
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid single crystal growth via guest displacement from host–guest complexes†
Mikayla L.
Horvath
 a,
Caylee E.
Jumbelic
a,
Rosemarie A.
Burynski
a,
M. Brody
Mistrot
a,
Robert D.
Pike
*b,
Brian J.
Smith
*a and
Hasan
Arslan
*a
a,
Caylee E.
Jumbelic
a,
Rosemarie A.
Burynski
a,
M. Brody
Mistrot
a,
Robert D.
Pike
*b,
Brian J.
Smith
*a and
Hasan
Arslan
*a
aDepartment of Chemistry, Bucknell University, Lewisburg, PA 17837, USA. E-mail: hasan.arslan@bucknell.edu; brian.smith@bucknell.edu
bDepartment of Chemistry, William & Mary, Williamsburg, VA 23185, USA. E-mail: rdpike@wm.edu
First published on 14th September 2023
Abstract
The preparation of single crystals of sparingly soluble polycyclic aromatic compounds in MeCN is facilitated by solubilizing ionic host–guest complexation under otherwise poor solvent conditions. The guest is then crystallized within minutes through controlled guest displacement from the host via competitor equilibria, or over days through direct crystallization of the host–guest complex itself.
Managing competing intermolecular interactions is central to successful molecular crystal growth and the preparation of high-quality single crystals. There are a multitude of crystal growth mechanisms, and attractive intermolecular forces, such as hydrogen bonding, halogen bonding, and π–π interactions govern many such processes.1–3 For planar compounds with large π surfaces known as polycyclic aromatic compounds (PACs),4,5 characterization of the crystalline form is a powerful technique to determine structural properties. Although there are crystallization approaches directly from the solid form, such as melt crystallization and sublimation, solution crystal growth remains the most common method for large PACs, due to their high melting and sublimation temperatures.6 However, PACs are also only sparingly soluble in many solvents,7 and as a result, crystal formation can pose a challenge if the compound cannot be taken into solution at practical volumes and concentrations.
Traditional solution-based single crystal growth methods rely on bringing a dissolved species to supersaturation beyond the metastable zone by transforming the solvent environment, such as by evaporation, heating/cooling, or slow diffusion of an antisolvent through vapor or layering (Fig. 1a–d).1,6 For sparingly soluble PACs, preparation of an initial concentrated stock is plagued by low solubility, and heat sensitive compounds can limit the use of significant temperature increases. Herein, to complement these existing methods we describe a single crystal growth approach that instead harnesses low solubility by utilizing reversible host–guest interactions and competitive guest displacement (Fig. 1e).
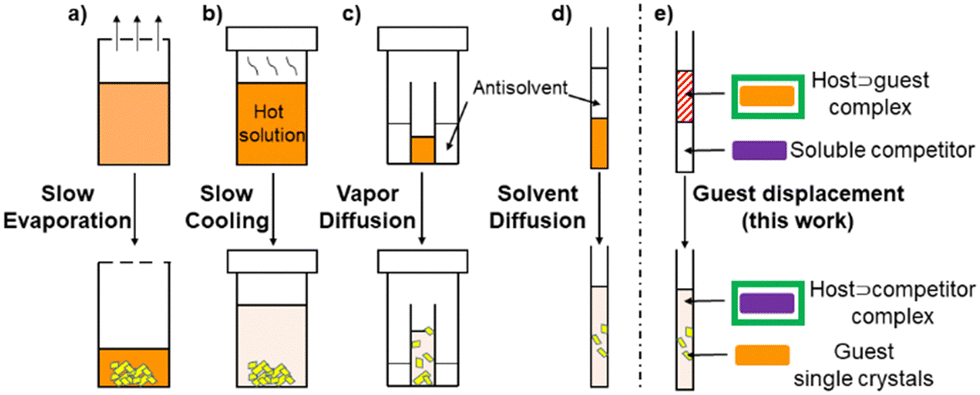 | ||
| Fig. 1 Various methods used to grow single crystals from solution. (a) Slow evaporation of solvent, (b) slow cooling of a hot solution, (c) vapor diffusion of an antisolvent using a saturated solution of the analyte, (d) diffusion of an antisolvent layered on top of the analyte solution, and (e) displacement of the guest encapsulated in a soluble macrocyclic host by a competitor (this work). | ||
Cocrystallization by taking advantage of supramolecular interactions have been a staple of crystal engineering since the early 1990s, employing a broad range of hosts and cages including macrocycles, cavitands, and cryptophanes.8–11 Host–guest interactions, in particular, use the interplay between intermolecular forces and shape complementarity to co-crystalize the species of interest. Recently, metal–organic frameworks have been employed as crystalline sponges, where the solid-state framework serves as the host for guests which diffuse into the material over time, yielding a single-crystal-to-single-crystal transformation.12,13 Success has also been had co-crystallizing with bulky chaperone compounds such as tetraaryladamantane.14,15 These approaches work well for guest substrates that are either liquid or readily soluble. In these cases, the structure of the guest is determined from crystals of the entire host–guest complex.8,16 Polycationic host molecules like cyclobis(paraquat-p-phenylene) (CBPQT4+) and its related analogs have been shown to bind a wide array of PACs, and the structures of their host–guest complexes were determined using single crystal X-ray crystallography.17 However, the solving of such structures can be challenging, due to disorder often arising from guest positioning within the host, as well as counterions and solvent within the crystal. In all these approaches, cocrystallization also only provides structural information about the guest within the host, not the free guest itself. In contrast, the crystal structure of an independent guest substrate provides information about intermolecular interactions and packing, which are essential to understanding material properties including electronic properties18,19 and mechanical strength.20
Our approach employs host encapsulation for poor solubility guests as a temporary reservoir which subsequently releases the guest for crystal growth. This is achieved via guest displacement (GuD), where a second guest species is introduced as a competitor for host binding. To demonstrate this approach, novel modular model substrate A-Me (Fig. 2) was prepared, a bisimine species readily synthesized from 9,10-anthracenedicarboxaldehyde (ADC) and p-toluidine using Schiff base reaction (ESI†). Due to its large π surface, A-Me is sparingly soluble in acetonitrile. Using traditional single crystal growth methods, A-Me produced only nanocrystalline powders (slow cooling in MeCN) or solutions that were too dilute for antisolvent driven approaches (slow vapor diffusion of iPr2O into saturated MeCN solutions). These issues are addressed through reversible binding with the cationic supramolecular host ExBox4+, a rectangular relatively rigid tetracationic macrocycle that is structurally well suited to bind PAHs and PACs.21 In this way, A-Me is dissolved in the otherwise poor solvent MeCN as a supramolecular ionic host–guest complex using soluble ExBox·4TFSI (Fig. 2 and ESI†). Following binding, the remaining excess undissolved A-Me is removed, and a solution of a second guest-competitor (pyrene, ca. 1 equiv. relative to ExBox4+) is introduced. This induces an equilibria-controlled displacement of the A-Me guest from the ExBox4+ host and subsequent crystallization of the free guest from the resulting supersaturation conditions. Initial crystals form rapidly upon the addition of a solution of pyrene in MeCN, while ExBox4+ serves as a soluble reservoir for additional A-Me release during crystallization, producing large, diffraction quality single crystals. The crystal structure of A-Me, previously unreported, confirms the expected compound free of host (Fig. 3a–c). Moreover, crystals of the released guest provide packing information, including an anthracene moiety spacing of 3.32 Å, and the tolyl group plane at an 85° angle to anthracene, likely as a result of prominent CH/π interactions between neighboring molecules.22 Notable interactions include those between anthracene C–H and imine atoms, measuring 2.69 Å for C–H/N distance and ca. 2.80 Å for C–H/C interactions, as well as an edge-to-face interaction between an anthracene C–H bond and the neighboring tolyl group at 2.98 Å.
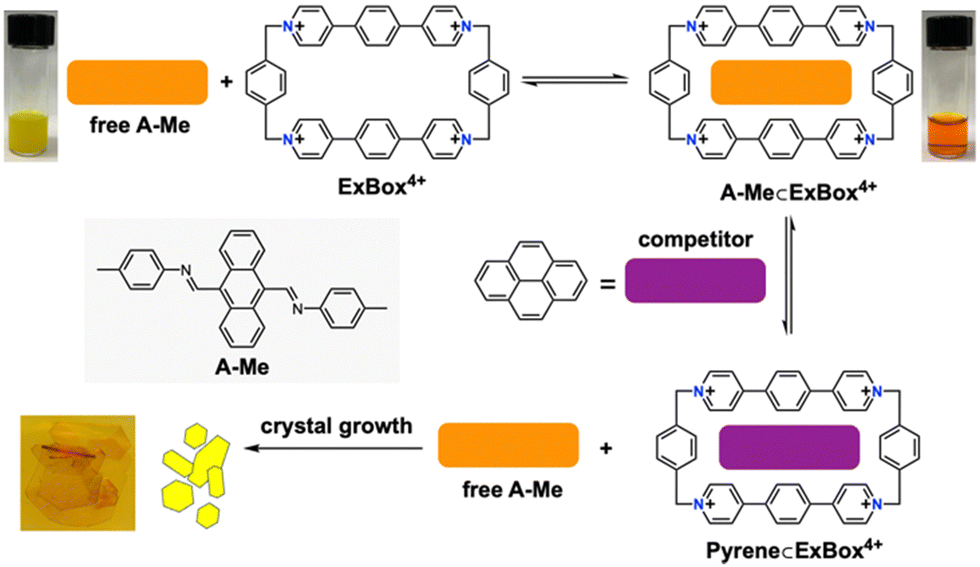 | ||
| Fig. 2 Host-mediated single crystal growth of an anthracene bisimine guest A-Me. As a competitor guest (pyrene) diffuses in slowly, A-Me is released from ExBox4+ and rapidly begins crystalizing. Further growth of crystals is sustained by the continuous dissociation of the host–guest complex and the release of the guest A-Me from ExBox4+ host. | ||
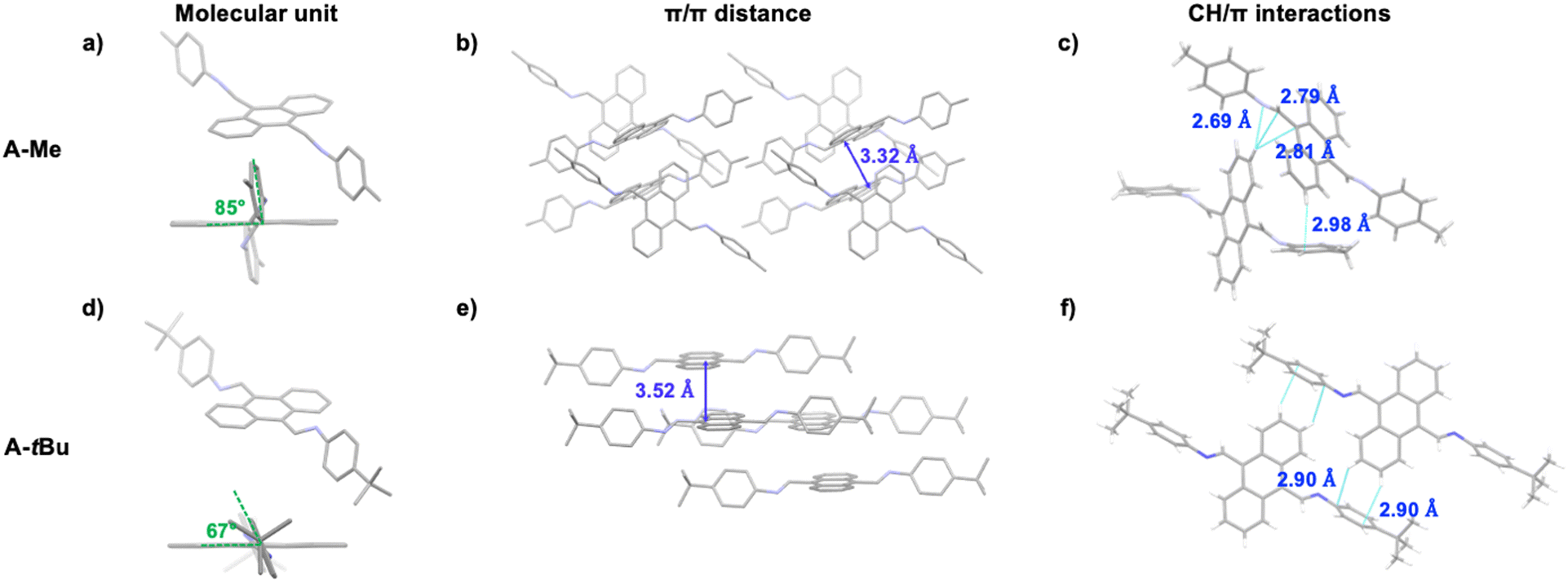 | ||
| Fig. 3 Single crystal structures of (a)–(c) A-Me, and (d)–(f) A-tBu. Both crystals were grown through controlled release of the guests from their host–guest complexes with ExBox4+ using guest displacement (GuD) via pyrene. | ||
Employing the modular anthracene substrate, crystals of A-tBu (Table 1) were similarly prepared via GuD crystallization (Fig. 3d–f). Despite the similarity in molecular structure, the packing of A-tBu was notably different from A-Me, including an increase in the spacing between anthracene moieties to 3.52 Å. The 4-tert-butylphenyl group plane is at a 67° angle to anthracene, which again is attributed to edge-to-face interactions in the crystal, between C–H bonds of anthracene and the π surface of 4-tert-butylphenyl groups. The GuD crystallization of A-tBu was also evaluated over time via optical microscopy (ESI,† Fig. S25 and accompanying video). Upon addition of ca. 3 equivalents of pyrene relative to ExBox4+, crystal formation of rectangular prisms is observed within the first minute, with the majority of growth occurring within the first 15 minutes. Further growth was minimal after 40 minutes, with many crystals displaying dimensions ca. 0.3 mm.
|
|
|||||
|---|---|---|---|---|---|
| Substrate | c sat (mM) | c tot (mM) | S r | K a/103 (M−1) | Crystal |
| All solutions were prepared in MeCN.a Total ExBox4+ concentration, c(ExBox·4TFSI)tot = 5 mM.b Sr = ctot/csat.c Binding constant was determined by UV-vis spectroscopy titration.d Crystals were small, thin, and highly twinned along the thin dimension.e Binding constant determined by 1H-NMR titration is 10.2 ± 1.0 × 103 M−1.f No crystal formation was observed.g Binding constant was determined by 1H-NMR titration. | |||||
| A-tBu | 0.13 ± 0.03 | 3.32 ± 0.24 | 26.0 ± 5.6 | 13.8 ± 4.2 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| BPA | 0.15 ± 0.03 | 2.25 ± 0.38 | 15.0 ± 4.0 | 4.8 ± 1.9 | Pbc 23 |
| A-H | 0.32 ± 0.13 | 4.05 ± 0.44 | 12.6 ± 5.1 | 6.6 ± 0.5c | |
| A-Me | 0.37 ± 0.04 | 4.22 ± 0.11 | 11.5 ± 1.3 | 9.2 ± 1.8e | P21/c |
| Anthrarufin | 0.52 ± 0.03 | 2.97 ± 0.22 | 5.7 ± 0.5 | 1.9 ± 0.4 | P21/c24 |
| Alizarin | 2.70 ± 0.07 | 5.60 ± 0.08 | 2.1 ± 0.1 | 0.5 ± 0.1 | |
| AQ | 3.12 ± 0.23 | 6.26 ± 0.29 | 2.0 ± 0.2 | 0.5 ± 0.2 | P21/c25 |
| DCAQ | 3.07 ± 0.14 | 5.93 ± 0.73 | 1.9 ± 0.3 | 0.4 ± 0.3 | P2/c26 |
| ADC | 4.91 ± 0.86 | 8.60 ± 1.49 | 1.7 ± 0.4 | 2.3 ± 0.3g | |
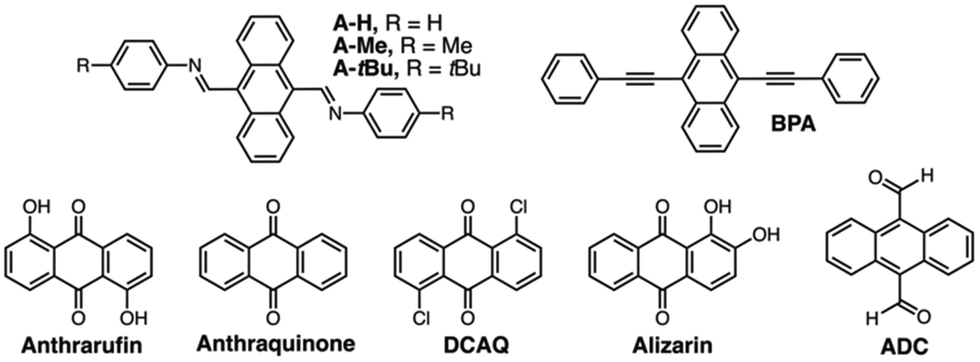
To investigate the scope of GuD crystallization with ExBox4+, a series of anthracene and anthraquinone derivatives were evaluated (Table 1 and ESI†). Solubility of each guest both with and without host were determined, as well as binding constants (Ka). For the three novel bisimine systems, modifying terminal groups modulated both sterics and overall solubility of the free guest. All guests studied contained three fused planar rings for binding within the pocket of ExBox4+, whose length is perfectly suited to accommodate these compounds. Despite having similar geometries, this group of compounds exhibit a wide range of both solubilities and binding constants in MeCN, and the effect of these variables on supersaturation and crystal growth was evaluated. Using a 5 mM solution of ExBox·4TFSI in MeCN, 1H-NMR spectra confirm host–guest complexation by downfield shifts of guest protons in the presence of ExBox4+ (ESI†), with protons that occupy the molecular cavity experiencing the greatest shifts. Pyrene, which acts as the competitor guest, has a Ka = 10.1 ± 1.1 × 103 M−1 for binding ExBox4+.21 This value is comparable to or greater than the Ka values for the substrates tested, enabling pyrene to effectively displace these guests from ExBox4+ and induce crystal growth. At 84 ± 10 mM, the solubility of pyrene in MeCN was also found to be at least an order of magnitude higher than the substrates studied. To describe the solubilizing effect of ExBox4+, we calculate a solubility ratio, Sr, the total concentration of the guest in the presence of the host relative to guest solubility without host, ctot/csat (for a detailed discussion, see ESI†). These Sr ranged from 1.7-fold increase to 26-fold increase in guest effective concentration.
For seven of the nine systems examined, GuD with pyrene yielded crystalline materials, and six were amenable to single crystal diffraction. Similar to A-tBu, most crystal growth was rapid, typically beginning within minutes of adding the competitor, and the majority of growth observed within hours. In the case of the four previously reported crystal structures,23–26 unit cell parameters were measured from diffraction data and confirmed to match literature values. In the case of A-H, crystals diffracted only weakly. In general, compounds with higher Sr crystallized well, and the only two that did not crystallize, alizarin and ADC, had some of the lowest Sr values due in part to their high intrinsic solubility. These results suggest GuD crystallization is best suited for guests in low solubility conditions. We attribute this behavior primarily to the solubilizing effect of the host complexation, Sr, where the large difference in solubility with and without host increases the effective supersaturation of the guest following displacement with pyrene. The binding constant Ka is an insufficient value alone to predict effectiveness of this approach, as it does not account for the intrinsic solubility of the guest. In the case of ADC (and pyrene itself), relatively high solubility in MeCN minimizes the overall impact of host complexation on species dissolution within the solvent. A more predictive measure of successful crystal growth is Sr, as this parameter provides a direct evaluation of the quasi-supersaturation of the guest via host complexation. To facilitate the comparison between substrates, Table 1 is organized by decreasing Sr values. Based on this, we hypothesize GuD crystallization has a higher propensity for success when Sr ratios are beyond ca. 2. For the systems studied, the increase in solubility upon addition of the host is consistent, suggesting that the extent of host binding is comparable, and that the free guest intrinsic solubility (csat) is the main determining factor. The optimal performance with low solubility guests is ideal for PAC substrates, as these systems are more likely to have issues with crystallization using traditional methods.
In addition to GuD crystallization, the solubilizing effect of ExBox4+ also allows for structural determination through more traditional cocrystallization methods. In the case of A-tBu and A-Me, single crystals of the host–guest complex were obtained via vapor diffusion of diisopropyl ether antisolvent (Fig. 4a and b). In the case of A-H, single crystals of the host–guest complex were also obtained, directly from a one-pot synthesis of A-H in the presence of ExBox4+ (Fig. 4c). This highlights the robustness of the system, and the complementary approach of host–guest complex cocrystallization, albeit at longer time scales (days versus hours). An analysis of the orientation of the guests suggests the anthracene unit occupies the host cavity in similar orientations. Aniline benzene rings in A-H are more perpendicular to the plane of anthracene compared to the other two compounds, as a result of edge-to-face interactions between this ring and a neighboring ExBox4+ host. Unlike free guest crystals, the host–guest crystals contain interstitial species, such as solvent, additives, and counterions, due to packing of the large inclusion complex. Even though the host–guest complex structure does not provide information on free guest packing, it is helpful in elucidating molecular structure when growing high-quality single crystals of the pure guest is challenging.
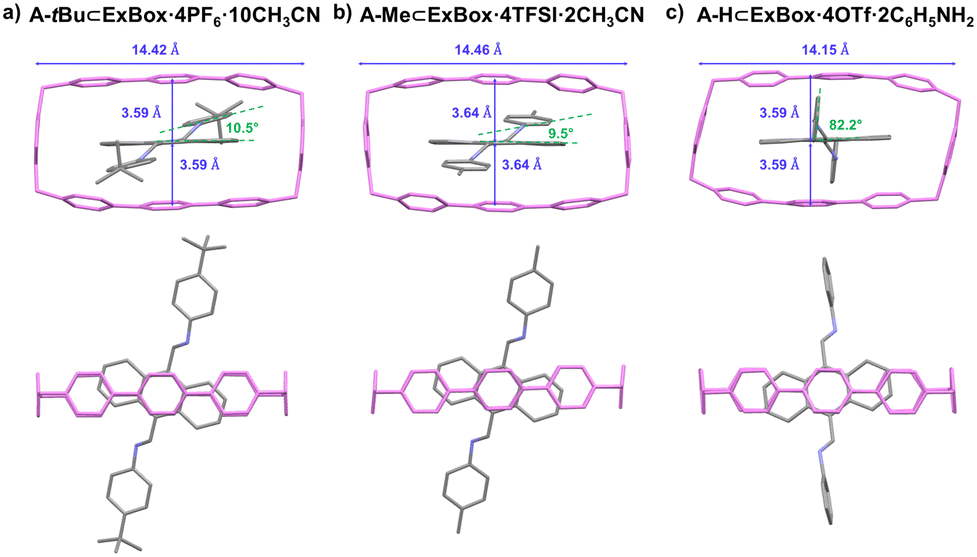 | ||
| Fig. 4 Crystal structures of host–guest complexes of A-R in ExBox4+. All crystals were grown by vapor diffusion of iPr2O into the solutions containing A-R⊂ExBox·4X complexes in MeCN. Hydrogens, counterions, solvent molecules, and interstitial aniline in the case of A-H⊂ExBox·4OTf·2C6H5NH2, are omitted for clarity. | ||
In summary, GuD crystallization is a promising approach for small molecule crystal growth. Our methodology of using transient host–guest complexation sidesteps limitations of sparingly soluble conditions for PAC substrates and provides a new tool of controlled guest release to facilitate rapid single crystal growth under ambient conditions. We believe this proof-of-concept study can be extended to other compounds with low solubility, and the substrate and solvent scope can be expanded with hosts of different shape and size, such as pillar[n]arenes27 or metallocages.28 Finally, the approach also allows for more traditional crystallization via nucleation of the host–guest complex itself. This type of rapid and robust crystal growth can prove useful in both academic and industrial settings as it allows quick screening of a large number of samples, making it attractive for robotics and combinatorial applications. We anticipate this approach will have a high impact in the crystal growth and engineering communities.
This work was supported by the National Science Foundation (NSF) under Grant No. (DMR/RUI 1904170). M. L. H. was partially supported by Henry Luce Foundation through the Clare Boothe Luce Program for Women in STEM. This research made use of an Agilent 6560 mass spectrometer that was acquired with support from the NSF under Grant No (CHE/MRI-2018547). We thank Dr Morgan R. Olsen for help with high-resolution mass spectrometry and Dr Joe Moore for help with optical microscopy.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. P. Metherall, R. C. Carroll, S. J. Coles, M. J. Hall and M. R. Probert, Chem. Soc. Rev., 2023, 52, 1995–2010 RSC.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286–2300 RSC.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed.
- C. Achten and J. T. Andersson, Polycyclic Aromat. Compd., 2015, 35, 177–186 CrossRef CAS PubMed.
- Handbook of Industrial Crystallization, ed. A. S. Myerson, D. Erdemir and A. Y. Lee, Cambridge University Press, Cambridge, 3rd edn, 2019 Search PubMed.
- D. A. Edwards, R. G. Luthy and Z. Liu, Environ. Sci. Technol., 1991, 25, 127–133 CrossRef CAS.
- J.-R. Wu, G. Wu, D. Li and Y.-W. Yang, Angew. Chem., Int. Ed., 2023, 62, e202218142 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- A. S. Tayi, A. K. Shveyd, A. C.-H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS PubMed.
- C. M. Kane, O. Ugono, L. J. Barbour and K. T. Holman, Chem. Mater., 2015, 27, 7337–7354 CrossRef CAS.
- N. Zigon, V. Duplan, N. Wada and M. Fujita, Angew. Chem., Int. Ed., 2021, 60, 25204–25222 CrossRef CAS PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- F. Krupp, W. Frey and C. Richert, Angew. Chem., Int. Ed., 2020, 59, 15875–15879 CrossRef CAS PubMed.
- A. Schwenger, W. Frey and C. Richert, Chem. – Eur. J., 2015, 21, 8781–8789 CrossRef CAS PubMed.
- K. Rissanen, Chem. Soc. Rev., 2017, 46, 2638–2648 RSC.
- E. J. Dale, N. A. Vermeulen, M. Juríček, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- M. Mahl, M. A. Niyas, K. Shoyama and F. Würthner, Nat. Chem., 2022, 14, 457–462 CrossRef CAS PubMed.
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern, R. Jakubas, S. Hong, V. P. Dravid and J. F. Stoddart, Nat. Commun., 2016, 7, 13108 CrossRef CAS PubMed.
- J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS PubMed.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, M. Rasras and P. Naumov, Angew. Chem., Int. Ed., 2021, 60, 26151–26157 CrossRef CAS PubMed.
- G.-Q. Wei, Y. Yu, M.-P. Zhuo, X.-D. Wang and L.-S. Liao, J. Mater. Chem. C, 2020, 8, 11916–11921 RSC.
- P.-W. Wang, J. Appl. Crystallogr., 1979, 12, 239 CrossRef CAS.
- Y.-J. Chen, S.-C. Yang, C.-C. Tsai, K.-C. Chang, W.-H. Chuang, W.-L. Chu, V. Kovalev and W.-S. Chung, Chem. – Asian J., 2015, 10, 1025–1034 CrossRef CAS PubMed.
- K. Yang, Y. Pei, J. Wen and Z. Pei, Chem. Commun., 2016, 52, 9316–9326 RSC.
- X.-Q. Wang, Q.-H. Ling, W. Wang and L. Xu, Mater. Chem. Front., 2020, 4, 3190–3200 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of ExBox·4TFSI, A-H, A-Me, and A-tBu, procedure for single crystal growth, X-ray crystallography data, measurement of binding constants and solubility ratios, nuclear magnetic resonance spectra for host–guest complexation. CCDC 2277355–2277359. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03406b |
| This journal is © The Royal Society of Chemistry 2023 |