DOI:
10.1039/D1SC06060K
(Edge Article)
Chem. Sci., 2022,
13, 2591-2603
Solid-state 17O NMR study of α-D-glucose: exploring new frontiers in isotopic labeling, sensitivity enhancement, and NMR crystallography†
Received
2nd November 2021
, Accepted 31st December 2021
First published on 3rd January 2022
Abstract
We report synthesis and solid-state 17O NMR characterization of α-D-glucose for which all six oxygen atoms are site-specifically 17O-labeled. Solid-state 17O NMR spectra were recorded for α-D-glucose/NaCl/H2O (2/1/1) cocrystals under static and magic-angle-spinning (MAS) conditions at five moderate, high, and ultrahigh magnetic fields: 14.1, 16.4, 18.8, 21.1, and 35.2 T. Complete 17O chemical shift (CS) and quadrupolar coupling (QC) tensors were determined for each of the six oxygen-containing functional groups in α-D-glucose. Paramagnetic Cu(II) doping was found to significantly shorten the spin–lattice relaxation times for both 1H and 17O nuclei in these compounds. A combination of the paramagnetic Cu(II) doping, new CPMAS CryoProbe technology, and apodization weighted sampling led to a sensitivity boost for solid-state 17O NMR by a factor of 6–8, which made it possible to acquire high-quality 2D 17O multiple-quantum (MQ) MAS spectra for carbohydrate compounds. The unprecedented spectral resolution offered by 2D 17O MQMAS spectra permitted detection of a key structural difference for a single hydrogen bond between two types of crystallographically distinct α-D-glucose molecules. This work represents the first case where all oxygen-containing functional groups in a carbohydrate molecule are site-specifically 17O-labeled and fully characterized by solid-state 17O NMR. Gauge Including Projector Augmented Waves (GIPAW) DFT calculations were performed to aid 17O and 13C NMR signal assignments for a complex crystal structure where there are six crystallographically distinct α-D-glucose molecules in the asymmetric unit.
Introduction
The element of oxygen is a key constituent of organic and biological molecules. Oxygen-containing functional groups are often directly involved in chemical reactions including biological transformation such as enzyme catalysis. While NMR spectroscopy is a powerful technique for structural elucidation of organic and biological molecules, most NMR studies are based on detection of signals from hydrogen, carbon, nitrogen, and phosphorus atoms. While it is highly desirable to add oxygen to the list of nuclear probes available for NMR studies, two major obstacles have made it difficult to characterize NMR signals from oxygen atoms. First, the NMR-active oxygen isotope, 17O, has an exceedingly low natural abundance (0.037%). Thus, it is usually necessary to prepare 17O-enriched molecular systems in order to boost NMR detectability. This 17O-labeling process can be a difficult task. Second, 17O has a quadrupolar nucleus (I = 5/2), which often gives rise to significantly broader NMR signals than those commonly encountered from other more NMR-friendly spin-1/2 nuclei such as 1H, 13C and 15N. This quadrupole line broadening is a major roadblock to 17O NMR applications in terms of spectral resolution. Over the last two decades, however, significant progress has been made in demonstrating 17O NMR as a viable tool to study organic and biological molecules in both solution and the solid state.1–7 For 17O NMR studies of biological molecules, in particular, some important developments have occurred in recent years. Zhu et al.8 showed that it is possible to obtain solid-state 17O NMR spectra from protein–ligand complexes where the ligand molecules are site-specifically 17O-labeled. Tang et al.9 applied this approach to study hydrogen-bonding interactions around the “oxyanion hole” in several acyl-enzymes. Zhu et al.10,11 demonstrated a technique known as quadrupole-central-transition (QCT) NMR in obtaining high-resolution 17O NMR spectra for biological macromolecules undergoing slow tumbling motion in aqueous solution. Young et al.12 applied the 17O QCT method to monitor the formation of enzymatic intermediates of tryptophan synthase under active catalysis. Recently, Paulino et al.13 reported a comprehensive 17O solid-state NMR study of the water–carbonyl interactions in gramicidin A ion channel. The latest advancement in the field was the work by Lin et al.14 where they demonstrated a general approach to incorporate 17O isotopes into recombinant proteins and reported solid-state 17O NMR spectra for yeast ubiquitin.
In addition to the abovementioned new applications, there have also been recent developments in solid-state 17O NMR methodology. One particular area of interest is concerned with heteronuclear correlation solid-state NMR spectroscopy between 17O and other nuclei such as 1H, 13C, and 15N.15–19 For example, Hung et al.19 reported a new 3D D-RINEPT/DARR OCC experiment where overlapping 17O NMR signals can be completely separated in the 13C dimension. Another highly promising direction is to use dynamic nuclear polarization (DNP) to enhance 17O NMR signals for organic and biological molecules.20–23 Currently, most DNP-enhanced 17O NMR studies were performed at low or moderate magnetic fields (≤14.1 T) to study inorganic materials; it would be highly beneficial for the study of organic and biological molecules if DNP for 17O becomes feasible at higher magnetic fields.24
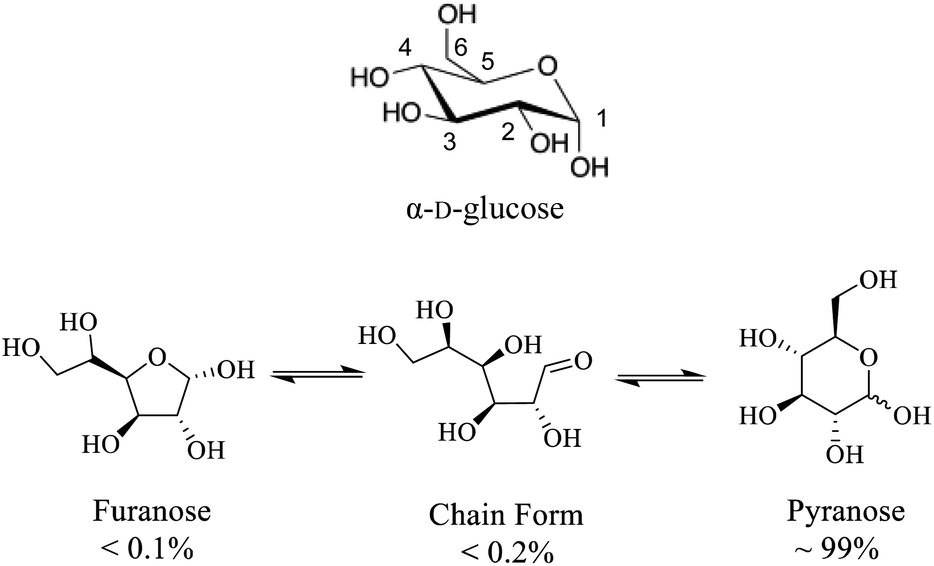
While fundamental 17O NMR data on chemical shift (CS) and electric-field-gradient (EFG) tensors have been reported for many oxygen-containing organic functional groups, there are still many unexplored classes of organic compounds for which little is known about their 17O NMR tensor properties. One notable example is concerned with carbohydrates. Carbohydrates are an important class of oxygen-rich organic molecules of biological significance. However, solid-state 17O NMR studies dealing with carbohydrate molecules are very rare in the literature. Sefzik et al.25 reported the first solid-state 17O NMR study of several protected carbohydrate compounds. Yamada et al.26 obtained the solid-state 17O NMR signal for the O6 atom of D-glucosamine. More recently, Hung et al.19 reported 2D and 3D 13C–17O heteronuclear correlation solid-state NMR spectra of [1-13C,17O]-α/β-D-glucose. Also relevant are two 17O QCT NMR studies by Shen et al.27 and by Gan et al.28 where 17O-labeled D-glucose samples were examined with the aid of high magnetic fields. One major challenge in solid-state 17O NMR studies of carbohydrates is the synthesis of 17O-labeled target compounds. To further explore synthetic procedures and solid-state 17O NMR for unprotected carbohydrate compounds, we selected D-glucose as an initial target (Scheme 1). In this work, we report synthesis of a total of six site-specifically 17O-labeled D-glucose compounds and their full solid-state 17O NMR characterization. For the latter part, because crystallization of D-glucose into a pure anomeric form (α or β) often encounters low yields, we decided to prepare all solid samples of D-glucose in the form of a D-glucose/NaCl/H2O (2/1/1) cocrystal. This cocrystal is known to contain exclusively α-D-glucose and can be readily prepared in crystalline form with near 100% yields.29–31 Throughout this work, we will use “α-D-glucose” as a shorthand name for the α-D-glucose/NaCl/H2O (2/1/1) cocrystal.
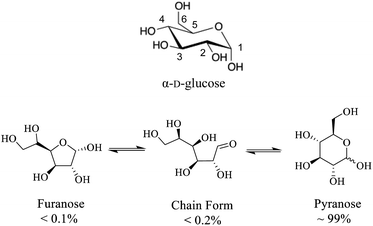 |
| | Scheme 1 (Top) Molecular structure of α-D-glucose where carbon atoms are numbered. (Bottom) different D-glucose tautomers present in aqueous solution. | |
Another objective of the present work is to demonstrate utilization of the current state-of-the-art solid-state 17O NMR technologies achieving unprecedented sensitivity and spectral resolution for organic and biological molecules. To this end, we explore the following three areas. First, we perform solid-state 17O NMR at multiple magnetic fields including an ultrahigh magnetic field of 35.2 T.28 Second, we investigate the effect of paramagnetic doping in shortening spin-relaxation times for 17O nuclei so that fast data acquisition might be possible. Third, we test the sensitivity enhancement for solid-state 17O NMR applications using a new CPMAS CryoProbe.32
Experimental section
Synthesis of sitespecifically 17O-labeled D-glucose compounds
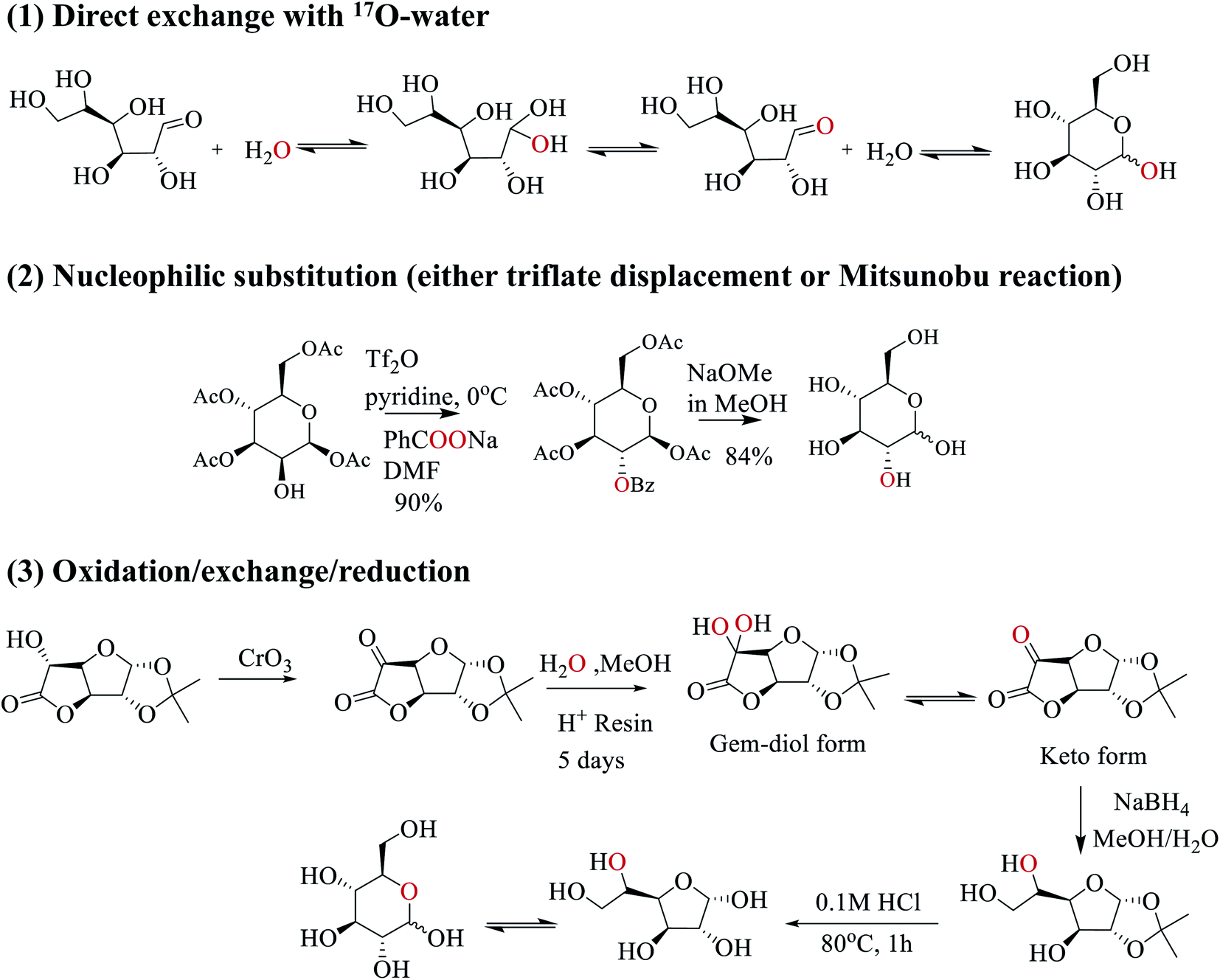
In this work, we employed three strategies to synthesize site-specifically 17O-labeled D-glucose compounds; see Scheme 2. First, the anomeric O1 atom in D-glucose can be readily 17O-labeled by a simple exchange with 17O-water.33–35 This exchange occurs through the hydration/dehydration process of the aldehyde functional group in the open chain glucose tautomer. Second, for both primary and secondary hydroxyl groups (O2, O3, O4, O6), 17O isotopes can be incorporated into glucose by SN2 nucleophilic substitution (via either triflate displacement route or Mitsunobu reaction) from appropriate starting epimers.36 In this case, either sodium [1,2-17O2]benzoate (triflate displacement reaction) or [1,2-17O2]benzoic acid (Mitsunobu reaction) can be used as the source of 17O. For example, as shown in Scheme 2, in pyridine at 0 °C, the O2 atom of [1,3,4,6-acetyl]-D-mannose is first functionalized with triflate anhydride, followed by the triflate displacement reaction in DMF with sodium [1,2-17O2]benzoate. Subsequent removal of the protecting groups gives [2-17O]-D-glucose. Third, for 17O-labeling of the O5 atom, we utilize a combined oxidation/exchange/reduction method starting from 1,2-O-isopropylidene-D-glucofuranurono-6,3-lactone as illustrated in Scheme 2. After oxidation of the OH group by chromium trioxide,3717O-labels are introduced onto the keto group from 17O-water via an acid-catalyzed hydration/dehydration process (or keto/gem-diol exchange). Then, reduction with NaBH4 converts the keto group back to the hydroxyl group.38 Finally, removal of protecting groups allows the furanose/pyranose equilibrium to occur, producing [5-17O]-D-glucose.39 Full details of the synthetic procedures and compound characterization are provided in the ESI.†
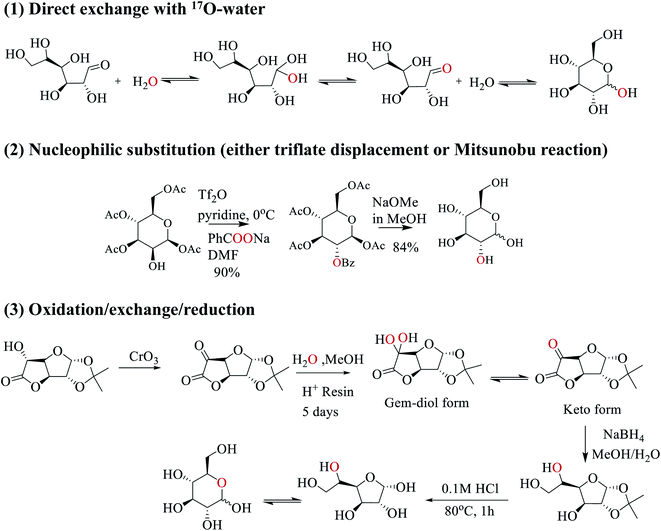 |
| | Scheme 2 Three synthetic strategies used in this work to prepare site-specifically 17O-labeled D-glucose. | |
Preparation of solid samples
As mentioned above, because crystallization of D-glucose into the pure α (or β) form is often associated with low yields, we prepared all solid samples of 17O-labeled D-glucose as a D-glucose/NaCl/H2O (2/1/1) cocrystal where all D-glucose molecules are in the α-form.29 The D-glucose/NaCl/H2O (2/1/1) cocrystal was readily prepared by adding solid NaCl to aqueous solution of D-glucose followed by lyophilization. A solid sample was prepared as an equal molar mixture of [3-17O]-D-glucose, [5-17O]-D-glucose, and [6-17O]-D-glucose. This sample was denoted as [3/5/6-17O]-α-D-glucose in this work. Because the three compounds may have different levels of 17O-enrichment, the mixing process was monitored by solution 17O NMR; see ESI.† As a result, the level 17O enrichment in this [3/5/6-17O]-α-D-glucose sample was only about 10%. The integrity of all solid samples was checked by acquiring solid-state 13C CPMAS NMR spectra; all spectra are provided in ESI. Solid samples with paramagnetic Cu(II) dopants were prepared in the following fashion. To 2 mL of H2O was first added 15 mg of solid Na2[Cu(EDTA)2] to give a clear blue solution, followed by addition of 150 mg D-glucose/NaCl/H2O (2/1/1) cocrystal. The solution turned greenish when solids were fully dissolved. The solution was then dried under a stream of N2 until it became a syrup. Addition of 2 mL of absolute ethanol induced crystallization. After removal of the supernatant, solids were dried in air. Cu(II)-doped solid samples displayed the same solid-state 13C CPMAS NMR spectra as regular D-glucose/NaCl/H2O cocrystals; see ESI.†
Solid-state NMR
Solid-state 17O and 13C CPMAS NMR data at 14.1 T were collected on a Bruker Avance-600 NMR spectrometer at Queen's University. For static 17O NMR experiments, a Bruker 4 mm HX MAS probe was used. The 90° pulse width for the 17O central-transition (CT) was 2.0 μs. 1H decoupling with 60 kHz rf field was applied during data acquisition in the static experiments. Solid-state 17O NMR experiments at 21.1 T were performed on a Bruker Avance-II 900 NMR spectrometer at the National Ultrahigh Field NMR Facility for Solids (Ottawa, Ontario, Canada). A Hahn-echo sequence was used for acquiring solid-state 17O NMR spectra under both MAS and static conditions to eliminate probe ringing artifacts. For MAS experiments, a 3.2 mm Bruker HX MAS probe was used where the effective 90° pulse width for the 17O CT was 1.0 μs. For static experiments, a homebuilt 5 mm solenoid probe was used with powder samples packed into 5 mm Teflon tubes to reduce background signals. On this solenoid probe, the 90° pulse width for the 17O CT was 2.0 μs. 1H decoupling with 75 kHz rf field was applied during data acquisition. A liquid H2O sample was used for both rf power calibration and 17O chemical shift referencing (δ = 0 ppm). All spectral simulations were performed with DMfit.40
Solid-state 17O NMR experiments at 35.2 T were carried out on the 36 T series-connected hybrid (SCH) magnet28 at the National High Magnetic Field Laboratory (NHMFL, Tallahassee, Florida, USA) with a Bruker Avance NEO console. A single-resonance 3.2 mm MAS probe with an external field regulation circuit designed and constructed at the NHMFL was used with pencil-type ZrO2 rotors spinning at a MAS frequency of 16 kHz. A Hahn-echo sequence was used with 5 and 10 μs pulses (with 16.7 kHz rf field) and a recycle delay of 0.1 s.
Solid-state 17O and 13C NMR experiments were also performed on a Bruker NEO-800 (18.8 T) at the Bruker application lab (Fällanden, Switzerland) with a broadband 3.2 mm CPMAS CryoProbe. The sample spinning was 15 kHz. The 17O rf field was about 64 kHz, which gave an effective 90° pulse of 1.3 μs for the CT. The 1H decoupling field was 83 kHz. An apodization weighted sampling (AWS) scheme41 was used for collecting 2D 17O shifted-echo 3QMAS data. For the 13C refocused INADEQUATE experiment, the 13C 90° pulse was 5.0 μs. The spectral width in the F1 dimension was 7.5 kHz. A frequency swept TPPM 1H decoupling (83 kHz) scheme was applied during data acquisition.
Computational details
All quantum chemical calculations were performed using the CASTEP code42 (version 2019) together with BIOVIA's Materials Studio. CASTEP employs DFT using the plane-wave pseudopotential approach. The generalized gradient approximation with either the Perdew–Burke–Ernzerhof43 or revised Perdew–Burke–Ernzerhof (rPBE)44 exchange correlation functionals was chosen. First, geometry optimization was performed employing the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm together with OTFG on-the-fly ultrasoft pseudopotentials (version 2017R2), a cut-off energy of 598.7 eV and a k-point grid with a maximum separation of 0.071 Å−1. We also tested the treatment of dispersion interactions by using the two-body force-field method of Grimme (D2) (ref. 45) with a re-parameterized damping function (s6 = 1.0; d = 3.25 or d = 5.0)46,47 in geometry optimizations. Subsequently, the NMR parameters were calculated using the Gauge Including Projector Augmented Waves (GIPAW) method implemented in the NMR module of CASTEP.48–50 In this work, a total of four sets of GIPAW DFT computations were performed and they are denoted as: (1) PBE, (2) rPBE, (3) rPBE-D2 (d = 3.25), and (4) rPBE-D2 (d = 5.0). However, because these four methods produced essentially the same results, we will focus on the results obtained with the PBE method and report the complete results from all four methods in the ESI.†
Results and discussion
Determination of 17O NMR tensors in α-D-glucose
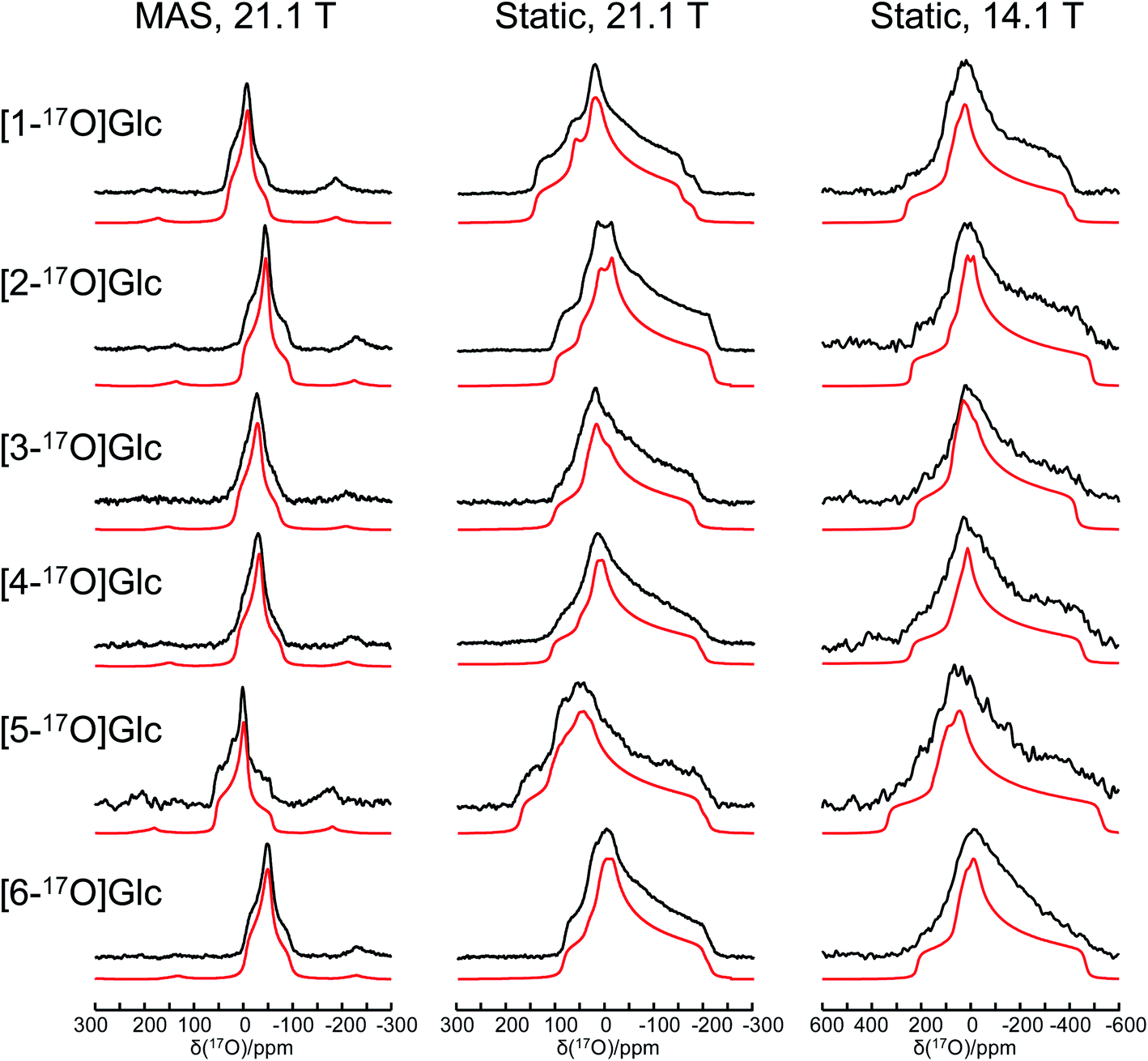
Fig. 1 shows the solid-state 17O NMR spectra obtained for all six site-specifically 17O-labeled D-glucose compounds. In each 17O MAS NMR spectrum, a well-defined powder line shape was observed, which is known to arise from the second-order quadrupole interaction. In general, second-order quadrupole interactions are inversely proportional to the applied magnetic field. However, as seen from Fig. 1, even at 21.1 T, second-order quadrupole interactions cause a line broadening on the order of 100 ppm. This is because the oxygen-containing functional groups in D-glucose (hydroxyl and ether groups) are known to experience rather large 17O nuclear quadrupole interactions. It is also immediately clear that the relatively small 17O chemical shift variations among the six oxygen-containing groups in D-glucose can be easily obscured by such second-order quadrupole broadenings (vide infra). In each case, an analysis of the observed powder line shape obtained under MAS conditions allowed us to obtain three 17O NMR parameters: δiso, CQ, and ηQ. Complete experimental results are listed in Table 1.
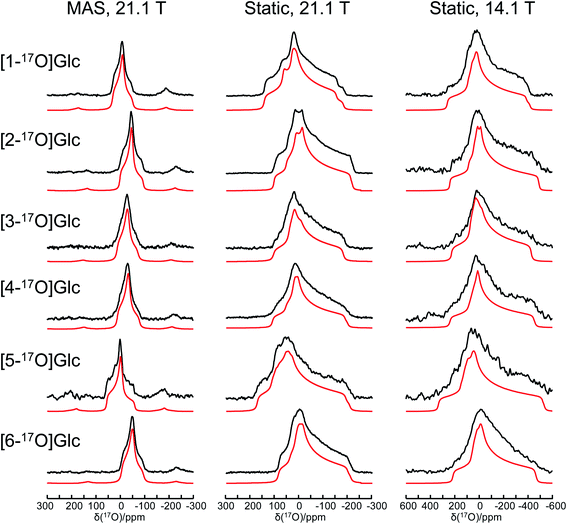 |
| | Fig. 1 Experimental (black traces) and simulated (red traces) solid-state 17O NMR spectra for a total of six site-specifically 17O-labeled α-D-glucose compounds under the MAS (22 kHz sample spinning) and static conditions at 21.1 and 14.1 T. The 17O NMR parameters used in the spectral simulations are summarized in Table 1. The same set of parameters were used for each compound to simulate spectra obtained at two magnetic fields. Detailed acquisition parameters are given in the ESI.† | |
Table 1 Experimental 17O NMR tensor parameters obtained for α-D-glucose from a spectral analysis of data presented in Fig. 1. The uncertainties in experimental values of δiso, δii (i = 1, 2, 3), CQ, and ηQ are estimated to be ±2 ppm, ±10 ppm, ±0.2 MHz, and ±0.2, respectively
| Atom |
δ
iso/ppm |
δ
11/ppm |
δ
22/ppm |
δ
33/ppm |
∣CQ∣/MHz |
η
Q
|
| O1 |
32 |
72 |
22 |
2 |
8.4 |
1.0 |
| O2 |
2 |
27 |
−6 |
−12 |
9.1 |
1.0 |
| O3 |
14 |
34 |
12 |
−5 |
8.8 |
0.9 |
| O4 |
13 |
33 |
6 |
−2 |
8.9 |
1.0 |
| O5 |
56 |
96 |
56 |
16 |
9.9 |
1.0 |
| O6 |
−5 |
20 |
−5 |
−30 |
9.0 |
0.9 |
When the solid-state 17O NMR experiments are performed for stationary (non-spinning) powder samples, even broader powder line shapes are observed, as also seen from Fig. 1. At 14.1 T, each static powder line shape spans about 700 ppm, which is reduced to roughly 300 ppm at 21.1 T. This is because now both 17O CS and QC tensors contribute to the static powder line shape. The interplay between the two NMR tensors is responsible for the observed field dependence of the static 17O NMR spectra. From an analysis of these static powder line shapes, we were able to obtain the principal components of the 17O CS tensor and their relative orientations with respect to the 17O QC tensor. All experimental 17O NMR tensor parameters determined for the six oxygen atoms in α-D-glucose are summarized in Table 1. In general, the values of |CQ(17O)| found in α-D-glucose are about 8–10 MHz with ηQ close to 1. These parameters are similar to those previously reported for protected carbohydrate compounds,25D-glucosamine,26 and several other related functional groups such as hemiacetal/hemiketal,51,52 gem-diol,53 hydroxyl,54 and phenolic groups.55–57 Because the six oxygen-containing functional groups in α-D-glucose are very similar, their 17O isotropic chemical shifts, δiso(17O), are found within a small range of 60 ppm. Nonetheless, there is a general trend in the observed δiso(17O) values: O5 (C–O–C part of a cyclic hemiacetal) > O1 (C–OH part of a cyclic hemiacetal) > O2, O3, O4 (secondary alcohol groups) > O6 (a primary alcohol group). These agree with previous solution 17O NMR studies58,59 as well as with our own measurements for the 17O-labeled D-glucose compounds in aqueous solution (see ESI†). Compared with the 17O NMR parameters found for crystalline hydrates,60–64 the values of |CQ(17O)| for the alcohol and ether groups in α-D-glucose are somewhat larger, but the spans of the 17O CS tensors are comparable.
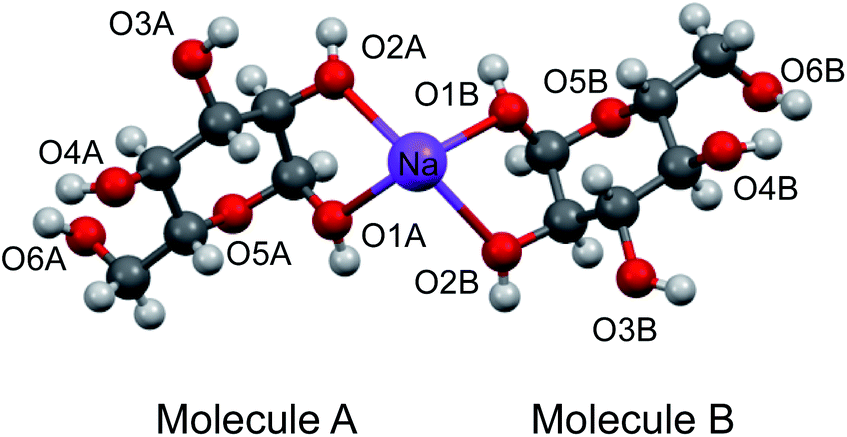
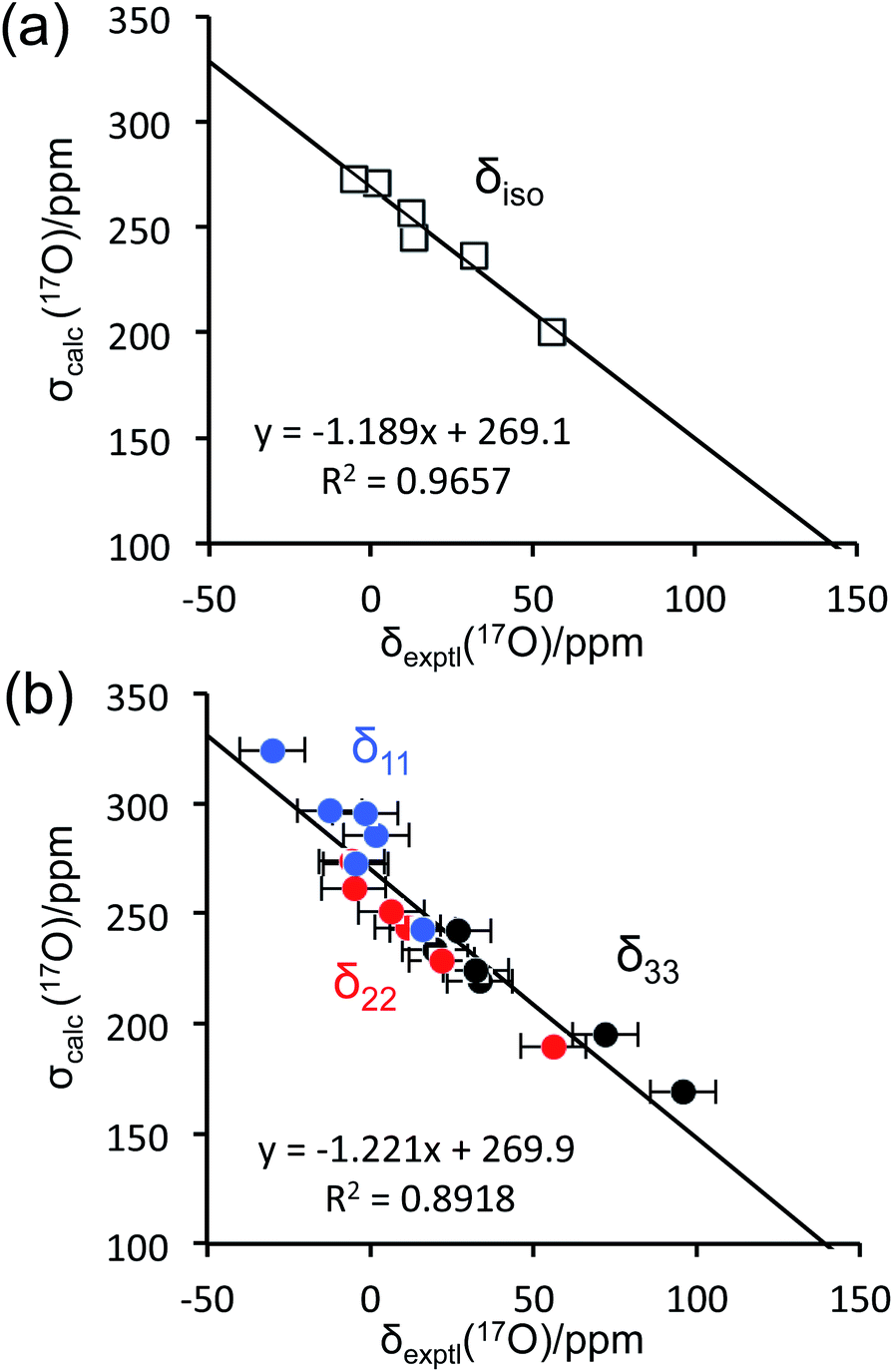
To aid the interpretation of experimentally determined 17O NMR tensor parameters, we performed extensive GIPAW DFT computations. As mentioned earlier, the choice of making D-glucose/NaCl/H2O cocrystal for solid-state 17O NMR experiments was based on the considerations for having a pure anomeric form and easy preparation of crystalline samples. Now this turns into a computational challenge, because the D-glucose/NaCl/H2O cocrystal has a very large unit cell (trigonal space group P31, a = 16.836 Å, c = 17.013 Å, V = 4176 Å3, Z = 9) that contains six crystallographically distinct glucose molecules in the asymmetric unit.29 Careful examination of the crystal structure reveals that the six crystallographically unique D-glucose molecules form three “dimers” via Na+ chelation with the O1 and O2 atoms, as depicted in Fig. 2. Furthermore, the asymmetric unit contains three water molecules, each involved in hydrogen bonding with both two symmetry-related D-glucose molecules and one Cl− ion. In the original crystal structure,29 one of the water molecules was missing a single hydrogen atom, which was added back into the structure before the geometry was optimized using DFT. As a result, all three water molecules have a similar hydrogen-bonding environment (see Fig. S6 in the ESI†). The GIPAW DFT results obtained with the PBE method for 17O NMR parameters are listed in Table 2; complete GIPAW DFT results from all four methods are given in the ESI.† It can be seen from Table 2 that all six crystallographically independent D-glucose molecules have similar 17O NMR parameters (vide infra). Thus, within the spectral resolution limit of the 1D 17O MAS spectra, we can assume just one 17O NMR signal for each oxygen position. For this reason, Fig. 3 shows comparison between experimental 17O CS tensor parameters and “averaged” GIPAW DFT results (averaged over the six crystallographically independent glucose molecules in the asymmetric unit). Because the 17O chemical shift anisotropies are rather small in glucose, the agreement seen in Fig. 3 is clearly satisfactory. Since the 17O QC tensor parameters do not show much variation, we will not examine them further, except to note that the GIPAW DFT calculations are consistent with the experimental results.
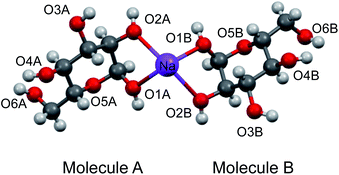 |
| | Fig. 2 Partial crystal structure of D-glucose/NaCl/H2O (2/1/1) cocrystal29 displaying one of the three Na+-chelated glucose “dimers” in the asymmetric unit. While the two α-D-glucose molecules within each dimer, A and B, are crystallographically distinct, they are nonetheless related by an approximate two-fold axis perpendicular to the page plane. The two axial ligands to complete the octahedron geometry around the Na+ ion are O4 atoms from neighboring glucose “dimers”, but are omitted for clarity. | |
Table 2 GIPAW DFT results on 17O NMR parameters computed with the PBE method for the six crystallographically distinct α-D-glucose molecules in the asymmetric unit of D-glucose/NaCl/H2O (2/1/1) cocrystal
| Mol |
Atoma |
Atomb |
σ
iso/ppm |
σ
11/ppm |
σ
22/ppm |
σ
33/ppm |
C
Q/MHz |
η
Q
|
|
Atomic numbering according to Scheme 1.
Atomic numbering in the original crystal structure (CCDC 1281434.cif).29
|
| A1 |
O1 |
O4 |
234.4 |
193.4 |
226.1 |
283.6 |
−9.166 |
0.84 |
| O2 |
O5 |
271.7 |
240.6 |
275.1 |
299.5 |
9.965 |
0.98 |
| O3 |
O6 |
248.6 |
229.2 |
241.5 |
275.2 |
9.514 |
0.99 |
| O4 |
O7 |
260.6 |
229.2 |
256.0 |
296.6 |
9.915 |
0.96 |
| O5 |
O8 |
199.4 |
166.8 |
189.2 |
242.3 |
10.95 |
0.86 |
| O6 |
O9 |
281.0 |
248.1 |
269.4 |
325.3 |
9.88 |
0.90 |
| A2 |
O1 |
O22 |
236.5 |
194.2 |
228.3 |
287.1 |
−9.196 |
0.89 |
| O2 |
O23 |
272.3 |
242.1 |
275.8 |
298.9 |
10.06 |
0.96 |
| O3 |
O24 |
249.0 |
228.9 |
242.2 |
276.1 |
9.579 |
0.99 |
| O4 |
O25 |
260.2 |
228.5 |
255.5 |
296.5 |
9.923 |
0.96 |
| O5 |
O26 |
198.4 |
166.2 |
188.9 |
240.0 |
10.91 |
0.87 |
| O6 |
O27 |
281.3 |
251.2 |
268.8 |
324 |
9.982 |
0.90 |
| A3 |
O1 |
O34 |
236.6 |
195.2 |
229.3 |
285.4 |
−9.168 |
0.86 |
| O2 |
O35 |
271.8 |
241.1 |
274.4 |
299.8 |
9.947 |
0.98 |
| O3 |
O36 |
248.8 |
229.0 |
241.7 |
275.6 |
9.571 |
0.98 |
| O4 |
O37 |
260.6 |
228.7 |
256.4 |
296.7 |
9.922 |
0.96 |
| O5 |
O38 |
199.5 |
167.4 |
188.8 |
242.3 |
10.96 |
0.86 |
| O6 |
O39 |
280.9 |
248.5 |
269.4 |
324.9 |
9.834 |
0.91 |
| B1 |
O1 |
O10 |
238.4 |
196.0 |
231.3 |
288.0 |
−9.162 |
0.93 |
| O2 |
O11 |
269.8 |
243.5 |
272.2 |
293.8 |
9.964 |
0.97 |
| O3 |
O12 |
241.8 |
209.2 |
246.9 |
269.4 |
10.11 |
0.89 |
| O4 |
O13 |
253.2 |
219.5 |
245.2 |
294.9 |
10.22 |
0.92 |
| O5 |
O14 |
201.5 |
171.5 |
190.2 |
242.8 |
10.89 |
0.89 |
| O6 |
O15 |
265.3 |
218.9 |
253.4 |
323.6 |
9.806 |
0.92 |
| B2 |
O1 |
O16 |
237.0 |
196.7 |
229.9 |
284.5 |
−9.109 |
0.89 |
| O2 |
O17 |
269.3 |
241.8 |
271.7 |
294.5 |
9.870 |
0.99 |
| O3 |
O18 |
241.0 |
209.2 |
245.1 |
268.6 |
10.01 |
0.90 |
| O4 |
O19 |
253.6 |
219.9 |
246.3 |
294.7 |
10.21 |
0.91 |
| O5 |
O20 |
202.7 |
171.7 |
190.8 |
245.4 |
10.92 |
0.89 |
| O6 |
O21 |
264.2 |
215.3 |
253.9 |
323.3 |
9.673 |
0.92 |
| B3 |
O1 |
O28 |
236.3 |
195.0 |
228.0 |
286.0 |
−9.153 |
0.91 |
| O2 |
O29 |
269.3 |
242.8 |
272.1 |
292.9 |
9.993 |
0.97 |
| O3 |
O30 |
241.4 |
208.9 |
245.1 |
270.2 |
10.02 |
0.90 |
| O4 |
O31 |
252.9 |
219.6 |
244.2 |
294.9 |
10.20 |
0.92 |
| O5 |
O32 |
201.1 |
170.4 |
190.6 |
242.4 |
10.88 |
0.88 |
| O6 |
O33 |
265.7 |
220.0 |
252.5 |
324.7 |
9.891 |
0.91 |
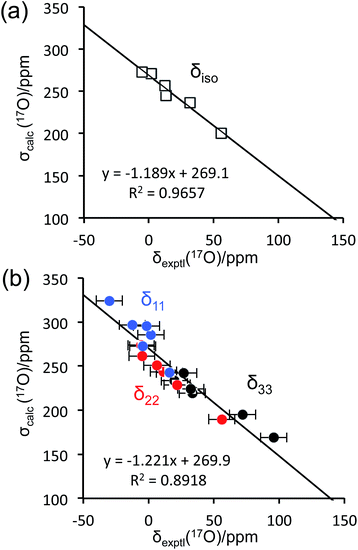 |
| | Fig. 3 Comparison between experimental 17O chemical shifts (δ) and GIPAW DFT computed magnetic shielding values (σ) for the α-D-glucose/NaCl/H2O cocrystal: (a) isotropic values; (b) principal CS tensor components. The root mean square errors (RMSE) are: (a), 4.5 ppm; (b), 12.9 ppm. | |
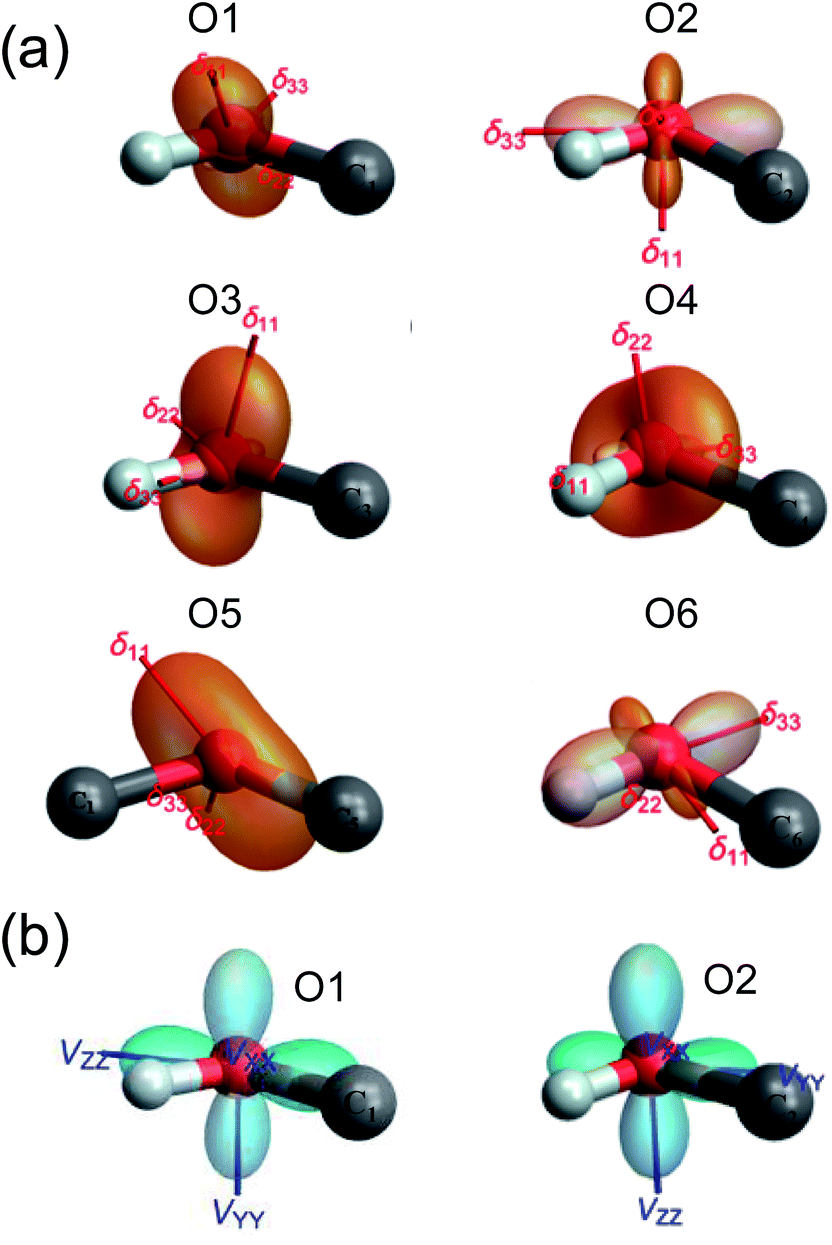
GIPAW DFT computations also yielded further information about the 17O NMR tensor orientations in the molecular frame. In Fig. 4, we used TensorView65 to display the ovaloid representation of the 17O CS and QC tensors for the six oxygen sites in α-D-glucose. Two general types of orientation were found for the 17O CS tensors, as seen in Fig. 4(a). For O1, O2, and O3, the direction of δ11 appears to be almost perpendicular to the H–O–C plane. For O4, O5, and O6, however, it is the δ22 component that is perpendicular the H–O–C or C–O–C plane. In all cases, δ33 lies approximately parallel to the H–O or C–O bonds. Because the 17O chemical shift anisotropies in α-D-glucose are generally small, it is difficult to detect any other general trends. Unlike the 17O CS tensors, the 17O QC tensors for the O–H and C–O–C groups in α-D-glucose were found to be invariant. Although Fig. 4(b) displays two types of QC tensor orientations in the molecular frame, the two seemingly different orientations are essentially the same. This is because the largest QC tensor or EFG tensor component (Vzz) is defined according to its absolute value so that |Vzz| ≥ |Vyy| ≥ |Vxx|. Because all the C–O–H and C–O–C groups in α-D-glucose exhibit ηQ ≈ 1, the two high tensor components in each case would have very similar magnitudes but opposite signs. The one with the negative sign lies in-plane being perpendicular to the bisector of the C–O–H or C–O–C angle, whereas the one with the positive sign is perpendicular to the C–O–H or C–O–C plane. The smallest QC or EFG tensor component bisects the C–O–H or C–O–C angle; but because this component is always very small for ηQ ≈ 1, it is hardly seen in the ovaloid representation shown in Fig. 4(b). If the tensor component with the negative sign is of slightly greater magnitude, its direction is defined as Vzz, so CQ(17O) < 0. The 17O QC tensor for O1 was found to belong to this case. On the other hand, if the component with the positive sign is larger, CQ(17O) > 0. The 17O QC tensors for the O2, O3, O4, O5 and O6 atoms in α-D-glucose were found to belong to this case. However, if the individual tensor components in the molecular frame are directly compared, the two 17O QC tensor orientations shown in Fig. 4(b) are rather similar. The link between the tensor orientation and the sign of CQ(17O) was recently explained with the concept of valence p-orbital population anisotropy (VPPA).66 Since the 17O QC tensor is invariant with respect to the molecular frame for the C–O–H and C–O–C groups, it is worth pointing out that one can use the 17O QC tensor as an internal reference to link the 17O CS tensor to the molecular frame once the relative orientation between the QC and CS tensors were experimentally determined.
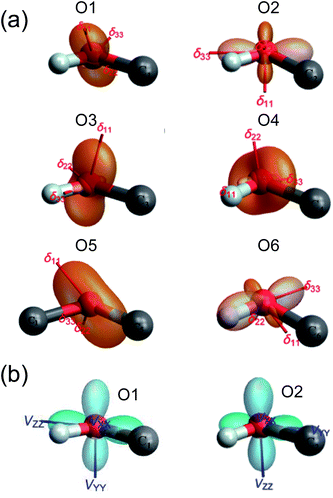 |
| | Fig. 4 The ovaloid representation of computed 17O CS (a) and QC (b) tensor orientations in the molecular frame of α-D-glucose. In (b), the 17O QC tensor orientations for O3, O4, O5, and O6 are the same as that shown for O2. See text for discussion. | |
Solid-state 17O NMR at high magnetic fields
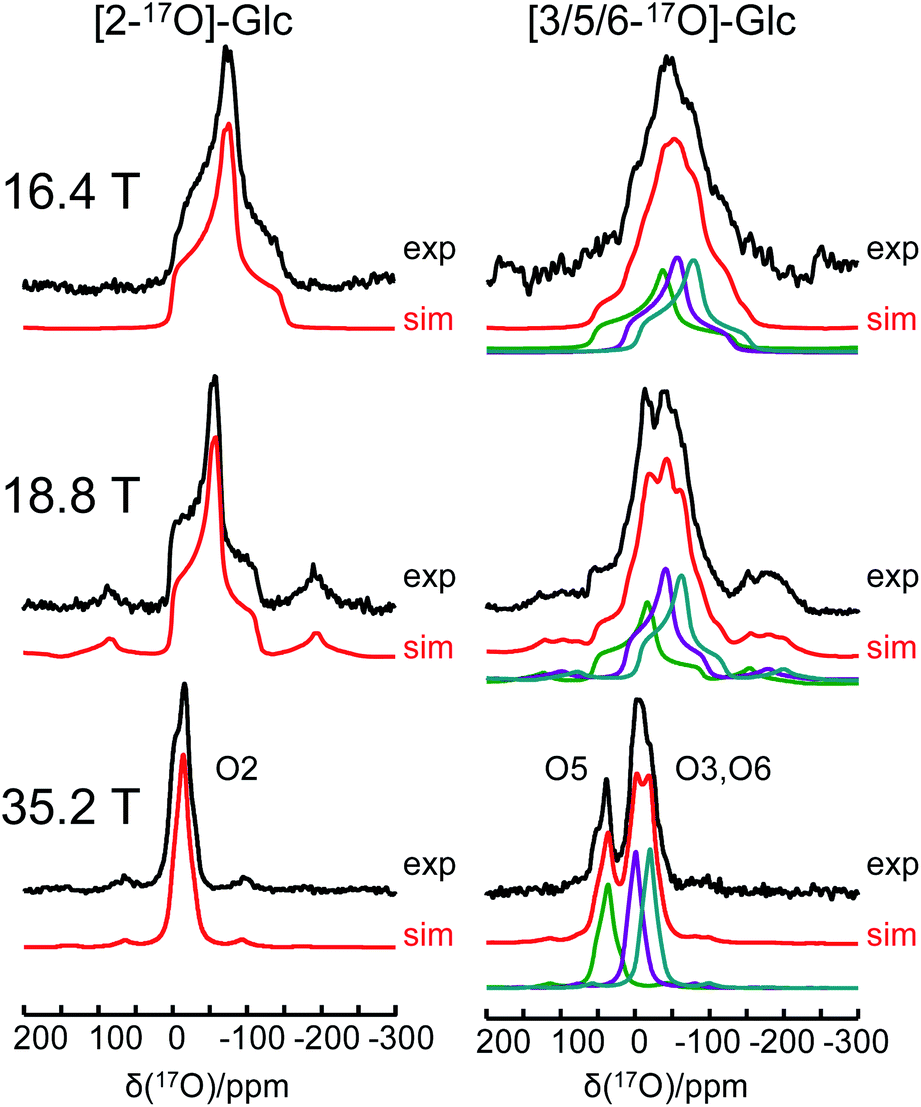
One of the major challenges in solid-state 17O NMR studies of carbohydrate compounds is that all oxygen-containing functional groups are either hydroxyl or ether groups. As a result, they exhibit very similar 17O NMR parameters. For example, the 17O isotropic chemical shifts for the six oxygen sites in α-D-glucose, given in Table 1, are within a narrow range of 60 ppm. If multiple oxygen sites are simultaneously 17O-labeled, it could be very difficult to resolve their 17O NMR signals because each signal would be significantly broadened by the second-order quadrupole interaction. Since the second-order quadrupole interaction is inversely proportional to the applied magnetic field, it is often advantageous to perform solid-state 17O NMR experiments at the highest possible magnetic field. To test the limit of this brute-force approach, we obtained 17O MAS NMR spectra for [2-17O]-α-D-glucose and [3/5/6-17O]-α-D-glucose at three magnetic fields, 16.4, 18.8, and 35.2 T. As seen from Fig. 5, the 17O NMR signals are progressively sharpened as the applied magnetic field strength increases. For example, at 16.4 T, the line width at the base of the signal for [2-17O]-α-D-glucose is 200 ppm. This line width is reduced to approximate 50 ppm at 35.2 T. Remarkably, at 35.2 T, the three oxygen sites in [3/5/6-17O]-α-D-glucose become partially resolved. It is also interesting to compare the 17O MAS NMR spectrum for [3/5/6-17O]-α-D-glucose shown in Fig. 5 with the 17O QCT spectrum reported by Gan et al.28 for the same compound in the slow motion regime. In isotropic liquids, the 17O NMR signals are broadened by the intrinsic transverse (T2) spin relaxation. In the slow motion regime (ω0τc ≫ 1), the quadrupole relaxation becomes multi-exponential with only the slow quadrupole relaxation component corresponding to the central transition being detected in the 17O QCT spectra. At 35.2 T, the 17O QCT signals are significantly narrower than the MAS signals. One additional benefit for studying carbohydrates at ultrahigh magnetic fields is that the oxygen sites in carbohydrates exhibit rather small 17O chemical shift anisotropies (CSAs). As seen from Fig. 5, no significant spinning sidebands are observed at 35.2 T. In contrast, 17O MAS NMR signals obtained at 35.2 T from protein backbone oxygen atoms display many spinning sidebands.14 Because the cross-relaxation between CSA and second-order quadrupole interactions becomes more important at high magnetic fields, 17O QCT spectra will display higher resolution for carbohydrates (with small CSAs) than for proteins (with large CSAs).67
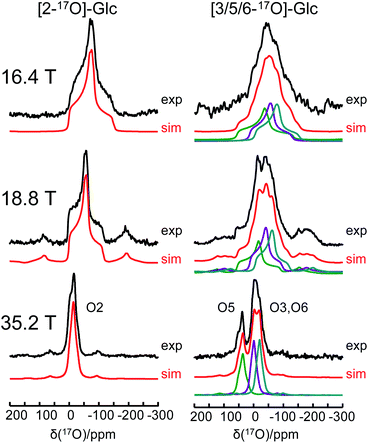 |
| | Fig. 5 Experimental (black traces) and simulated (red traces) 17O MAS NMR spectra of [2-17O]-α-D-glucose and [3/5/6-17O]-α-D-glucose at three magnetic fields. | |
Combination of paramagnetic doping and CPMAS CryoProbe technology
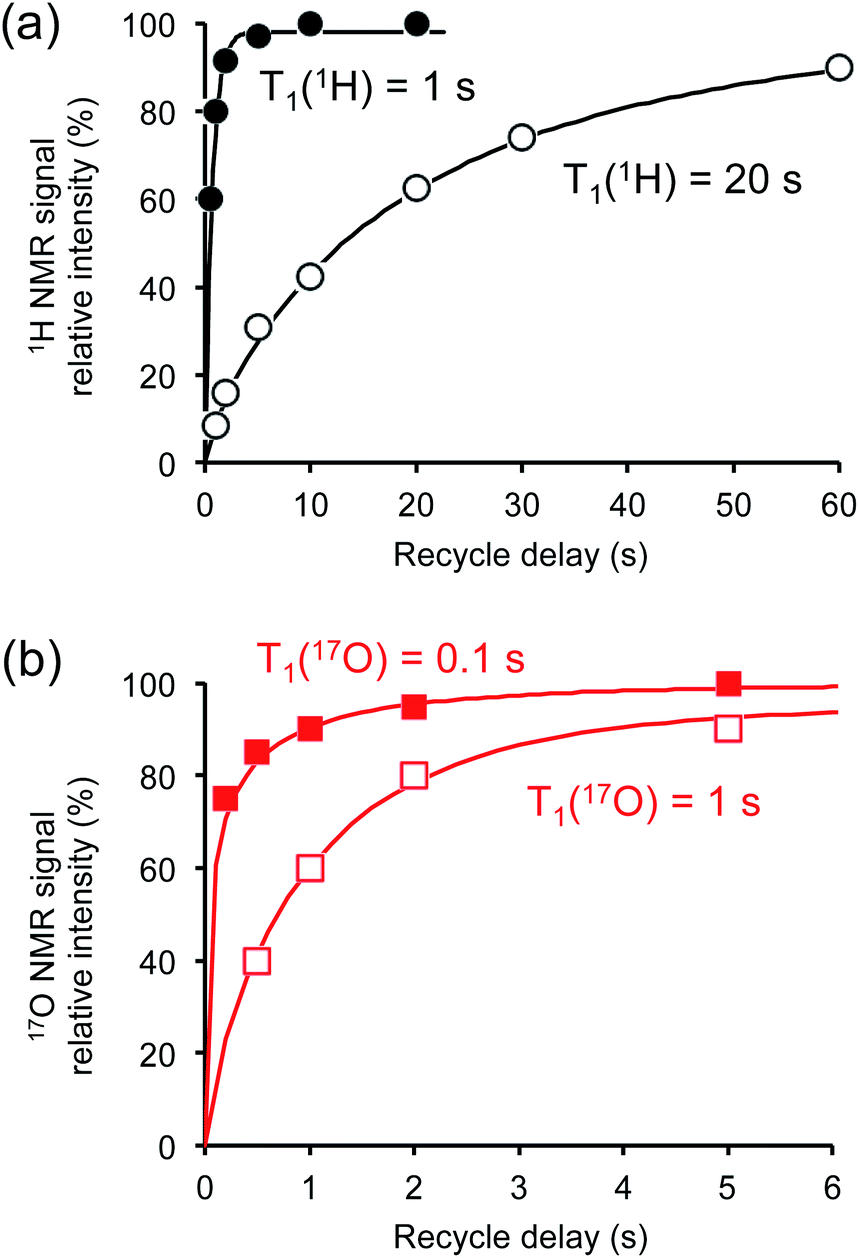
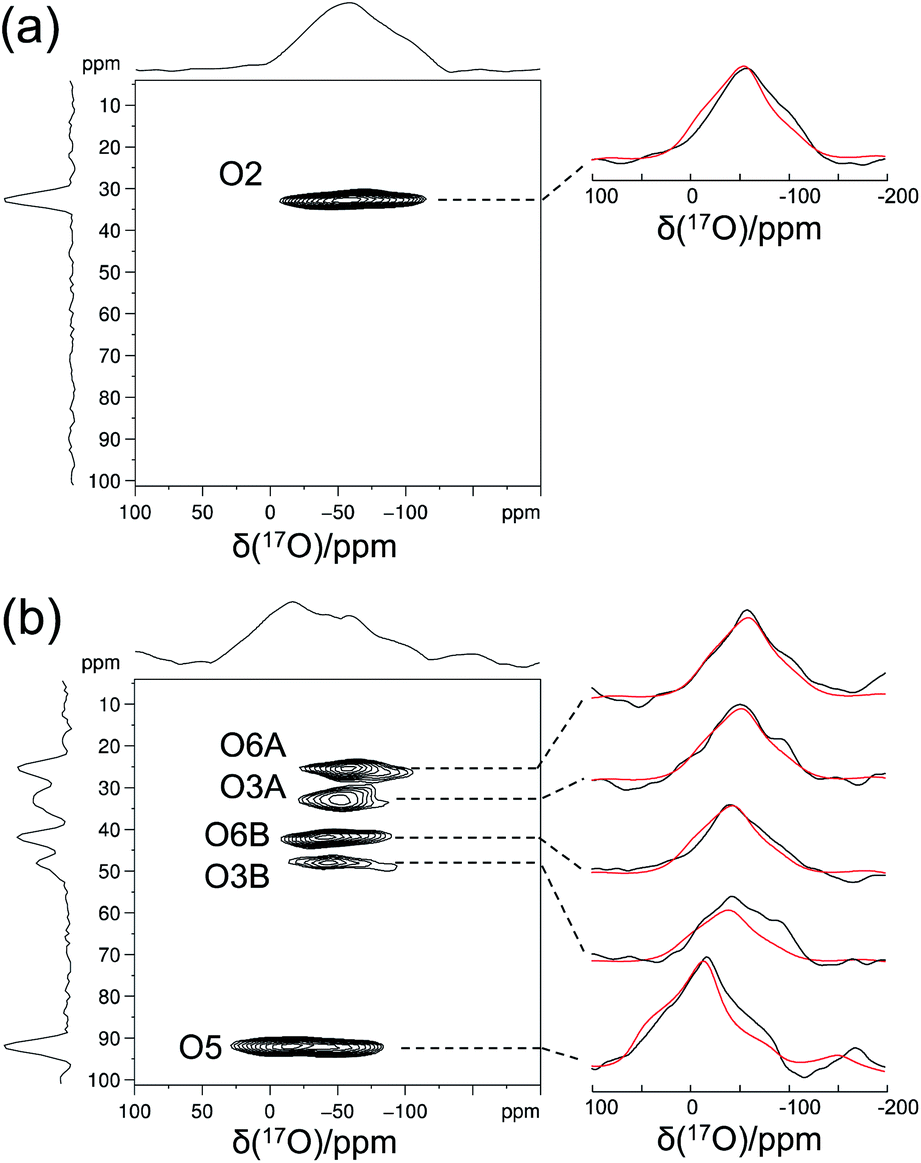
Crystalline D-glucose is known for its exceedingly long T1(1H) values. It was observed that T1(17O) values are also long for D-glucose compounds, hindering rapid repetition of 17O data acquisition. One common approach that has been widely employed in cross polarization (CP)-based solid-state 13C NMR studies is to add paramagnetic Cu(II) dopants to shorten T1(1H).68–70 In this work, we hypothesized that the same paramagnetic doping approach might be useful for 17O NMR studies as well. To this end, we prepared two D-glucose/NaCl/H2O cocrystal samples, [2-17O]-α-D-glucose and [3/5/6-17O]-α-D-glucose, each containing 10% (w/w) Na2[Cu(EDTA)2]. Fig. 6 shows the effects of paramagnetic doping on the 1H and 17O NMR signals of [2-17O]-α-D-glucose. We found that paramagnetic doping at the 10% (w/w) level shortens the T1(1H) and T1(17O) values in [2-17O]-α-D-glucose by about 20 and 10 times, respectively. The [3/5/6-17O]-α-D-glucose cocrystal sample containing Cu(II)-EDTA exhibited similar results. This shortening of T1(17O) allowed more rapid data acquisition, effectively enhancing the sensitivity by approximately a factor of 2. However, the reduction of T1(17O) alone is still insufficient when performing more demanding experiments such as 2D 17O multiple-quantum MAS71,72 for the α-D-glucose samples prepared in this work. To further increase sensitivity, we combined the paramagnetic doping with new CPMAS CryoProbe technology. It has recently been shown that a CPMAS CryoProbe provides a 3–4 times higher sensitivity for detecting 13C and 15N nuclei compared to a conventional MAS probe.32 After the submission of this work, we learned that Michaelis and co-workers73 also obtained some preliminary solid-state 17O NMR data using the CPMAS CryoProbe. For acquiring 17O MAS spectra for the α-D-glucose compounds, we found that the combination of paramagnetic doping and CPMAS CryoProbe yielded a sensitivity gain by a factor of 6–8. Fig. 7 shows the 2D 17O 3QMAS spectra obtained for [2-17O]-α-D-glucose and [3/5/6-17O]-α-D-glucose samples doped with Cu–EDTA. This is the first time that 2D 17O 3QMAS spectra are reported for carbohydrate compounds. It should be emphasized that the level of 17O enrichment in the [3/5/6-17O]-α-D-glucose sample was only about 10%. Thus, the observed sensitivity shown in Fig. 7 is quite remarkable. Interestingly, whereas each of the O3 and O6 signals appears to split into two signals, no signal splitting was observed for the O2 and O5 signals (vide infra). We were able to fit the F2-slice spectra and obtained the following 17O NMR parameters: O2, δiso = 2 ppm, CQ = 9.1 MHz, ηQ = 1.0; O3A, δiso = 6 ppm, CQ = 8.8 MHz, ηQ = 0.9; O3B, δiso = 12 ppm, CQ = 8.8 MHz, ηQ = 0.9; O6A, δiso = −6 ppm, CQ = 8.8 MHz, ηQ = 0.9; O6B, δiso = 8 ppm, CQ = 8.8 MHz, ηQ = 0.9; O5, δiso = 56 ppm, CQ = 9.9 MHz, ηQ = 1.0. These values are also confirmed by the signal positions in the isotropic dimension of the 17O 3QMAS spectrum; see ESI.† As expected, the 17O NMR parameters for O2 and O5 are identical to those extracted from 1D MAS spectra as listed in Table 1. For O3 and O6, in contrast, the unprecedented spectral resolution offered by 2D 17O 3QMAS spectra revealed finer spectral details. We will further discuss these new details in the next section.
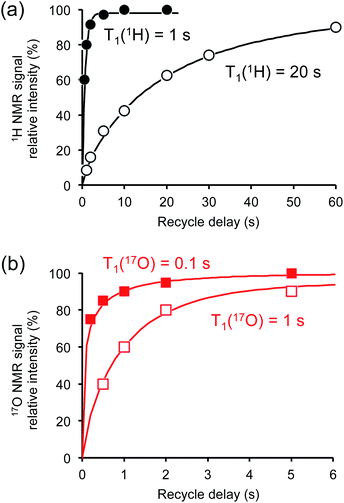 |
| | Fig. 6 Effects of paramagnetic Cu(II)–EDTA doping on (a) 1H and (b) 17O spin–lattice relaxation times. All measurements were carried out at 14.1 T for the [2-17O]-D-glucose/NaCl/H2O cocrystal with (closed symbols) and without (open symbols) added Cu–EDTA. | |
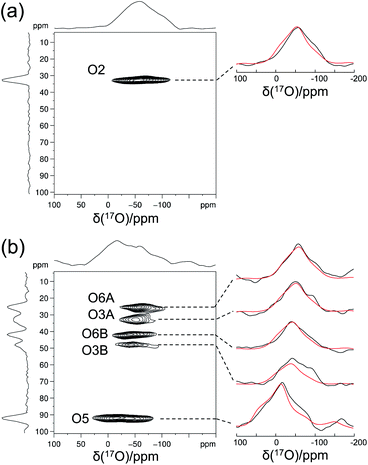 |
| | Fig. 7 2D 17O 3QMAS spectra of (a) [2-17O]-α-D-glucose and (b) [3/5/6-17O]-α-D-glucose obtained at 18.8 T with a Bruker 3.2 mm CPMAS CryoProbe. The sample spinning frequency was 15 kHz. Each sample contains 10% (w/w) Cu–EDTA. Experimental (black traces) and simulated (red traces) F2-slice spectra are displayed on the side. Data acquisition parameters are: (a), recycle delay 1 s, 48 t1 increments (with apodization weighted sampling, n0 = 1536 transients), total experimental time 10 h; (b) recycle delay 1 s, 32 t1 increments (with apodization weighted sampling, n0 = 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 transients), total experimental time 6 days. 680 transients), total experimental time 6 days. | |
Further 17O and 13C NMR signal assignments
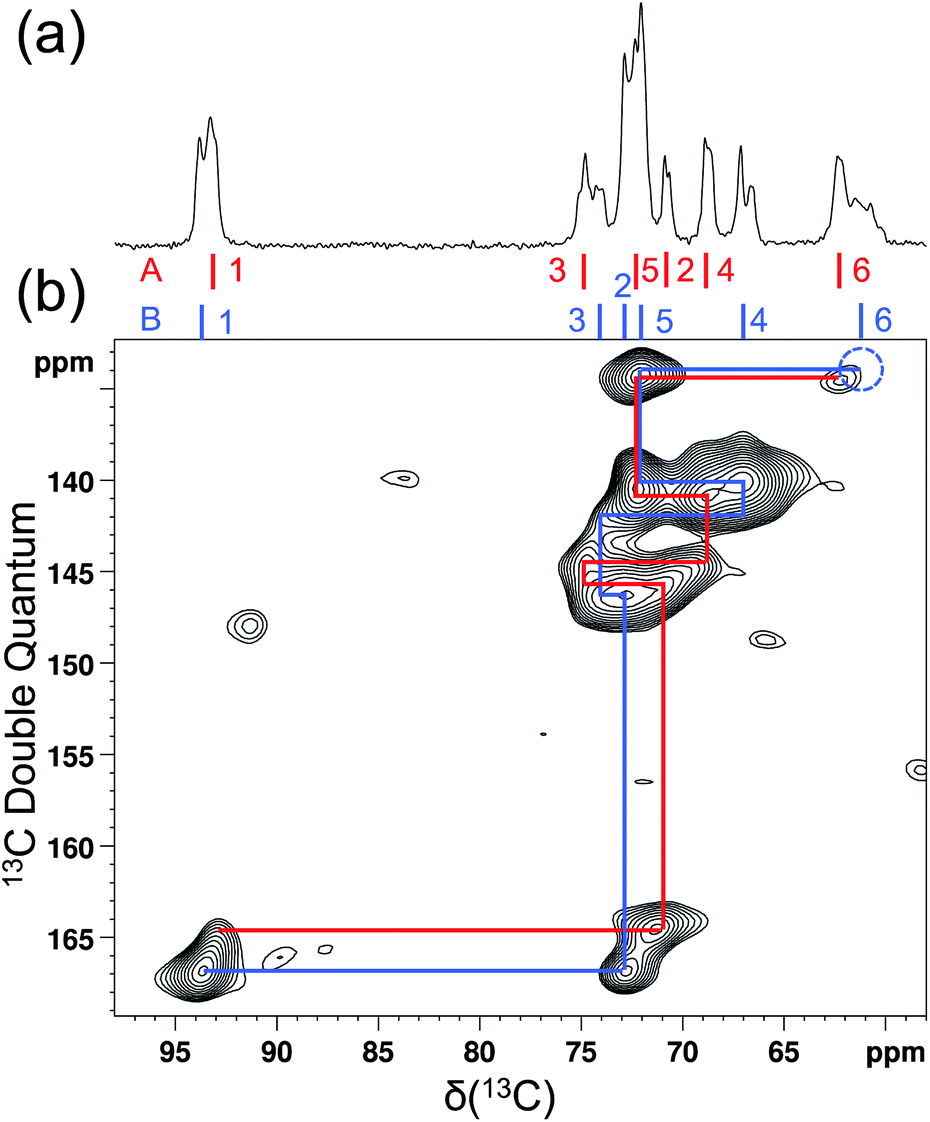
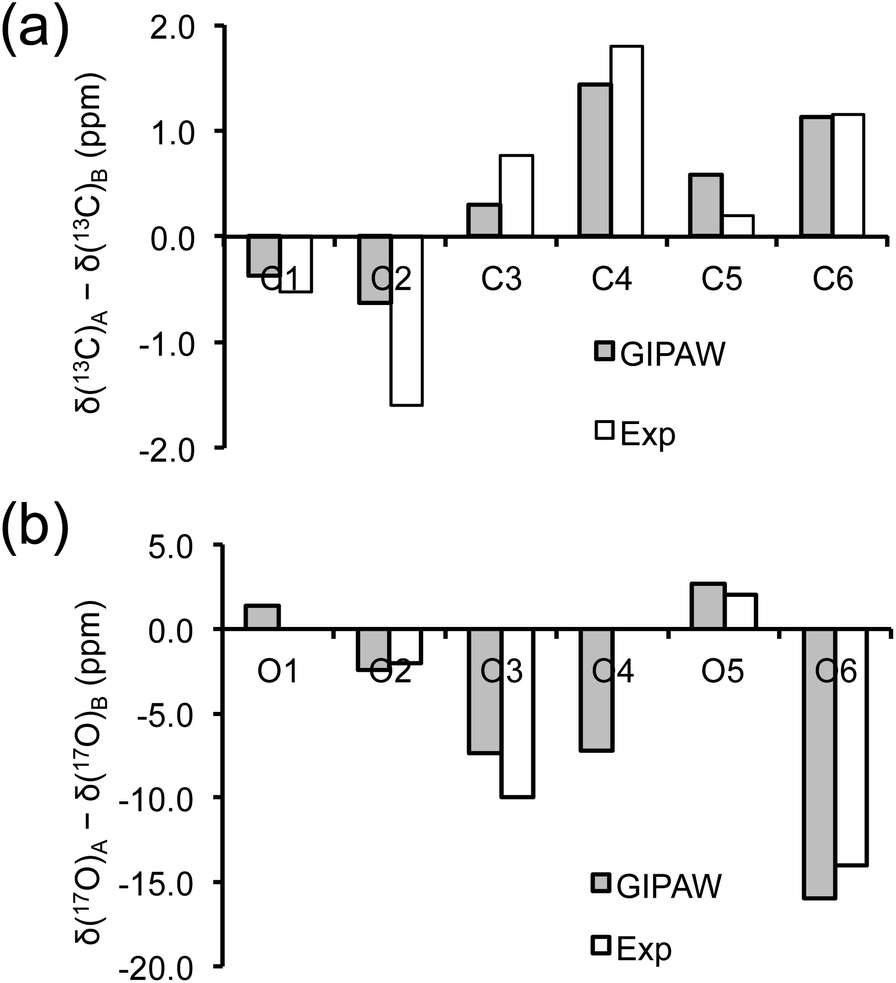
As mentioned earlier, there are six crystallographically independent glucose molecules in the asymmetric unit of D-glucose/NaCl/H2O cocrystal. Thus, in principle, there should be six 17O NMR signals for each oxygen atom in this compound. However, the six crystallographically independent glucose molecules form three Na+-chelated glucose “dimers” with very similar structures. For this reason, the two different signals observed for each of the O3 and O6 groups in the 2D 17O 3QMAS spectrum shown in Fig. 7 can be attributed to the two types of α-D-glucose molecules, A and B, within each Na+-chelated glucose “dimer”. This also implies that the difference among the three “dimers” cannot be detected with the current spectral resolution. The tentative signal assignments shown in Fig. 7 were based on the GIPAW DFT calculations listed in Table 2. To further confirm this hypothesis, we decided to fully assign the solid-state 13C NMR signals for the same α-D-glucose sample. To this end, we obtained a 2D refocused INADEQUATE NMR spectrum at the 13C natural-abundance isotope level for the same compound using the CPMAS CryoProbe. As seen from Fig. 8, a similar signal “doubling” was indeed observed for each carbon atom. Fig. 8 also shows the 13C NMR signal assignment for Molecules A and B, based on GIPAW DFT results for 13C chemical shifts (provided in the ESI). In fact, in the 1D 13C CPMAS spectrum shown in Fig. 8, there are also hints that smaller resonance splittings beyond the signal “doubling” are also present for C1, C2A, C3, C4, and C6B. Unfortunately, within the currently achievable spectral resolution, it is not possible to resolve all six 13C NMR signals for each site. So, for now we focus on the chemical shift differences between Molecules A and B within the glucose “dimer”. Clearly, for different carbon sites, the 13C chemical shift differences between Molecules A and B show different patterns. We will further examine these patterns for all the carbon and oxygen atoms in α-D-glucose. Fig. 9 shows a comparison between experimental and GIPAW DFT results with the PBE method for both 13C and 17O chemical shifts; complete GIPAW DFT results from all four methods are provided in the ESI.† The observed general agreement between experiment and computation suggests that the reported signal assignment is quite reasonable. Now we can understand why no “doubling” or “splitting” was observed for the O2 and O5 signals in the 17O 3QMAS spectra shown in Fig. 7. As seen from Fig. 9, the GIPAW DFT calculations predict that the 17O chemical shift difference between Molecules A and B is indeed rather small for O2 and O5 (<3 ppm). It is also evident from Fig. 9 that the 17O chemical shift is a much more sensitive probe than the 13C chemical shift to any structural variation. In practice, however, the generally lower spectral resolution encountered in 17O NMR often makes it difficult to fully utilize such sensitivity. On the other hand, it is also not difficult to imagine that, in some cases, the superior sensitivity of 17O NMR to molecular structure and chemical bonding can produce information that is unobtainable by 13C NMR. Ideally, one should utilize all available magnetically-active nuclei in a molecular system as a general approach of “NMR crystallography”.74
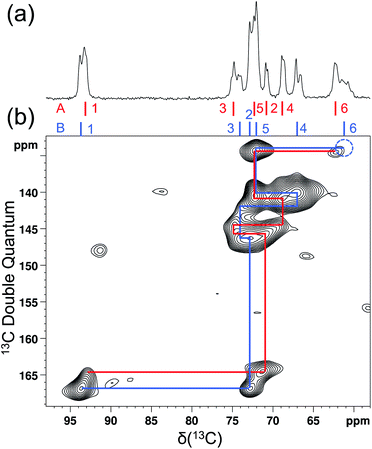 |
| | Fig. 8 Natural abundance 13C (a) 1D CPMAS and (b) 2D refocused INADEQUATE NMR spectra of [2-17O]-α-D-glucose doped with 10% (w/w) Cu–EDTA. The dotted blue circle indicates the absence of the C6B signal due to its relatively short T2 (3.7 ms). Both spectra were obtained at 18.8 T with a Bruker 3.2 mm CPMAS CryoProbe. The sample spinning frequency was 15 kHz. Data acquisition parameters are: (a), 1D CP/MAS, contact time 3 ms, recycle time 3.7 s, 16 transients; (b), 2D refocused INADEQUATE, recycle delay 2 s, 1536 transients per t1, 34 t1 increments, J-evolution period of 3.99 ms, total experimental time 30 h. | |
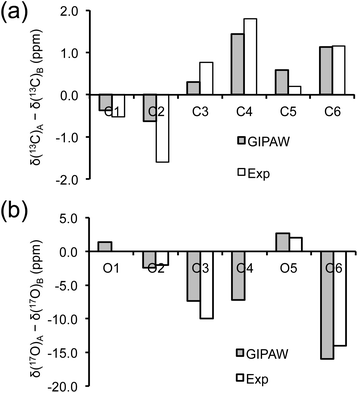 |
| | Fig. 9 Comparison between observed and GIPAW DFT calculated (a) 13C and (b) 17O chemical shift differences between Molecules A and B in α-D-glucose. In (b), because the line width observed in the 3Q isotropic dimension for the O2 and O5 3QMAS signals was about 5 ppm, the upper limit of any potential signal splittings for O2 and O5 was estimated to be 2 ppm. | |
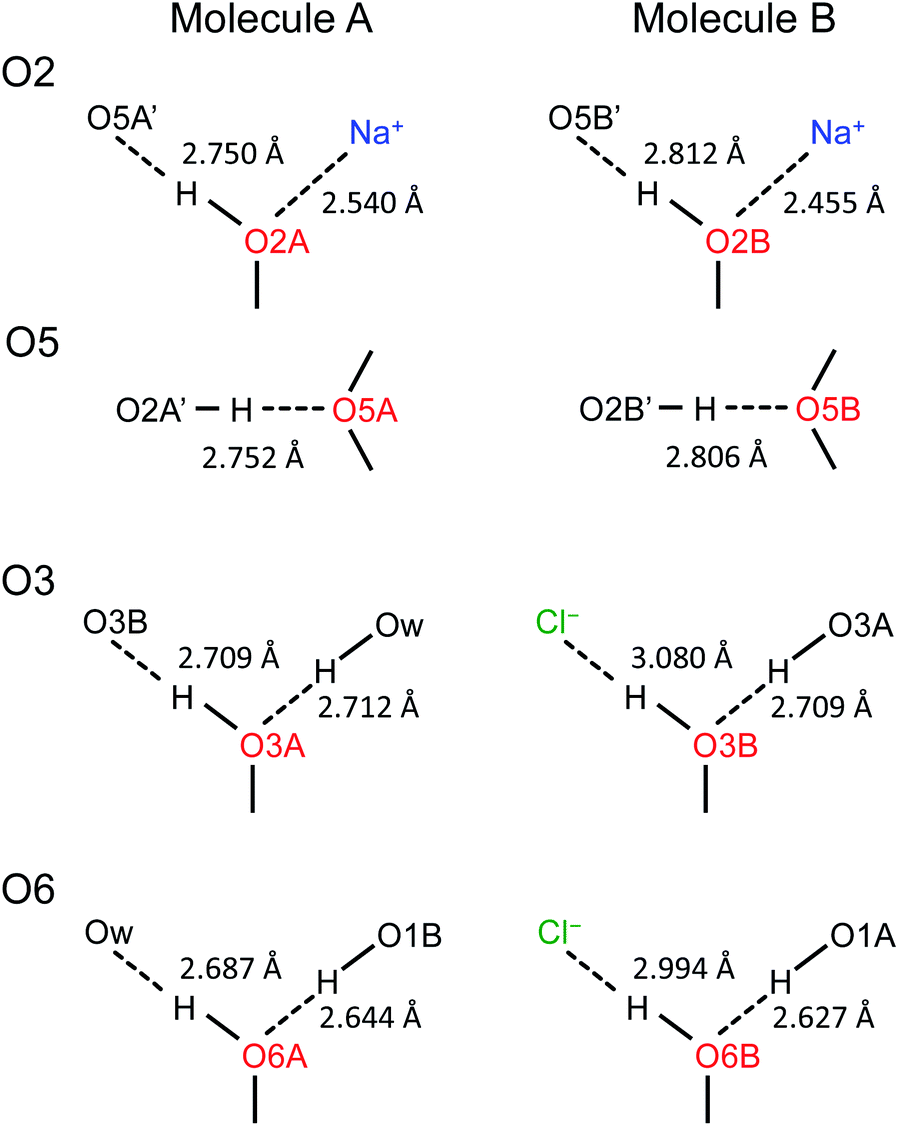
Now, what are the reasons for the 17O chemical shift differences between Molecules A and B to show the patterns displayed in Fig. 9? Why do the O2 and O5 atoms between Molecules A and B exhibit very similar 17O chemical shifts (within 2 ppm), but the O3 and O6 atoms have so different values (by more than 10 ppm)? To link the structural features to these spectral characteristics, we will need to further examine the crystal structure of the D-glucose/NaCl/H2O cocrystal. Fig. 10 summarizes the hydrogen-bonding and ion-coordination environments around the O2, O5, O3 and O6 atoms in Molecules A and B. Clearly, the O2 and O5 atoms have essentially the same hydrogen-bonding and ion-coordination environments between Molecules A and B. In both Molecules A and B, the O2 atom forms a hydrogen bond of the O–H⋯O type and is also coordinated to a Na+ ion. In sharp contrast, the O3 and O6 atoms display quite different hydrogen-bonding environments between Molecules A and B. As seen from Fig. 10, the key structural difference is the replacement of a neutral O–H⋯O hydrogen bond in Molecule A by a stronger ionic O–H⋯Cl− hydrogen bond in Molecule B. Thus, both O3 and O6 experience stronger hydrogen-bonding environments in Molecule B than in Molecule A. For example, the hydrogen bond enthalpies for the H–O–H⋯OH2 and H–O–H⋯Cl− dimers are 3.6 and 13.5 kcal mol−1, respectively.75 While hydrogen-bonding effects on 17O NMR parameters are well known for carbonyl compounds,76–86 data on hydroxyl and ether functional groups are scarce in the literature. The best-known case is that the 17O NMR signal from gaseous H2O was found at δ(17O) = −36.1 ppm with respect to that from liquid H2O, δ(17O) = 0 ppm.87–89 This means that hydrogen-bonding interactions would cause deshielding on the 17O nucleus of the O–H group (thus increase in the δ(17O) value). This general trend was first firmly established by Reuben90 in the study of solvent effects on 17O chemical shifts. The same trend was also observed for the hydronium ion H3O+ in the solid state.91 In the present case of the D-glucose/NaCl/H2O cocrystal, because the O3 and O6 atoms are involved in stronger hydrogen-bonding interactions in B than in A, the values of δ(17O)A − δ(17O)B are negative for both O3 and O6, as seen from Fig. 10. Thus, the unprecedented resolution in the 2D 17O 3QMAS spectra allowed us to detect a subtle structural difference between the two crystallographically distinct molecules. More specifically, we found that replacement of a neutral O–H⋯O hydrogen bond by a stronger ionic O–H⋯Cl− hydrogen bond causes an increase in δ(17O) by ca. 10–14 ppm. Once again, this finding illustrates the remarkable sensitivity of 17O NMR parameters to hydrogen bonding interactions. Interestingly, the GIPAW DFT calculations showed that the protons attached to O3B and O6B are also significantly deshielded by 2–3 ppm, due to the stronger hydrogen bonding, than the corresponding protons attached to O3A and O6A.
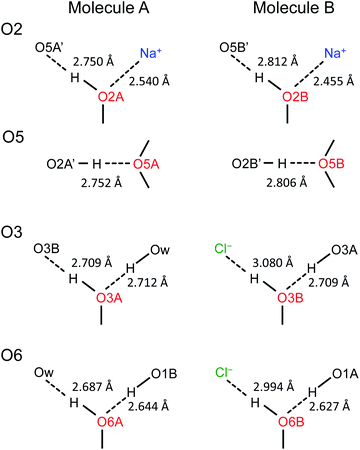 |
| | Fig. 10 Comparison of hydrogen-bonding environments around the O2, O5, O3, and O6 atoms between Molecules A and B in the D-glucose/NaCl/H2O cocrystal. Distances between the two heavy atoms in each hydrogen bond are listed. | |
Conclusions
We have carried out a comprehensive solid-state 17O NMR study for α-D-glucose. In this work, a total of six site-specifically 17O-labeled α-D-glucose compounds were synthesized. The 17O CS and QC tensors were determined for each of the six oxygen sites in α-D-glucose from an analysis of solid-state 17O NMR spectra obtained at multiple magnetic fields. This is the first case where all oxygen-containing functional groups in a carbohydrate molecule are site-specifically 17O-labeled and have their 17O NMR tensors fully characterized. We found that paramagnetic Cu(II) doping can significantly shorten the T1(17O) values for solid α-D-glucose samples, making it possible to rapidly collect 17O NMR data. By combining the paramagnetic doping effect with the new CPMAS CryoProbe technology and apodization weighted sampling at high magnetic fields, we have achieved a significant sensitivity boost that allowed us to obtain the first set of 17O 3QMAS spectra ever reported for carbohydrate compounds. The unprecedented resolution offered by 2D 17O 3QMAS spectra permitted the detection of a subtle structural difference for a single hydrogen bond between two types of crystallographically distinct D-glucose molecules. With the aid of GIPAW DFT calculations, all observed 17O and 13C NMR signals were assigned to the two groups of crystallographically distinct α-D-glucose molecules. This combined 17O and 13C solid-state NMR approach adds a new dimension to the field of “NMR crystallography”. Successful synthesis of site-specifically 17O-labeled D-glucose also paves the way for researchers to consider 17O NMR as a new spectroscopic tool in glucose-related research, which can range from glucose binding proteins to glucose metabolism of live cells. In a broader context, this work demonstrates that continuing advancement of solid-state 17O NMR spectroscopy has begun to open the door for studying many biological molecules that are usually considered too difficult for 17O NMR spectroscopy. It is about time to add 17O to the NMR toolbox for probing organic and biological molecules.
Author contributions
J. Shen synthesized the 17O-labeled compounds and recorded solid-state 17O NMR data at 14.1 T. VT obtained solid-state 17O NMR spectra at 21.1 T. J. Struppe, AH, and MM acquired the solid-state 17O and 13C NMR data at 18.8 T with the CPMAS CryoProbe. IH and ZG conducted solid-state 17O NMR experiments at 35.2 T. AB performed the GIPAW DFT calculations. GW designed the project, supervised the work, obtained solid-state 17O NMR data at 16.4 T and 13C CPMAS data at 14.1 T, and interpreted the results. All authors contributed to the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh Field NMR Facility for Solids (Ottawa, Canada), a national research facility funded by a consortium of Canadian universities, National Research Council Canada, and Bruker BioSpin (http://nmr900.ca). A portion of this work was performed at the National High Magnetic Field Laboratory (NHMFL), which is supported by the National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida. The SCH magnet and NMR instrumentation were supported by NSF (DMR-1039938 and DMR-0603042) and NIH P41 GM122698. We thank Dr Xianqi Kong for assistance at the early stage of this project.
References
- G. Wu, Biochem. Cell Biol., 1998, 76, 429–442 CrossRef CAS PubMed.
- V. Lemaitre, M. E. Smith and A. Watts, Solid State Nucl. Magn. Reson., 2004, 26, 215–235 CrossRef CAS PubMed.
- G. Wu, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 118–169 CrossRef CAS.
- A. Wong and F. Poli, Annu. Rep. NMR Spectrosc., 2014, 83, 145–220 CrossRef.
- G. Wu, Solid State Nucl. Magn. Reson., 2016, 73, 1–14 CrossRef CAS PubMed.
- G. Wu, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 135–191 CrossRef CAS PubMed.
- J. Palmer and G. Wu, Annu. Rep. NMR Spectrosc., 2021, 103, 1–46 CrossRef.
- J. Zhu, E. Ye, V. Terskikh and G. Wu, Angew. Chem., Int. Ed., 2010, 49, 8399–8402 CrossRef CAS PubMed.
- A. W. Tang, X. Kong, V. Terskikh and G. Wu, J. Phys. Chem. B, 2016, 120, 11142–11150 CrossRef CAS PubMed.
- J. Zhu, I. C. M. Kwan and G. Wu, J. Am. Chem. Soc., 2009, 131, 14206–14207 CrossRef CAS PubMed.
- J. Zhu and G. Wu, J. Am. Chem. Soc., 2011, 133, 920–932 CrossRef CAS PubMed.
- R. P. Young, B. G. Caulkins, D. Borchardt, D. N. Bulloch, C. K. Larive, M. F. Dunn and L. J. Mueller, Angew. Chem., Int. Ed., 2016, 55, 1350–1354 CrossRef CAS PubMed.
- J. Paulino, M. Yi, I. Hung, Z. Gan, X. Wang, E. Y. Chekmenev, H.-X. Zhou and T. A. Cross, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11908–11915 CrossRef CAS PubMed.
- B. Lin, I. Hung, Z. Gan, P. H. Chien, H. L. Spencer, S. P. Smith and G. Wu, ChemBioChem, 2021, 22, 826–829 CrossRef CAS PubMed.
- I. Hung, A. C. Uldry, J. Becker-Baldus, A. L. Webber, A. Wong, M. E. Smith, S. A. Joyce, J. R. Yates, C. J. Pickard, R. Dupree and S. P. Brown, J. Am. Chem. Soc., 2009, 131, 1820–1834 CrossRef CAS PubMed.
- E. G. Keeler, V. K. Michaelis, M. T. Colvin, I. Hung, P. L. Gor’kov, T. A. Cross, Z. Gan and R. G. Griffin, J. Am. Chem. Soc., 2017, 139, 17953–17963 CrossRef CAS PubMed.
- A. Venkatesh, M. P. Hanrahan and A. J. Rossini, Solid State Nucl. Magn. Reson., 2017, 84, 171–181 CrossRef CAS PubMed.
- S. L. Carnahan, B. J. Lampkin, P. Naik, M. P. Hanrahan, I. I. Slowing, B. VanVeller, G. Wu and A. J. Rossini, J. Am. Chem. Soc., 2019, 141, 441–450 CrossRef CAS PubMed.
- I. Hung, Z. Gan and G. Wu, J. Phys. Chem. Lett., 2021, 12, 8897–8902 CrossRef CAS PubMed.
- V. K. Michaelis, E. Markhasin, E. Daviso, J. Herzfeld and R. G. Griffin, J. Phys. Chem. Lett., 2012, 3, 2030–2034 CrossRef CAS PubMed.
- V. K. Michaelis, B. Corzilius, A. A. Smith and R. G. Griffin, J. Phys. Chem. B, 2013, 117, 14894–14906 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- F. A. Perras, T. Kobayashi and M. Pruski, J. Am. Chem. Soc., 2015, 137, 8336–8339 CrossRef CAS PubMed.
- N. J. Brownbill, D. Gajan, A. Lesage, L. Emsley and F. Blanc, Chem. Commun., 2017, 53, 2563–2566 RSC.
- T. H. Sefzik, J. B. Houseknecht, T. M. Clark, S. Prasad, T. L. Lowary, Z. Gan and P. J. Grandinetti, Chem. Phys. Lett., 2007, 434, 312–315 CrossRef CAS.
- K. Yamada, Y. Yamaguchi, Y. Uekusa, K. Aoki, I. Shimada, T. Yamaguchi and K. Kato, Chem. Phys. Lett., 2020, 749, 137455 CrossRef CAS.
- J. Shen, V. Terskikh and G. Wu, J. Phys. Chem. Lett., 2016, 7, 3412–3418 CrossRef CAS PubMed.
- Z. Gan, I. Hung, X. Wang, J. Paulino, G. Wu, I. M. Litvak, P. L. Gor'kov, W. W. Brey, P. Lendi, J. L. Schiano, M. D. Bird, I. R. Dixon, J. Toth, G. S. Boebinger and T. A. Cross, J. Magn. Reson., 2017, 284, 125–136 CrossRef CAS PubMed.
- G. Ferguson, B. Kaitner, B. E. Connett and D. F. Rendle, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 479–484 CrossRef.
- H. Oertling, CrystEngComm, 2016, 18, 1676–1692 RSC.
- H. Oertling, C. Besnard, T. Alzieu, M. Wissenmeyer, C. Vinay, J. Mahieux and R. Fumeaux, Cryst. Growth Des., 2017, 17, 262–270 CrossRef CAS.
- A. Hassan, C. M. Quinn, J. Struppe, I. V. Sergeyev, C. Zhang, C. Guo, B. Runge, T. Theint, H. H. Dao, C. P. Jaroniec, M. Berbon, A. Lends, B. Habenstein, A. Loquet, R. Kuemmerle, B. Perrone, A. M. Gronenborn and T. Polenova, J. Magn. Reson., 2020, 311, 106680 CrossRef CAS PubMed.
- D. Rittenberg and C. Graff, J. Am. Chem. Soc., 1958, 80, 3370–3372 CrossRef CAS.
- J. M. Risley and R. L. Van Etten, Biochemistry, 1982, 21, 6360–6365 CrossRef CAS PubMed.
- T. L. Mega, S. Cortes and R. L. Van Etten, J. Org. Chem., 1990, 55, 522–528 CrossRef CAS.
- F. W. D'Souz and T. L. Lowary, J. Org. Chem., 1998, 63, 3166–3167 CrossRef.
- K. Ochi and K. Okui, Chem. Pharm. Bull., 1974, 22, 2223–2230 CrossRef CAS.
- W. Mackie and A. S. Perlin, Can. J. Chem., 1965, 43, 2921–2924 CrossRef CAS.
- N. Yamauchi and K. Kakinuma, J. Org. Chem., 1995, 60, 5614–5619 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- B. Simon and H. Köstler, J. Biomol. NMR, 2019, 73, 155–165 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B, 1999, 59, 7413–7421 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. T. Holmes and R. W. Schurko, J. Phys. Chem. C, 2018, 122, 1809–1820 CrossRef CAS.
- S. T. Holmes, O. G. Engl, M. N. Srnec, J. D. Madura, R. Quiñones, J. K. Harper, R. W. Schurko and R. J. Iuliucci, J. Phys. Chem. A, 2020, 124, 3109–3119 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B, 2001, 61, 245101 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B, 2007, 76, 024401 CrossRef.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed.
- X. Kong, Y. Dai and G. Wu, Solid State Nucl. Magn. Reson., 2017, 84, 59–64 CrossRef CAS PubMed.
- G. J. Rees, S. P. Day, K. E. Barnsley, D. Iuga, J. R. Yates, J. D. Wallis and J. V. Hanna, Phys. Chem. Chem. Phys., 2020, 22, 3400–3413 RSC.
- J. Zhu, A. J. Geris and G. Wu, Phys. Chem. Chem. Phys., 2009, 11, 6972–6980 RSC.
- L. J. Rowlands, A. Marks, J. M. Sanderson and R. V. Law, Chem. Commun., 2020, 56, 14499–14502 RSC.
- S. Dong, K. Yamada and G. Wu, Z. Naturforsch., A: Phys. Sci., 2000, 55, 21–28 CrossRef CAS.
- J. Zhu, J. Y. C. Lau and G. Wu, J. Phys. Chem. B, 2010, 114, 11681–11688 CrossRef CAS PubMed.
- X. Kong, M. Shan, V. Terskikh, I. Hung, Z. Gan and G. Wu, J. Phys. Chem. B, 2013, 117, 9643–9654 CrossRef CAS PubMed.
- I. P. Gerothanassis, J. Lauterwein and N. Sheppard, J. Magn. Reson., 1982, 48, 431–446 CAS.
- J. Schulte and J. Lauterwein, J. Magn. Reson., Ser. A, 1993, 101, 95–97 CrossRef CAS.
- Q. W. Zhang, H. M. Zhang, M. G. Usha and R. J. Wittebort, Solid State Nucl. Magn. Reson., 1996, 7, 147–154 CrossRef CAS PubMed.
- G. Wu, D. Rovnyak, P. C. Huang and R. G. Griffin, Chem. Phys. Lett., 1997, 277, 79–83 CrossRef CAS.
- Q. Zhang, E. Y. Chekmenev and R. J. Wittebort, J. Am. Chem. Soc., 2003, 125, 9140–9146 CrossRef CAS PubMed.
- V. K. Michaelis, E. G. Keeler, T. C. Ong, K. N. Craigen, S. Penzel, J. E. C. Wren, S. Kroeker and R. G. Griffin, J. Phys. Chem. B, 2015, 119, 8024–8036 CrossRef CAS PubMed.
- S. Nour, C. M. Widdifield, L. Kobera, K. M. N. Burgess, D. Errulat, V. V. Terskikh and D. L. Bryce, Can. J. Chem., 2016, 94, 189–197 CrossRef CAS.
- R. P. Young, C. R. Lewis, C. Yang, L. Wang, J. K. Harper and L. J. Mueller, Magn. Reson. Chem., 2019, 57, 211–223 CrossRef CAS PubMed.
- A. Rinald and G. Wu, J. Phys. Chem. A, 2020, 124, 1176–1186 CrossRef CAS PubMed.
- J. Shen, V. Terskikh, X. Wang, I. Hung, Z. Gan and G. Wu, J. Phys. Chem. B, 2018, 122, 4813–4820 CrossRef CAS PubMed.
- N. P. Wickramasinghe, M. Kotecha, A. Samoson, J. Past and Y. Ishii, J. Magn. Reson., 2007, 184, 350–356 CrossRef CAS PubMed.
- R. Linser, V. Chevelkov, A. Diehl and B. Reif, J. Magn. Reson., 2007, 189, 209–216 CrossRef CAS PubMed.
- S. Parthasarathy, Y. Nishiyama and Y. Ishii, Acc. Chem. Res., 2013, 46, 2127–2135 CrossRef CAS PubMed.
- L. Frydman and J. S. Harwood, J. Am. Chem. Soc., 1995, 117, 5367–5368 CrossRef CAS.
- A. Medek, J. S. Harwood and L. Frydman, J. Am. Chem. Soc., 1995, 117, 12779–12787 CrossRef CAS.
- M. Ha, S. Nader, S. Pawsey, J. Struppe, M. Monette, S. S. Mansy, J. Boekhoven and V. K. Michaelis, J. Phys. Chem. B, 2021, 125, 11916–11926 CrossRef CAS PubMed.
- A. Wong, M. E. Smith, V. Terskikh and G. Wu, Can. J. Chem., 2011, 89, 1087–1094 CrossRef.
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- S. Kuroki, A. Takahashi, I. Ando, A. Shoji and T. Ozaki, J. Mol. Struct., 1994, 323, 197–208 CrossRef CAS.
- K. Yamauchi, S. Kuroki, I. Ando, T. Ozaki and A. Shoji, Chem. Phys. Lett., 1999, 302, 331–336 CrossRef CAS.
- S. Dong, R. Ida and G. Wu, J. Phys. Chem. A, 2000, 104, 11194–11202 CrossRef CAS.
- G. Wu, K. Yamada, S. Dong and H. Grondey, J. Am. Chem. Soc., 2000, 122, 4215–4216 CrossRef CAS.
- K. Yamada, S. Dong and G. Wu, J. Am. Chem. Soc., 2000, 122, 11602–11609 CrossRef CAS.
- G. Wu, S. Dong, R. Ida and N. Reen, J. Am. Chem. Soc., 2002, 124, 1768–1777 CrossRef CAS PubMed.
- A. Wong, K. J. Pike, R. Jenkins, G. J. Clarkson, T. Anupõld, A. P. Howes, D. H. G. Crout, A. Samoson, R. Dupree and M. E. Smith, J. Phys. Chem. A, 2006, 110, 1824–1835 CrossRef CAS PubMed.
- I. C. M. Kwan, X. Mo and G. Wu, J. Am. Chem. Soc., 2007, 129, 2398–2407 CrossRef CAS PubMed.
- K. Yamauchi, M. Okonogi, H. Kurosu, M. Tansho, H. Shimizu, T. Gullion and T. Asakura, J. Magn. Reson., 2008, 190, 327–332 CrossRef CAS PubMed.
- X. Kong, A. Brinkmann, V. Terskikh, R. E. Wasylishen, G. M. Bernard, Z. Duan, Q. Wu and G. Wu, J. Phys. Chem. B, 2016, 120, 11692–11704 CrossRef CAS PubMed.
- J. Lu, I. Hung, A. Brinkmann, Z. Gan, X. Kong and G. Wu, Angew. Chem., Int. Ed., 2017, 56, 6166–6170 CrossRef CAS PubMed.
- A. E. Florin and M. Alei, J. Chem. Phys., 1967, 47, 4268–4269 CrossRef CAS.
- W. T. Raynes, Mol. Phys., 1983, 49, 443–447 CrossRef CAS.
- R. E. Wasylishen, S. Mooibroek and J. B. Macdonald, J. Chem. Phys., 1984, 81, 1057–1059 CrossRef CAS.
- J. Reuben, J. Am. Chem. Soc., 1969, 91, 5725–5729 CrossRef CAS.
- G. Wu, A. Hook, S. Dong and K. Yamada, J. Phys. Chem. A, 2000, 104, 4102–4107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis and characterization of 17O-labeled D-glucose; solution 17O NMR spectra of all D-glucose compounds; 13C CP/MAS NMR spectra of all solid α-D-glucose/NaCl/H2O cocrystal samples; complete GIPAW DFT results for 17O and 13C NMR parameters (PDF). See DOI: 10.1039/d1sc06060k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Jochem
Struppe
b,
Jochem
Struppe