the free mass transfer of O2 from the atmosphere to the reaction boundary, while the strong hydrophobicity of the CB layer (contact angle of 136.1° in the inset of Fig. S2†) ensures unimpeded O2 capture at the three-phase interface. Therefore, this electrode configuration can endow the H2O2 production process with an interesting self-sustained aeration characteristic driven by a hydrophobicity-controlled O2 concentration gradient between the reaction interface and the atmosphere. As a result, the significant energy consumption (even exceeding 50% of the total cost) associated with aeration facilities in conventional submerged or gas diffusion electrode systems is effectively avoided.15,22The H2O2 production performance of ADC was then evaluated (Fig. 2a). Obviously, the accumulation of H2O2 increased linearly with the reaction time for different current densities (5–100 mA cm−2). And the final H2O2 concentration corresponding to each measurement (60 min) was also observed to increase linearly with the applied current density (R2 = 0.995, inset in Fig. 2c), with a maximum of 1261.9 mg L−1 at 100 mA cm−2. This indicated that the 2e− ORR process was no longer limited by the O2 mass transfer but only controlled by the electron transfer within the scope of the investigation. Generally, CE and EEC were considered as the key parameters to determine the economic viability of an electrochemical system applied toward H2O2 production.4 As revealed in Fig. 2b, although the increase in the current density yielded a decrease in CE (from 104.9% at 5 mA cm−2 to 85.3% at 100 mA cm−2), the performance is still satisfactory because CE is always higher than 85% or even higher than 90% in the range of current density less than 45 mA cm−2. The CE exceeding the theoretical maximum value at 5 mA cm−2 may be due to the amplification of small sampling errors by the CE calculation method (eqn (1)). And the observed decrease in CE at higher current densities can be ascribed to the promotion of parasitic reactions involving either H2O2 decomposition (via anodic oxidation and cathodic reduction) or the hydrogen evolution reaction (eqn (5)–(7)). For EEC (Fig. 2c), in contrast, it exhibited a positive correlation with the applied current density due to the increased cell voltage. Nevertheless, the EEC of the ADC system at 5 mA cm−2 (5.3 kW h kg−1) was noted to be very competitive with that of previously reported H2O2 electrosynthesis processes (3.7–53.9 kW h kg−1, Table S1†). Furthermore, although the application of a current density of 100 mA cm−2 (44.2 kW h kg−1) resulted in an 8.3-fold increase in EEC over the value at 5 mA cm−2 (5.3 kW h kg−1), the corresponding H2O2 production (1261.9 mg L−1) was 16.2-fold higher than that at 5 mA cm−2 (77.7 mg L−1). Therefore, ADC with cheap and easy preparation is highly desirable for the subsequent HEF process, due to its stable H2O2 supply and excellent process efficiency with respect to both yield and energy consumption.
| H2O2 → O2 + 2H+ + 2e− | (5) |
| H2O2 + 2H+ + 2e− → 2H2O | (6) |
| 2H+ + 2e− → H2 | (7) |
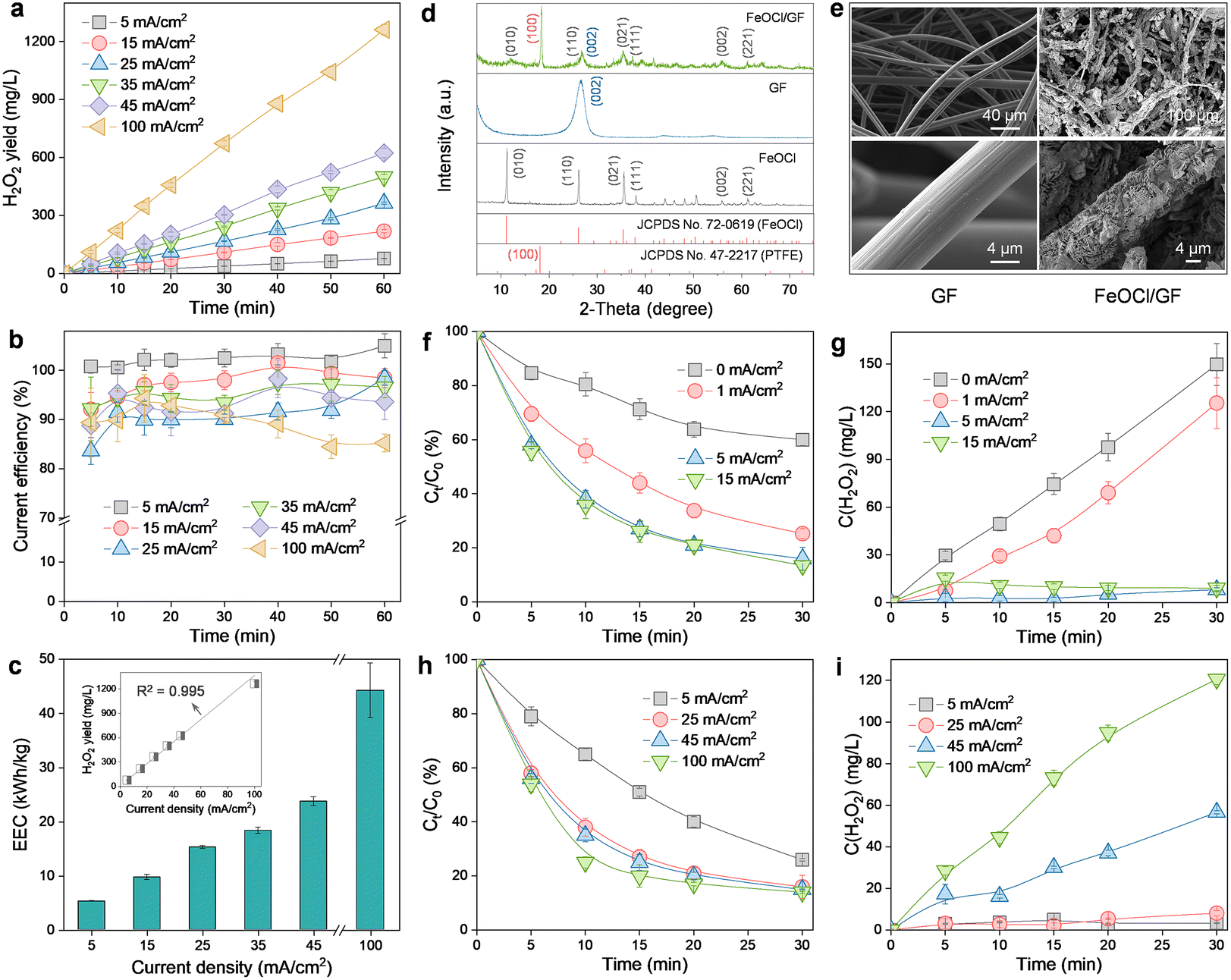 | ||
| Fig. 2 Physicochemical properties and catalytic performance of different cathodes. Effect of current density on the (a) yield, (b) current efficiency, and (c) energy consumption of the H2O2 production at ADC; (d) XRD patterns of FeOCl, GF, and FeOCl/GF; and (e) SEM images of GF and FeOCl/GF; effect of current density on the OTC degradation performance of the dual-cathode HEF system and the corresponding H2O2 concentration evolution (conditions: [OTC] = 20 mg L−1 and pH = 4): (f) and (g) FeOCl/GF, (h) and (i) ADC. | ||
The physicochemical properties of the catalytic cathode were also characterized by different techniques. The diffraction peaks related to GF ((002))9 and FeOCl ((010), (110), (021), (111), (002), and (221))23 were observed in the X-ray diffractometer (XRD) pattern of FeOCl/GF (Fig. 2d), indicating the successful formation of the composite. Note that the diffraction peak corresponding to the (010) plane of FeOCl in the FeOCl/GF composite was found to be shifted to higher angles compared to the FeOCl sample, which may be attributed to the interaction between the metal and the carbon substrate that induces narrowing of the interlayer spacing of FeOCl through structural modulation. According to the low-magnification SEM images, pristine GF presents a porous network structure composed of carbon fibers with smooth surfaces, while FeOCl/GF fibers reveal a rather rough surface morphology (Fig. 2e). High-magnification SEM images confirm that the rough transition on the carbon fiber surface is caused by the omnidirectional coverage of sheet-like FeOCl. In previously reported single-cathode HEF systems, the fine-tuning of the solid catalyst growth process on the surface of the catalytic cathode, although challenging, is necessary and mandatory to simultaneously expose carbon and metal sites for the realization of the intended bifunctionality (i.e., sequential 2e− ORR and Fenton activation).6 However, the catalytic cathode in the present work only exhibits activation function for H2O2, which allows high loading of FeOCl on GF to thoroughly stimulate the catalytic potential of the composite cathode. The brighter colors corresponding to Fe, O, and Cl elements compared to the C element in the energy dispersive spectroscopy (EDS) results strongly demonstrate the rather high FeOCl loading of the synthesized FeOCl/GF composite cathode (Fig. S3†). In addition, a PTFE post-treatment process was also performed on this catalytic cathode to alleviate the risk of catalyst shedding and leaching exacerbated by high loading, relying on the bonding and hydrophobic protection effects of PTFE.9 A characteristic peak indexed to the (100) plane of PTFE was observed in the XRD pattern of the FeOCl/GF composite, suggesting the successful functionalization of PTFE.9
The efficacy of the FeOCl/GF catalytic cathode was then investigated by fixing the current density applied to the ADC at 25 mA cm−2. It can be found that the degradation of OTC is limited when the FeOCl/GF cathode was not supplied with current (Fig. 2f). This may be because the collision probability of the catalyst and H2O2 in this filter configuration is much lower than that of the typical HEF process with the catalyst suspended. More importantly, the integrated catalysts of the composite cathode are always exposed to the strong oxidizing microenvironment (aggressive reactive oxygen species), leading to the rapid depletion of low-valent active Fe(II) and the passivation of the catalytic cathode.13 This can be confirmed by the concentration evolution of H2O2, as the accumulated concentration of H2O2 in the dual-cathode system with no current applied to the FeOCl/GF cathode is only slightly lower than that in the ADC single-cathode system (Fig. S4†). Interestingly, the OTC degradation kinetics were accelerated once a constant current was applied to the catalytic cathode, indicating a significant improvement in the activation of the catalytic cathode to H2O2. This is indeed the case as demonstrated in Fig. 2g that the accumulated concentration of H2O2 decreased rapidly as the current was applied, and even almost no residual H2O2 was detected when the catalytic cathode was operated with a current density higher than 5 mA cm−2. In this work, the Fe(II)/Fe(III) redox cycling on the surface of the composite cathode can be kept quasi-reversible with the assistance of an electric field (as evidenced by the relative Fe(II) content on the FeOCl/GF cathode surface increasing from 46.4% before the reaction to 47.3% after the reaction, Fig. S5†),13,24 which accelerates catalytic activation of ADC-produced H2O2 and thus the degradation of OTC. However, a further increase in the current density will lead to a decrease in the Fe(III) reduction. This can be explained by the promotion of side reactions, like the hydrogen evolution reaction, which not only competed with the electroreduction of Fe(III), but more seriously, caused the hydrolysis of active iron to inert hydrous oxyhydroxides due to the local alkalinization near the catalyst.16 For this reason, as the current density applied to the FeOCl/GF cathode increased up to 15 mA cm−2, the OTC degradation efficiency did not change significantly compared to 5 mA cm−2 (Fig. 2f). Note that the above discussions all exclude Fe homogeneous oxidation contributions because the concentration of total Fe leached from the catalytic cathode is too low (1.4 mg L−1vs. 68.0 mg L−1 for FeOCl/GF without PTFE post-treatment, Fig. S6†) to drive significant degradation of OTC (Fig. S7†).
The effect of the ADC on the reactivity of the dual-cathode HEF was also investigated by fixing the operating current density of the catalytic cathode at 5 mA cm−2 (Fig. 2h). For comparison with the conventional single-cathode HEF process, the current density applied to the ADC was adjusted to the same 5 mA cm−2 as the catalytic cathode. A reduced OTC degradation rate was observed compared with a current density of 25 mA cm−2, which may be attributed to the fact that the concentration of H2O2 produced by the ADC was too low to keep up with the demand of the FeOCl/GF cathode. This is well supported by the evolution of the H2O2 concentration in Fig. 2i, with almost no H2O2 accumulation for ADC current densities in the range of 5 to 25 mA cm−2. It has been recently reported that a single-cathode HEF system based on the same FeOCl/GF catalytic cathode can afford a degradation rate constant (k value) of 0.05629 min−1 for 100 mL of OTC solution (10 mg L−1) at 5 mA cm−2,15 while the k value of the present dual-cathode HEF system was established to be 0.04505 min−1 (Fig. S8†) for 300 mL of OTC solution (20 mg L−1) under the same operating conditions (i.e., 5 mA cm−2 for both ADC and FeOCl/GF cathodes). By comparing these results, one can conclude that the dual-cathode HEF system developed herein was highly competitive as it allowed a 6-fold higher total pollutant treatment (6 mg vs. 1 mg) with only a 1.2-fold lower k value (0.04505 min−1vs. 0.05629 min−1) compared to the single-cathode HEF system. Therefore, this study demonstrated that the separation of the composite cathode-based HEF process into independent H2O2 production and activation processes may be highly desirable as it enabled the two individual subprocesses to be performed optimally. Of course, attention must be paid to the additional electricity cost consumed by the FeOCl/GF electrode pair, and rational system optimization to improve the economics of the dual-cathode HEF process, which is necessary for future research. It is also worth noting that increasing the working current density of the ADC can further accelerate the degradation of OTC, but at the expense of excessive H2O2 residue in the effluent (especially >100 mg L−1 at 100 mA cm−2). This trade-off can be overcome by adding a UV radiation module after the dual-cathode HEF process, which will be discussed in detail in the next section.
3.2. Process efficiency of the dual-cathode HEF and UV tandem system
A matter scarcely addressed in previous HEF processes based on in situ H2O2 electrosynthesis is the examination of the residual H2O2 concentration. Most reports have been confined solely to the analysis of pollutant removal, without considering the fate of unreacted H2O2 remaining in the bulk solution after treatment. The idea of the present study is to integrate a low-power UV module at the outlet of the dual-cathode HEF facility to eliminate the risk of the H2O2 residue. In addition, the purpose of the following experiments also involves evaluating the efficacy of the coupled process by comparing OTC degradation and H2O2 concentration evolution achieved by different configurations (i.e., UV, H2O2, UV/H2O2, HEF, and HEF/UV). Experiments were carried out by fixing the optimum current density of the FeOCl/GF catalytic cathode at 5 mA cm−2 (as demonstrated in section 3.1) but varying the current density of the ADC (5–100 mA cm−2).Fig. 3 shows the decay of the OTC concentration over time in different systems and the corresponding H2O2 concentration evolution. Regardless of the current density applied on the ADC, H2O2 alone can hardly oxidize OTC with almost negligible k values due to its weak oxidizing ability (Fig. 3a, d, and g). This is evidenced by the insignificant difference in the accumulation of H2O2 using the same ADC in the presence of pollutants (Fig. 3b, e, and h) versus the absence of pollutants (Fig. 2a). Although OTC can be degraded by direct photolysis in the UV system, its efficiency is still limited and unsatisfactory (Fig. 3a, d, and g). After incorporating UV radiation and/or FeOCl/GF catalytic cathode into the H2O2 system (i.e., HEF/UV, UV/H2O2, and HEF), the concentration of OTC decreased significantly due to the production of oxidant species homogeneously in the bulk solution (photolytic homolysis of H2O2 by UV radiation, eqn (8))25 and/or heterogeneously at the surface of the catalytic cathode (Fenton activation of H2O2 by FeOCl/GF, eqn (9)). These phenomena were consistent with the greatly attenuated H2O2 accumulation in HEF/UV, UV/H2O2, and HEF systems compared with the H2O2 standalone system in Fig. 3b, e, and h. Furthermore, it is worth noting that the H2O2 consumption rate of the HEF/UV tandem system was found to be close to 100% for all operating current densities of the ADC, since there was almost no H2O2 residue in the treated solution. For the UV/H2O2 and HEF systems, however, a relatively high percentage of H2O2 (especially 36.4% (416.4 mg L−1) for UV/H2O2 and 16.7% (201.8 mg L−1) for HEF at 100 mA cm−2) did not participate in the treatment process after 60 min of reaction. These results suggest that the technology proposed in this study is highly efficient for further improving the oxidation capacity of the dual-cathode HEF process without generating H2O2-containing effluent.
| H2O2 + hv → ˙OH | (8) |
| Fe(II) + H2O2 → Fe(III) + ˙OH + OH− | (9) |
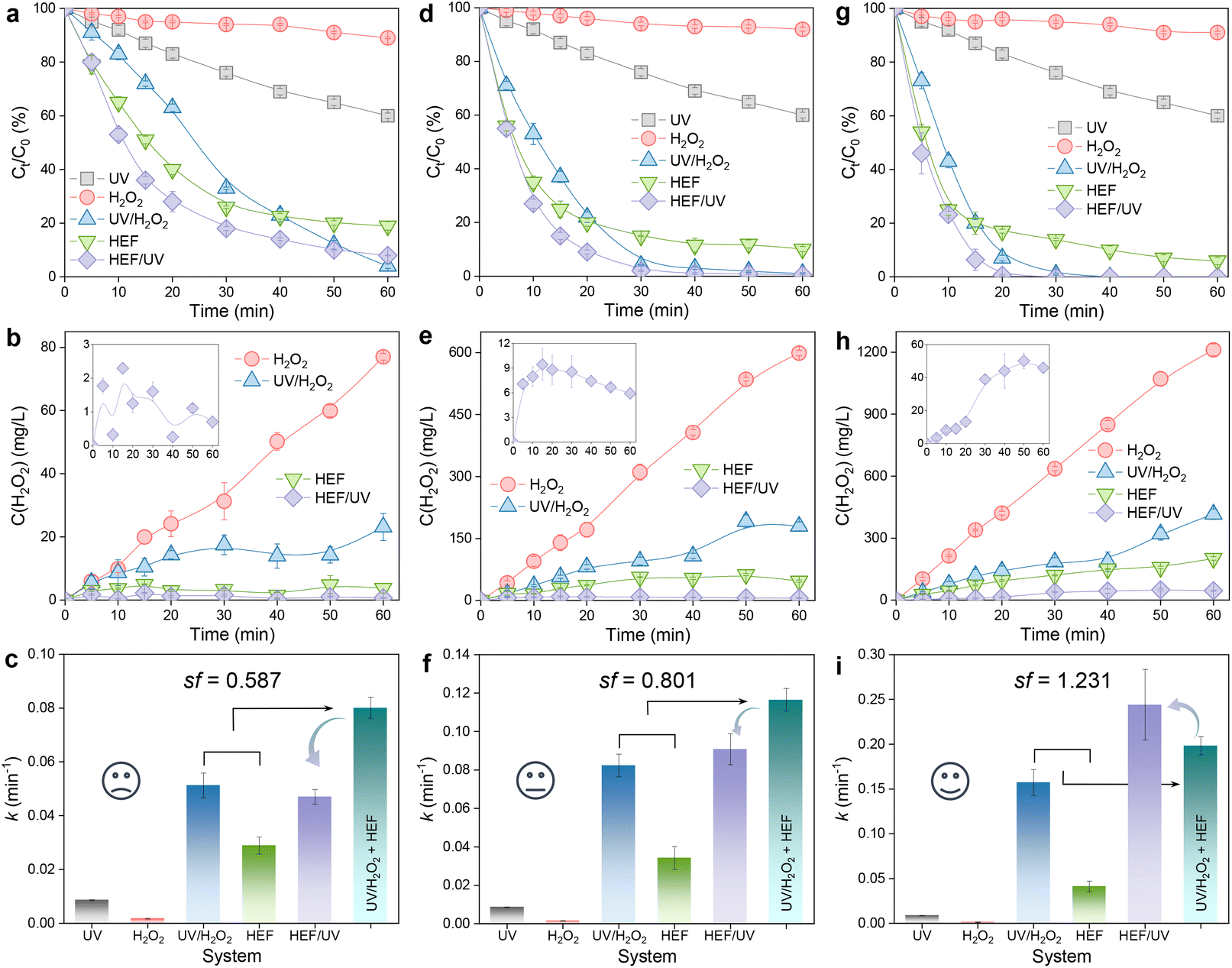 | ||
| Fig. 3 Investigation of the reaction characteristics of each sub-process in the tandem system. Effect of different ADC operating current densities on process efficiency: (a–c) 5 mA cm−2, (d–f) 45 mA cm−2, and (g–i) 100 mA cm−2. OTC degradation performance in different systems (first row), corresponding H2O2 concentration evolution (second row), and comparison of apparent rate constant k (third row) (conditions: [OTC] = 20 mg L−1, pH = 4, IFeOCl/GF = 5 mA cm−2, and UV = 6 W). | ||
In addition, the reaction kinetics of different independent or coupled processes were carefully analyzed, and accordingly the sf values at different operating current densities were obtained. Interestingly, as presented in Fig. 3c, f, and i, the sf value of the tandem system increases with the current density applied to the ADC and is >1 at 100 mA cm−2. This means that the k value corresponding to the HEF/UV configuration is higher than the sum of the individual HEF and UV/H2O2 processes, indicating that the tandem of dual-cathode HEF and UV radiation within the same system leads to synergistic acceleration of OTC degradation. Whereas when the current density is less than 100 mA cm−2, the sf value <1 suggests that there seems to be some unfavorable interaction between HEF and UV in the tandem system. Therefore, the following discussion will emphasize the differences in the reaction characteristics at different current densities to gain mechanistic insights into the electrooxidation process of the HEF/UV tandem system.
At 5 mA cm−2, the OTC degradation in the dual-cathode HEF and UV tandem systems was faster than the HEF or UV/H2O2 standalone systems before 50 min (Fig. 3a), due to the additional free radicals generated by UV decomposition of the residual trace H2O2 (3.7 mg L−1, Fig. 3b) in the HEF effluent. After 50 min, however, the degradation rate of OTC gradually slowed down and was even worse than that of the UV/H2O2 system, which resulted in the k value of the tandem system being lower than that of the UV/H2O2 standalone system (Fig. 3c). Therefore, the sf value at a current density of 5 mA cm−2 cannot be greater than 1, according to the calculation principle (eqn (4)). Regarding the extremely low sf value (0.587) under these conditions, it can be explained by the following two aspects. For one thing, the residual concentration of H2O2 in the solution after HEF treatment in the tandem system was too low to fully exert the efficiency of the subsequent UV/H2O2 system. The similar OTC degradation trends of the HEF standalone system and HEF/UV system also indicated that the HEF subprocess in the coupled process mainly contributed to OTC degradation under these conditions. For another, the UV/H2O2 system achieved a higher OTC degradation rate (0.05126 min−1vs. 0.02885 min−1, Fig. 3c) with less H2O2 consumption (69.9% vs. 95.1%, Fig. 3b) compared to the HEF system, implying the ineffective decomposition of H2O2 in the HEF module. This is evidenced by the fact that FeOCl/GF cathode always exhibited a significantly faster H2O2 decomposition rate than the UV lamp at different initial concentrations of H2O2 (Fig. S9†). According to the well-established reaction pathways in the literature,5 there is actually a competition between different species in the influent (pollutant molecules and H2O2) for the reaction with ˙OH generated heterogeneously on the FeOCl/GF surface (eqn (10)–(12)).7 The relatively high concentration of OTC in the early stage of the reaction makes the degradation reaction between pollutant molecules and free radicals (mainly ˙OH) in the system dominate. However, as the OTC concentration decays to a critical value (at 50 min), the quenching reaction between H2O2 in the flowing solution and the locally high concentration of surface ˙OH at the FeOCl/GF catalytic cathode instead dominates, which in turn deteriorates the OTC degradation efficiency. A similar phenomenon to the wasting reaction between H2O2 and ˙OH affects the treatment performance of EAOPs was also observed by Sánchez-Montes et al.19 and Dong et al.26 Therefore, it can be concluded that the low sf value at 5 mA cm−2 is mainly attributed to the wasting reaction of reactive oxygen species inherent in the dual-cathode HEF process, which cannot be mitigated after the introduction of the UV module.
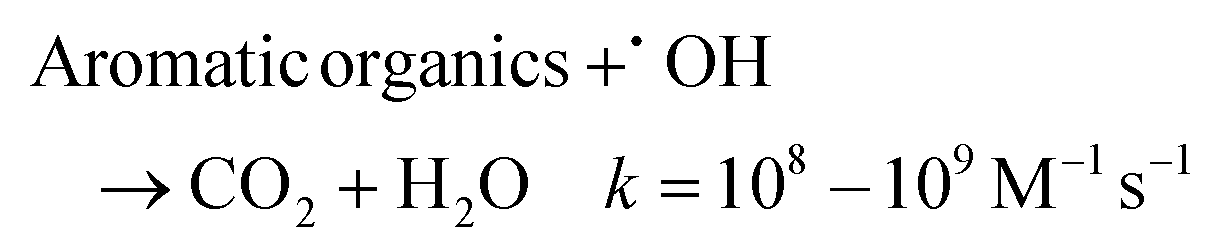 | (10) |
| H2O2 + ˙OH → H2O + HO2˙ k = 2.7 × 107 M−1 s−1 | (11) |
| HO2˙ + ˙OH → H2O + O2 k = 7.1 × 109 M−1 s−1 | (12) |
Different from 5 mA cm−2, the OTC degradation of the HEF/UV system at 45 mA cm−2 was significantly faster than each standalone system throughout the entire reaction period (Fig. 3d), and the corresponding k value was also higher than each standalone system (Fig. 3f). This is because the residual H2O2 concentration in the effluent from the HEF unit at 45 mA cm−2 exceeds 11 times that at 5 mA cm−2 (47.1 mg L−1vs. 3.7 mg L−1), which allows more homogeneous ˙OH to be generated in the subsequent UV module for oxidative degradation. Nevertheless, the sf value under these conditions was noticed to be still less than 1 (0.801), indicating that the self-scavenging reaction between H2O2 and heterogeneous ˙OH on the surface of the catalytic cathode still occurred. This can be confirmed by the decrease of H2O2 concentration after 20 min of reaction in the HEF/UV system (inset of Fig. 3e). Encouragingly, the sf value was greater than 1 upon increasing the current density of the ADC to 100 mA cm−2 (Fig. 3i). According to the kinetic constant information of the EF chain reaction established by Brillas et al.,7 the rate constant of the ˙OH recombination reaction (eqn (13)) was determined to be two orders of magnitude higher than that of the aforementioned self-scavenging reaction (eqn (11)). Based on this, it could be speculated that due to the further increase in the relative content of homogeneous ˙OH in the UV unit (by activating a higher concentration of residual H2O2 in the HEF effluent at 100 mA cm−2, Fig. 3h), the dimerization reaction between these homogeneous ˙OH and heterogeneous ˙OH generated at the FeOCl/GF cathode might be promoted, thereby decreasing the wasting reaction of H2O2 with heterogeneous ˙OH. More importantly, H2O2, the dimerization product of homogeneous ˙OH and heterogeneous ˙OH, would flow back into the UV irradiation module with higher H2O2 activation efficiency and be converted into ˙OH again to participate in the OTC degradation process. The gradual increase of the accumulated concentration of H2O2 in the HEF/UV system (inset of Fig. 3h) can also support the above speculation, which is completely different from the situations of 5 and 45 mA cm−2 (inset of Fig. 3b and e). Note that the faster accumulation of H2O2 after 20 min may be related to the conversion of the dominant reaction in the system from the degradation reaction between pollutant molecules and ˙OH to the dimerization reaction between ˙OH (since OTC has been completely degraded at 20 min, Fig. 3g).
| ˙OH + ˙OH → H2O2 k = 6.0 × 109 M−1 s−1 | (13) |
Additionally, this interesting synergistic effect between dual-cathode HEF and UV was noted independent of the initial pH of the treatment solution. As illustrated in Fig. S10,† under the optimal working current density justified above (i.e., 5 mA cm−2 for FeOCl/GF and 100 mA cm−2 for ADC), the pH range was set from 4 to 8 to investigate the influence of initial pH on the oxidation capacity of the HEF/UV tandem system. Similar to the results at pH 4, the OTC degradation efficiency of the HEF/UV tandem system at pH 6 and 8 was significantly superior to that of various isolated systems, and the sf values at pH 6 (1.515) and 8 (1.467) were both greater than 1 and higher than that at pH 4 (1.231). This can be because the increased residual H2O2 in the HEF effluent at pH 6 and 8 would promote the generation of homogeneous ˙OH in the UV module, thus further weakening the wasting reaction (Fig. S10b, e, and h†). Notably, the highest sf (1.515) and k (0.2978 min−1) values corresponding to the HEF/UV tandem system were achieved at a solution pH of 6, which was considered to be a compromise between the different reaction characteristics of each subprocess in terms of initial solution pH (see the discussion shown in Fig. S10 and S11† for detailed reasons). This finding indicates that the HEF/UV tandem system is of great practical value since the majority of the effluents from industrial companies are at circumneutral pH.
3.3. Identification of reactive oxygen species responsible for OTC removal
The above discussion on the removal of OTC was based on the radical pathway dominated by ˙OH. However, an increasing number of recent studies have reported that in addition to ˙OH, there may be other active species-controlled radical pathways, or even nonradical pathways, during EAOP treatment.17,27–29 In view of this, EPR tests were implemented in different systems to determine the generated reactive oxygen species. When DMPO was used as a spin trapping agent (Fig. 4a), a weak quadruple peak signal for DMPO-˙OH (intensity ratio 1:2:2:1) appeared in the UV system, which may be caused by the ionization of H2O molecules by UV radiation (eqn (14)).30 With the coupling of the ADC (i.e., the UV/H2O2 system), the photolytic activation of H2O2 by UV radiation resulted in a significant increase in the peak intensity of the DMPO-˙OH signal. A similar phenomenon in the HEF system can be attributed to the Fenton activation of H2O2 at the FeOCl/GF catalytic cathode. The production of the highest concentration of ˙OH was achieved in the HEF/UV tandem system due to the synergistic effect between these two subprocesses. Importantly, the signal intensity of DMPO-˙OH did not change significantly as the reaction proceeded (5 min to 10 min), indicating that ˙OH was continuously generated in this tandem system to compensate for the self-quenching effect caused by its short lifetime.31 Note that the DMPO-SO4˙− signal was also detected in those systems equipped with the UV module, although the signal intensity was generally weaker than that of ˙OH. In a previous study of the photocatalytic oxidation reaction, Liu et al. proved that minor SO42− ions can be directly excited to convert into SO4˙− under UV irradiation (eqn (15)).32 This could also explain why no DMPO-SO4˙− signal was observed in the HEF standalone system.| H2O + hv → ˙OH + H+ | (14) |
| SO42− + hv → SO4˙− | (15) |
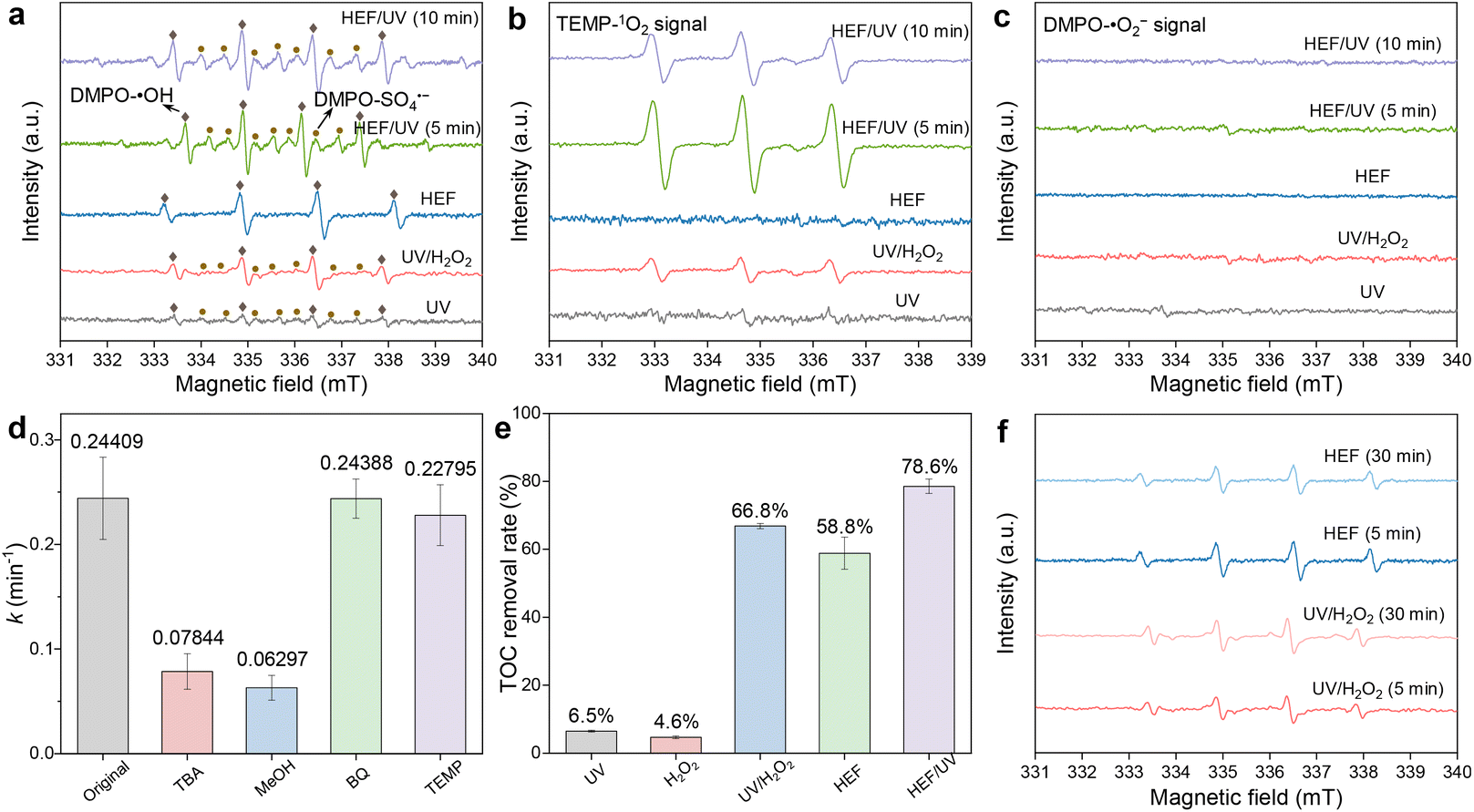 | ||
| Fig. 4 Identification of active species formed in the tandem system. EPR spectra of (a) DMPO-˙OH (DMPO-SO4˙−), (b) TEMP-1O2, and (c) DMPO-˙O2− in different systems (conditions: [DMPO] = 200 mM, [TEMP] = 25 mM, pH = 4, IADC = 100 mA cm−2, IFeOCl/GF = 5 mA cm−2, and UV = 6 W); (d) effects of TBA (200 mM), MeOH (200 mM), BQ (5 mM), and TEMP (2.5 mM) on OTC degradation kinetics in HEF/UV tandem systems; (e) TOC removal rate of different systems; and (f) EPR spectra of DMPO-˙OH in the UV/H2O2 and HEF systems at different reaction times. | ||
For the case using TEMP as the spin trapping agent (Fig. 4b), the 3-line TEMP-1O2 signal was detected in all systems except for HEF, with the order of peak intensity as follows: HEF/UV > UV/H2O2 > UV. Regarding the generation path of 1O2, although most previous studies emphasized that ˙O2− as an intermediate is critical for the conversion of 1O2,33 it may be trivial in our work. This was evidenced by the failure to detect the DMPO-˙O2− signal in all systems (Fig. 4c). Similar to SO4˙−, the generation of 1O2 in the present system may also be closely related to UV radiation. When the solution flows through the UV lamp module, both dissolved oxygen and ˙OH are likely to be excited and transformed into 1O2 (eqn (16) and (17)), as demonstrated by Jin et al.34 and Guo et al.27 Accordingly, the observed stronger TEMP-1O2 signals in the UV/H2O2 and HEF/UV systems compared to the UV standalone system could be interpreted as the elevated concentration of homogeneous ˙OH, and the additional dissolved oxygen contributed by the anodic oxygen evolution reaction during electrolysis, respectively. Note, however, that the concentration of 1O2 in the HEF/UV system was found to gradually decrease (Fig. 4b) and remained stable after 20 min (Fig. S12†), due to the consumption of dissolved oxygen at the ADC and FeOCl/GF cathode via the ORR.
| O2 + hv → 1O2 | (16) |
| 4˙OH + hv → 1O2 + 2H2O | (17) |
In order to distinguish the relative contributions of the abovementioned different reactive oxygen species in the OTC degradation process, chemical quenching experiments were also performed with the HEF/UV system as a representative. As shown in Fig. 4d, TBA, BQ, and TEMP were chosen as the scavengers for ˙OH, ˙O2−, and 1O2, respectively.15 And MeOH was used to simultaneously quench ˙OH and SO4˙−.17 Apparently, both TBA and MeOH could strongly inhibit OTC degradation in the HEF/UV system, with MeOH-induced inhibition slightly higher than TBA. This indicated that both ˙OH and SO4˙− were involved in the degradation process of OTC. Whereas the contribution of SO4˙− may be negligible, as evidenced by the fact that increasing the Na2SO4 concentrations did not significantly improve the process efficiency. When the Na2SO4 concentration was even higher than 100 mM, the degradation rate was almost unchanged (Fig. S13†). Consistent with the absence of DMPO-˙O2− signal in the EPR results, the addition of BQ hardly affected the degradation of OTC, revealing that ˙O2− was not involved in the oxidation process. Despite the obvious TEMP-1O2 signal in the EPR spectrum, the degradation kinetics of OTC decreased only slightly from 0.24409 min−1 to 0.22795 min−1 in the presence of TEMP, which may be due to the rapid deterioration of the 1O2 production rate with the reaction time (Fig. S12†). Moreover, similar results to the above can still be observed in Fig. S14† when changing the doses of different quenchers. Note that the contribution of potential chlorine radicals (˙Cl) and high-valent iron species (Fe(IV), Fe(V), and Fe(VI)) can also be excluded, since no corresponding signals (DMPO-˙Cl) were detected in all EPR spectra (Fig. 4) and addition of dimethyl sulfoxide (DMSO) (a quencher for high-valent iron species) did not significantly affect the degradation kinetics of OTC (Fig. S15†). Overall, these facts reflect the contribution of ˙OH, SO4˙−, and 1O2 to the degradation of OTC, with ˙OH playing a dominant role. This is also supported by the fact that the mineralization rates of OTC in different systems (Fig. 4e) almost coincide with the order of intensity of DMPO-˙OH. The contradiction between the mineralization rate (Fig. 4e) and the DMPO-˙OH signal intensity (Fig. 4a) of the UV/H2O2 system and HEF system may be related to the difference in the evolution of ˙OH production with reaction time. As depicted in Fig. 4f, the UV/H2O2 system contributed to a gradually enhanced ˙OH concentration with the reaction time, while the HEF system showed an opposite trend. This is precisely the result of the scavenging of ˙OH by H2O2 in the later stage of the dual-cathode HEF reaction discussed in section 3.2.
3.4. Assessment of practical application potential
The practical application potential of the constructed HEF/UV tandem system was evaluated from multiple perspectives. Fig. 5a shows that refractory organic compounds with different chemical structures (tetracycline (TC), ranitidine (RNTD), and sulfamethoxazole (SMX)) can all be effectively degraded in this HEF/UV process, demonstrating its versatility and universality. In addition, the effect of real water matrices (tap water, river water, and lake water) was also evaluated. The OTC degradation rate decreased to varying degrees in this tandem system compared to the deionized water background, as shown in Fig. 5b. This phenomenon occurred because of the presence of complex inorganic anions (especially Cl−) and dissolved organic matter in natural waters (see Table S2† for water quality parameters of different real water matrices), which were well established as ˙OH scavengers and will strongly affect the degradation efficiency of organic molecules.35 Despite the reduced performance, it is still acceptable because the complete degradation of OTC within 60 min was achieved in different natural waters.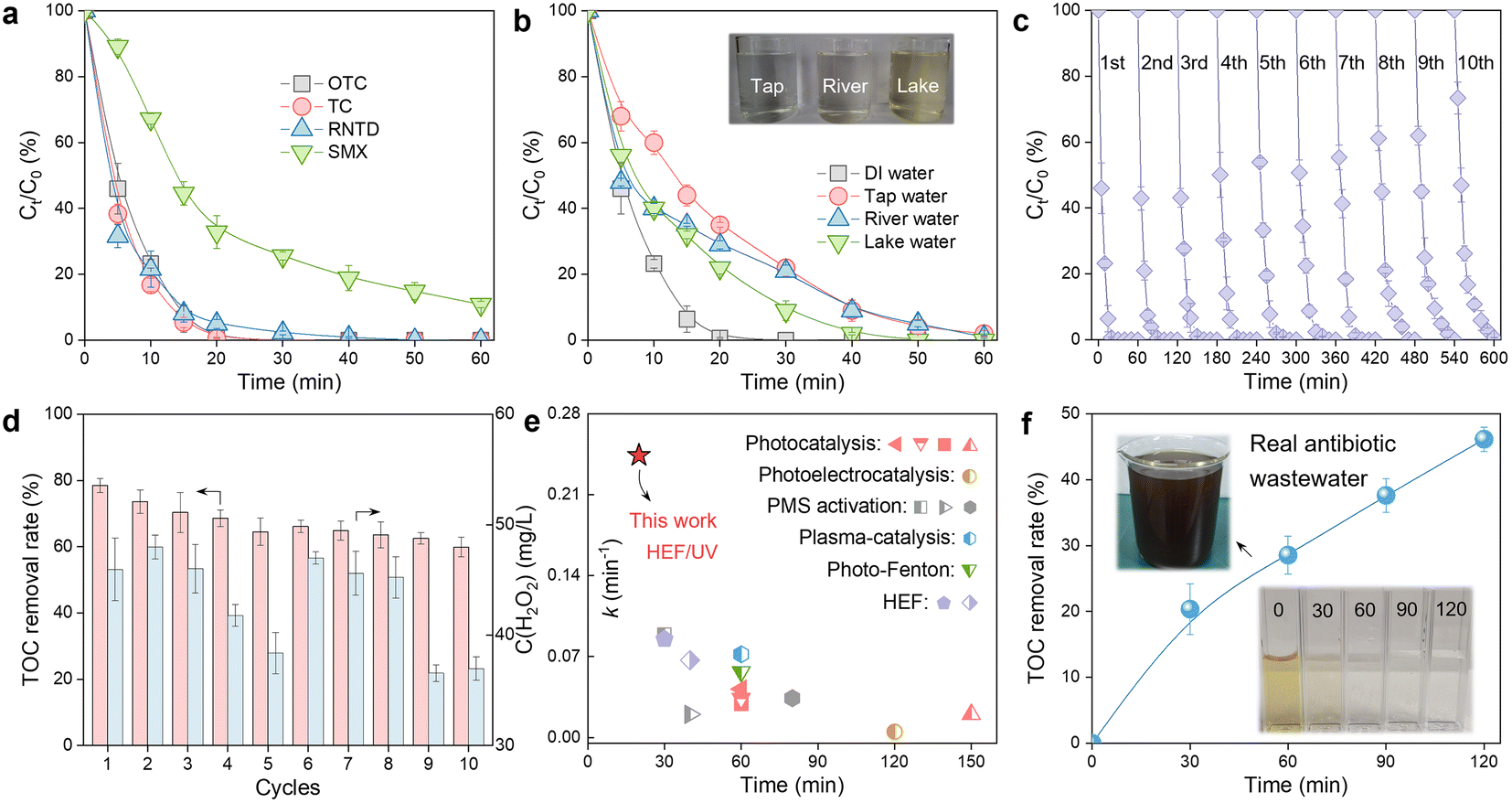 | ||
| Fig. 5 Practicability of the dual-cathode HEF and UV radiation tandem system. Evaluation of the universality of the HEF/UV tandem system in terms of (a) contaminant species and (b) natural water matrix background; durability of the HEF/UV tandem system over 10 consecutive runs: (c) OTC degradation and (d) TOC removal rate and H2O2 residue (conditions: [OTC] = 20 mg L−1, pH = 4, IADC = 100 mA cm−2, IFeOCl/GF = 5 mA cm−2, and UV = 6 W); (e) comparison of OTC degradation performance between different AOP technologies reported in the literature and the HEF/UV tandem system in this study; and (f) treatment of real antibiotic wastewater using the HEF/UV tandem system (conditions: IADC = 100 mA cm−2, IFeOCl/GF = 5 mA cm−2, and UV = 6 W). | ||
Service life is critical to the large-scale application of water treatment technologies, for which successive cycles of OTC degradation experiments in the HEF/UV system were performed (Fig. 5c and d). Both the degradation kinetics and mineralization rate of OTC showed a slight decrease after 10 runs. Considering the robust durability of the ADC electrode and UV lamp, the fading of the oxidation capacity of this tandem system was identified to originate from the gradual leaching of Fe ions from the surface of the FeOCl/GF catalytic cathode (Fig. S16,† ∼1.2 mg in each run). Therefore, the development of innovative and practical fabrication strategies to enhance the stability of catalytic cathodes under realistic harsh electrolysis conditions is suggested to be a priority for future research. Fig. 5d also shows the residual H2O2 concentration corresponding to each cycle, and only about 40 mg L−1 of unactivated H2O2 was detected in the HEF/UV effluent. Meanwhile, although nearly half of the degradation intermediates were predicted to be more toxic than OTC in the early stages of remediation, this HEF/UV tandem system still enabled a detoxification process for OTC as the oxidative conversion of these intermediates proceeded (Fig. S17, S18 and Tables S3, S4†). These abovementioned characteristics allowed to greatly reduce the environmental risk of OTC-containing sewage to aquatic ecosystems. Nonetheless, follow-up studies are recommended to examine the intermittent power supply mode (i.e., interruption of electric current when OTC is almost completely degraded at 20 min) to further eliminate the H2O2 residue and minimize energy consumption.
To further illustrate the superiority of the HEF/UV tandem system, a comparison of the k values for OTC degradation with other advanced oxidation-based technologies was carried out. As shown in Fig. 5e, HEF/UV probably represents the state-of-the-art catalytic performance with corresponding k values 2.7 to 48.4-fold higher than other systems,6,15,36–45 suggesting the significant synergy between electro- and photo-catalysis. In view of the excellent performance of the HEF/UV tandem system, its feasibility for real industrial antibiotic wastewater treatment was evaluated. Wastewater samples were obtained from an antibiotic pharmaceutical factory (CP group, Zhengzhou, China), and the main water quality parameters are shown in Table S5.† Note that this real antibiotic wastewater is characterized by high salinity (conductivity up to 5030 μS cm−1), for which the treatment process was performed without any supporting electrolyte addition. After 120 min, the treated wastewater became clear and transparent with a TOC removal rate of nearly 50%, even without pre-adjustment of the solution pH (Fig. 5f). Endowed with the aforementioned merits, the proposed HEF/UV tandem system may be regarded as a powerful solution for distributed water pollution remediation, due to its reagent- (i.e., catalyst dosing and oxygen aeration) and oxidant-free philosophy of green chemistry.
4. Conclusions
In summary, a coupled system of a dual-cathode HEF process in series with UV radiation was developed for efficient distributed water pollution control. In the dual-cathode electrolysis unit, H2O2 was first efficiently produced in situ at the ADC without aeration and then rapidly converted into active free radicals at the highly loaded FeOCl/GF catalytic cathode. The optimal operating current densities of the two cathodes were experimentally verified to be different (100 mA cm−2 at ADC and 5 mA cm−2 at FeOCl/GF), thus enabling simultaneous efficient 2e− ORR, H2O2 activation and Fe(III) electroreduction. Importantly, the underutilized H2O2 remaining in the HEF effluent will continue to be activated in the UV irradiation module, resulting in the elimination of the risk of oxidant residues and helping in further improving the degradation efficiency. The current density applied to the ADC has a significant impact on the synergy between the electro- and photo-catalysis subprocesses in the HEF/UV tandem system. For IADC < 100 mA cm−2, the deployment of the UV module cannot eliminate the wasting reaction of ˙OH heterogeneously produced at the FeOCl/GF cathode with H2O2 produced at the ADC, while for IADC ≥ 100 mA cm−2, UV photolysis of relatively high concentrations of H2O2 in the HEF effluent will generate considerably homogeneous ˙OH, which can outcompete the above wasting reactions by dimerization with heterogeneous ˙OH at the FeOCl/GF cathode. The HEF/UV tandem strategy proposed in the present work offers an efficient, green and residual oxidant-free wastewater treatment solution, which is promising especially for distributed wastewater treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (52170041), the Tsinghua SIGS Start-up Funding (QD2020002N), the Cross-disciplinary Research and Innovation Fund (JC2022006), the Key Research and Development Program of Zhejiang Province (2023C03148) and the Committee of Science and Technology Innovation of Shenzhen (JCYJ20190813163401660) is gratefully acknowledged.References
- B. C. Hodges, E. L. Cates and J. H. Kim, Nat. Nanotechnol., 2018, 13, 642–650 CrossRef CAS PubMed
.
- J. M. Barazesh, T. Hennebel, J. T. Jasper and D. L. Sedlak, Environ. Sci. Technol., 2015, 49, 7391–7399 CrossRef CAS PubMed
- I. Sirés and E. Brillas, Curr. Opin. Electrochem., 2021, 27, 100686 CrossRef
- S. O. Ganiyu, M. Zhou and C. A. Martínez-Huitle, Appl. Catal., B, 2018, 235, 103–129 CrossRef CAS
- C. Trellu, M. Rivallin, S. Cerneaux, C. Coetsier, C. Causserand, M. A. Oturan and M. Cretin, Chem. Eng. J., 2020, 400, 125936 CrossRef CAS
- L. Cui, X. Zhao, H. Xie and Z. Zhang, ACS Catal., 2022, 12, 13334–13348 CrossRef CAS
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed
- S. Cheng, C. Shen, H. Zheng, F. Liu and A. Li, Appl. Catal., B, 2020, 269, 118785 CrossRef CAS
- L. Cui, Z. Li, Q. Li, M. Chen, W. Jing and X. Gu, Chem. Eng. J., 2021, 420, 127666 CrossRef CAS
- F. Xiao, Z. Wang, J. Fan, T. Majima, H. Zhao and G. Zhao, Angew. Chem., Int. Ed., 2021, 60, 10375–10383 CrossRef CAS PubMed
- P. Cao, X. Quan, K. Zhao, S. Chen, H. Yu and Y. Su, Environ. Sci. Technol., 2020, 54, 12662–12672 CrossRef CAS PubMed
- Q. Zhang, M. Zhou, X. Du, P. Su, W. Fu and G. Song, Chem. Eng. J., 2022, 429, 132436 CrossRef CAS
- S. Cheng, H. Zheng, C. Shen, B. Jiang, F. Liu and A. Li, Adv. Funct. Mater., 2021, 31, 2106311 CrossRef CAS
- P. Su, M. Zhou, G. Ren, X. Lu, X. Du and G. Song, J. Mater. Chem. A, 2019, 7, 24408–24419 RSC
- L. Cui, M. Sun and Z. Zhang, Chem. Eng. J., 2022, 450, 138263 CrossRef CAS
- F. Deng, S. Li, Y. Cao, M. A. Fang, J. Qu, Z. Chen and S. Qiu, J. Power Sources, 2020, 466, 228342 CrossRef CAS
- Q. Zhang, H. Yin, P. Su, W. Fu, G. Song and M. Zhou, Chem. Eng. J., 2022, 444, 136590 CrossRef CAS
- D. Wang, J. Hu, B. Liu, H. Hou, J. Yang, Y. Li, Y. Zhu, S. Liang and K. Xiao, J. Hazard. Mater., 2021, 412, 125269 CrossRef CAS PubMed
- I. Sánchez-Montes, G. O. S. Santos, T. O. Silva, R. Colombo and M. R. V. Lanza, J. Cleaner Prod., 2023, 392, 136242 CrossRef
- D. Sánchez-Quiles and A. Tovar-Sánchez, Environ. Sci. Technol., 2014, 48, 9037–9042 CrossRef PubMed
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustain., 2021, 4, 233–241 CrossRef PubMed
- Q. Zhang, M. Zhou, G. Ren, Y. Li, Y. Li and X. Du, Nat. Commun., 2020, 11, 1731 CrossRef CAS
- M. Sun, C. Chu, F. Geng, X. Lu, J. Qu, J. Crittenden, M. Elimelech and J.-H. Kim, Environ. Sci. Technol. Lett., 2018, 5, 186–191 CrossRef CAS
- S. O. Ganiyu, T. X. Huong Le, M. Bechelany, G. Esposito, E. D. van Hullebusch, M. A. Oturan and M. Cretin, J. Mater. Chem. A, 2017, 5, 3655–3666 RSC
- C. H. Han, H. D. Park, S. B. Kim, V. Yargeau, J. W. Choi, S. H. Lee and J. A. Park, Water Res., 2020, 172, 115514 CrossRef CAS PubMed
- H. Dong, B. Dong, L. Sun, Z. Chi, M. Wang and H. Yu, Chem. Eng. J., 2020, 390, 124650 CrossRef CAS
- D. Guo, Y. Liu, H. Ji, C. C. Wang, B. Chen, C. Shen, F. Li, Y. Wang, P. Lu and W. Liu, Environ. Sci. Technol., 2021, 55, 4045–4053 CrossRef CAS PubMed
- X. Du, P. Su, W. Fu, Q. Zhang and M. Zhou, Electrochim. Acta, 2022, 412, 140122 CrossRef CAS
- P. Su, W. Fu, Z. Hu, J. Jing and M. Zhou, Appl. Catal., B, 2022, 313, 121457 CrossRef CAS
- S. Pan, X. Guo, R. Li, H. Hu, J. Yuan, B. Liu, S. Hei and Y. Zhang, Sep. Purif. Technol., 2022, 300, 121909 CrossRef CAS
- Q. V. Ly, L. Cui, M. B. Asif, W. Khan, L. D. Nghiem, Y. Hwang and Z. Zhang, Water Res., 2023, 230, 119577 CrossRef CAS PubMed
- Z. Liu, W. Hao, C. Yuan, W. Ruan, W. Jiang and F. Teng, J. Water Process Eng., 2022, 48, 102912 CrossRef
- Y. Zhao, M. Sun, X. Wang, C. Wang, D. Lu, W. Ma, S. A. Kube, J. Ma and M. Elimelech, Nat. Commun., 2020, 11, 6228 CrossRef CAS PubMed
- Y. Jin, Y. Shi, Z. Chen, R. Chen, X. Chen, X. Zheng, Y. Liu and R. Ding, Appl. Catal., B, 2020, 267, 118730 CrossRef CAS
- Y. Q. Gao, J. Zhang, C. Li, F. X. Tian and N. Y. Gao, Chemosphere, 2020, 243, 125325 CrossRef CAS PubMed
- M. Li, J.-F. Yan, Z.-X. Zhang, W. Han, S.-Q. Zhou, K. L. Yeung and C.-H. Mo, Environ. Sci.: Nano, 2022, 9, 4214–4232 RSC
- Y. Chen, R. Yin, L. Zeng, W. Guo and M. Zhu, J. Hazard. Mater., 2021, 412, 125256 CrossRef CAS
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, X. Li, B. Li, W. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CrossRef CAS
- Y. Liu, X. Wang, Q. Sun, M. Yuan, Z. Sun, S. Xia and J. Zhao, J. Hazard. Mater., 2022, 424, 127387 CrossRef CAS PubMed
- D. Liu, M. Li, X. Li, F. Ren, P. Sun and L. Zhou, Chem. Eng. J., 2020, 387, 124008 CrossRef CAS
- H. Guo, Y. Wang, X. Yao, Y. Zhang, Z. Li, S. Pan, J. Han, L. Xu, W. Qiao, J. Li and H. Wang, Chem. Eng. J., 2021, 425, 130614 CrossRef CAS
- J. Qin, S. Ye, K. Yan and J. Zhang, J. Colloid Interface Sci., 2022, 607, 1936–1943 CrossRef CAS PubMed
- J. Ni, D. Liu, W. Wang, A. Wang, J. Jia, J. Tian and Z. Xing, Chem. Eng. J., 2021, 419, 129969 CrossRef CAS
- W. Zhao, Q. Dong, C. Sun, D. Xia, H. Huang, G. Yang, G. Wang and D. Y. C. Leung, Chem. Eng. J., 2021, 409, 128185 CrossRef CAS
- H. Y. Liu, C. G. Niu, H. Guo, C. Liang, D. W. Huang, L. Zhang, Y. Y. Yang and L. Li, J. Colloid Interface Sci., 2020, 576, 264–279 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01653f |
| This journal is © The Royal Society of Chemistry 2023 |