
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent applications of organic cages in sensing and separation processes in solution
Sonia
La Cognata
 and
Valeria
Amendola
*
and
Valeria
Amendola
*
Department of Chemistry, University of Pavia, Viale Taramelli 12, Pavia, I-27100, Italy. E-mail: valeria.amendola@unipv.it
First published on 25th October 2023
Abstract
Organic cages are three-dimensional polycyclic compounds of great interest in the scientific community due to their unique features, which generally include simple synthesis based on the dynamic covalent chemistry strategies, structural tunability and high selectivity. In this feature article, we present the advances over the last ten years in the application of organic cages as chemosensors or components in chemosensing devices for the determination of analytes (pollutants, analytes of biological interest) in complex aqueous media including wine, fruit juice, urine. Details on the recent applications of organic cages as selective (back-)extractants or masking agents for potential applications in relevant separation processes, such as the plutonium and uranium recovery by extraction, are also provided. Over the last ten years, organic cages with permanent porosity in the liquid and solid states have been highly appreciated as porous materials able to discriminate molecules of different sizes. These features, combined with good solvent processability and film-forming tendency, have proved useful in the fabrication of membranes for gas separation, solvent nanofiltration and water remediation processes. An overview of the recent applications of organic cages in membrane separation technologies is given.
 Sonia La Cognata | Sonia La Cognata is Research fellow at the University of Pavia, where she obtained her PhD in Chemical and Pharmaceutical Sciences and related Industrial Innovation in 2021 under the supervision of V. Amendola. During her PhD, Sonia's research focused on the application of cage-like molecules in molecular recognition, sensing and extraction processes. In the last three years, her research concerned the development of cage-based materials for the separation of gas mixtures within the project MOCA, sponsored by the Cariplo Foundation (grant no. 2019–2090). (Scopus ID: 57200437479). |
 Valeria Amendola | Valeria Amendola is Associate Professor at the Department of Chemistry of the University of Pavia, Italy. Her research interests are in the supramolecular chemistry field, and are especially focused on the development of molecular cages for the capturing of target pollutants in contaminated media. Since 2020, she is the Responsible of the research project MOCA (Metal Organic frameworks and organic CAges for highly selective gas separation membranes and heavy metal capture devices) sponsored by the Cariplo Foundation (grant no. 2019–2090). Her research activity has produced 95 publications with about 5400 total citations (h-index: 36). (Scopus ID: 6601991229). |
Introduction
Organic cages are a class of molecular hosts that exhibit a three-dimensional structure with an internal cavity enclosed between at least two organic building blocks, corresponding to the cage roof and floor, joined together by two or more linkers, representing the bars.1–3 This classification excludes monocyclic hosts like cyclodextrins or calix[n]arenes, and metal–organic cages or polyhedra (MOCs and MOPs, respectively)4–6 containing metal ions as constitutional components.Since the initial works by Park, Simmons and Lehn1b,c in the late 1960s, researchers have explored the design and synthesis of cages with varying degrees of flexibility and responsiveness. The development of novel organic cages, which generally benefits from dynamic covalent chemistry (DCC) strategies,7,8 has been raising significant interest in the past few years due to the potential application of this type of receptors in various fields, from supramolecular chemistry in solution (e.g. host–guest chemistry, sensing,9,10 mechanically interlocked molecules11 and molecular machines12) to catalysis and material science.3
The selectivity of organic cages in molecular recognition relies on the establishment of multiple non-covalent interactions (e.g. hydrogen or halogen bonding, π–π, ion–π, ionic, hydrophobic, metal–ligand) with the trapped guest, also operating in a synergistic manner.1 One of the key features of cage-like receptors is their structural diversity and tunability. By incorporating functional groups within the cage structure, these hosts can be modified to selectively trap and release specific guests in solution, making them useful for sensing and separation processes. First examples of cages, called cryptands,13–16 were synthesized and reported by Lehn in 1968–1969. These systems contained two tertiary amine bridgeheads linked together by three polyether chains. Similarly to crown ethers, cryptands showed a high selectivity for s-block cations, originating from the size matching between the cage cavity and the included spherical guest. Protonation of cages containing amino groups in the framework, e.g. azacryptands,14 enabled the successful recognition of anions in aqueous media through electrostatic and H-bonding interactions. As for cations, also for anionic or neutral guests, the selectivity relies on the complementary size and shape, and on the interaction sites matching between the guest and the cage cavity.1,14–17 For systems supposed to operate in aqueous matrixes, the cage design should take into account: pH effects, nature of potentially interfering species, solubility and hydrophobicity of both the cage and guest. In particular, for organic hosts that undergo multiple acid–base equilibria, the cavity shape can dramatically change under protonation, hence the binding affinity is strongly affected by the pH in solution. Transition metal cations present within the cavity, if contributing to the binding, can improve the selectivity in guest recognition and associated applications (e.g. chemosensing, extraction of pollutants from contaminated solutions). This is particularly evident in bistren cages, which can coordinate two Cu(II) cations and an ambidentate anion, as bridging ligand, within the cavity, leading to a stable cascade complex.1,14 With respect to MOCs and MOPs, metal ions here do not have any structural role: their function is mainly expressed in the binding of the guest inside the cavity or in the chemosensing process or, at least, in the synthesis of the cage structure as templating agents. Due to the importance of cavity effects on the cage selectivity, it is crucial that any structural change – e.g. made to convert the cage into an extracting agent or a chemosensor – does not affect the internal structure of the cavity and its binding capacity.
Over the last ten years, organic cages have also raised attention as nanoporous materials for sorption and separation processes. Despite the difficulty of obtaining porous materials from the assembling of simple molecules because of the frequent loss of porosity upon desolvation, numerous examples of organic cages with a high porosity in the solid (or liquid18) state are now available in the literature.19,20 These materials, which are generally called porous organic cages (POCs),3,19,21,22 have been proposed as selective sorbents, and have been investigated in membranes for e.g. gas separation and ion transport.23,24 Since the seminal works by Cooper,21 Mastalerz25 and Zhang,26 considerable progress has been made in the development of novel POCs, which are stable both physically and chemically,27 and present high permeability and selectivity. For applications in separation membranes,28 organic cages have been highly appreciated for the ability of discriminating molecules of different sizes, solvent processability29 and film-forming tendency. However, issues such as scaling up and robustness still pose challenges for the implementation of organic cages-based membranes beyond the lab-scale. With the aim of improving their stability in aqueous solution, organic cages have been utilized as monomers and building blocks30 in the fabrication of polymers and COFs, respectively.31 Crosslinked polycage membranes, for instance, impregnated with metal (Pd) nanoclusters, can effectively separate and catalytically decompose organic pollutants (e.g. azo dyes) in aqueous solution.32
In this feature article, we present the advances over the last ten years in the application of organic cages (i) as chemosensors or components in chemosensing devices for the determination of analytes (pollutants, analytes of biological interest) in complex aqueous media, such as wine, fruit juice, urine; (ii) as selective (back-)extractants or masking agents for potential applications in relevant processes, such as the plutonium and uranium recovery by extraction (PUREX). Finally, we will provide an overview of the uses of cages in membrane technologies for separation and solvent nanofiltration. The limitations and scope of these systems will be also discussed.
Chemosensors and sensing devices
Chemosensors are compounds capable of detecting specific analytes by undergoing a detectable change of their physico-chemical (e.g. optical) properties.33,34 Due to their high selectivity for target substrates, organic cages are excellent candidates for sensing. In addition, thanks to their good dispersibility and water solubility, organic cages have great potential for biological applications,35 such as bioimaging and therapeutics.9,36–38 For the conversion of a cage-like host into a chemosensor, a mechanism to signal the binding is required.39 In the case of fluorescent cages,40,41 this mechanism can involve e.g. photoinduced electron or energy transfer, (twisted)-intramolecular charge transfer, chelation enhanced fluorescence (CHEF), aggregation-induced/enhanced emission or quenching, encapsulation induced emission (EIE), etc.34,42–44 In several examples of the recent literature, the signalling process involves a combination of different and independent types of responses. There are also cases in which the signalling unit participates directly in the recognition process.A successful strategy to obtain a chemosensor consists in placing the signalling unit in close proximity to the cage cavity. This is obtained, for instance, by modifying the covalent skeleton of the cage, replacing one of the bars with a fluorophore or chromophore. This approach, which has been known for a long time, has been employed over the last ten years in the sensing of various types of analytes: pollutants, biomolecules (e.g. choline),45,46 explosives (e.g. picric acid,47 nitroaromatics48) both in solution (although rarely in water) and solid state.34,49
A few years ago, our group investigated a series of protonated bistren cryptands50 as potential hosts for the β-emitting 99TcO4− and its non-radioactive surrogate, ReO4−. The pertechnetate anion is the stable form of technetium in water. As a potential pollutant, this radioactive species causes concern because of long lifetime (>105 years), high solubility in water and environmental mobility.
Our study51 identified the hexaprotonated cryptand 1 (Fig. 1) as an excellent host for pertechnetate and perrhenate in acidic aqueous solution, as shown by the high 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 association constants: logK = 5.49(1) and 5.22(1) for 99TcO4− and ReO4−, respectively, measured at pH 2 in 0.1 mol L−1 CF3SO3Na (0.01 mol L−1 CF3SO3H). Anion inclusion into the cage cavity was confirmed by X-ray diffraction (XRD) studies on single crystals. The high stability of these inclusion complexes is attributable to the multiple H-bonding interactions (both direct and water-mediated), between the ammonium groups and the guest oxygen atoms, and to the size-matching between the anion and cavity.
:1 association constants: logK = 5.49(1) and 5.22(1) for 99TcO4− and ReO4−, respectively, measured at pH 2 in 0.1 mol L−1 CF3SO3Na (0.01 mol L−1 CF3SO3H). Anion inclusion into the cage cavity was confirmed by X-ray diffraction (XRD) studies on single crystals. The high stability of these inclusion complexes is attributable to the multiple H-bonding interactions (both direct and water-mediated), between the ammonium groups and the guest oxygen atoms, and to the size-matching between the anion and cavity.
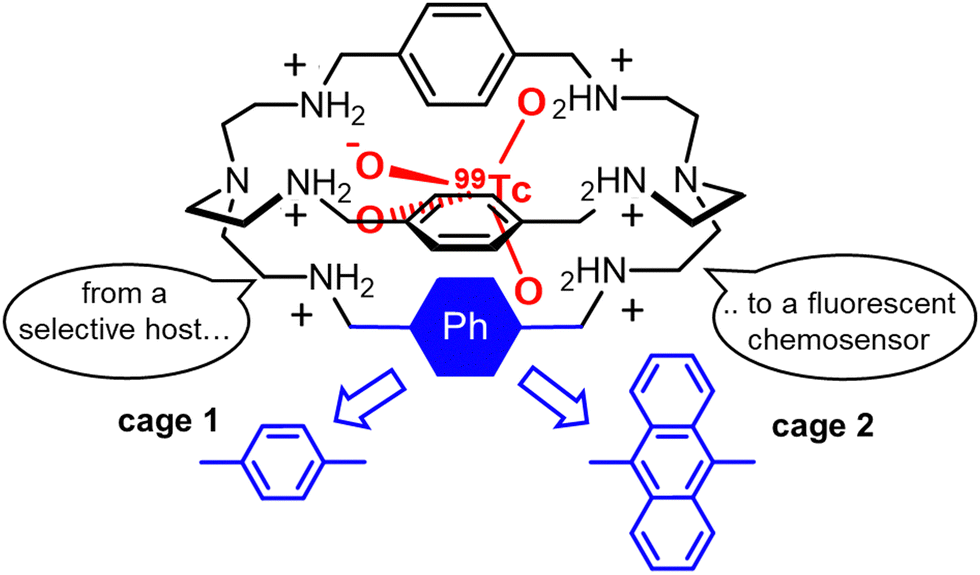 | ||
| Fig. 1 Schematized structures of the hexaprotonated bistren cages 1 and 2, employed in the capture and sensing of the pertechnetate anion in aqueous solution. | ||
The protonation of the six secondary amines is crucial for the recognition process. In fact, a partial protonation would promote the establishment of intramolecular H-bonds between the ammonium groups and the free amines on the same cage, which would result in a deformation of the cavity with a loss of selectivity for the guest.
The conversion of cage 1 into a fluorescent chemosensor for 99TcO4− was accomplished by replacing one of the 1,4-xylyl groups with a fluorophore, e.g. the 1,9-anthracenyl fragment52 (see cage 2 in Fig. 1). This structural change does not interfere with the affinity trend: anion binding studies actually confirmed the selectivity for 99TcO4−. At pH 2 in aqueous solution, the hexaprotonated 2 cage is fully fluorescent and displays the typical emission of the anthracenyl fluorophore (λmax = 425 nm). In these conditions, a significant quenching was observed on addition of either 99TcO4− or ReO4− to the cage. In contrast, no effects were observed with ClO4−, NO3−, Cl− and Br−, beside the formation of stable complexes between these anions and the cage. The quenching by 99TcO4− and ReO4− can be interpreted as a consequence of the higher basicity of these two anions (pKa = 0.32 and −1.25 for HTcO4 and HReO4, respectively) compared to the others investigated (pKa = −7, −9 for HCl, HBr, respectively). A higher anion basicity can actually favour proton transfer processes from the ammonium groups of the hexaprotonated 2 cage to the included anion, with subsequent quenching of the anthracene emission by photo-induced electron transfer from the resulting free amines.52
Even if cage 2 enables the detection of 99TcO4− at micromolar concentration in water, its application in a complex matrix is strongly limited by the presence of potential masking agents and competitors. This issue was addressed by our group,53 by replacing the anthracenyl unit with the chiral R-BINOL chromophore. With respect to chemosensor 2, the hexaprotonated chiroptical cage 3 (see Fig. 2) was also effective in presence of biological ingredients and sweeteners, absorbing in the UV-vis range of interest, and in presence of mixed competing anions. In particular, the cage proved useful in the sensing of ReO4−, as surrogate for 99TcO4−, in both the artificial urine medium and fruit juice.
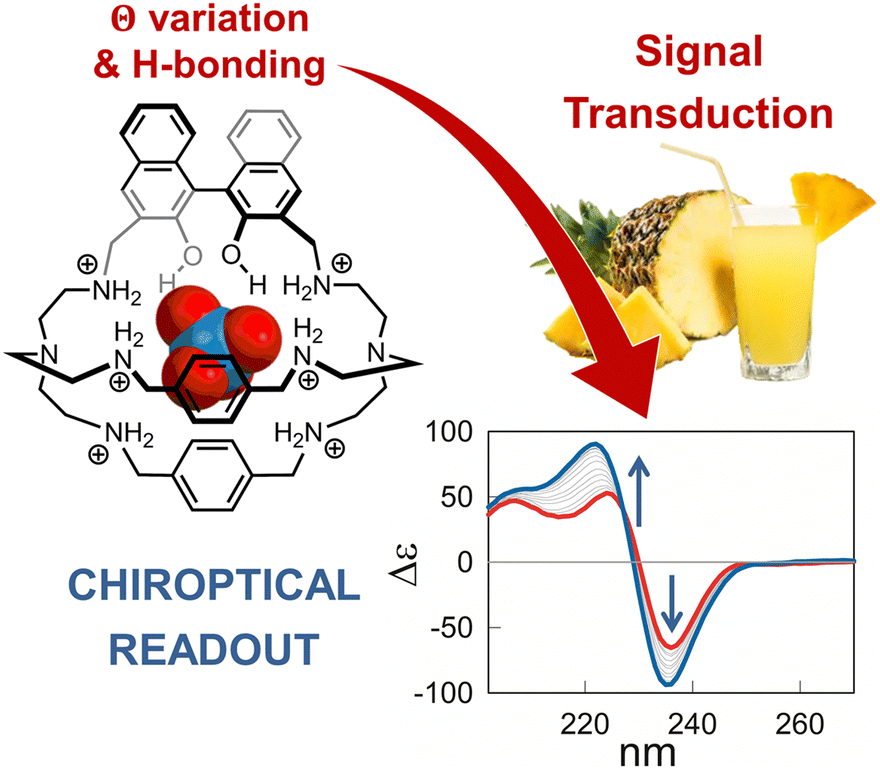 | ||
| Fig. 2 Schematized structure of the inclusion complex formed by the hexaprotonated cage 3 and perrhenate in aqueous solution. The cage was applied in the chiroptical sensing of this target anion (as surrogate for pertechnetate) in complex aqueous media, such as fruit juice and the artificial urine medium. Reproduced from Chem. Commun., 2022, 58, 3897.53 | ||
The chiroptical sensing mechanism resides in a change of the dihedral angle in R-BINOL, that accompanies the binding and produces a remarkable CD response. The participation of the BINOL O–H groups in the binding was confirmed by studies in silico and, experimentally, by comparing the binding constants of cage 3 with those determined for the methylated analogue host, 3-Me. Interestingly, BINOL methylation not only reduced the anion binding tendencies of the cage, but also cancelled its chiroptical response to the guest. Perrhenate encapsulation also promoted the partial quenching of the BINOL emission, therefore cage 3 provided a dual-channel response to the guest.
In the literature, there are dozens of examples of cascade complexes formed by bistren cages, incorporating two metal cations and a bridging ligand. In these systems, selectivity is determined by the good correspondence between the bite length of the anion (e.g. the −O⋯O− distance in −OOC–(CH2)n–COO−) and the separation between the metal centres in the cage cavity.54 For instance, the separation between Cu(II) ions in [Cu2(4)]4+ (Fig. 3) makes this host suitable for the recognition of terephthalate55 in aqueous solution at neutral pH. In these conditions, [Cu2(4)]4+ also shows strong affinity for trans,trans-muconate (t,t-MA), i.e. an urinary biomarker of benzene featuring the same bite length as terephthalate.56 This property was exploited by our group to develop a portable device for the sensing of t,t-MA in urine samples. The quantification of urinary t,t-MA, in fact, is one of the recommended methods for monitoring the occupational exposure to benzene, a well-known human carcinogen still employed in the industry.
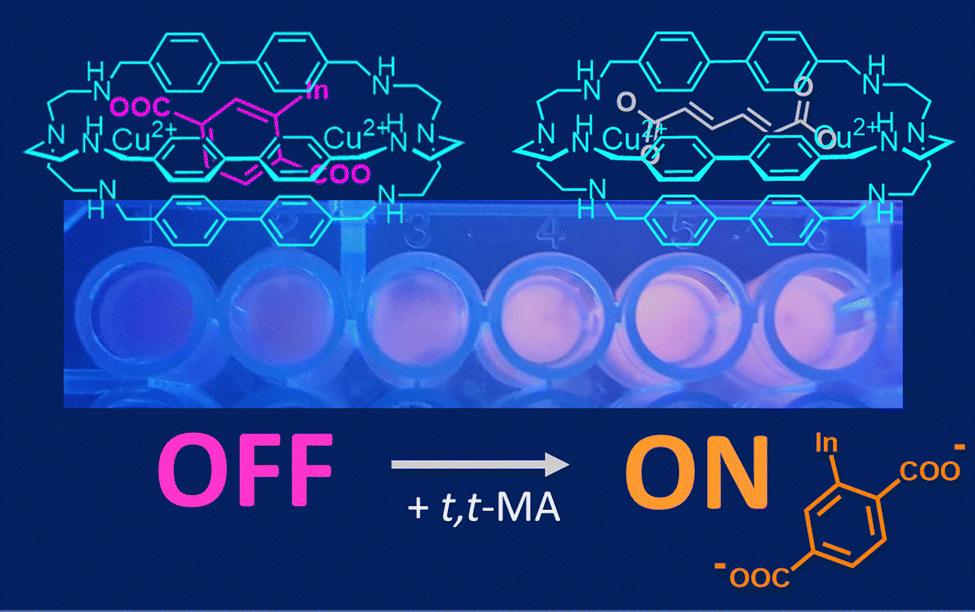 | ||
| Fig. 3 Chemosensing device, based on [Cu2(4)]4+ and 6-TAMRA (as selective host and fluorescent indicator, respectively), employed in the quantification of t,t-MA in spiked urine samples. Reproduced and adapted from New J. Chem., 2018, 42, 15460.56 | ||
For hosts like [Cu2(4)]4+, the conversion into a chemosensor is conveniently obtained using the indicator displacement approach. In particular, when an (e.g. coloured, fluorescent) indicator is bound to a cryptate, and a substrate with a higher affinity for the receptor is added to the solution, the indicator is displaced from the cavity and its release is accompanied by a detectable signal (colour/emission changes).34,57 When fluorescent dyes are employed, the displacement is typically monitored using a fluorimeter. However, if the dye is coloured, its displacement can be also monitored using a smartphone provided with a suitable colour-picking application.34b As a matter of fact, the RGB values recorded by the smartphone camera can be correlated to the analyte concentration. This approach was applied by our group in the determination of t,t-MA in real samples. In particular, a solution containing both the [Cu2(4)]4+ cryptate (2.3 nmol) and the coloured 6-TAMRA indicator (0.23 nmol) was absorbed on silica gel into the wells of an ELISA microplate (Fig. 3). After the addition of human urine spiked with t,t-MA into the wells, the microplate was exposed to a UV-lamp (λ = 366 nm, 16 W) and the analyte concentration was determined by recording the RGB values with a smartphone. A good correlation was found between the recorded R-index and the concentration of urinary t,t-MA at the occupational level. In collaboration with Martínez-Màñez,58 a nanoprobe based on gated mesoporous silica was also developed. In particular, silica nanoparticles were loaded with the sulforhodamine B dye (SRh B) and capped with the inclusion complex formed by [Cu2(4)]4+ and a terephthalic acid derivative, grafted onto the external surface of nanoparticles. In this system, the sensing mechanism was based on the displacement of terephthalate by urinary t,t-MA. The consequent emission enhancement due to pore opening and SRh B release was detected using a fluorimeter. Compared to the smartphone-based device described previously, this nanoprobe is less portable, but features a high sensitivity without pretreatment and a low limit of detection (0.017 mmol L−1) in spiked urine.
Over the last years, the sensing of biologically relevant dicarboxylates in real matrixes has raised considerable interest among the scientific community. Zonta59a recently reported a series of tris(pyridylmethyl)amine (TPMA)-based supramolecular cages, containing Zn(II) ions, able to selectively capture dicarboxylic acids in wine. These supramolecular systems can also amplify the circular dichroism (CD) signal of the encapsulated chiral guest (e.g.L-tartaric acid), thus providing a chiroptical response to the guest inclusion.
This peculiar feature was employed in the quantification of tartaric acid in wines, and in the discrimination of different matrixes through principal component analysis (PCA) of the raw CD data.59,60 Zonta showed that the stereodynamic TPMA units in a supramolecular cage are capable of inverting their helicity according to the length of the included guest, thus transducing the achiral information (i.e. the guest size) into different diastereomeric states with opposite chiroptical absorptions. This result paves the way to the development of new methods for the transduction of the chemical information into a chiroptical response.61 A similar behaviour was observed in a flexible and adaptive cage formed by two 1,3,5-triacylbenzene caps connected by three diarylthiourea-based arms.62 This organic cage showed a strong affinity for tricarboxylates. The guest encapsulation locked the cage in a helical conformation, producing a gearing-like chirality transmission mechanism. Hence, the cage became “chiral”, generating racemates upon guest inclusion.
Tetraphenylethene-based fluorescent derivatives (TPEs), with a cage-like covalent structure,63 have been widely employed in biological applications, such as for the detection of 3-nitrotyrosine (i.e. a crucial biomarker of chronic kidney disease) in serum.64 TPE-based systems are actually characterized by peculiar features: the propeller-like P/M rotational conformation of the phenyl rings which can be modified by the guest binding, and the excellent photophysical properties due to aggregation-induced emission (AIE) and circularly polarized luminescence (CPL).65 A TPE-based octacationic cage63 has recently been proposed for the recognition of polycyclic aromatic hydrocarbons and water-soluble dyes in water and organic solvents. The obtained host–guest complexes showed an improved AIE-based emission, a larger excitation–emission gap and a longer emission lifetime compared to the free cage. This molecular host was also employed in the recognition of enantiopure deoxynucleotides in water. The resulting cage-deoxynucleotide complexes showed excellent chiroptical (i.e. CD and CPL) properties thanks to the adaptive chirality of the cage induced by the guest binding. TPE-based triangular prismatic enantiomeric structures,66 with mirror symmetrical CD and CPL signals, showed enhanced fluorescence as a result of both chiral induction and the restriction of the phenylene rotation by geometrical constraints. Changes in fluorescence and CPL, from blue to yellow, were observed in the presence of trace acids (see Fig. 4). This feature makes these molecules suitable for the quantitative detection of acids in common organic solvents. Chiral emissive cages were also proposed as porous materials for the separation of organic racemates.67
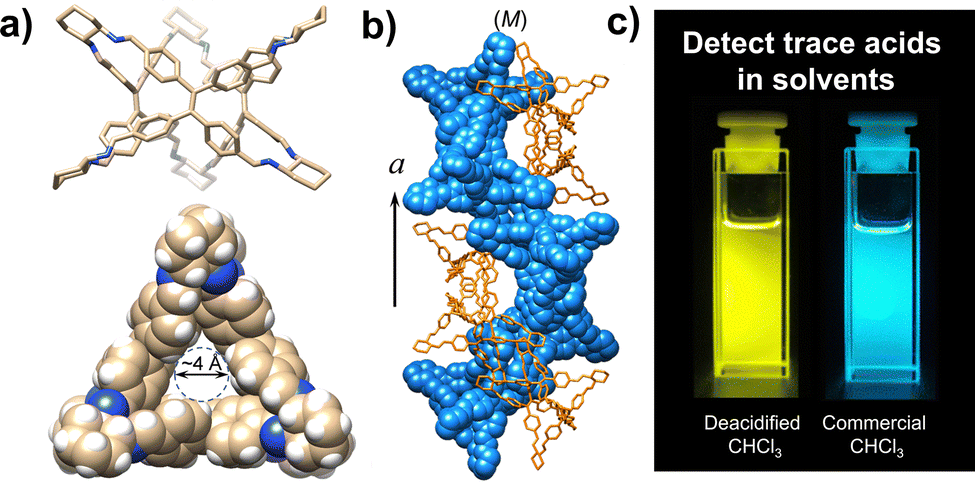 | ||
| Fig. 4 (a) Views of TPE-based triangular prismatic enantiomers, self-assembling into supramolecular double helices, (b) changes in the colour of the emission from blue to yellow in response to traces of acids in organic solvents, (c) reproduced and adapted with permission from J. Mater. Chem. C, 2022, 10, 15394.66 | ||
Extracting and masking agents
The selective encapsulation into a cage cavity allows for the separation of target species from mixtures also in presence of potential competitors. Organic cages can be also designed to selectively facilitate or prevent the transfer of a substrate between immiscible solvents.As schematized in Fig. 5, cage-like hosts can be used as extractants to transfer hydrophobic or hydrophilic substrates between two phases. For applications as masking agents, the formation of a stable complex with the cage is employed to stabilize the substrate in the original phase, thus preventing its extraction.
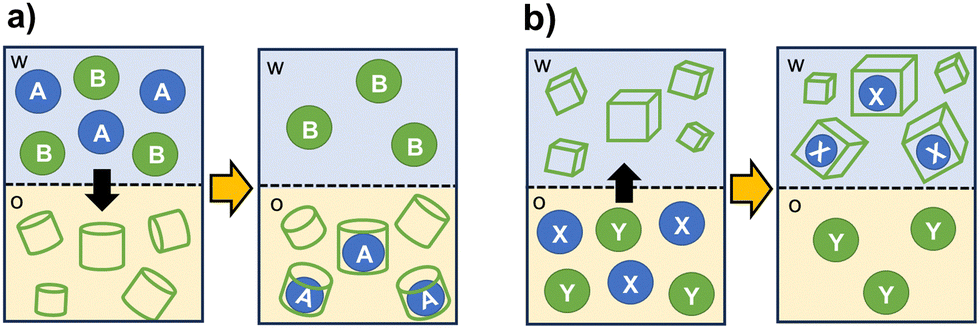 | ||
| Fig. 5 Schematized solvent extraction processes involving organic cages as extractants: (a) selective extraction of compound A from the aqueous phase (w), that contains potential competitors or masking agents (B), into the organic phase (o); (b) selective extraction of compound X from the organic phase (o), containing Y as competing species or masking agent, into the aqueous phase (w). | ||
In the recent years, one of the extraction processes in which cryptands have revealed remarkable potentialities is the separation of technetium and rhenium from aqueous mixtures.68 The selective extraction from contaminated matrixes (either liquid or solid), in which these elements are contained as oxoanions at low concentration, is very challenging. Within this context, cage-like hosts, such as the metal–organic tetrahedra by Nitschke,6 can give an important contribution. Gale69 recently showed that the solid–liquid (or liquid–liquid) extraction of either perrhenate or pertechnetate from aqueous solution is achievable with an organic phase containing a selective superphane in chloroform. Noticeably, the target anion was recognized and transferred into the receiving phase also in presence of abundant competing species (e.g. phosphate, nitrate, molybdate, etc.). The application of the superphane as extractant was successful even for low concentrations of the anion (ppb level) in the original phase.
Over the last decade, organic cages with a high selectivity for pertechnetate have also been investigated for potential applications in spent nuclear fuel treatment. 99Tc can indeed contaminate the final products of the nuclear fuel cycle, and interfere with uranium and plutonium extraction and reprocessing. As a consequence, strategies for the removal of technetium as pertechnetate, also based on the direct application of molecular hosts in the extraction process, are of great interest. However, for application in the PUREX process, selectivity is a necessary but not sufficient requirement: the cage must also be soluble and chemically resistant in aqueous nitric acid medium. In these conditions, the cage should immobilize pertechnetate in the aqueous phase, thus preventing its extraction into the organic solvent also in presence of potential competitors.70
A few years ago, the protonated cage 1 was tested by Tamain as a potential masking agent for technetium in nitric acid medium, in presence of uranium.70,71 Liquid–liquid extraction experiments were conducted on aqueous phases of 1 (40 mmol L−1 in 0.5 mol L−1 HNO3), containing Tc (or Re) and U (0.3 g L−1 and 30 g L−1, respectively). As receiving phase, a solution of N-methyl-N-octyl-(2-ethyl)hexanamide in isane† (MOEHA, 1.4 mol L−1) was employed. MOEHA is an extractant for U(VI) and Pu(IV), already utilized on spent nuclear fuel solutions. These experiments showed that pertechnetate encapsulation by the protonated cage 1 significantly reduced the extraction of the anion into isane. In particular, using 1 at 40 mmol L−1 concentration in the aqueous phase (0.5 mol L−1 HNO3), the distribution ratio for Tc (DTc, defined as the ratio of Tc concentration into the isane phase versus the concentration in the aqueous phase at equilibrium) resulted five-fold decreased, compared to the experiment conducted without the cage. The application in real conditions is however restricted by the poor solubility of the cage at concentrations of nitric acid >0.5 mol L−1. These solubility issues were addressed by incorporating hydrophilic groups in the cage framework. In collaboration with Marie,71 our group synthesized a series of soluble cryptands containing polar groups (e.g. hydroxyl groups or polyoxyethylene chains) on the p-xylyl spacers (see cages 5–7 in Fig. 6). These novel cages were tested in extraction processes, conducted in the conditions employed for 1 (aqueous phase: 0.3 g L−1 Tc, 30 g L−1 U in 0.5 mol L−1 HNO3; organic phase: 1.4 mol L−1 MOEHA in isane; T = 298 K). These tests confirmed the improved solubility of the cages in nitric acid, accompanied by an unvarying affinity for pertechnetate compared to 1. Interestingly, with cages 5–7 (100 mmol L−1 M in 0.5 mol L−1 HNO3), DTc resulted divided by a factor of fifteen compared to the reference without the cage, with an extraction efficiency improvement of 3 times compared to 1. These new ligands were also successfully tested as scrubbing agents, to selectively back extract technetium from an organic phase containing this element and U. For these experiments, Tc and U were firstly extracted in organic solvent (aqueous phase: 0.3 g L−1 Tc, 30 g L−1 U in 0.5 mol L−1 HNO3; organic phase: 1.4 mol L−1 MOEHA in isane; T = 298 K). The collected organic phases, containing both Tc and U, were then contacted with an aqueous solution (1 mol L−1 HNO3) of the cage, see the scheme in Fig. 6. The back-extraction of Tc was successfully achieved: with cages 6–7 (200 mmol L−1), DTc resulted twelve-fold decreased compared to the reference without the cage.
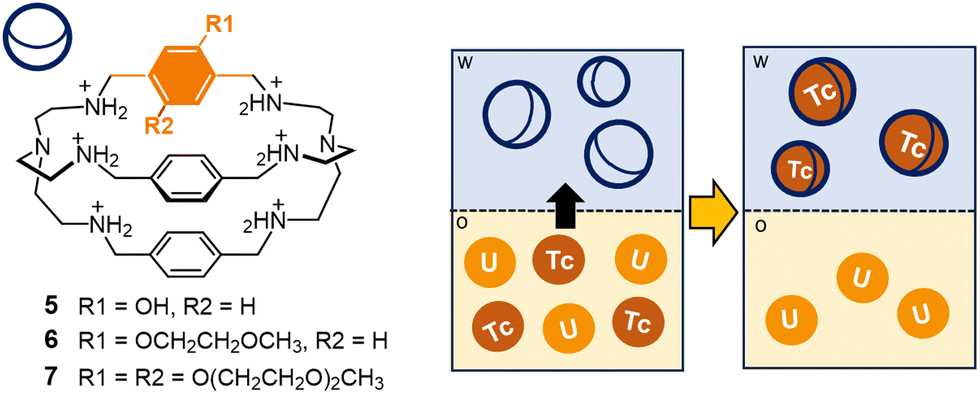 | ||
| Fig. 6 Schematized solvent extraction process involving cages 5–7 as extractants: in particular, the cages are employed in the selective back-extraction of Tc (as pertechnetate) from an organic phase containing U in isane.† Successful extraction of Tc was achieved using cages 6–7 as extractants into the aqueous phase (1 mol L−1 HNO3). | ||
Another relevant issue, addressable using cage-like receptors, is represented by the extraction of alkali halides from aqueous solutions or solid media. In particular, the ditopic macrobicyclic host by Delgado, containing a dibenzofuran spacer and an isophthalamide head unit, showed potential in the liquid–liquid extraction of potassium halides from neutral aqueous phases into chloroform.72 Among alkali halides, lithium salts are of special importance due to their applications in various areas, from pharmaceuticals to lithium-ion batteries.73 Due to the worldwide shortage of lithium, selective materials and hosts for the recovery and purification of lithium salts by extraction, especially of LiCl from used-batteries, are receiving considerable interest. However, the high hydration energies of both Li+ (−475 kJ mol−1) and Cl− (−340 kJ mol−1) make LiCl extraction very challenging. This issue can be overcome using ion-pair receptors with strong affinity for both Li+ and Cl− ions. A series of calix[4]pyrroles (see the example in Fig. 7) have been reported by Sessler as the first hosts capable of stabilizing LiCl in the solid state, without the participation of a water molecule between cation and anion.73 These systems can actually capture and extract the LiCl ion-pair from NaCl/KCl solid mixtures (containing 1% LiCl) with 100% selectivity. They can also extract LiCl from aqueous solutions into nitromethane (or chloroform).
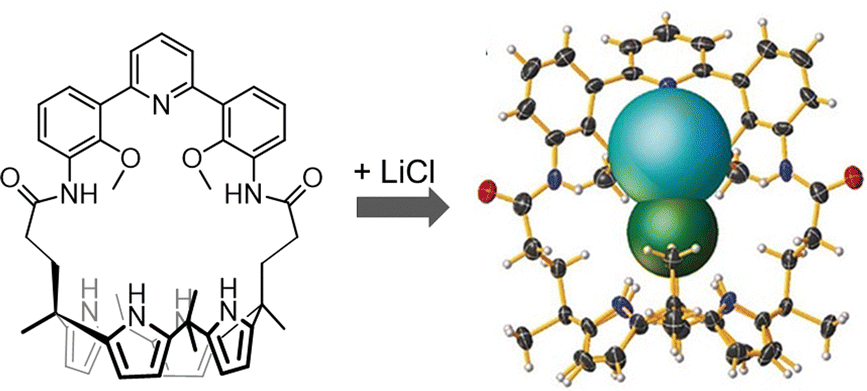 | ||
| Fig. 7 Calix[4]pyrrole employed by Sessler et al. in the selective binding extraction of the LiCl ion-pair. Reproduced and adapted with permission from Angew. Chem., Int. Ed., 2018, 57, 11924.73 | ||
The ditopic cage-like hosts, described previously, are capable of entrapping both the cation and anion, stabilizing the ion-pair into the organic phase. However, in other reported examples, only one of the two ions of the pair is effectively bound into the cage cavity.74 For instance, in a recent paper by Flood,75 a triazole-based organic cage was proposed as extractant for Cl− from water into dichloromethane (see 8 in Fig. 8). The high extraction efficiency for chloride is correlated to the selectivity of the cage for this guest. Chloride is actually encapsulated into the cage cavity and stabilized by nine CH⋯Cl− H-bonds: six from the 1,2,3-triazole units and three from the phenylenes. This is reflected in the high value of the association constant in dichloromethane: logK = 17. Significantly lower extraction efficiencies were found for more loosely bound anions, such as bromide, while no extraction was observed for iodide. Counter-ions are not entrapped into the cage cavity. Na+ is actually extracted with a higher efficiency than other alkali metal ions, due to the strong stability of the ion-pair between Na+ and the [8(Cl)]− complex. Interestingly, cage 8 also showed anticorrosion properties: a film of the cage, deposited on a sample of mild steel, could make Cl− inaccessible and protect steel from corrosion. This was demonstrated by immersing the coated steel sample into a brine solution for two weeks.75
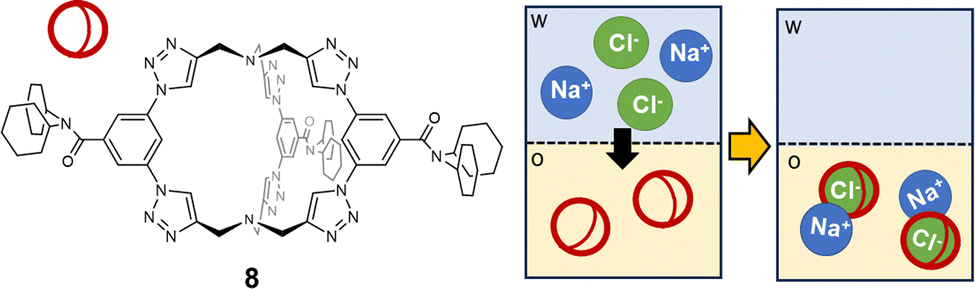 | ||
| Fig. 8 1,2,3-Triazole-based organic cage employed by Flood et al.75 as a selective extractant for chloride from water (w) into dichloromethane (o). In the organic solvent, chloride is encapsulated into the cage cavity, and the [8(Cl)]− complex forms a stable ion-pair with Na+ as counter-ion. | ||
In the systems by Sessler and Flood, the extraction efficiency was correlated to the binding affinity, and both depended on the stabilization of the anion by multiple H-bonding interactions. This strategy proved effective in the anti-Hofmeister extraction of spherical monocharged guests, such as halides. In the case of multicharged and highly hydrated anions, such as polycarboxylates, high extraction efficiency and selectivity are achievable using dimetallic cryptates. The modulation of spacers length in cryptands can actually improve the selectivity for the target guest, while the presence of hydrophobic groups on the host structure can enhance its solubility in non-polar solvents. Finally, the strong affinity of a dimetallic cryptate for the included guest can stabilize this latter into the organic phase, compensating for loss due to dehydration energy. As a proof of concept, the [Cu2(9)]4+ cryptate was employed by our group76 as an extractant for dicarboxylates from water into dichloromethane, using succinate as model anion (Fig. 9). The water-soluble analogue host, [Cu2(10)]4+, is actually known in the literature77 for its selectivity towards rigid dicarboxylates with two/three C-atoms between the –COO− groups, such as succinate and fumarate. [Cu2(10)]4+ can also distinguish fumarate from the geometric isomer, maleate. The sensing of fumarate in neutral aqueous solution was achieved using the indicator-displacement approach, with 5-carboxyfluorescein as the fluorescent indicator and [Cu2(10)]4+ as receptor (0.1 and 5 μmol L−1, respectively, in 0.050 mol L−1 HEPES buffer at pH 7).76 On the other hand, the [Cu2(9)]4+ cryptate, rendered soluble in dichloromethane by the hexyl-chains on the naphthyl spacers, showed good potentialities as extractant for the succinate anion from neutral water. For the solvent extraction studies, we employed solutions of the cryptate (0.2 mmol L−1) in dichloromethane, stirred in contact with an aqueous phase containing succinic acid (1 mmol L−1 in HEPES buffer, 0.050 mol L−1 at pH 7). As expected, succinate was quantitatively extracted from the aqueous phase into dichloromethane, as demonstrated by monitoring the changes in the UV-vis spectrum of the cryptate in the organic phase, before and after contacting with solutions of succinate (see Fig. 9). These spectral changes are consistent with the binding of succinate into the cryptate cavity (in 1:1 molar ratio), as bridging ligand between the copper ions. The extraction efficiency was further confirmed by following the decrease of succinate concentration in the aqueous phase by HPLC-UV and 1H-NMR.
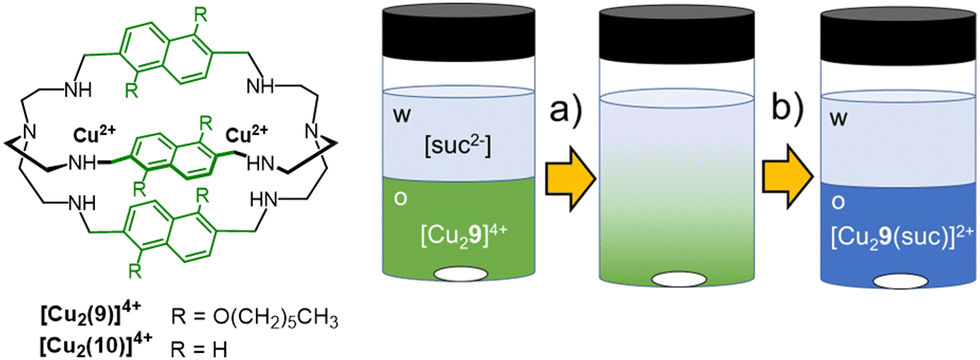 | ||
| Fig. 9 Schematized structures of the dicopper cryptates of cages 9 and 10, and application of [Cu2(9)]4+ as extractant for succinate76 from aqueous solutions (0.05 mol L−1 HEPES buffer, pH = 7) into dichloromethane. The transfer of succinate into the organic phase and its binding into the cryptate cavity are accompanied by a change of colour in the organic phase, from green to blue. | ||
Organic cages as adsorbents and membranes
Beside applications in solution, organic cages have recently been considered attractive in the condensed phase as porous liquids or solid materials for sorption processes. However, for applications as selective sorbents to be effective, the cages should display rigid framework, shape-persistent cavity and undergo non-efficient packing in the solid state. Porous organic cages (POCs) that meet these requirements have been proposed for the selective sorption of carbon dioxide,21,25,78,79 sulphur dioxide,80 hydrocarbons,81,82 perfluorocarbons83 (Fig. 10), iodine vapours,84,85 and as solid phases for gaseous mixtures separation.86–92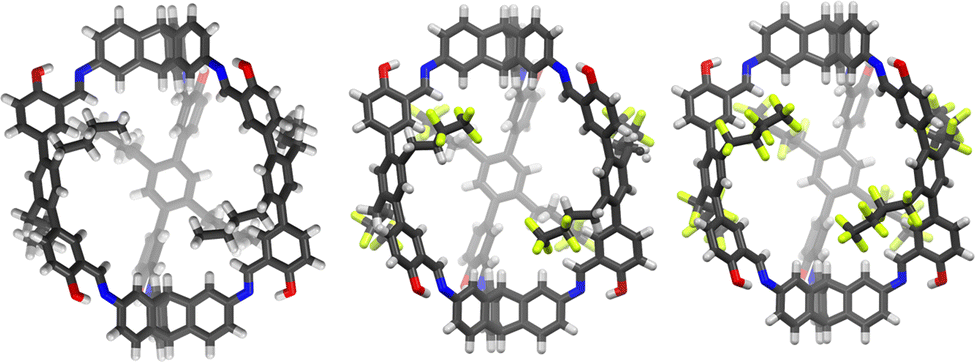 | ||
| Fig. 10 Single-crystal X-ray structures of the organic cages (CCDC codes 2150479, 2150480, 2150481) for the sorption of perfluorocarbons, proposed by Mastalerz et al. in Adv. Mater., 2022, 2202290.83 | ||
A solid phase able to both capture and sense CO2 was also obtained by polymerizing a TPE-based POC. However, when compared to polymer networks or covalent organic frameworks, solid phases constituted by assemblies of discrete molecules have the advantage of being more soluble and solution processable. Thanks to these features, cages can be evenly mixed with polymers, leading to mixed-matrix membranes (MMMs)93 by solution casting. MMMs – incorporating cages94–96 as filler materials into a polymer matrix (Fig. 11) – have been proposed to address the orthogonal (permeability vs. selectivity) issue of membrane separation technologies. These MMMs can actually combine the selectivity of fillers with the scalability and permeability of polymers, thus boosting the membrane separation performance.94 POC-based MMMs have been applied successfully in the size-sieving separation of relevant gaseous mixtures,87,95,96e.g. C2H2/CO2, CO2/N2, CO2/CH4, and in the capture of iodine vapours.85
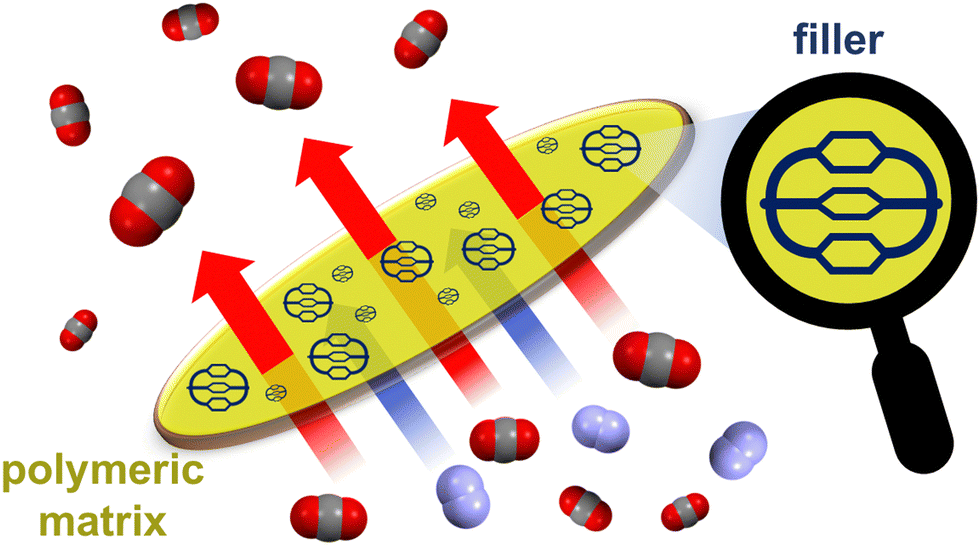 | ||
| Fig. 11 Schematized CO2/N2 separation using a mixed-matrix membrane, which contains an organic cage filler embedded into a polymeric matrix. | ||
In several instances, the cage molecules crystallize into the polymer matrix, thus producing a discrete particle phase inside the membrane.97 The lack of real integration between polymer and cages is unfortunately detrimental to the MMM performance. To overcome this risk, it is necessary to reduce the interactions between individual cages and promote the amorphous packing in the solid phase.
This goal was achieved by introducing different and/or bulky functionalities on the cage structures. Porous materials obtained by mixing POCs with different groups at the cage vertices showed greater solubility and solution-processability98 than the individual non-scrambled compounds. In addition, these molecularly-mixed polyimine POCs, embedded into a polymer matrix, could effectively produce homogenous MMMs with improved gas separation performance.
Solution-processable organic cages were deposited as thin-layer films on solid supports (Fig. 12), and employed in separation processes based on size, shape, and polarity. Films of chiral POCs (such as the CC3 cage by Cooper et al., shown in Fig. 12b), deposited on the internal walls of standard capillary columns, were applied as stationary phase in gas chromatography (GC) and electrochromatography for the separation of chiral alcohols.99,100 When porous solid supports (e.g. alumina, polyacrylonitrile) were concerned, thin-film membranes of the POCs were prepared by either spin-coating,101 or contra-diffusion growth,102 and solvothermal synthesis.103 Thin-film composite (TFC) membranes98 were also fabricated using mixtures of scrambled POCs and matrix-forming polymer (Matrimid), deposited onto crosslinked polyimide supports. These membranes were utilized for a range of molecular sieving applications, such as gas separation and organic solvent nanofiltration (OSN).104 As shown by Cooper in his seminal work on POCs, one of the peculiar features of these molecular materials is the possibility to change their porosity (and therefore their selectivity) by switching their crystalline phase between different polymorphs. This property was recently exploited to fabricate membranes for graded molecular sieving. In particular, crystalline membranes of scrambled POCs were grown on a polyacrylonitrile support by interfacial synthesis. The membrane pore size was then modified by changing the methanol/water ratio, and therefore by switching the POC between two crystalline polymorphs. These switchable membranes showed high permeances for a range of organic solvents and high rejection of organic dyes.
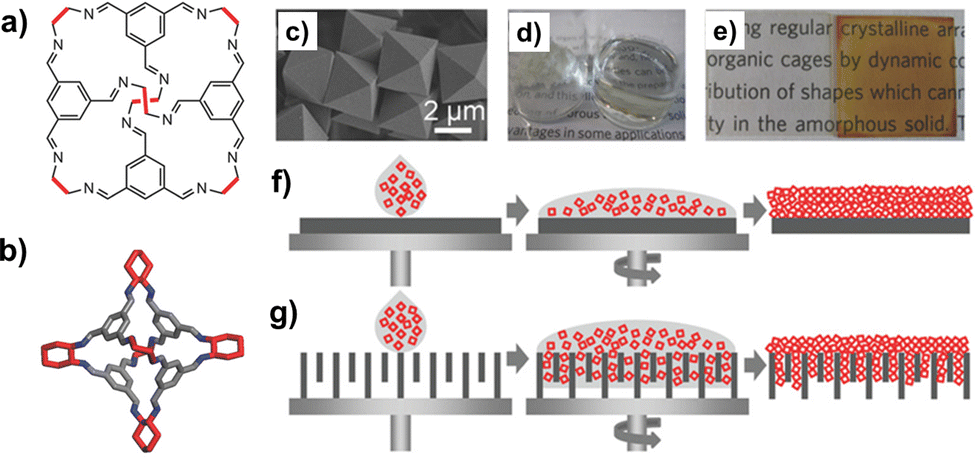 | ||
| Fig. 12 (a) Schematized structure of the POCs investigated by Cooper et al., including the CC3 cage (b) obtained by condensation of the 1,3,5-triformylbenzene with the R,R-1,2-diaminocyclohexane. (c) Single crystals of the CC3 cage; (d) vials containing CC3 as either solid or in solution; (e) pictures of CC3-films spin-coated on glass, before (left) and after (right) exposure to iodine. Schematized deposition of POCs on either (f) non-porous or (g) porous supports by spin-coating. Reproduced and adapted with permission from Adv. Mater., 2016, 28, 2629–2637.98 | ||
The good solution-processability and film-forming properties of cages can be utilized in the fabrication of dense films and self-standing membranes‡‡ of various thickness (between 30 nm and 100 μm). For instance, our group fabricated mechanically robust membranes of a chiral organic cage105 (Fig. 13a), simply by casting chloroform solutions of the cage in a Teflon dish. The obtained membranes resulted homogeneous, transparent, stable for months, and could be investigated through gas permeation and tensile tests.
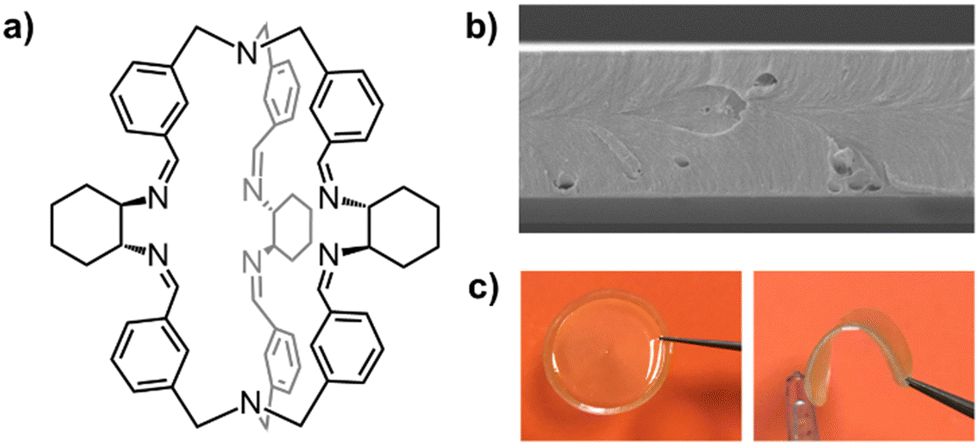 | ||
| Fig. 13 (a) Schematized structure of a chiral cage employed in the preparation of mechanically robust self-standing membranes with various thickness; (b) SEM image (25 K×) of the cross-section of a thin membrane obtained from the neat cage (thickness: 4.3 μm); (c) pictures of a ∼70 μm thick membrane of the same cage (20 mm diameter). Reproduced and adapted with permission from Chem. – Eur. J., 2023, e202301437.105 | ||
The deposition of selective cages as transparent films on glass or quartz slides105 opens perspectives for their application in sensing processes. For instance, in the seminal work by Cooper,98 the adsorption of iodine vapours by a thin layer of POC (100 nm), spin-coated on a glass slide, was evidenced by the development of a dark brown coloration (Fig. 12e), and of a strong band around 400 nm in the UV-vis spectrum. The intensity of this band is correlated to the quantity of iodine adsorbed in the film. More recently, Mastalerz et al.106 obtained thin films of a chemically stable acidochromic cage by drop-casting onto glass. Very interestingly, the cage films could be switched in colour and fluorescence, from pale yellow to fluorescent red (and vice versa), by exposing the glass to vapours of either hydrochloric acid or ammonia. Thanks to the chemical stability of the cage structure, this reversible colour switching could be repeated for several cycles without decomposition.
Outlook and conclusions
Cage-like molecules have been conceived by the supramolecular chemists for capturing, sensing and/or separating chemical species in solution. As shown in this review, the high selectivity for the target guest makes these receptors suitable for the sensing of analytes in complex aqueous media, or as extractants/masking agents in solvent extraction processes. Recent studies have also demonstrated the possibility of using cages in technological applications, including as anticorrosion agents for steel.75Around fifteen years ago, under the influence of material science before and then of membrane technology, organic cages also began to arouse interest as nanoporous materials for gas sorption and separation processes. Experiencing a range of new challenges – primarily related to maintaining the material's porosity after solvent removal – the design of porous organic cages has followed an independent path from that of the traditional host–guest chemistry. Over the last decade, the growing interest in membrane separation technologies has boosted the application of porous cages as fillers in MMMs, or as film-forming materials for self-supporting/composite membranes. Although for now this film-forming ability of cages has been little exploited in the development of chemosensing devices,106 our belief is that the widespread awareness of the potential of these molecular systems will foster their use in point-of-care diagnostics (e.g. as functional materials in portable/miniaturized sensing devices). The utilization of high-throughput technologies107 in the discovery of nanoporous cages will surely speed up their development, boosting their implementation in solvents nanofiltration, desalination, catalysis,108,109 ion transport and exchange,102,110 technologies for water remediation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Cariplo Foundation (MOCA project, Bando Economia Circolare 2019, grant no. 2019–2090) is gratefully acknowledged for sponsorship. We also acknowledge the support from the Ministero dell’Università e della Ricerca (MUR) and the University of Pavia through the program “Dipartimenti di Eccellenza 2023–2027”.Notes and references
- (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, Germany, 1995 CrossRef; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, 2nd edn, Wiley, Chichester, UK, 2009 Search PubMed; (c) C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS; (d) B. Dietrich, J.-M. Lehn and J.-P. Sauvage, Tetrahedron Lett., 1969, 10, 2889–2892 CrossRef.
- (a) G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef PubMed; (b) K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed.
- X. Yang, Z. Ullah, J. F. Stoddart and C. T. Yavuz, Chem. Rev., 2023, 123, 4602–4634 CrossRef CAS PubMed.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- Z. Yang, F. Esteve, C. Antheaume and J. M. Lehn, Chem. Sci., 2023, 6631–6642 RSC.
- D. A. Mc Naughton, M. Fares, G. Picci, P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2021, 427, 213573 CrossRef CAS.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2018, 119, 322–477 CrossRef PubMed.
- (a) S. La Cognata, A. Miljkovic, R. Mobili, G. Bergamaschi and V. Amendola, ChemPlusChem, 2020, 85, 1145–1155 CrossRef CAS PubMed; (b) A. B. Aletti, A. Miljkovic, L. Toma, R. Bruno, D. Armentano, T. Gunnlaugsson, G. Bergamaschi and V. Amendola, J. Org. Chem., 2019, 84(7), 4221–4228 CrossRef CAS PubMed; (c) V. Amendola, A. Miljkovic, L. Legnani, L. Toma, D. Dondi and S. Lazzaroni, Inorg. Chem., 2018, 57, 3540–3547 CrossRef CAS PubMed.
- M. Zhang, X. Yan, F. Huang, Z. Niu and H. W. Gibson, Acc. Chem. Res., 2014, 47, 1995–2005 CrossRef CAS PubMed.
- J. M. Lehn, Acc. Chem. Res., 1978, 11(2), 49–57 CrossRef CAS.
- (a) L. Fabbrizzi, Cryptands and Cryptates, World Scientific Publishing Europe Ltd, London, 2018 CrossRef; (b) G. Alibrandi, V. Amendola, G. Bergamaschi, L. Fabbrizzi and M. Licchelli, Org. Biomol. Chem., 2015, 13, 3510–3524 RSC.
- V. Amendola, G. Bergamaschi and A. Miljkovic, Supramol. Chem., 2018, 30, 236–242 CrossRef CAS.
- (a) S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 RSC; (b) K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- V. Amendola, M. Boiocchi, L. Fabbrizzi and N. Fusco, Eur. J. Org. Chem., 2011, 6434–6444 CrossRef CAS.
- B. D. Egleston, A. Mroz, K. E. Jelfs and R. L. Greenaway, Chem. Sci., 2022, 13, 5042–5054 RSC.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- T. D. Bennett, F. X. Coudert, S. L. James and A. I. Cooper, Nat. Mater., 2021, 20, 1179–1187 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- T. Kunde, T. Pausch and B. M. Schmidt, Eur. J. Org. Chem., 2021, 5844–5856 CrossRef CAS.
- D. Hu, J. Zhang and M. Liu, Chem. Commun., 2022, 58, 11333–11346 RSC.
- S. Yu, M. Yang, Y. Liu and M. Liu, Mater. Chem. Front., 2023, 7, 3560–3575 RSC.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed.
- Y. Jin, B. A. Voss, R. D. Noble and W. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6348–6351 CrossRef CAS PubMed.
- A. S. Bhat, S. M. Elbert, W. S. Zhang, F. Rominger, M. Dieckmann, R. R. Schröder and M. Mastalerz, Angew. Chem., Int. Ed., 2019, 58, 8819–8823 CrossRef CAS PubMed.
- H. Wang, M. Wang, X. Liang, J. Yuan, H. Yang, S. Wang, Y. Ren, H. Wu, F. Pan and Z. Jiang, Chem. Soc. Rev., 2021, 50, 5468–5516 RSC.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- Q. H. Zhu, G. H. Zhang, L. Zhang, S. L. Wang, J. Fu, Y. H. Wang, L. Ma, L. He and G. H. Tao, J. Am. Chem. Soc., 2023, 145, 6177–6183 CrossRef CAS PubMed.
- C. Liu, Y. Jin, Z. Yu, L. Gong, H. Wang, B. Yu, W. Zhang and J. Jiang, J. Am. Chem. Soc., 2022, 144, 12390–12399 CrossRef CAS PubMed.
- X. Li, W. Lin, V. Sharma, R. Gorecki, M. Ghosh, B. A. Moosa, S. Aristizabal, S. Hong, N. M. Khashab and S. P. Nunes, Nat. Commun., 2023, 14, 3112 CrossRef CAS PubMed.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS PubMed.
- (a) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed; (b) K. Cantrell, M. M. Erenas, I. de Orbe-Paya and L. F. Capitan-Vallvey, Anal. Chem., 2010, 82, 531–542 CrossRef CAS PubMed.
- L. Tapia, I. Alfonso and J. Solà, Org. Biomol. Chem., 2021, 19, 9527–9540 RSC.
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- A. B. Aletti, D. M. Gillen and T. Gunnlaugsson, Coord. Chem. Rev., 2018, 354, 98–120 CrossRef CAS.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2021, 50, 2737–2763 RSC.
- K. Hua, Y. Y. An, Y. Y. Wang and Y. F. Han, Chem. – Eur. J., 2020, 26, 7190–7193 CrossRef CAS PubMed.
- S. S. R. Namashivaya, A. S. Oshchepkov, H. Ding, S. Förster, V. N. Khrustalev and E. A. Kataev, Org. Lett., 2019, 21, 8746–8750 CrossRef CAS PubMed.
- (a) E. A. Kataev, Chem. Commun., 2023, 59, 1717–1727 RSC; (b) B. S. Morozov, S. S. R. Namashivaya, M. A. Zakharko, A. S. Oshchepkov and E. A. Kataev, ChemistryOpen, 2020, 9, 171–175 CrossRef CAS PubMed.
- B. Daly, J. Ling and A. P. De Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- H. Niu, J. Liu, H. M. O’Connor, T. Gunnlaugsson, T. D. James and H. Zhang, Chem. Soc. Rev., 2023, 52, 2322–2357 RSC.
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef PubMed.
- N. Fantozzi, R. Pétuya, A. Insuasty, G. S. Vallverdu, E. Genin, D. Bégué, A. Martinez, S. Pinet and I. Gosse, Org. Lett., 2023, 25, 2444–2449 CrossRef CAS PubMed.
- Ł. Szyszka, M. Górecki, P. Cmoch and S. Jarosz, J. Org. Chem., 2021, 86, 5129–5141 CrossRef PubMed.
- K. Acharyya and P. S. Mukherjee, Chem. Commun., 2014, 50, 15788–15791 RSC.
- K. Acharyya and P. S. Mukherjee, Chem. – Eur. J., 2015, 21, 6823–6831 CrossRef CAS PubMed.
- V. Kumar, H. Kim, B. Pandey, T. D. James, J. Yoon and E. V. Anslyn, Chem. Soc. Rev., 2022, 52, 663–704 RSC.
- V. Amendola, G. Alberti, G. Bergamaschi, R. Biesuz, M. Boiocchi, S. Ferrito and F. P. Schmidtchen, Eur. J. Inorg. Chem., 2012, 3410–3417 CrossRef CAS.
- R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772–9776 CrossRef CAS PubMed.
- V. Amendola, G. Bergamaschi, M. Boiocchi, R. Alberto and H. Braband, Chem. Sci., 2014, 5, 1820–1826 RSC.
- R. Mobili, G. Preda, S. La Cognata, L. Toma, D. Pasini and V. Amendola, Chem. Commun., 2022, 58, 3897–3900 RSC.
- A. Miljkovic, S. La Cognata, G. Bergamaschi, M. Freccero, A. Poggi and V. Amendola, Molecules, 2020, 25, 1733–1744 CrossRef CAS PubMed.
- V. Amendola, M. Bonizzoni, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, F. Sancenón and A. Taglietti, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS.
- D. Merli, S. La Cognata, F. Balduzzi, A. Miljkovic, L. Toma and V. Amendola, New J. Chem., 2018, 42, 15460–15465 RSC.
- P. Mateus, R. Delgado, V. André and M. Teresa Duarte, Org. Biomol. Chem., 2015, 13, 834–842 RSC.
- M. Domínguez, J. F. Blandez, B. Lozano-Torres, C. de la Torre, M. Licchelli, C. Mangano, V. Amendola, F. Sancenón and R. Martínez-Máñez, Chem. – Eur. J., 2021, 27, 1306–1310 CrossRef PubMed.
- (a) F. Begato, G. Licini and C. Zonta, Angew. Chem., Int. Ed., 2023, e202311153 Search PubMed; (b) C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3523–3528 RSC.
- C. Bravin, G. Mason, G. Licini and C. Zonta, J. Am. Chem. Soc., 2019, 141, 11963–11969 CrossRef CAS PubMed.
- F. Begato, R. Penasa, G. Licini and C. Zonta, ACS Sens., 2022, 7, 1390–1394 CrossRef CAS PubMed.
- Q. P. Hu, H. Zhou, T. Y. Huang, Y. F. Ao, D. X. Wang and Q. Q. Wang, J. Am. Chem. Soc., 2022, 144, 6180–6184 CrossRef CAS PubMed.
- H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., Int. Ed., 2020, 59, 10101–10110 CrossRef CAS PubMed.
- (a) W. T. Dou, C. Y. Yang, L. R. Hu, B. Song, T. Jin, P. P. Jia, X. Ji, F. Zheng, H. B. Yang and L. Xu, ACS Mater. Lett., 2023, 5, 1061–1082 CrossRef CAS; (b) S. La Cognata, D. Armentano, N. Marchesi, P. Grisoli, A. Pascale, M. Kieffer, A. Taglietti, A. P. Davis and V. Amendola, Chemistry, 2022, 4, 855–864 CrossRef CAS.
- H. T. Feng, Y. X. Yuan, J. Bin Xiong, Y. S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- L. Bian, M. Tang, J. Liu, Y. Liang, L. Wu and Z. Liu, J. Mater. Chem. C, 2022, 10, 15394–15399 RSC.
- Y. L. Sun, Z. Wang, H. Ma, Q. P. Zhang, B. Bin Yang, X. Meng, Y. Zhang and C. Zhang, Chem. Commun., 2022, 59, 302–305 RSC.
- D. Banerjee, D. Kim, M. J. Schweiger, A. A. Kruger and P. K. Thallapally, Chem. Soc. Rev., 2016, 45, 2724–2739 RSC.
- W. Zhou, A. Li, P. A. Gale and Q. He, Cell Rep. Phys. Sci., 2022, 3, 100875 CrossRef CAS.
- A. Thevenet, C. Marie, C. Tamain, V. Amendola, A. Miljkovic, D. Guillaumont, N. Boubals and P. Guilbaud, Dalton Trans., 2020, 49, 1446–1455 RSC.
- A. Thevenet, A. Miljkovic, S. La Cognata, C. Marie, C. Tamain, N. Boubals, C. Mangano, V. Amendola and P. Guilbaud, Dalton Trans., 2021, 50, 1620–1630 RSC.
- N. Bernier, S. Carvalho, F. Li, R. Delgado and V. Félix, J. Org. Chem., 2009, 74, 4819–4827 CrossRef CAS PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- A. Brown, T. Bunchuay, C. G. Crane, N. G. White, A. L. Thompson and P. D. Beer, Chem. – Eur. J., 2018, 24, 10434–10442 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
- S. La Cognata, R. Mobili, F. Merlo, A. Speltini, M. Boiocchi, T. Recca, L. J. Maher and V. Amendola, ACS Omega, 2020, 5, 26573–26582 CrossRef CAS PubMed.
- G. Y. Xie, L. Jiang and T. B. Lu, Dalton Trans., 2013, 42, 14092–14099 RSC.
- Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS PubMed.
- T. Kunde, E. Nieland, H. V. Schröder, C. A. Schalley and B. M. Schmidt, Chem. Commun., 2020, 56, 4761–4764 RSC.
- E. Martínez-Ahumada, D. He, V. Berryman, A. López-Olvera, M. Hernandez, V. Jancik, V. Martis, M. A. Vera, E. Lima, D. J. Parker, A. I. Cooper, I. A. Ibarra and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 17556–17563 CrossRef PubMed.
- Z. Wang, N. Sikdar, S. Q. Wang, X. Li, M. Yu, X. H. Bu, Z. Chang, X. Zou, Y. Chen, P. Cheng, K. Yu, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 9408–9414 CrossRef CAS PubMed.
- S. Fang, M. Wang, Y. Wu, Q. H. Guo, E. Li, H. Li and F. Huang, Chem. Sci., 2022, 13, 6254–6261 RSC.
- K. Tian, S. M. Elbert, X. Y. Hu, T. Kirschbaum, W. S. Zhang, F. Rominger, R. R. Schröder and M. Mastalerz, Adv. Mater., 2022, 34, 2202290 CrossRef CAS PubMed.
- D. Luo, Y. He, J. Tian, J. L. Sessler and X. Chi, J. Am. Chem. Soc., 2022, 144, 113–117 CrossRef CAS PubMed.
- X. J. Zhao, S. H. Liu and J. K. Sun, Chem. – Eur. J., 2022, 28, e2022011 CrossRef PubMed.
- S. La Cognata, R. Mobili, C. Milanese, M. Boiocchi, M. Gaboardi, D. Armentano, J. C. Jansen, M. Monteleone, A. R. Antonangelo, M. Carta and V. Amendola, Chem. – Eur. J., 2022, 28, e202201631 CrossRef CAS PubMed.
- K. Krishnan, J. M. Crawford, P. K. Thallapally and M. A. Carreon, Ind. Eng. Chem. Res., 2022, 61, 10547–10553 CrossRef CAS.
- J. Liu, L. Li, Z. Zhao and F. Yu, ACS Sustainable Chem. Eng., 2021, 9, 14890–14899 CrossRef CAS.
- M. Liu, L. Zhang, M. A. Little, V. Kapil, M. Ceriotti, S. Yang, L. Ding, D. L. Holden, R. Balderas-Xicohténcatl, D. He, R. Clowes, S. Y. Chong, G. Schütz, L. Chen, M. Hirscher and A. I. Cooper, Science, 2019, 366, 613–620 CrossRef CAS PubMed.
- K. Su, W. Wang, S. Du, C. Ji and D. Yuan, Nat. Commun., 2021, 12, 2–8 CrossRef PubMed.
- R. Epsztein, R. M. Du Chanois, C. L. Ritt, A. Noy and M. Elimelech, Nat. Nanotechnol., 2020, 15, 426–436 CrossRef CAS PubMed.
- J. M. Lucero and M. A. Carreon, ACS Appl. Mater. Interfaces, 2020, 12, 32182–32188 CrossRef CAS PubMed.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292–9310 CrossRef CAS PubMed.
- M. Monteleone, R. Mobili, C. Milanese, E. Esposito, A. Fuoco, S. La Cognata, V. Amendola and J. C. Jansen, Molecules, 2021, 26, 1–16 CrossRef PubMed.
- Q. Zhang, H. Li, S. Chen, J. Duan and W. Jin, J. Membr. Sci., 2020, 611, 118288 CrossRef CAS.
- X. Kong and J. Liu, J. Phys. Chem. C, 2019, 123, 15113–15121 CrossRef CAS.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253–1256 CrossRef CAS PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Adv. Mater., 2016, 28, 2629–2637 CrossRef CAS PubMed.
- Z. M. Wang, Y. Y. Cui, C. X. Yang and X. P. Yan, ACS Appl. Nano Mater., 2020, 3, 479–485 CrossRef CAS.
- D. X. Cui, Y. Geng, J. N. Kou, G. G. Shan, C. Y. Sun, K. H. Zhang, X. L. Wang and Z. M. Su, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- G. Zhu, F. Zhang, M. P. Rivera, X. Hu, G. Zhang, C. W. Jones and R. P. Lively, Angew. Chem., Int. Ed., 2019, 58, 2638–2643 CrossRef CAS PubMed.
- T. Xu, B. Wu, Y. Li, Y. Zhu, F. Sheng, L. Ge, X. Li and T. Xu, Ind. Eng. Chem. Res., 2023, 62, 717–724 CrossRef CAS.
- M. A. Carreon, ACS Mater. Lett., 2022, 4, 868–873 CrossRef CAS.
- A. He, Z. Jiang, Y. Wu, H. Hussain, J. Rawle, M. E. Briggs, M. A. Little, A. G. Livingston and A. I. Cooper, Nat. Mater., 2022, 21, 463–470 CrossRef CAS PubMed.
- R. Mobili, S. La Cognata, M. Marcello, M. Longo, A. Fuoco, S. A. Serapian, B. Vigani, C. Milanese, D. Armentano, J. C. Jansen and V. Amendola, Chem. – Eur. J., 2023, e202301437 CrossRef CAS PubMed.
- P. E. Alexandre, W. S. Zhang, F. Rominger, S. M. Elbert, R. R. Schröder and M. Mastalerz, Angew. Chem., Int. Ed., 2020, 59, 19675–19679 CrossRef CAS PubMed.
- R. L. Greenaway, V. Santolini, M. J. Bennison, B. M. Alston, C. J. Pugh, M. A. Little, M. Miklitz, E. G. B. Eden-Rump, R. Clowes, A. Shakil, H. J. Cuthbertson, H. Armstrong, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- P. Bhandari and P. S. Mukherjee, ACS Catal., 2023, 13, 6126–6143 CrossRef CAS.
- N. Luo, Y. F. Ao, D. X. Wang and Q. Q. Wang, Angew. Chem., Int. Ed., 2021, 60, 20650–20655 CrossRef CAS PubMed.
- H. Li, Y. Huang, Y. Zhang, X. Zhang, L. Zhao, W. Bao, X. Cai, K. Zhang, H. Zhao, B. Yi, L. Su, A. K. Cheetham, S. Jiang and J. Xie, Nano Lett., 2022, 22, 2030–2037 CrossRef CAS PubMed.
Footnotes |
| † Isoparaffinic solvent. |
| ‡ Without the aid of solid support nor matrix-forming polymers. |
| This journal is © The Royal Society of Chemistry 2023 |