
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural variation, magnetism and single-source deposition of lanthanide-containing polyoxotitanates†
Rosa
Müller
,
Olivia
Georghiades
,
Joshua D.
Bocarsly
,
Farheen N.
Sayed
 ,
Víctor
Riesgo-González
,
Andrew D.
Bond
,
Clare P.
Grey
and
Dominic S.
Wright
*
,
Víctor
Riesgo-González
,
Andrew D.
Bond
,
Clare P.
Grey
and
Dominic S.
Wright
*
The Yusuf Hamied Department of Chemistry, Cambridge University, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: dsw1000@cam.ac.uk; Tel: +44 (0)1223 763122
First published on 15th September 2023
Abstract
Heterometal-containing polyoxotitanates (POTs) are much-studied single-source precursors (SSPs) for doped TiO2. In this work the properties of a wide range of lanthanide-containing POTs are studied to assess their potential use as SSPs for Ln–Ti hybrid oxides. The novel cage compounds [{Ti2O(OEt)8}(EtOH·LnCl)]2 (Ln = Sm, Gd, Tb, Dy, Ho, Tm and Yb) are structurally characterised. The magnetic properties of the Ln = Dy and Ho compounds were characterised using SQUID magnetometry—in both cases, there is evidence of significant uniaxial magnetic anisotropy, but magnetic relaxation is fast and therefore no single-molecule magnetic properties are observed. Upon decomposition lanthanide-doped anatase (Ln = La) or titania/LnTi–oxide mixtures are obtained, which show efficient stabilisation of the catalytically active anatase phase up to high temperatures, making the materials of potential interest for applications in photocatalysis.
1. Introduction
Titanium dioxide is a versatile, abundant and non-toxic high band gap semiconductor used in a multitude of applications.1 Its photocatalytic activity means that it has found important uses in photovoltaics, sensors, photochromic devices, and as a photocatalyst in water splitting.2 Due to the wide band gap of titanium dioxide (3.20 eV direct band gap for anatase)3 many of these technologies are limited to the absorption of UV light. However, the introduction of dopant metal ions reduces the band gap due to the introduction of additional states above the valence band, which leads to higher efficiencies using ambient light.4 In addition to first-row transition metals such as Fe, Co or Cu, f-block elements (lanthanides) are also suitable dopants, and lead to enhanced properties of the resulting titania materials.5 Early lanthanides (La, Ce) are reported to stabilise the thermodynamically less stable, but highly catalytically active, anatase phase of TiO2 by suppressing the thermal anatase-to-rutile phase transition.6 Furthermore the presence of hard lanthanide ions promotes interactions with Lewis basic functionalities (such as acids, aldehydes or alcohols) which are present in organic pollutants.7 Hence, the resulting materials are promising candidates for photocatalytic degradation applications.When the Ln ratio is increased, a series of mixed-metal oxides of general formulae Ln2Ti2O7 can also be formed. These compounds can adopt closely related pyrochlore or fluorite structures which differ in the extent of cation mixing, where the stability window of the ‘ideal’ pyrochlores (A2B2O7, in which no cation mixing is present) can be predicted using the radius ratio of the cations (rA/rB).9a It is well established, however, that the degree of cation and anion mixing in these species is very dependent on synthetic conditions employed.9b Pyrochlores have a range of desirable properties depending on their composition, and have found uses as luminescence materials, catalysts, nuclear waste immobilization materials and as ionic and electrical conductors. For simplicity, here we will use the term ‘pyrochlore-like’ generically to describe compounds of formula Ln2Ti2O7 in the text.
Pyrochlore-like Ln2Ti2O7 compounds can have a range of desirable chemical and physical properties. Owing to their non-centrosymmetric structures La2Ti2O7 and Nd2Ti2O7 are ferroelectric materials analogous to Cd2Nb2O7 and are also suitable components for high-temperature microwave dielectric materials.10 Due to the increasing number of unpaired electrons, pyrochlore-like compounds containing later lanthanides show unusual antiferromagnetic (Ln = Eu–Er) and ferromagnetic (Ln = Yb) behaviour as well as abnormal luminescence (Ln = Eu) and oxygen-ion conductivity (Ln = Gd).11
Common synthetic strategies to prepare lanthanide-containing titania include sol–gel methods and high-temperature sintering of Ln2O3/TiO2 mixtures.8 Given the large impact of the metal-dopant level on the properties of TiO2-based materials a high level of stoichiometric control is required in their synthesis. The use of molecular single-source precursors (SSPs) allows precise control over dopant concentration, distribution and connectivity in the bulk materials.12 A class of suitable SSPs for this purpose are polyoxotitanates (POTs) of the general form [TixOy(OR)zMn] which have been widely studied as SSPs for transition metal-doped titania.13 We have previously reported a series of lanthanide-containing POTs obtained via solvothermal reactions of Ti(OEt)4 with LnCl3 in ethanol. The high-nuclearity cage arrangement [Ti28O38(OEt)40H2LnCl] (Ti28Ln, Fig. 1a) was obtained for the early lanthanides (Ln = La, Ce), a structure whose TiO core offers a high coordination number for the lanthanide ions.14 In the case of Ce, further analysis of the reaction mixture showed the formation of two smaller cages, [Ti8O7(OEt)21(EtOH·Ce)] (Ti8Ln) and [{Ti2O(OEt)8}(EtOH·CeCl)]2 (Ti4Ln2) (Fig. 1b and c, respectively).15 However, moving along the lanthanide series to the slightly smaller lanthanides Nd and Eu, only the Ti4Ln2 cages were obtained.16 This trend indicates the dependency of the preferred structure on the coordination geometry and the ionic radius of the lanthanide ion, which is a well-known feature of various classes of Ln-compounds.17 Interestingly, for Er this trend towards lower nuclearity cages is disrupted, as the main reaction product is the Ti8Er-cage.18 Decomposition of the Ce and Eu-containing cages as SSPs gives either Ln-doped TiO2 for low Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ln ratios (28:1 and 8:1, i.e.Ti28Ln and Ti8Ln), or TiO2-stabilised Ln2Ti2O7 for higher ratios (2:1, i.e.Ti4Ln2).
:Ln ratios (28:1 and 8:1, i.e.Ti28Ln and Ti8Ln), or TiO2-stabilised Ln2Ti2O7 for higher ratios (2:1, i.e.Ti4Ln2).
 | ||
| Fig. 1 The three different structural types (a) Ti28Ln, (b) Ti8Ln and (c) Ti4Ln2 of ethoxy-substituted Ln–POTs, with ligands omitted for clarity. A consistent colour scheme of Ln = pink, Ti = blue, O = red, Cl = green is used throughout this paper. Structural data were obtained from the CSD (Ti28La = DEJQUG, Ti8Ln = ORIFAY, Ti4Eu2 = CURJEG). For (b), the structure containing Ce (Ti8Ce = ZIWYAI) has a further EtOH molecule coordinated to the lanthanide centre. | ||
In this work we explore the structural variation of alkoxy-substituted Ln–POTs with a broader range of lanthanide ions [Ln = Sm (Ti4Sm2), Gd (Ti4Gd2), Tb (Ti4Tb2), Dy (Ti4Dy2), Ho (Ti4Ho2), Tm (Ti4Tm2) and Yb (Ti4Yb2)] and their applications as SSPs for the respective Ln2Ti2O7 pyrochlore-like materials. We identify and quantify the phases formed upon the hydrolytic decomposition of each of the cages using synchrotron powder X-ray diffraction and compare the results to those obtained for the literature-known compounds Ti28La, Ti4Ce2, Ti4Nd2, Ti4Eu2 and Ti8Er. Some unexpected solid-state products, namely Nd4Ti9O24 from the Nd cage Ti4Nd2 and Ce(IV) oxides from the Ce cage Ti4Ce2, are identified. Furthermore, the magnetic properties of Ti4Dy2 and Ti4Ho2 are investigated to assess the possibility of single-molecule magnetism (SMM). This study provides a better understanding of the nature of these cages as SSPs and highlights their potentially diverse applications in materials synthesis.
2. Results and discussion
Synthesis of Ln–POTs
The desired Ln–POTs were obtained via solvothermal reactions of Ti(OEt)4 with the respective Ln(III)Cl3 salts (Ln = Sm, Gd, Tb, Dy, Ho, Tm and Yb) in anhydrous ethanol at 180 °C in an autoclave, following a procedure described in our previous paper.15 All the products were shown to be of the type Ti4Ln2, [{Ti2O(OEt)8}(EtOH·LnCl)]2 by powder and single-crystal XRD. Due to the paramagnetic nature of the cages NMR spectroscopy was not of any value in their characterisation. In addition, mass spectrometry only revealed minor fragments together with presumed hydrolysis products. In general, yields decreased with decreasing ionic radius of the Ln3+ ion, which can potentially be attributed to an increase in structural strain in accommodating the Ln3+ ions (see the structural discussion below). The formation of solid products was only observed for Ti(OEt)4:LnCl3 reaction stoichiometries as high as 5:1. When Ti(OEt)4 was replaced with Ti(OiPr)4 using ethanol as the reaction solvent the same ethoxy-substituted compounds were formed, illustrating significant ligand exchange at the cage periphery with the solvent. Interestingly, no major contamination with biproducts such as [Ti16O16(OEt)32] was observed in any of these reactions, as observed in our previous report.14
An influence of the anion in LnX3 on the resulting cage structure has been described for similar solvothermal reactions.19 We find that this is also the case here. For example, the reaction of Eu(OAc)3 with Ti(OEt)4 gives a cage isostructural with [Ti4O(OEt)15Cu(Cl)] (CSD: EWONIO), as confirmed by the crystallographic unit-cell parameters. This structural type has previously been reported for the ethoxy-cages of first-row transition metals using MCl2 precursors (M = Co, Zn, Fe, Cu).13
Single-crystal X-ray structures
The reaction conditions were modified to investigate formation of the different cage types shown in Fig. 1. Eight new cages [{Ti2O(OEt)8}(EtOH·LnCl)]2 [Ln = Sm (Ti4Sm2), Gd (Ti4Gd2), Tb (Ti4Tb2), Dy (Ti4Dy2), Ho (Ti4Ho2), Tm (Ti4Tm2), Yb (Ti4Yb2)] were structurally characterised in the current study. The analogous structures of Ti4Ce2, Ti4Nd2 and Ti4Eu2 have been reported previously.15,16 The molecular structure of all of these cages is centrosymmetric (Fig. 2), with the asymmetric unit containing two face-sharing TiO6 octahedra and with the Ln3+ ion situated in a “pocket” above two faces (Fig. 3). Two {Ti2O(OEt)8}(EtOH·LnCl) subunits come together so that Ln3+ ion in one unit bridges an edge of a TiO6 octahedron of the other. The Ln3+ ions adopt overall a distorted 8-coordinate geometry (ESI, Fig. S4†). | ||
| Fig. 2 Single-crystal X-ray structure of [{Ti2O(OEt)8}(EtOH·SmCl)]2 (Ti4Sm2) with hydrogen atoms omitted for clarity. The other novel compounds (Ti4Gd2, Ti4Tb2, Ti4Dy2, Ti4Ho2, Ti4Er2, Ti4Tm2 and Ti4Yb2) reported in this work are isostructural. (Ln = pink, Ti = blue, O = red, Cl = green). Full details of the data collections, refinements, and bond lengths and angles can be found in the ESI.† | ||
 | ||
| Fig. 3 Schematic representation of the structural units in Ti4Ln2. Ln3+ sits above two faces of the face-sharing TiO6 octahedra, and pairs of these units come together with Ln3+ bridging an edge of the neighbouring TiO6 octahedron. | ||
The fact that the {Ti2O(OEt)8} fragments in the Ti4Ln2 cage are held together solely by the bridging Ln3+ ions (they are not directly linked through Ti–O bonds) provides a flexible molecular geometry. To quantify the structural strain imposed on the TiO6 octahedra across the series, we considered the continuous shape measure (CShM) relative to a centred regular octahedron.20 A CShM value of zero indicates perfect octahedral symmetry, while an increasing value indicates increasing deviation from regularity (ESI, Tables S2, S3 and S7†). It is evident that the TiO6 octahedron involved in the edge-sharing contact with Ln3+ is more distorted than the other TiO6 octahedron in the face-sharing pair, and its CShM value also increases more rapidly across the series. The degree of structural strain in the {Ti2O(OEt)8} fragments increases as Ln3+ gets smaller, but the molecular flexibility is evidently sufficient to permit formation of this cage type across the entire Ln series.
The previously reported Ti8Ln (Ln = Ce or Er) cages contain the same face-sharing {Ti2O(OEt)8} fragments as Ti4Ln2, but they adopt a “tetrahedral” arrangement around a single Ln3+ ion (Fig. 4).15 The location of Ln3+ relative to the face-sharing TiO6 octahedra is equivalent to that in Ti4Ln2. The existence of these common {Ti2O(OEt)8} fragments in both Ti4Ln2 and Ti8Ln suggest that these small units are likely to exist as precursors in solution. In Ti8Ln, however, the {Ti2O(OEt)8} fragments are directly linked through four further TiO6 octahedra in an edge-sharing arrangement. Hence, the Ti8O28 framework in Ti8Ln presents an inherently less flexible binding pocket for Ln3+. In the Ti8Ce cage, Ce3+ is accommodated with further coordination by an EtOH molecule, giving 9-coordination overall. In Ti8Er, Er3+ is 8-coordinate with no further coordination. The CShM values for the TiO6 octahedra in Ti8Ln vary little between Ln = Ce and Er, consistent with the picture of an inherently less flexible framework.
 | ||
| Fig. 4 Polyhedral representation of the Ti8Ln cage (Ln = Er, CSD ORIFAY). Carbon and hydrogen atoms are omitted for clarity. Ln3+ is located between two pairs of face-sharing TiO6 octahedra making a “tetrahedral” arrangement, further linked through four edge-sharing TiO6 octahedra. | ||
It was shown previously that for Ln = Ce all three structural types are obtained upon fractional crystallisation, with the large Ti28Ce cluster crystallising first, followed by Ti8Ce and Ti4Ce2, which could be separated by hand on the basis of their difference in colour.15 The presence of all three cage types for Ce suggests either simultaneous formation of all three products during the reaction or a dynamic equilibrium between them in solution. Consistent with the latter proposition, we have found in the current work that when the reaction mixture is cooled rapidly down to −30 °C (instead of slowly from 180 °C), the yellow crystalline product is exclusively the Ti4Ce2 cage (Fig. S2†). However, for Ln = La only the largest cage type is obtained (Ti28La). Nd and later lanthanides appear to form the smallest Ti4Ln2 type cluster exclusively, with no indication of Ti8Ln cage formation even after storing the reaction mixture at −30 °C for several weeks. The late lanthanide Er is an exception to this trend, as it forms Ti8Er as the main product. The preference for Er to adopt the Ti8Er cage is consistent with size selectivity, which is optimal for Er3+ and is also able to accommodate Ce3+ with further EtOH coordination. Moving across the lanthanide series, the elements between Ce and Er may be too small to permit 9-coordination but too large to permit 8-coordination.
Within the larger Ti28Ln cages (Ln = La, Ce), the TiO6 octahedra adopt corner- and edge-sharing arrangements, with some additional 7-coordinate pentagonal–bipyramidal sites also present. Face-sharing TiO6 octahedra are not seen. The Ln3+ ions are coordinated by a planar Ti6(OEt)6 metallocrown fragment, giving a seven-coordinate, hexagonal-based pyramidal geometry. Again, the Ti28O39 cage is directly connected, and therefore is expected to show limited flexibility in its Ln3+ binding site. The arrangement is presumably unstable for the smaller Ln3+ ions after Ce3+, leading to preference for the smaller Ti8Ln and Ti4Ln2 cages.
Magnetic measurements
Ln(III) compounds with axial symmetry have often been observed to show single-molecule magnet behaviour, characterised by slow magnetic relaxation arising from a bistable magnetic ground state with a significant barrier to relaxation (Ueff). Therefore, the clusters reported here, which feature approximate axial symmetry along the Ln3+–Ln3+ axis, could be of potential interest for single-molecule magnetism.21Our studies focused on the two rare earths with the highest total magnetic moments, Dy3+ (f9, J = 15/2, a Kramers ion) and Ho3+ (f10, a non-Kramer's ion, J = 8). In particular, Dy3+ is known to show strong magnetic anisotropy and a bistable magnetic ground state owing to its Kramers nature, and therefore is the most likely lanthanide ion to result in single molecule magnet behaviour.22 The Ti4+ is d0, and therefore not expected to contribute to the magnetic moment in these compounds. DC magnetisation vs. field/temperature and AC susceptibility measurements were carried out for powdered samples of Ti4Dy2 and Ti4Ho2 using SQUID magnetometry. Plots of χ(T)*T show that both compounds saturate to a constant value around 17.1 emu K Oe−1 mol−1, corresponding to an effective magnetic moment μeff = 11.7μB (Fig. 5a). The theoretical effective moments for Dy3+ and Ho3+ may be estimated according to  , where gJ is the Landé g-factor (1.33 for Dy3+ and 1.25μB for Ho3+). This gives expected moments of 10.63μB for Dy3+ and 10.60μB for Ho3+, in reasonable agreement with the experiment. Even better agreement can be produced by assuming slightly larger g-values of 1.46 for Dy3+ and 1.38 for Ho3+. At lower temperature, the χ(T)*T for Ti4Ho2 decreases smoothly, indicating likely antiferromagnetic exchange interactions between the nearby Ho3+ ions. Ti4Dy2 shows more complicated behaviour at low temperature, which may arise from a combination of magnetic exchange and magnetic anisotropy.
, where gJ is the Landé g-factor (1.33 for Dy3+ and 1.25μB for Ho3+). This gives expected moments of 10.63μB for Dy3+ and 10.60μB for Ho3+, in reasonable agreement with the experiment. Even better agreement can be produced by assuming slightly larger g-values of 1.46 for Dy3+ and 1.38 for Ho3+. At lower temperature, the χ(T)*T for Ti4Ho2 decreases smoothly, indicating likely antiferromagnetic exchange interactions between the nearby Ho3+ ions. Ti4Dy2 shows more complicated behaviour at low temperature, which may arise from a combination of magnetic exchange and magnetic anisotropy.
 | ||
| Fig. 5 (a) χT vs. temperature plots for Ti4Ho2 and Ti4Dy2 (powder), showing a room temperature effective moment μeff of about 11.7μB per lanthanide for both complexes. (b) Magnetisation vs. field hysteresis loops taken between 70 and −70 kOe (sweep rate = 100 Oe s−1) at 1.8 K, showing a saturated moment of μsat = 7.6μB and 5.5μB, respectively. Neither compound shows measurable hysteresis. (c) Magnetisation vs. temperature for both compounds, measured under both zero-field-cooled (ZFC) and field-cooled (FC) conditions in an applied field of 200 Oe. Neither compound shows a measurable difference between the ZFC and FC curves, indicating fast magnetic relaxation across the temperature range. In (a) and (c), susceptibility is expressed per mol of Ln3+ (rather than mol of Ln2Ti4 clusters). | ||
However, comparison of field-cooled (FC) and zero-field cooled (ZFC) magnetisation vs. temperature curves show no splitting in either compound (Fig. 5c), implying the absence of significant magnetic memory or slow magnetic relaxation. Similarly, magnetic hysteresis loops (M(H)) collected at 1.8 K show no measurable hysteresis (Fig. 5b). Interestingly, the Ti4Dy2M(H) shows sharp saturation behaviour with a saturated moment μsat of 5.5μB, which is half that of the expected value for free (isotropic) Dy3+ ions (μsat = g*J = 11.0μB). This halving is commonly seen in powder samples of Dy3+ compounds with strong uniaxial anisotropy, where powder averaging of the randomly oriented Ising-like spins reduces the moment by ½ compared to the free-spin value. However, Ti4Dy2 does not appear to satisfy this condition as the molecular Dy…Dy vectors are not aligned in the same direction in the lattice (Fig. 6). The Ti4Ho2 compound shows a saturated moment around 7.6μB, somewhat lower than the free spin value of 11.04μB.
 | ||
| Fig. 6 Packing of Ti4Dy2 within the unit cell, in which the Dy…Dy axes are not aligned in the same direction (Ln = pink, Ti = blue, O = red, Cl = green). | ||
To examine the possibility of slow magnetic relaxation further, we performed AC susceptibility measurements at temperatures between 1.8 K and 10 K and frequencies between 10−1 Hz and 103 Hz. For both Ti4Dy2 and Ti4Ho2 no resonance was observed (ESI, Fig. S8†). The magnetic relaxation time is therefore assumed to be faster than 0.001 s, and neither compound can be considered to be single-molecule magnets. A possible reason for this is the long Ln⋯Ln distance (ca. 3.676 Å) and the resulting changes to the crystal field in Ti4Ln2.23 While we have not measured the magnetic properties of the other lanthanide clusters, the lack of slow magnetic relaxation in these two clusters (especially Ti4Dy2) implies that the other members are unlikely to show slow magnetic relaxation.
Decomposition studies
The decomposition of the previously reported and novel Ln–POT cages prepared in the current study was analysed using two different strategies. In the first, the crystalline compounds were heated to 800 °C under an inert atmosphere of N2, and their decomposition was monitored via thermogravimetric analysis (TGA). A distinct process of mass loss is observed for each structural type, irrespective of the Ln3+ ion present (ESI, Fig. S9†). Fig. 7 shows representative TGA traces for the thermal decomposition of selected cages. | ||
| Fig. 7 TGA curves under nitrogen for the three different structural types of Ln–POTs. The measurements were carried out up to 800 °C at a heating rate of 10 °C min−1, but mass loss was found to be completed at 500 °C in all cases. | ||
For the smallest Ti4Ln2 cages decomposition starts at around 150 °C in all cases, with the total mass loss occurring at around 350 °C due to the loss of all ethoxy ligands. As this series of compounds already shows a condensation ratio (O:Ti) of 2:1 (the same as that in TiO2), this indicates the direct decomposition into titania. Although the decomposition of Ti28La commences at a similar temperature to that of the Ti4Ln2 cages a lower and more gradual total mass loss is seen, which is attributed to the lower condensation ratio of 1.4:1 (i.e., far off that of TiO2). The Ti8Er cage has an even lower ratio of 0.9:1 leading to greater thermal stability compared to Ti28La and the Ti4Ln2 cages, with decomposition commencing at a higher temperature of around 250 °C.13 EDX data confirm the retention of chloride in the decomposition products of the Ti4Ln2 cages. As no chloride ligands are present in the structure of Ti8Er a very different decomposition mechanism is likely. All the solid-state materials obtained by thermolysis under N2 at 800 °C were found to be amorphous by powder X-ray diffraction, which made further analysis of the phases difficult.
The second decomposition strategy involved hydrolytic treatment of the crystalline Ln–POT cages with a EtOH/H2O solution, followed by annealing of the powders formed under air. The thermal removal of organic residues proved to be essential for successful characterisation of the products. As indicated by elemental (C, H) analysis, heating to 500 °C (12 h) was found to be sufficient for this purpose. However, crystalline materials (essential for characterisation by powder X-ray diffraction) were only obtained at higher temperature (800 °C). Analysis of these crystalline materials by SEM (Scanning Electron Microscopy) shows sharp-edged particles with an average size of >100 μm (Fig. 8 and S11–S17†). The surfaces appear to be smooth but show surface features at higher magnification. Earlier reports have described the formation of agglomerates of nanoparticles by room-temperature hydrolysis of Ln–POTs under sonication conditions, which was not the case in the current study due to the different decomposition conditions.16
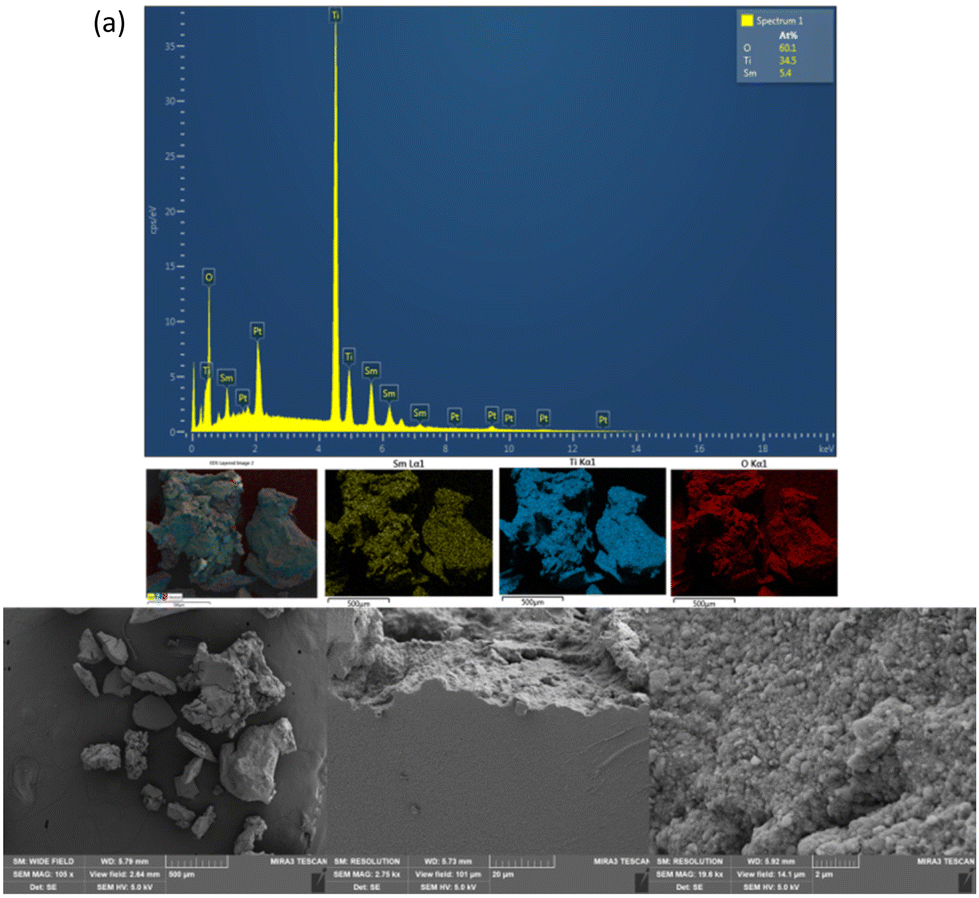 | ||
| Fig. 8 (a) EDX map spectrum recorded at 15 kV of Sm2Ti2O7/TiO2 after annealing to 800 °C, showing a uniform elemental distribution across the sample (Sm in yellow, Ti in blue, O in red). The sample was sputtered with Pt prior to measurement. (b) SEM images of the same sample taken at different viewing fields (2.5 mm, 100 μm and 15 μm) with a 5 kV electron beam. | ||
Analysis of the decomposition products obtained by this route (annealed to 800 °C) was carried out using Raman spectroscopy, synchrotron XRD and EDX. For the Ti4Ln2 series and Ti8Er the formation of TiO2 (anatase and rutile)/Ln2Ti2O7 (pyrochlore-like) mixtures is expected based on the limited previous reports.15,16,18 A varying degree of crystallinity across the series of the decomposition products leads to differences in resolution of the spectra. However, Raman spectra confirm the presence of anatase in all of the decomposition products of Ti4Ln2 and Ti8Er, with characteristic peaks at around 144, 197, 399, 516 and 639 cm−1 (ESI, Fig. S10†).24 Since the anatase phase exhibits an unusually high degree of thermal stability, low-level Ln-doping is assumed for all samples. In some cases (Ti4Ln2, Ln = Gd, Tb, Dy; Ti8Er) the appearance of a weak band at around 300 cm−1 was also observed which can be assigned to the O–Ln–O bending mode of the respective pyrochlore-like phases.25 For Ln = Ce, Nd, Tm (Ti4Ln2) and Ti8Er more complex spectra were obtained than for the other precursors, which it was assumed result from the presence of multiple crystalline phases.
To quantify the suggested compositions, high-resolution synchrotron powder diffraction patterns were measured and the data were analysed by Rietveld refinement (Table 1). For all samples apart from the decomposition products of Ti28La, Ti4Ce2 and Ti4Nd2, titania/pyrochlore-like mixtures were obtained, as expected from Raman studies. The ratios of anatase and rutile vary across all the decomposition products depending on the lanthanide. This indicates different degrees of stabilisation of the anatase phase caused by low-level doping with Ln3+. In general, a large ionic radius of Ln3+ is reported to efficiently inhibit the anatase-to-rutile transition through the formation of Ti–O–Ln bonds.5 Therefore, only the anatase phase is observed for the decomposition product of Ti28La. The decomposition of Ti4Ce2 is reported to give Ce2Ti2O7 (stabilised by a titania coating), which forms at 150 °C, but is found in the current study to decompose into CeIVO2 and CeIVTi2O6 (brannerite) upon heating to 800 °C.14 Further analysis of this monoclinic brannerite phase has shown that the stoichiometry is Ce0.975Ti2O5.95, in which oxygen vacancies at the O1 site lead to charge compensation through Ce4+ vacancies.27 No oxidation of Nd3+ occurs in the decomposition of Ti4Nd2, but the large ionic radius impedes the formation of a high-symmetry pyrochlore-like structure, thus orthorhombic Nd4Ti9O24 is formed (Fig. 9).5
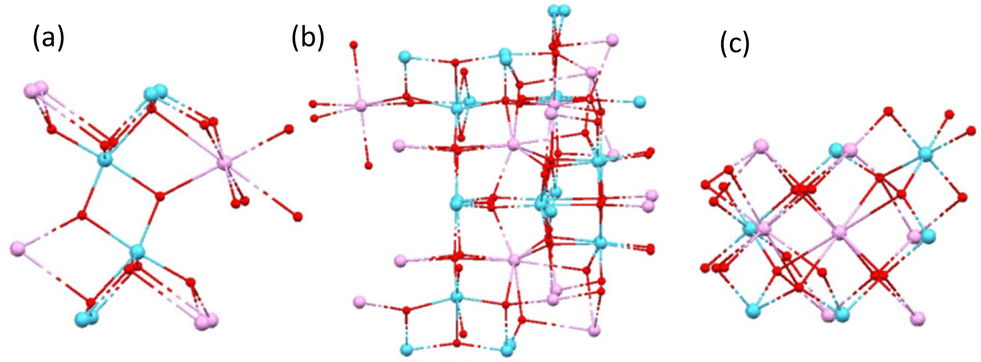 | ||
Fig. 9 Crystal structures of (a) Ce0.975Ti2O5.95, (b) Nd4Ti9O24 and (c) Sm2Ti2O7. In each case the Ln3+ ions are coordinated by eight oxygen ions. The space group changes from monoclinic C2/m for brannerite to orthorhombic face-centred Fddd for the Nd–Ti oxide and then cubic face-centred Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m for the pyrochlores. Structural data were obtained from the ICSD (193469, 72316, 24208). (Ln = pink, Ti = blue, O = red). m for the pyrochlores. Structural data were obtained from the ICSD (193469, 72316, 24208). (Ln = pink, Ti = blue, O = red). | ||
| Ln | Rietveld refinement | Ln (at%) | Ti (at%) | O (at%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Anatase (wt%) | Rutile (wt%) | LnxTiyOz (wt%) | Calcd | EDX | Calcd | EDX | Calcd | EDX | |
| a From Ti28La. b From Ti4Ln2. c From Ti8Er. d The remaining 12.3% is CeO2. | |||||||||
| Laa | 100 | 0 | 0 | 0 | 0.3 | 33.3 | 33.3 | 66.7 | 66.7 |
| Ceb,d | 0 | 17.7 | 70.0 | 12.8 | 6.4 | 12.8 | 15.7 | 72.3 | 78.0 |
| Ndb | 65.7 | 0 | 34.3 | 3.7 | 3.0 | 30.0 | 26.3 | 65.6 | 70.7 |
| Smb | 11.2 | 23.4 | 65.4 | 11.9 | 4.5 | 23.3 | 28.8 | 64.5 | 66.8 |
| Eub | 0 | 0 | 100.0 | 18.2 | 14.4 | 18.2 | 17.4 | 63.6 | 68.3 |
| Gdb | 74.2 | 0 | 25.8 | 4.7 | 3.6 | 29.2 | 23.5 | 65.4 | 73.0 |
| Tbb | 45.3 | 9.4 | 45.3 | 8.2 | 3.6 | 26.3 | 17.7 | 64.9 | 78.5 |
| Dyb | 0 | 23.1 | 76.9 | 14.0 | 9.5 | 21.6 | 18.8 | 64.2 | 71.8 |
| Hob | 62.2 | 0 | 37.8 | 6.9 | 3.8 | 27.4 | 24.4 | 65.7 | 71.8 |
| Erc | 82.9 | 3.9 | 13.2 | 2.4 | 2.1 | 31.0 | 30.7 | 65.7 | 67.2 |
| Tmb | 22.5 | 22.6 | 54.9 | 10.0 | 8.5 | 24.9 | 19.3 | 64.7 | 72.3 |
| Ybb | 57.5 | 8.0 | 34.5 | 6.3 | 5.5 | 27.9 | 25.0 | 65.2 | 65.5 |
Previous studies suggest that Nd4Ti9O24 consists of a 9:2 ratio of TiO2:Nd2O3, which is usually formed by a Nd2Ti2O7 intermediate in high-temperature solid-state reactions.28 For the rest of the Ti4Ln2 series (Ln = Sm–Yb) cubic pyrochlore-like Ln2Ti2O7 were formed in varying degrees, depending on the lanthanide. In general, partial site-mixing between the Ti and Ln positions is observed in Ln2Ti2O7 with Ti4+ occupying some of the Ln3+ sites and vice versa (Table S8†), suggesting variable formation of the fluorite phases. However, a particularly low pyrochlore content (13.2 wt%) with no site-mixing is observed in the decomposition product of Ti8Er, which is attributed to the Ti:Er stoichiometry in the precursor and the resulting preference to form Er-doped anatase (82.9 wt%).
EDX analysis of all the decomposition products shows a uniform elemental distribution across the series of lanthanides (Fig. 8, ESI Figs. S18 and S19†). Atomic percentages of Ln, Ti and O were obtained from multiple map and point spectra per sample. The mean results were compared to those calculated from the composition obtained from the Rietveld refinements (Table 1). In general, the calculated and experimental percentages match reasonably well, however, in some cases the oxygen contents lie significantly above the values obtained from the Rietveld refinements, leading to underestimation of the metal content by EDX. It has previously been reported that particles obtained by decomposition of Ln–POTs have a higher concentration of TiO2 on their surfaces, which could account for the low Ln content measured in some cases since EDX is a surface technique.15 In addition, since EDX measurements only have limited detection accuracy for light elements, the oxygen percentages determined here should be interpreted with care.
Band gap determination
The band gaps of the materials produced by decomposition of the Ln–POTs at 800 °C in air were estimated by linear extrapolation of the solid-state UV-Vis spectra (Fig. 10b).29 The results were compared to the HOMO–LUMO gaps of the Ln–POT cages obtained from both solution (Fig. 10a) and solid-state (ESI Fig. S22†) UV-Vis spectra (Table 2). The results obtained by linear extrapolation of the cages in the solid state and of the solid decomposition products are similar to those found using Tauc plots, with the values from direct extrapolation being generally higher by ca. 0.2 eV (ESI, Table S9†). Similar observations were made previously in our comparison of bandgap determination using direct extrapolation or Tauc plots.29 It can be noted that the best fits of the data for the decomposition products are obtained in Tauc plots with an exponent of ½, suggesting direct (allowed) band gap behaviour. | ||
| Fig. 10 (a) Solution-state UV-Vis spectra for the series of Ln–POTs in DCM. The samples were prepared under an inert atmosphere using anhydrous DCM (0.033 mg mL−1). (b) Solid-state UV-Vis reflectance spectra for the hydrolytically decomposed materials. In both cases the spectra were normalised to allow better comparison of the influence of different lanthanides on the optical properties of the sample. | ||
| Ln | HOMO–LUMO gap solution-state (eV) | HOMO–LUMO gap solid-state (eV) | Solid-state band gap (eV) |
|---|---|---|---|
| a From Ti28La. b From Ti4Ln2. c From Ti8Er. d Not enough sample was obtained. | |||
| Laa | 4.40 | 3.09 | 3.15 |
| Ceb | 4.29 | 2.95 | 2.38 |
| Ndb | 4.16 | 3.53 | 2.99 |
| Smb | 4.21 | 3.42 | 3.03 |
| Eub | 4.14 | 3.51 | 2.99 |
| Gdb | 4.10 | 3.44 | 3.08 |
| Tbb | 4.14 | 3.37 | 2.81 |
| Dyb | 4.10 | 3.36 | 2.98 |
| Hob | 4.23 | 3.52 | 3.05 |
| Erc | 4.26 | 3.44 | 2.95 |
| Tmb | —d | —d | 2.94 |
| Ybb | 4.07 | 3.27 | 3.17 |
As shown in Fig. 10a the absorption maxima of the Ln–POTs in DCM are blue-shifted compared to the starting material Ti(OEt)4 (245 → 229 nm).30 The larger Ti28La cluster gives a high HOMO–LUMO gap of around 4.40 eV, whereas the smaller Ti4Ln2 cages and Ti8Er show slightly lower values. For the decomposed materials a broad UV-Vis absorption band around 300–400 nm is observed, which is attributed to the O(p) → Ti(d) charge-transfer transition in titania (Fig. 10b). Additional small peaks in the visible range depend on the electronic structure of the Ln3+ ions and their f–f transitions. When moving from discrete molecular energy levels (POTs) to the solid-state materials a general reduction of the band gap is observed. Most solid-state samples show band gaps similar to those of rutile and anatase. A large red shift is obtained for the decomposition product of Ti4Ce2, which is in agreement with literature.31 The HOMO–LUMO gaps obtained for the cages in the solid state lie between those from solution-state measurements and the band gaps of the decomposed materials. It is unclear, however, to what extent hydrolysis is responsible for this as the cages in the solid state could not be measured under inert atmosphere.
Conclusions
Following on from earlier reports on selected Ln–POTs,14–16,18 in this work we have investigated an expanded series of Ln–POTs and their potential use as single-source precursors for lanthanide/titanium oxide materials. Structural analysis of the cages showed a dependency of the nuclearity on both the size of the Ln3+ ion and the reaction conditions. Assessment of the magnetism of Ti4 Dy2 and Ti4 Ho2 shows that both compounds are not single molecule magnets. Upon hydrolytic decomposition of the cages Ln2Ti2O7 pyrochlore-like compounds were obtained for Ti8Ln and Ti4Ln2-type POTs, while lower Ln:Ti ratios (Ti28La) formed lanthanide-doped titania. The presence of lanthanide ions led to efficient inhibition of the anatase-to-rutile transition at temperatures up to 800 °C. For Ce and Nd-containing precursors annealing of the hydrolytically decomposed samples led to the formation of different phases (Ce(IV)-oxidation products and orthorhombic Nd4Ti9O24, respectively). Only for the decomposition product of Ti4Ce2 at 800 °C in air (consisting of CeIVO2 and CeIVTi2O6) was a significant reduction of the band gap observed from that of titania (3.20 → 2.38 eV).
Overall, this work shows that single-source Ln–POTs have promising applications in the synthesis of pyrochlore-like composite materials. Analysis of the photocatalytic properties of the solid-state Ln–Ti oxide decomposition products would be of interest in future studies to assess the impact of different forms of lanthanide-doping further.
Experimental section
General remarks
All experimental procedures were carried out under a dry inert atmosphere of N2 with the aid of a vacuum-line and glovebox (Saffron, type-α) unless specified otherwise. Starting materials and dry alcohols were purchased from suppliers (Fischer Scientific, Alpha Aesar, Merck) and used as received. Lanthanide(III) chlorides were obtained as anhydrous salts. Elemental analysis was performed using a PerkinElmer 240 Elemental Analyser, with samples (1–2 mg) being sealed in one-piece tin tubes in the glovebox before analysis. IR spectra were acquired on a Thermo Scientific Nicolet iS50 FT-IR in ATR mode. TGA data were acquired on a Mettler Toledo TGA/DSC. SEM/EDX measurements were carried out on a Tescan MIRA3 SEM under vacuum. The samples were deposited on carbon tape and sputtered with 10 nm Pt prior to measurement. UV-Vis spectra were measured using a VARIAN Cary 50 Bio UV-Visible spectrometer (solution) or an Aligent Cary 60 UV-Vis spectrophotometer with external light source/detector (solid state). Solution UV samples were prepared under nitrogen in a glovebox. Raman spectroscopy was carried out using a Renishaw inVia Raman Microscope (532 nm laser source). Magnetic measurements were acquired using a Quantum Design cryogen-free Magnetic Property Measurement System (MPMS). Standard powder sample holders (brass) were used. Samples were maintained under a nitrogen atmosphere throughout the measurements. Single-crystal X-ray diffraction data were collected using a Bruker D8-QUEST PHOTON-III (Incoatec IμS Cu microsource) diffractometer. The temperature was held at 180 K using an Oxford Cryosystem N2 cryostat. Structures were solved using SHELXT32 and refined using SHELXL.33 In-house pXRD analysis was carried out on a Panalytical XPert Pro diffractometer (using Cu radiation, 1.5406 Å). High resolution powder XRD data were collected at the Diamond Light Source Synchrotron i11 beamline using a wavelength of 0.82443 Å. In-house powder XRD samples were protected from ambient air by being sandwiched between two Kapton sheets. High-resolution synchrotron pXRD samples were prepared in glass cannulae and sealed with glue. Single-crystal X-ray data has been deposited with the Cambridge Crystallographic Data Base (deposition numbers 2283715–2283722†).Ln–POT synthesis
The La, Ce, Nd, Eu and Er-containing POTs were synthesised as described in the literature for reasons of completeness.14–16,18Under a dry inert atmosphere of N2, Ti(OEt)4 (3.5 mL, 16.7 mmol) was added to a suspension of LnCl3 (1.7 mmol) in anhydrous ethanol (7 mL) and sealed in a 12 mL Teflon-lined autoclave. After heating to 150 °C for 3 days the reaction was slowly cooled to room temperature (1–10 °C min−1). The resulting solution was kept at −30 °C until a crystalline product had formed. After removal of the mother liquor the crystals were washed with anhydrous ethanol (2 × 3 mL) and n-pentane (1 × 3 mL) and dried under reduced pressure.
[{Ti 2 O(OEt) 8 }(EtOH·SmCl)] 2 pale yellow needles (21% wrt Sm). Elem. anal; calcd C, 30.7, H, 6.6. Found C, 30.8, H, 6.8. Important IR data (KBr, cm−1): 2968(s), 2922(s), 2863(s), 1376(vs), 1127(s), 1086(s), 1044(s), 888(s).
[{Ti 2 O(OEt) 8 }(EtOH·GdCl)] 2 colourless block-shaped crystals (16% wrt Gd). Elem. anal; calcd C, 30.4, H, 6.5. Found C, 30.5, H, 6.8. Important IR data (KBr, cm−1): 2966(s), 2921(s), 2862(s), 1627(w), 1376(vs), 1127(s), 1085(s), 1044(s), 890(s).
[{Ti 2 O(OEt) 8 }(EtOH·TbCl)] 2 colourless needles (12% wrt Tb). Elem. anal; calcd C, 30.3, H, 6.5. Found C, 29.6, H, 7.0. Important IR data (KBr, cm−1): 2967(s), 2922(s), 2862(s), 1376(vs), 1127(s), 1087(s), 1045(s), 892(s).
[{Ti 2 O(OEt) 8 }(EtOH·DyCl)] 2 pale blue block-shaped crystals (9% wrt Dy). Elem. anal; calcd C, 30.2, H, 6.5. Found C, 29.8, H, 6.6. Important IR data (KBr, cm−1): 2968(s), 2922(s), 2862(s), 1378(vs), 1127(s), 1085(s), 1045(s), 890(s).
[{Ti 2 O(OEt) 8 }(EtOH·HoCl)] 2 pink needles (10% wrt Ho). Elem. anal; calcd C, 30.1, H, 6.4. Found C, 28.6, H, 6.3. Important IR data (KBr, cm−1): 2969(s), 2922(s), 2862(s), 1378(vs), 1127(s), 1085(s), 1045(s), 890(s).
[{Ti 2 O(OEt) 8 }(EtOH·TmCl)] 2 colourless needles (1.2% wrt Tm). Elem. anal. unsuccessful due to insufficient amount of sample.
[{Ti 2 O(OEt) 8 }(EtOH·YbCl)] 2 colourless block-shaped crystals (11% wrt Yb). Elem. anal; calcd C, 29.8, H, 6.4. Found C, 29.4, H, 6.4. Important IR data (KBr, cm−1): 2967(s), 2923(s), 2861(s), 1377(vs), 1126(s), 1089(s), 1046(s), 892(s).
Hydrolytic decomposition
The obtained crystalline Ln–POTs (300–400 mg) were dissolved in DCM (5 mL) and added dropwise to a stirred solution of ethanol (10 mL) and water (10 mL). Stirring was continued overnight and after phase separation the organic layer was discarded. The white to off-white precipitate was dried under reduced pressure to give the initial products (150–200 mg). The amorphous powders were heated to temperatures between 350–800 °C to remove organic residues and induce crystallisation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Dr. Cheng Liu (Maxwell Centre Cambridge) for his technical support at the SQUID magnetometer and Dr Chloe Coates for her input on the Rietveld analysis. We also acknowledge I11 beamline for synchrotron XRD under the BAG proposal (CY28349). The authors would like to thank the Todd-Hamied fund (Cambridge) and the Cusanuswerk e.V. for financial support.References
- X. Chen and S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed; A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef PubMed; J. Disegi and H. Wyss, Orthopedics, 1989, 12, 75 CrossRef PubMed.
- H. Macleod, Thin film optical filters, MacMillan, New York, 1986, p. 370 CrossRef CAS; A. Linsebigler, G. Lu and J. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS; A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef; P. Moseley and B. Tofield, Solid state gas sensors, Adam Hilger, Bristol, 1987 CrossRef; A. Alivisatos, Science, 1996, 271, 933 CrossRef.
- H. Tang, K. Prasad, R. Sanjines, P. E. Schmid and F. Levy, J. Appl. Phys., 1994, 75, 2024 Search PubMed.
- K. E. Karakitsou and X. E. Verykios, J. Phys. Chem., 1993, 97, 1184 CrossRef CAS; V. Augugliaro, L. Palmisano, M. Schiavello and A. Scafani, J. Phys. Chem., 1991, 95, 274 Search PubMed.
- Y. Zhang, H. Zhang, Y. Xu and Y. Wang, J. Solid State Chem., 2004, 177, 3490 CrossRef CAS.
- S. Yuan, Q. Sheng, J. Zhang, F. Chen, M. Anpo and Q. Zhang, Microporous Mesoporous Mater., 2005, 79, 93 CrossRef CAS; T.-D. Nguyen-Phan, M. B. Song, E. J. Kim and E. W. Shin, Microporous Mesoporous Mater., 2009, 119, 290 CrossRef.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, Environ. Sci. Technol., 2001, 35, 1544 CrossRef CAS PubMed.
- Y.-H. Zhang and A. Reller, J. Mater. Chem., 2001, 11, 2537 RSC; M. Martos, B. Julián-López, J. V. Folgado, E. Cordoncillo and P. Escribano, Eur. J. Inorg. Chem., 2008, 3163 CrossRef CAS; L. H. Brixner, Inorg. Chem., 1964, 3, 1065 CrossRef.
- (a) M. A. Subramanian, G. Aravamudan and G. V. S. Rao, Prog. Solid State Chem., 1983, 15, 55 CrossRef CAS; (b) A. F. Fuentes, S. M. Montemayor, M. Maczka, M. Lang, R. C. Ewing and U. Amador, Inorg. Chem., 2018, 57, 12093 CrossRef CAS PubMed.
- A. V. Prasadarao, U. Selvaraj, S. Komarneni and A. S. Bhalla, J. Mater. Res., 1992, 7, 2859 CrossRef CAS.
- P. Berdowski and G. Blasse, J. Solid State Chem., 1986, 62, 317 CrossRef CAS; P. Dasgupta, Y. M. Jana, A. Nag Chattopadhyay, R. Higashinaka, Y. Maeno and D. Gosh, J. Phys. Chem. Solids, 2007, 68, 347 CrossRef; G. Luo, S. Hess and L. R. Corruccini, Phys. Lett. A, 2001, 291, 306 CrossRef; P. Bonville, S. Petit, I. Mirebeau, J. Robert, E. Lhotel and C. Paulsen, J. Phys.: Condens. Matter, 2013, 25, 275601 CrossRef PubMed; L.-J. Chang, S. Onoda, Y. Su, Y.-J. Kao, K.-D. Tsuei, Y. Yasui, K. Kakurai and M. R. Lees, Nat. Commun., 2012, 3, 992 CrossRef PubMed; T. Fennell, P. P. Deen, A. R. Wildes, K. Schmalzl, D. Prabhakaran, A. T. Boothroyd, R. J. Aldus, D. F. McMorrow and S. T. Bramwell, Science, 2009, 326, 415 CrossRef PubMed; D. J. P. Morris, D. A. Tennant, S. A. Grigera, B. Klemke, C. Castelnovo, R. Moessner, C. Czternasty, M. Meissner, K. C. Rule, J.-U. Hoffmann, K. Kiefer, S. Gerischer, D. Slobinsky and R. S. Perry, Science, 2009, 326, 411 CrossRef PubMed.
- M. Veith, J. Chem. Soc., Dalton Trans., 2002, 2405 RSC.
- S. Eslava, M. McPartlin, R. Thomson, J. Rawson and D. S. Wright, Inorg. Chem., 2010, 49, 11532 CrossRef CAS PubMed.
- Y. Lv, J. Willkomm, M. Leskes, A. Steiner, T. C. King, L. Gan, E. Reisner, P. T. Wood and D. S. Wright, Chem. – Eur. J., 2012, 18, 11867 CrossRef CAS PubMed.
- Y. Lv, M. Yao, J. P. Holgado, T. Roth, A. Steiner, L. Gan, R. M. Lambert and D. S. Wright, RSC Adv., 2013, 3, 13659 RSC.
- Y. Lv, Z. Cai, D. Yan, C. Su, W. Li, W. Chen, Z. Ren, Y. Wei, O. Mi, C. Zhang and D. S. Wright, RSC Adv., 2016, 6, 57 RSC.
- C. Artner, S. Kronister, M. Czakler and U. Schubert, Eur. J. Inorg. Chem., 2014, 5596 CrossRef CAS PubMed.
- Y. Lv, W. Du, Y. Ren, Z. Cai, K. Yu, C. Zhang, Z. Chen and D. S. Wright, Inorg. Chem. Front., 2016, 3, 1119 RSC.
- D.-F. Lu, Z.-F. Hing, J. Xie, X.-J. Kong, L.-S. Long and L.-S. Zheng, Inorg. Chem., 2017, 56, 12186 CrossRef CAS PubMed.
- I. Tuvi-Arad, G. Alon and D. Avnir, CoSyM: Continuous Symmetry Measures, 2018, https://csm.ouproj.org.il/molecule Search PubMed.
- D. Parker, E. A. Suturina, I. Kuprov and N. F. Chilton, Acc. Chem. Res., 2020, 53, 1520 CrossRef CAS PubMed.
- N. F. Chilton, Annu. Rev. Mater. Res., 2022, 52, 79 CrossRef.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198 CrossRef CAS PubMed.
- A. Li Bassi, D. Cattaneo, V. Russo, C. E. Bottani, E. Barborini, T. Mazza, P. Piseri, P. Milani, F. O. Ernst, K. Wegner and S. E. Pratsinis, J. Appl. Phys., 2005, 98, 74305 CrossRef.
- D. M. De los Santos, J. Navas, T. Aguilar, A. Sánchez-Coronilla, C. Fernández-Lorenzo, R. Alcántara, J. C. Pinero, G. Blanco and J. Martín-Calleja, Beilstein J. Nanotechnol., 2015, 6, 605 CrossRef CAS PubMed.
- A. Coelho, Topas, v4.1, Bruker AXS GmbH, Karlsruhe, Germany, 2003 Search PubMed.
- M. C. Stennett, C. L. Freeman, A. S. Gandy and N. C. Hyatt, J. Solid State Chem., 2012, 192, 172 CrossRef CAS.
- N. Hübner and R. Gruehn, Z. Anorg. Allg. Chem., 1992, 616, 86 CrossRef.
- Y. Lv, J. Cheng, P. D. Matthews, J. P. Holgado, J. Willkomm, M. Leskes, A. Steiner, D. Fenske, T. C. King, P. T. Wood, L. Gan, R. M. Lambert and D. S. Wright, Dalton Trans., 2014, 43, 8679 RSC.
- F. Babonneau, A. Leaustic and J. Livage, MRS Proc., 1988, 121, 317 CrossRef CAS.
- L. Kumaresana, A. Prabhua, M. Palanichamy, E. Arumugam and V. Murugesan, J. Hazard. Mater., 2011, 186, 1183 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, CShM analysis, Raman spectra, AC susceptibility data, EDX/SEM results and details of the Rietveld refinement. CCDC 2283715–2283722. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02553e |
| This journal is © The Royal Society of Chemistry 2023 |