
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterometathesis of diphosphanes (R2P–PR2) with dichalcogenides (R′E–ER′, E = O, S, Se, Te)†
Callum
Branfoot
 a,
Paul G.
Pringle
*a,
Natalie E.
Pridmore
a,
Tom A.
Young
b and
Duncan F.
Wass
c
a,
Paul G.
Pringle
*a,
Natalie E.
Pridmore
a,
Tom A.
Young
b and
Duncan F.
Wass
c
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: paul.pringle@bristol.ac.uk
bChemistry Research Laboratory, University of Oxford, Oxford OX1 3TA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
First published on 20th May 2022
Abstract
The reactions of R2P–PR2 with R′E–ER′, (where E = Se, S, O, Te) to give R2P–ER′ have been explored experimentally and computationally. The reaction of Ph2P–PPh2 with PhSe–SePh gives Ph2P–SePh (1) rapidly and quantitatively. The P–P/Se–Se reaction is inhibited by the addition of the radical scavenger TEMPO which is consistent with a radical mechanism for the heterometathesis reaction. Compound 1 has been fully characterised, including by X-ray crystallography. A range of other Ar2P–SeR (R = Ph, nBu or CH2CH2CO2H) have also been prepared and characterised. The reaction of 1 with [Mo(CO)4(nbd)] (nbd = norbornadiene) gives two products which, from their characteristic 31P NMR data, have been identified as cis-[Mo(CO)4(Ph2PSePh–P)2] (8) and the mixed-donor complex cis-[Mo(CO)4(Ph2P–SePh–P)(Ph2P–SePh–Se)] (9). It is deduced that the P and Se atoms in ligand 1 have comparable capacity to coordinate to Mo(0). The reaction of Ph2P–PPh2 with PhS–SPh gives Ph2P–SPh (2) quantitatively but no reaction was observed between Ph2P–PPh2 and PhTe–TePh. Heterometathesis between Ph2P–PPh2 and tBuO–OtBu does not occur thermally but has been observed under UV irradiation to give Ph2P–OtBu along with P(V) oxidation by-products. DFT calculations have been carried out to illuminate why heterometatheses with dichalcogenides R′E–ER′ occur readily when E = S and Se but not when E = O and Te. The calculations show that heterometathesis is predicted to be thermodynamically favourable for E = O, S and Se and unfavourable for E = Te. The fact that a metathesis reaction between Ph2P–PPh2 with tBuO–OtBu is not observed in the absence of UV radiation, is therefore due to kinetics.
Introduction
The chemistry of diphosphanes (R2P–PR2) has attracted considerable attention, and the high reactivity of the P–P bond has been exploited for the diphosphination of a wide range of unsaturated substrates, including alkenes,1–4 alkynes,4–8 1,3-dienes,9 arynes,10 CO2 and CS2.11 We recently reported that tetra-aryldiphosphanes readily undergo homometathesis reactions (Scheme 1) under ambient conditions, most likely via homolysis of the P–P bond and the generation of Ar2P˙ radicals.12 Also shown in Scheme 1 are diphosphane reactions with diatomic X2 that could be classed as examples of diphosphane heterometathesis reactions.13,14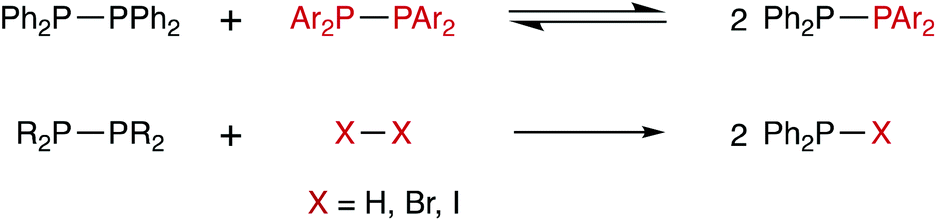 | ||
| Scheme 1 Metathesis reactions of diphosphanes. | ||
Here, we report our experimental and computational investigations of the heterometatheses shown in Scheme 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and show that this is an effective route for the synthesis of compounds of the type Ph2P–SeR.
15 and show that this is an effective route for the synthesis of compounds of the type Ph2P–SeR.
 | ||
| Scheme 2 | ||
Compounds containing P–S or P–Se bonds have been widely applied in organic synthesis as reagents for S or Se transfer (Lawesson's reagent16 and Woollins’ reagent17 respectively). Furthermore, P/S and P/Se compounds are of interest in their own right18–20 with potential applications ranging from semi-conductors to pesticides.21 However, to date, little attention has been given to compounds of the type R2P–SR or R2P–SeR which are amongst the simplest organophosphorus(III) compounds containing S or Se. Arbuzov reported the first examples of Ph2P–SR (R = alkyl) compounds over a century ago.22 In the 1960s, McLean reported the synthesis of Ph2P–SePh (1) and Ph2P–SPh (2) from Ph2PCl by the routes shown in Scheme 3, although no NMR spectroscopic data for these compounds were reported.23 More recently, Cui et al.24 prepared thioaniline derivative 3 similarly (Scheme 3).
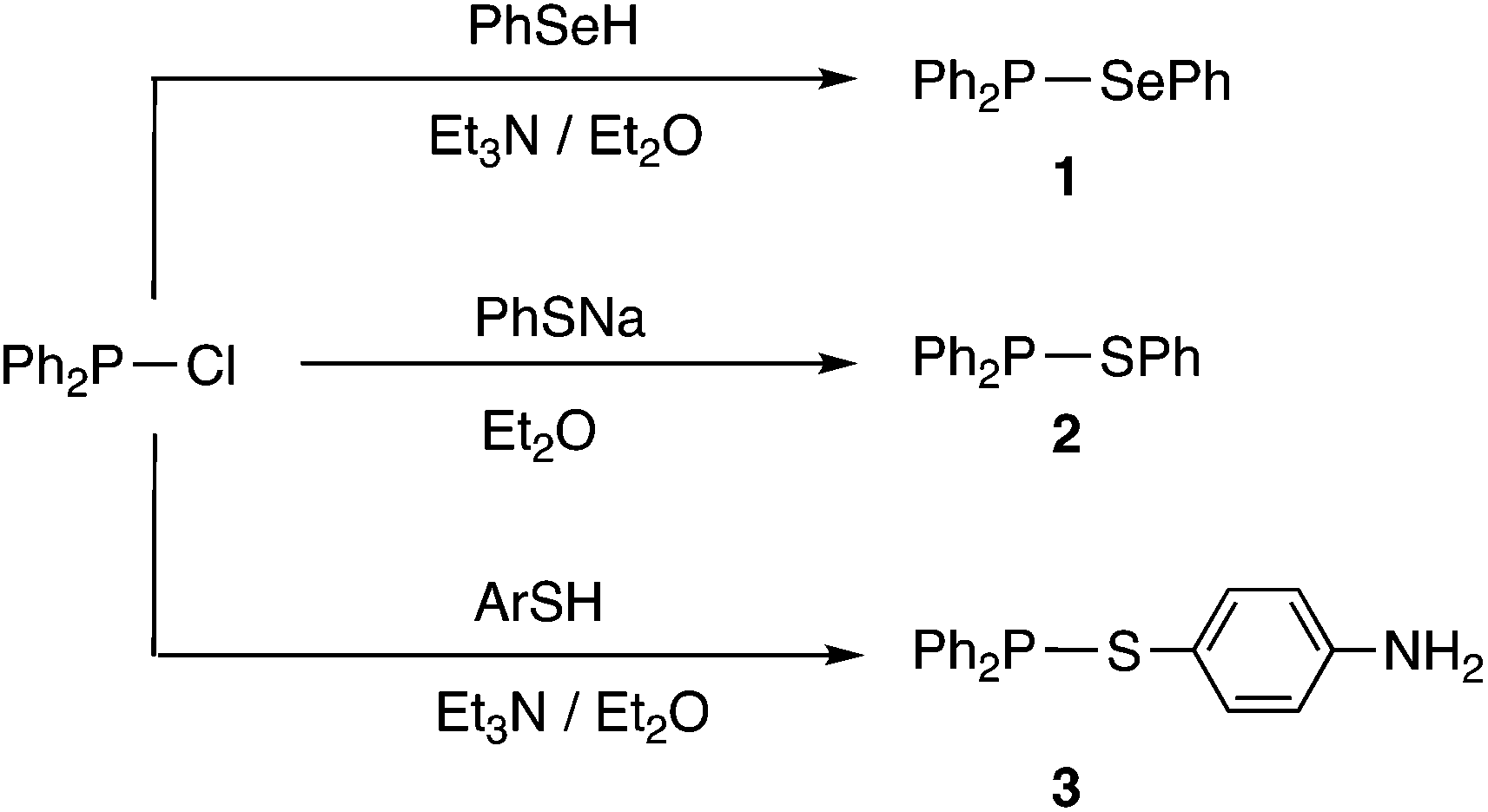 | ||
| Scheme 3 Previously reported Ar2P–ER compounds.23,24 | ||
Results and discussion
Diphosphane–diselenide metathesis
The reaction between equimolar quantities of tetraphenyldiphosphane and diphenyldiselenide in THF (Scheme 4) was monitored by 31P{1H} NMR spectroscopy. After 10 min, the signal for Ph2P–PPh2 at δ(P) −15.0 ppm had been replaced by a singlet at δ(P) +29.4 ppm with 77Se satellites (1JPSe = 229 Hz); the 77Se{1H} NMR spectrum is a doublet at δ(Se) 307.5 ppm (1JPSe = 229 Hz), consistent with the formation of the selanylphosphane, Ph2P–SePh (1). The product was isolated in 92% yield and further characterised by 1H and 13C{1H} NMR, and mass spectrometry (see ESI† for the data).25 Crystals of 1 were grown from CH2Cl2/n-hexane via vapour diffusion and its X-ray crystal structure determined (see Fig. 1).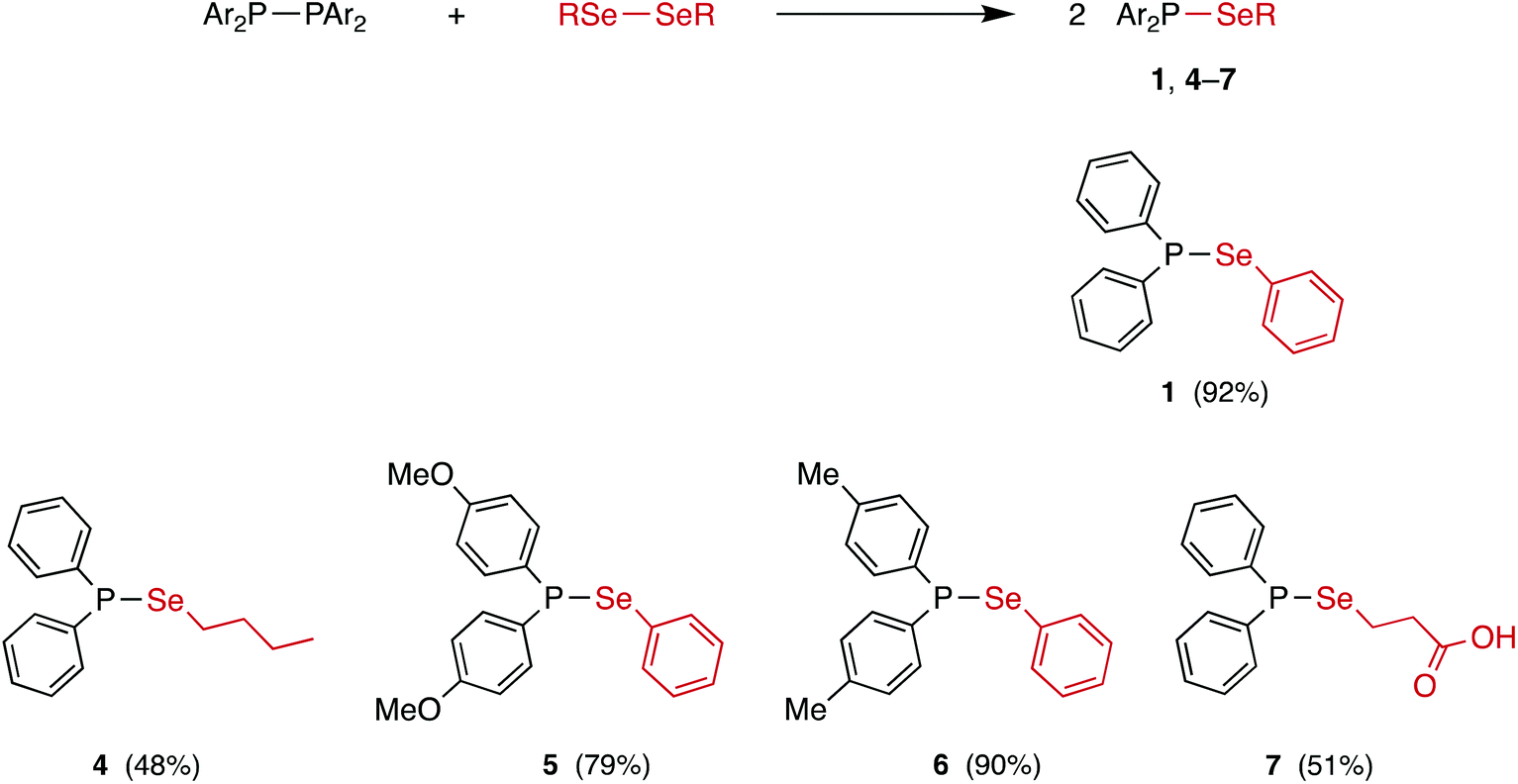 | ||
| Scheme 4 | ||
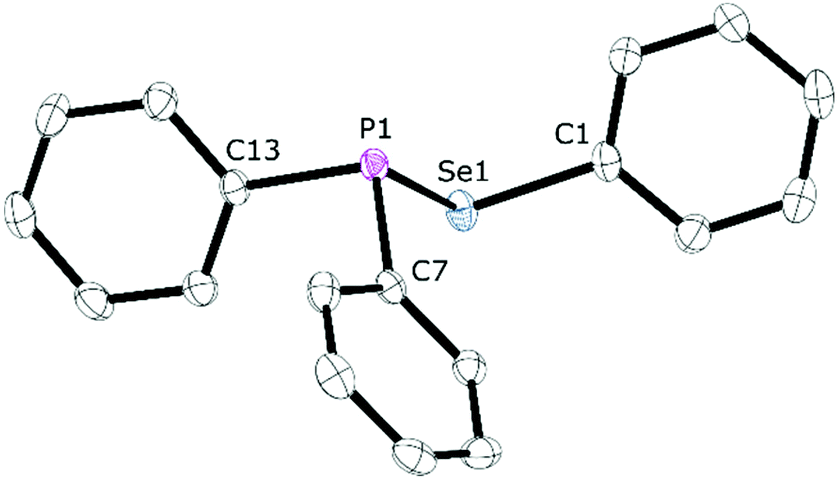 | ||
| Fig. 1 Thermal ellipsoid (50% probability) representation of the crystal structure of diphenyl(phenylselanyl)phosphane (1), hydrogen atoms omitted for clarity. Selected bond lengths (Å) and bond angles (°): P1–Se1 2.2620(5), P1–C7 1.8421(18), P1–C13 1.8343(18), Se1–C1 1.9316(18); C7–P1–Se1 103.62(6), C7–P1–C13 100.15(8), C13–P1–Se1 96.38(6), P1–Se1–C1 98.66(5). | ||
The P–Se bond length in 1 of 2.2620(5) Å is close to the mean average of 2.217 Å obtained from the 686 single crystal X-ray structures that feature a P–Se bond in the Cambridge Structural Database (CSD). There are no crystal structures in the CSD of compounds of the type Ar2P–SeAr′ (or Ar2P–SAr′) with which to compare 1, but the pyramidal geometry at P and bent geometry at Se are as expected.
The P2R4/Se2R2 heterometathesis reaction has been extended to the preparation of the new selanylphosphanes 4–7 (Scheme 4) by combination of the appropriate diphosphane and diselenide. Each of these compounds was isolated in good yield (quantitative yields observed by in situ31P{1H} NMR spectroscopy) as an oil or oily solid and then fully characterised by 31P{1H}, 77Se{1H}, 1H, 13C{1H} and APCI mass spectrometry (see ESI† for the data). The modest isolated yields of 4 and 7 (shown in Scheme 4) are due to losses during work-up, since the NMR yields were quantitative. These reactions were carried out in THF, CH2Cl2 or toluene and in no case was the solvent observed to affect the rate or outcome of the reaction, which in all cases had proceeded to completion within the time required (ca. 30 s) to obtain a 31P{1H} NMR spectrum of the reaction mixture. Of particular note is the carboxylic acid 7, which shows that the P–Se bond is tolerant of reactive functional groups. The synthesis of 1 in CDCl3 was repeated in an amberised NMR tube (to reduce the effect of photolysis) but again, complete conversion to 1 was observed within 30 s.
When the reaction of Ph2P–PPh2 with PhSe–SePh was carried out in the presence of 4 equiv. of TEMPO in THF, the heterometathesis reaction was inhibited: in the absence of TEMPO, complete conversion was observed within 1 min, while in the presence of TEMPO, the reaction proceeded to 54% conversion after 90 min. Other products are detected associated with the formation of Ph2P-TEMPO species (see ESI†). This is consistent with radicals being involved in the reaction, as we previously demonstrated in the homometathesis of diphosphanes.12 As a result, the radical chain mechanism shown in Scheme 5 is proposed. Diselenides are well-known to form RSe˙ radicals by photolytic cleavage of the weak Se–Se bond (BDE 41 kcal mol−1).26–32 It is therefore plausible that RSe–SeR cleavage to produce RSe˙ radicals initiates the radical chain process shown in Scheme 5.33
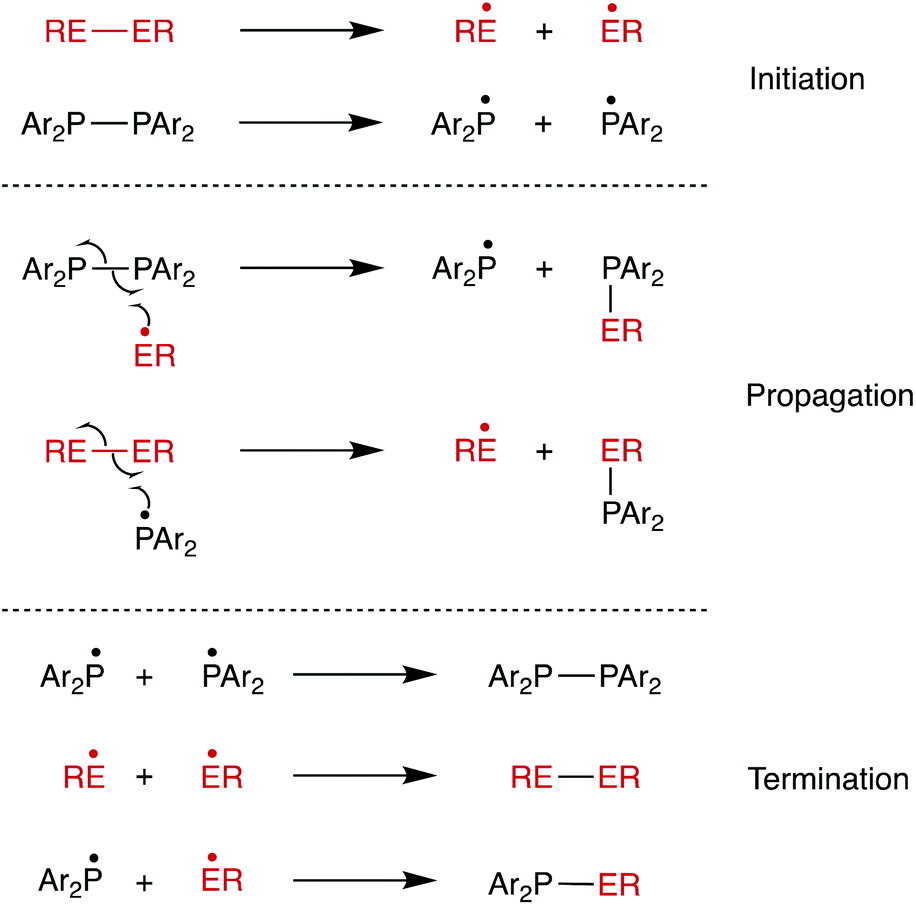 | ||
| Scheme 5 Radical chain mechanism proposed for diphosphane–dichalcogenide metathesis. | ||
McLean reported23 that Ph2P–SePh (1) is unstable with respect to isomerisation to Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se at 100 °C, over a period of 3–4 h. We have found that 1 is relatively stable under ambient conditions, which makes it easy to handle. Thus solid samples of 1 can be stored under argon at room temperature with <10% degradation observed (traces of Ph2PH detected) even after 4 months. When water was added to a solution of 1 in CDCl3 and the emulsion shaken under nitrogen, only very slow hydrolysis to Ph2P(O)H was observed: after 2 days, the hydrolysis mixture contained unreacted 1 (ca. 60%) along with a prominent signal (22%) at +32.5 ppm, consistent with the formation of Ph2P(O)OH;34 crystals that grew from this solution were shown to match the structure of Ph2P(O)OH, ‘DPPHIN’ in the CSD.
Se at 100 °C, over a period of 3–4 h. We have found that 1 is relatively stable under ambient conditions, which makes it easy to handle. Thus solid samples of 1 can be stored under argon at room temperature with <10% degradation observed (traces of Ph2PH detected) even after 4 months. When water was added to a solution of 1 in CDCl3 and the emulsion shaken under nitrogen, only very slow hydrolysis to Ph2P(O)H was observed: after 2 days, the hydrolysis mixture contained unreacted 1 (ca. 60%) along with a prominent signal (22%) at +32.5 ppm, consistent with the formation of Ph2P(O)OH;34 crystals that grew from this solution were shown to match the structure of Ph2P(O)OH, ‘DPPHIN’ in the CSD.
The observed kinetic stability of selanylphosphane 1 prompted us to explore its coordination chemistry with Mo(0). To the best of our knowledge, selanylphosphanes have not previously been used as ligands for transition metals. It was of interest to investigate which of the two potential donor atoms (P and Se) would bind to the metal in the complexes, since both are known to bind to metals in mixed P,Se ligands such as Ph2PCH2CH2SePh.35–37 Treatment of [Mo(CO)4(nbd)] (nbd = norbornadiene) with 2 equiv. of 1 in CD2Cl2, gave a mixture of two products in the ratio of ca. 4:1 according to 31P{1H} NMR spectroscopy (see Fig. 2), which have been assigned to the isomers of cis-[Mo(CO)4(1)2] (8 and 9 in Scheme 6) on the basis of their characteristic 31P NMR parameters.
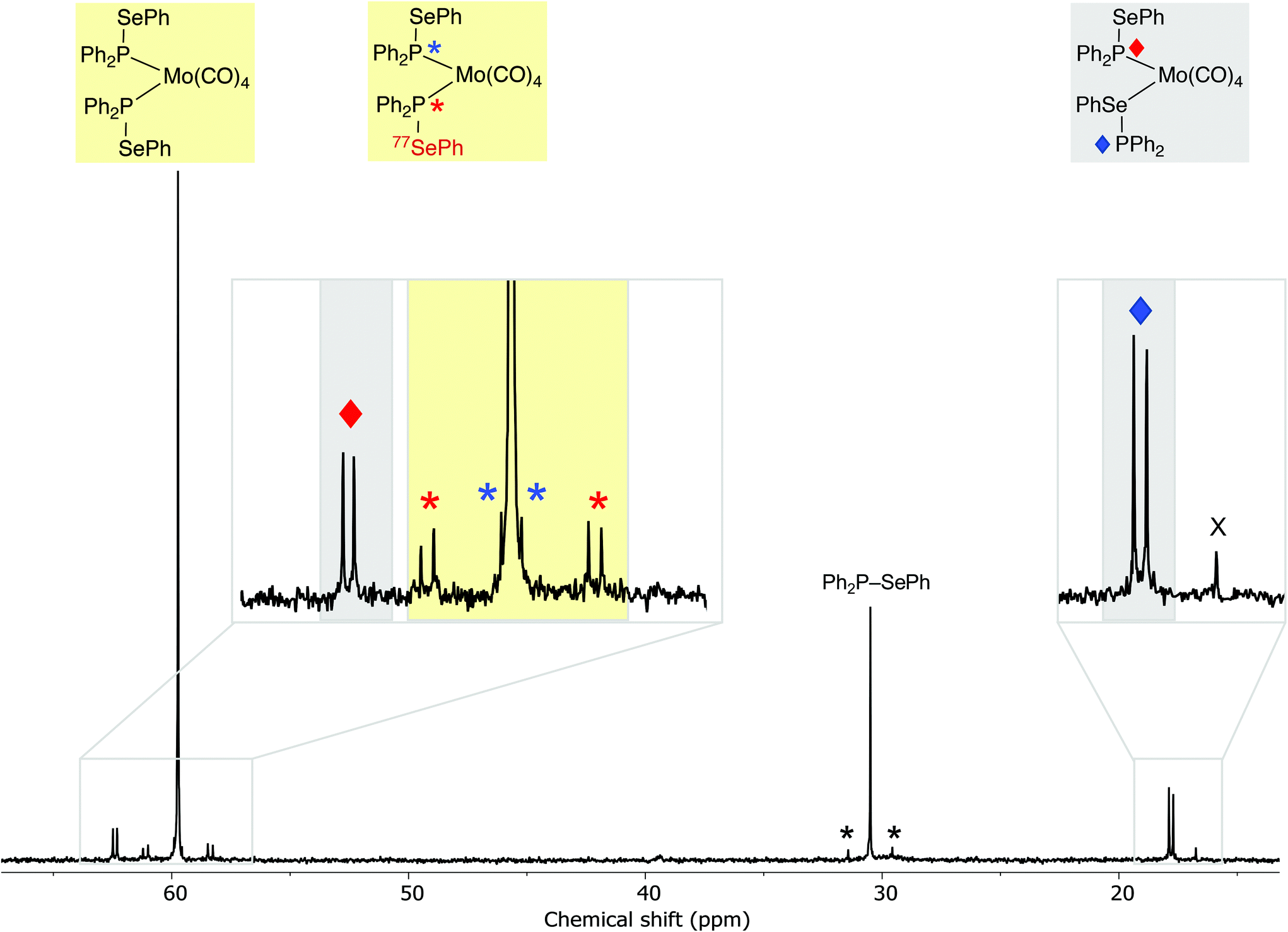 | ||
Fig. 2
31P{1H} NMR spectrum of the product mixture obtained from the reaction of Ph2P–SePh with [Mo(CO)4(nbd)]. The large singlet at ca. 60 ppm is assigned to the symmetrical P,P-complex 8 and the expansion shows the signals associated with the 77Se isotopologue of 8, which show satellite signals due to 1JPSe ( ) and 3JPSe ( ) and 3JPSe ( ). The doublets at ca. +62 ( ). The doublets at ca. +62 ( ) and +18 ( ) and +18 ( ) are assigned to 9, the P,Se-linkage isomer of 8 (77Se satellites obscured by the noise). The signal at ca. 31 ppm is for unreacted ligand 1 with 77Se satellites (*). The minor signal labelled X at ca. +17 ppm is unassigned. ) are assigned to 9, the P,Se-linkage isomer of 8 (77Se satellites obscured by the noise). The signal at ca. 31 ppm is for unreacted ligand 1 with 77Se satellites (*). The minor signal labelled X at ca. +17 ppm is unassigned. | ||
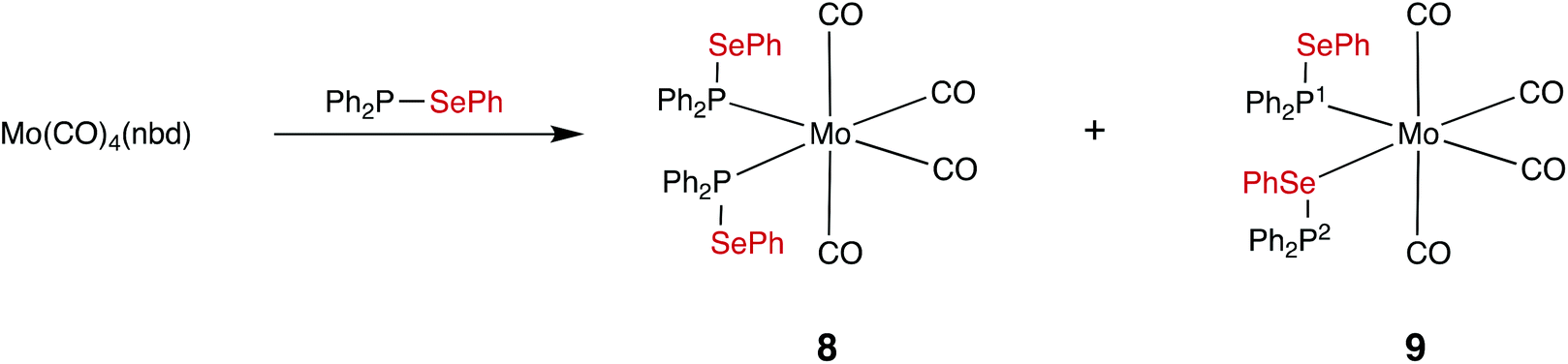 | ||
| Scheme 6 | ||
The major product is a singlet at δ(P) +59.7 ppm with outer 77Se satellites (1JPSe = 333 Hz) that are themselves split into doublets (2JPP = 25 Hz) and inner 77Se satellites (3JPSe = 40 Hz). This product is assigned the structure 8 where both ligands are P-bound cis on the Mo; this is supported by the large coordination chemical shift (ΔδP = +29.2) and the small 2JPP. The inequivalence observed in the satellites is a result of the loss of symmetry in the 77Se isotopologue (see Fig. 2).
The second product is assigned structure 9, containing one P-bound and one Se-bound ligand 1, on the basis of the 31P NMR data. Two doublets are observed at δ(P1) +62.4 ppm and δ(P2) +17.8 ppm (3JPP = 22 Hz). The ΔδP of +31.2 for P1 in 9 is similar to the ΔδP of the P in 8 and is therefore consistent with P1 being directly bound to Mo. The ΔδP of −12.7 ppm for P2 is assigned to the Se-bound ligand in 9. After the mixture of products was heated to 60 °C in CDCl3 for 3.5 h, complex 9 was the dominant species (ratio of complexes 9:8 was 5:1 according to 31P NMR spectroscopy, see ESI†). In the IR spectrum of the mixtures of 8 and 9, several absorptions in the range 1892–2027 cm−1 were evident, as expected for the ν(CO) bands for these cis complexes present. Solutions of the molybdenum complexes 8 and 9 in a CH2Cl2/water emulsion did not undergo any changes after 17 h, indicating good water tolerance.
The analogous complexation reaction of selanylphosphane 7 with [Mo(CO)4(nbd)] in CH2Cl2 produced a similar pattern of products according to 31P NMR spectroscopy (see ESI†): two products in the ratio 4:1, with a singlet at +50.8 ppm (with 77Se satellites) and two doublets at +54.0 and 17.7 ppm (3JPP = 21 Hz) consistent with the formation of analogues of 8 and 9.
The notable conclusion from the Mo coordination chemistry is that, for Ph2P–SeR, the PPh2 and SeR groups have comparable donor ability. The fine balance between the linkage isomers with P or Se coordination is likely a consequence of the donor atoms both being soft and the steric hindrance around the SeR being less than around the PPh2.
Diphosphane–disulfide metathesis
The reaction of tetraphenyldiphosphane with diphenyl disulfide or 4-aminophenyl disulfide gave the previously reported sulfanylphosphanes 2 and 3 respectively (see Scheme 7).23,24,38 Compared with the rapid diselenide reactions (Scheme 4), the disulfide analogues are sluggish, requiring up to 20 h to reach completion in an unagitated NMR tube. The reactions were significantly more rapid when they were stirred and were most rapid in chloroform (as with the diphosphane homometathesis).12 The conversions to 2 and 3 were apparently quantitative in the reactions monitored by 31P{1H} NMR spectroscopy but the isolated yields were modest (see Scheme 7); there were significant losses on the short alumina column used to purify the crude products (see ESI†).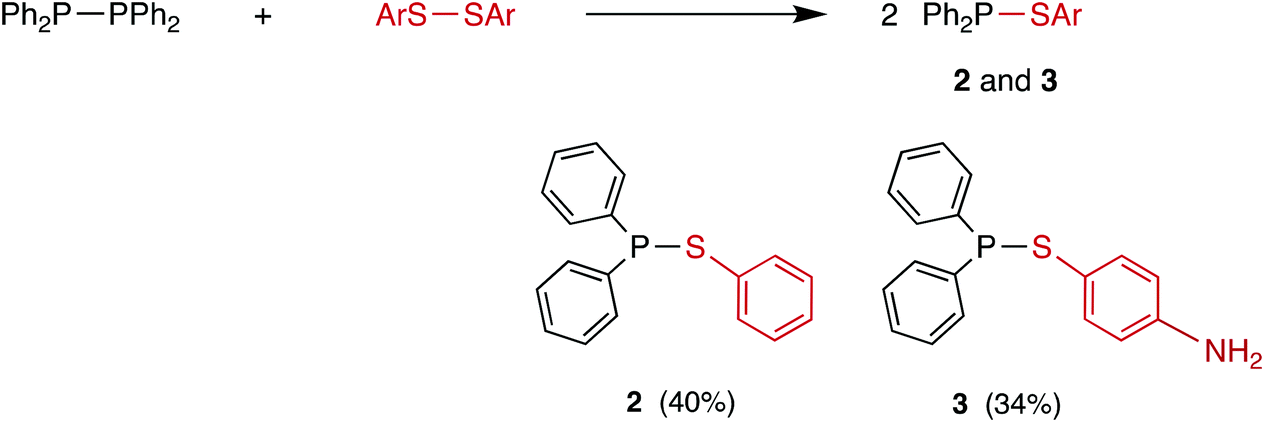 | ||
| Scheme 7 | ||
The diphosphane–disulfide metathesis was inhibited by the presence of TEMPO (4 equiv.), consistent with the reaction following the radical chain mechanism shown in Scheme 5, as for the diphosphane–diselenide process. The reaction of Ph2P–PPh2 with PhS–SPh in CDCl3 was followed by 31P{1H} NMR spectroscopy and 2 was formed in 34% yield after 1 h of reaction. When the same reaction was performed in the presence of 4 equivalents of TEMPO, 8% conversion to 2 was observed after 1 h (see ESI†). The higher rate observed with the diselenide than with the disulfide reflects the ease with which homolysis of the weak Se–Se bond takes place compared to the homolysis of either the S–S or P–P bonds.39–41
Attempted diphosphane–ditelluride and diphosphane–peroxide metatheses
When Ph2P–PPh2 and PhTe–TePh were mixed in C6D6, no heterometathesis reaction (Scheme 2) was observed even after 12 days. The compound Ph2P–TePh has not been previously reported, although its P(V) isomer Ph3PTe is readily prepared.42
Similarly, when Ph2P–PPh2 and tBuO–OtBu were mixed in toluene, no heterometathesis reaction (Scheme 8) to give the known17 compound Ph2P–OtBu was observed after 72 h. The only products detected by 31P{1H} NMR spectroscopy were the monoxide Ph2P(O)–PPh2 (12%: 2 doublets at +32.7 and −24.7, JPP = 218 Hz)43 and Ph2PH (12%, −40.2 ppm).12 Even when a large excess (∼15-fold) of tBuO–OtBu was used and the reaction mixture was heated to 60 °C, only oxidation products were observed and no heterometathesis product. However, when this reaction was repeated in C6D6 and irradiated with near-UV light, gradual formation of a new species was observed by 31P{1H} NMR spectroscopy characterised by a singlet at +87.1 ppm and assigned to the phosphinite Ph2P–OtBu on the basis of the chemical shift being consistent with the reported value.44 After the irradiation was continued for a further 1 h, a significant quantity of the phosphinate Ph2P(O)–OtBu (47% of the total 31P integral, see ESI†) was identified by a singlet at +25.7 ppm, in good agreement with literature data.45 The formation of this P(V) species was further supported by the detection of the phosphinate [M + Na]+ ion by mass spectrometry (297.1 m/z observed, 297.1 theoretical). In summary, there is evidence that under UV irradiation, the diphosphane/peroxide heterometathesis does indeed occur but that by-products associated with oxidation by tBuO–OtBu contaminate the product (see Scheme 8).
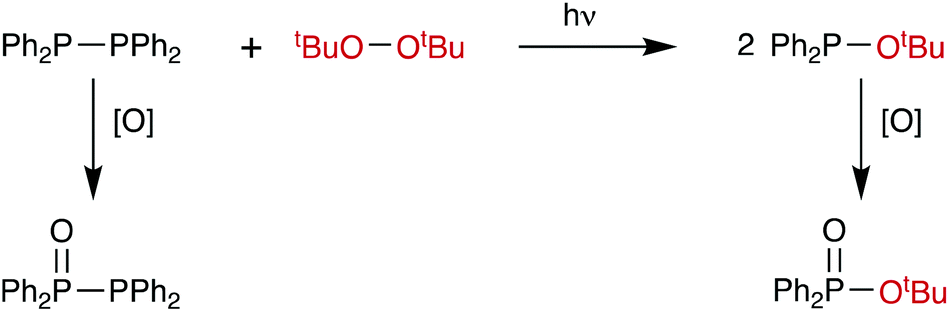 | ||
| Scheme 8 | ||
DFT study of heterometathesis reactions of diphosphanes
The general reaction that has been calculated is shown in Scheme 9 and the results are given in Table 1. It was anticipated that the results for the diphosphane metatheses with diatomic molecules Z2 would be the simplest to interpret. The DFT-calculated ΔE values for the addition of diatomic Z2 to R2P–PR2, where R = Ph (A), or Me (B), are presented as entries 1–10 of Table 1. The values of ΔH estimated using the mean bond strengths for P–P (51 kcal mol−1),46 Z–Z, and P–Z are also given in Table 1 and there is good agreement between ΔE and ΔH for Z = H, F, Cl, Br and I. The two consistent trends in the heterometathesis equilibria are: (i) ΔE becomes increasingly more favourable with increasing electronegativity of Z; (ii) Me2P–PMe2 metatheses are more favourable (by 6–10 kcal mol−1) than the corresponding Ph2P–PPh2 metatheses. The position of the equilibrium in Scheme 9 is determined by the relative Z–Z and P–Z bond energies, which in turn will be influenced by a combination of the following factors. (a) The very high H–H and very low F–F bond strengths dominate the explanation of why the metathesis equilibria with H2 and F2 are the least and most favourable respectively. (b) The electrostatic stabilising effect of the Pδ+–Z δ− dipole on the P–Z bond strength will increase with increasing electronegativity of Z. (c) The size of Z increases in the order F < Cl < Br < I and this will contribute to a lower P–Z bond strength (in that same order) due to increasingly poorer orbital overlap and increasing steric congestion. (d) The P–Z bond may be stronger in Me2P–Z than in Ph2P–Z, due to the lower steric hindrance of the PMe2 group and the greater +I inductive effect of the Me substituents stabilising the δ+ charge on the PMe2.47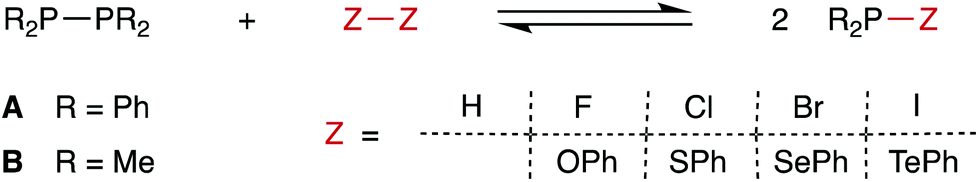 | ||
| Scheme 9 | ||
| Entry | Diphosphane | Z–Z | D(Z–Z) | Product | D(P–Z) | ΔH | ΔE |
|---|---|---|---|---|---|---|---|
| a All energies are given in kcal mol−1 and are calculated at STP (298 K, 1 atm). D(Z–Z) and D(P–Z) are the average bond dissociation energies given in ref. 46. | |||||||
| 1 | A | H2 | 104 | Ph2P–H | 77 | +1 | +6.8 |
| 2 | A | F2 | 37 | Ph2P–F | 117 | −146 | −131.2 |
| 3 | A | Cl2 | 58 | Ph2P–Cl | 79 | −49 | −44.1 |
| 4 | A | Br2 | 45 | Ph2P–Br | 65 | −34 | −28.9 |
| 5 | A | I2 | 36 | Ph2P–I | 51 | −15 | −10.9 |
| 6 | B | H2 | 104 | Me2P–H | 77 | +1 | +0.7 |
| 7 | B | F2 | 37 | Me2P–F | 117 | −146 | −141.3 |
| 8 | B | Cl2 | 58 | Me2P–Cl | 79 | −49 | −52.4 |
| 9 | B | Br2 | 45 | Me2P–Br | 65 | −34 | −36.6 |
| 10 | B | I2 | 36 | Me2P–I | 51 | −15 | −17.9 |
| 11 | A | t BuO–OtBu | 33 | Ph2P–OtBu | 84 | −84 | −69.4 |
| 12 | A | PhO–OPh | 33 | Ph2P–OPh | 84 | −84 | −71.7 |
| 13 | A | PhS–SPh | 51 | Ph2P–SPh | 55 | −8 | −4.2 |
| 14 | A | PhSe–SePh | 41 | Ph2P–SePh | — | — | −5.6 |
| 15 | A | PhTe–TePh | — | Ph2P–TePh | — | — | 7.4 |
The calculated thermodynamics of the metathesis reactions of Ph2P–PPh2 with PhE–EPh, where E = O, S, Se, or Te are given in Table 1, entries 12–15. These equilibria will depend on the relative PhE–EPh and Ph2P–EPh bond energies with potentially similar factors at play to those labelled (a)–(c) above, used for the diatomic Z2 reactions. However, in the case of the dichalcogenides, the PhO–OPh equilibrium (entry 12) is exceptional in being extremely favourable (ΔE of −69.4 kcal mol−1) as a consequence of the low O–O bond energy and the high P–O bond energy (due in part to the large Pδ+–Z δ− dipole). By contrast, for the other dichalcogenides (entries 13–15), the ΔE values are relatively small, reflecting the fact that the P–P, E–E and P–E bond energies are similar due to the similarity of the electronegativities and sizes of P, S, Se and Te. The calculated ΔE for the diselenide and disulfide reactions are both ca. −5 kcal mol−1 and therefore favour the formation of Ph2P–EPh, in agreement with the experimental observations. The calculated ΔE of +7 kcal mol−1 for the ditelluride/diphosphane metathesis disfavours the formation of Ph2P–TePh which is consistent with no reaction between Ph2P–PPh2 and PhTe–TePh being observed experimentally.
Although the tBuO–OtBu/Ph2P–PPh2 metathesis (entry 11, Table 1) is very strongly favoured energetically for the formation of Ph2P–OtBu, experimentally, the reaction (Scheme 8) only proceeded under UV irradiation and produced oxidation by-products; the lack of thermal reaction is therefore due to a kinetic barrier, possibly caused by the bulky tBu substituents.
Conclusion
The heterometathesis reactions of Ph2P–PPh2 with RE–ER to give Ph2P–ER proceed smoothly when E = S or Se but not when E = O or Te. We have shown that Ar2P–PAr2 with RSe–SeR gives a range of Ar2P–SeR compounds (1 and 4–7) in quantitative spectroscopic yields and 100% atom-economy. The reaction of Ph2P–PPh2 with ArS–SAr produces Ph2P–SAr, albeit more slowly than with the Se analogues. The inhibition by TEMPO of the heterometatheses of Ph2P–PPh2 with RE–ER (E = Se or S) suggests radical processes are involved in the mechanism. DFT calculations have shown that the heterometatheses with RE–ER are thermodynamically favourable when E = S or Se but not when E = Te, which aligns with the experimental observations. However, the calculations also suggest that the heterometathesis of diphosphanes with peroxides (i.e. E = O) would be very strongly favoured thermodynamically. The lack of thermal reaction between Ph2P–PPh2 with tBuO–OtBu is therefore due to kinetics. There is evidence of formation of Ph2P–OtBu when mixtures of Ph2P–PPh2 and tBuO–OtBu are photolysed but the reaction is complicated by the simultaneous formation of oxidation products. The selanylphosphane Ph2P–SePh (1) can be stored for weeks, largely unchanged at ambient temperatures and is only very slowly hydrolysed by water over several days. The ligand properties of 1 are of interest as it coordinates to Mo(0) to give products whose 31P parameters are consistent with coordination via the P and Se being competitive. This is an unusual example of linkage isomerism that is worthy of further study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Engineering and Physical Sciences Research Council through the EPSRC Centre for Doctoral Training in Advanced Composites for Innovation and Science (grant number EP/L0160208/1). We thank the Department of Chemistry at Oxford and the Centre for Computational Chemistry in Bristol for access to computing facilities.References
- H. Yorimitsu, Homolytic substitution at phosphorus for C-P bond formation in organic synthesis, Beilstein J. Org. Chem., 2013, 9, 1269–1277 CrossRef CAS PubMed.
- I. Hajdók, F. Lissner, M. Nieger, S. Strobel and D. Gudat, Diphosphination of Electron Poor Alkenes, Organometallics, 2009, 28, 1644–1651 CrossRef.
- N. Otomura, Y. Okugawa, K. Hirano and M. Miura, Bromine Cation Initiated vic-Diphosphination of Styrenes with Diphosphines under Photoredox Catalysis, Synthesis, 2018, 50, 3402–3407 CrossRef CAS.
- K. Hirano and M. Miura, Recent advances in diphosphination of alkynes and alkenes, Tetrahedron Lett., 2017, 58, 4317–4322 CrossRef CAS.
- D. L. Dodds, M. F. Haddow, A. G. Orpen, P. G. Pringle and G. Woodward, Stereospecific diphosphination of activated acetylenes: A general route to backbone-functionalized, chelating 1,2-diphosphinoethenes, Organometallics, 2006, 25, 5937–5945 CrossRef CAS.
- D. L. Dodds, J. Floure, M. Garland, M. F. Haddow, T. R. Leonard, C. L. McMullin, A. G. Orpen and P. G. Pringle, Diphosphanes derived from phobane and phosphatrioxa-adamantane: similarities, differences and anomalies, Dalton Trans., 2011, 40, 7137–7146 RSC.
- Y. Yamamoto, R. Tanaka, S. Kodama, A. Nomoto and A. Ogawa, Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization, Molecules, 2022, 27, 1284 CrossRef CAS PubMed.
- A. D. Gorman, J. A. Cross, R. A. Doyle, T. R. Leonard, P. G. Pringle and H. A. Sparkes, Phosphophosphidites Derived from BINOL, Eur. J. Inorg. Chem., 2019, 2019, 1633–1639 CrossRef CAS.
- N. Otomura, K. Hirano and M. Miura, Diphosphination of 1,3-Dienes with Diphosphines under Visible-Light-Promoted Photoredox Catalysis, Org. Lett., 2018, 20, 7965–7968 CrossRef CAS PubMed.
- Y. Okugawa, Y. Hayashi, S. Kawauchi, K. Hirano and M. Miura, Diphosphination of Arynes with Diphosphines, Org. Lett., 2018, 20, 3670–3673 CrossRef CAS PubMed.
- N. Szynkiewicz, L. Ponikiewski and R. Grubba, Diphosphination of CO2 and CS2 mediated by frustrated Lewis pairs-catalytic route to phosphanyl derivatives of formic and dithioformic acid, Chem. Commun., 2019, 55, 2928–2931 RSC.
- C. Branfoot, T. A. Young, D. F. Wass and P. G. Pringle, Radical-initiated P,P-metathesis reactions of diphosphanes: evidence from experimental and computational studies, Dalton Trans., 2021, 50, 7094–7104 RSC.
- S. J. Geier and D. W. Stephan, Rh-catalyzed P-P bond activation, Chem. Commun., 2008, 054, 99–101 RSC.
- H. Hacklin and G.-V. Röschenthaler, 1-Halogenphosphorinane: Darstellung Und Derivate, Phosphorus Sulfur Relat. Elem., 1988, 36, 165–169 CrossRef CAS.
- (a) The heterometathesis reaction between Ph2Bi–BiPh2 and PhE–EPh (E = S, Se, Te) to give Ph2Bi–EPh has been previously reported, see: F. Calderazzo, A. Morvillo, G. Pelizzi, R. Poli and F. Ungari, Reactivity of Molecules Containing Element–Element Bonds. 1. Nontransition Elements, Inorg. Chem., 1988, 27, 3730–3733 CrossRef CAS; (b) The reaction between Me2P–PMe2 and MeE–EMe produces equilibrium mixtures with the corresponding Me2P–EMe (E = S, Se, Te); see: A. J. Ashe III and E. G. Ludwig Jr., The Exchange Reactions of Tetramethylpnictogens with Dimethyldichalcogenides, J. Organomet. Chem., 1986, 308, 289–296 CrossRef.
- T. Ozturk, E. Ertas and O. Mert, Use of Lawesson's reagent in organic syntheses, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS PubMed.
- I. P. Gray, P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, A New Synthesis of (PhPSe2)2 (Woollins Reagent) and Its Use in the Synthesis of Novel P-Se Heterocycles, Chem. – Eur. J., 2005, 11, 6221–6227 CrossRef CAS PubMed.
- G. Hua, R. A. M. Randall, A. M. Z. Slawin and J. D. Woollins, Novel organo phosphorus-selenium heteroatom compounds from selenation of diamines, Tetrahedron, 2013, 69, 5299–5305 CrossRef CAS.
- L. Ascherl, A. Nordheider, K. S. A. Arachchige, D. B. Cordes, K. Karaghiosoff, M. Bühl, A. M. Z. Slawin and J. D. Woollins, The activation of Woollins’ reagent. Isolation of pyridine stabilised PhPSe2, Chem. Commun., 2014, 50, 6214–6216 RSC.
- G. Hua, Y. Li, A. M. Z. Slawin and J. D. Woollins, Unexpected four- and eight-membered organo P–Se heterocycles, Chem. Commun., 2007, 83, 1465–1467 RSC.
- R. Davies and L. Patel, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. Devillanova and W.-W. Du Mont, Royal Society of Chemistry, 2nd edn, 2013, pp. 238–306 Search PubMed.
- A. Arbuzov, J. Russ. Phys.-Chem. Soc., 1910, 42, 549 Search PubMed.
- R. A. N. McLean, Phenylthio- and Phenylseleno-Diphenyl Phosphine, Inorg. Nucl. Chem. Lett., 1969, 5, 745–747 CrossRef CAS.
- H. Cui, M. Hu, H. Wen, G. Chai, C. Ma, H. Chen and C. Chen, Efficient [FeFe] hydrogenase mimic dyads covalently linking to iridium photosensitizer for photocatalytic hydrogen evolution, Dalton Trans., 2012, 41, 13899–13907 RSC.
- P. J. Gates, Atmospheric pressure chemical ionisation mass spectrometry for the routine analysis of low molecular weight analytes, Eur. J. Mass Spectrom., 2021, 27, 13–28 CrossRef PubMed.
- R. Franzi and M. Geoffroy, Spin trapping identification of radical intermediates during photolysis of organoselenium compounds, J. Organomet. Chem., 1981, 218, 321–324 CrossRef CAS.
- T. G. Back and M. Vijaya Krishna, Free-Radical Additions of Diselenides to Dimethyl Acetylenedicarboxylate, Methyl Propiolate, and Dimethyl Maleate, J. Org. Chem., 1988, 53, 2533–2536 CrossRef CAS.
- D. H. Brown, R. J. Cross and D. Millington, Photochemical reactions between tertiary phosphines and organic diselenides, J. Chem. Soc., Dalton Trans., 1977, 159–161 RSC.
- M. M. Alam, O. Ito, Y. Koga and A. Ouchi, Laser-flash photolysis of naphthyl diselenides; reactivities of naphthylseleno radicals, Int. J. Chem. Kinet., 1998, 30, 193–200 CrossRef CAS.
- C. Liu, J. Xia, S. Ji, Z. Fan and H. Xu, Visible-light-induced metathesis reaction between diselenide and ditelluride, Chem. Commun., 2019, 55, 2813–2816 RSC.
- J. Xia, S. Ji and H. Xu, Diselenide covalent chemistry at the interface: stabilizing an asymmetric diselenide-containing polymer via micelle formation, Polym. Chem., 2016, 7, 6708–6713 RSC.
- S. Ji, W. Cao, Y. Yu and H. Xu, Dynamic diselenide bonds: exchange reaction induced by visible light without catalysis, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- The reaction of tetraphenyldiphosphane and dibenzyl diselenide gave the expected product Ph2P–SeBn, identified by a singlet in the 31P{1H} NMR spectrum in CDCl3 at +23.4 ppm, with 77Se satellites (1JPSe = 242 Hz). However, after 25 min this resonance accounts for only 50% of the 31P integral, with 37% residual tetraphenyldiphosphane and 13% formation of various other species (see ESI† for the spectrum). This contrasting reactivity is consistent with previous reports that photolysis of BnSe–SeBn preferentially leads to cleavage of the C–Se bond, due to the relative stability of the benzyl radical. The mixture of BnSe˙, Bn˙, and BnSeSe˙ radicals present would explain the low conversion to Ph2P–SeBn and the formation of the by-products. See: E. N. Deryagina, M. G. Voronkov and N. A. Korchevin, Selenium- and tellurium-centred radicals, Russ. Chem. Rev., 1993, 62, 1107–1117 CrossRef.
- J. Ash, H. Huang, P. Cordero and J. Y. Kang, Selective hydrolysis of phosphorus(V) compounds to form organophosphorus monoacids, Org. Biomol. Chem., 2021, 19, 6007–6014 RSC.
- T. J. Cunningham, M. R. J. Elsegood, P. F. Kelly, M. B. Smith and P. M. Staniland, Versatile routes to selenoether functionalized tertiary phosphines, Dalton Trans., 2010, 39, 5216–5218 RSC.
- A. M. Spokoyny, M. S. Rosen, P. A. Ulmann, C. Stern and C. A. Mirkin, Selective Formation of Heteroligated Pt(II) Complexes with Bidentate Phosphine-Thioether (P,S) and Phosphine-Selenoether (P,Se) Ligands via the Halide-Induced Ligand Rearrangement Reaction, Inorg. Chem., 2010, 49, 1577–1586 CrossRef CAS PubMed.
- M. S. Rosen, A. M. Spokoyny, C. W. Machan, C. Stern, A. Sarjeant and C. A. Mirkin, Chelating Effect as a Driving Force for the Selective Formation of Heteroligated Pt(II) Complexes with Bidentate Phosphino-Chalcoether Ligands, Inorg. Chem., 2011, 50, 1411–1419 CrossRef CAS PubMed.
- T. Shirai, S. Kawaguchi, A. Nomoto and A. Ogawa, Photoinduced highly selective thiophosphination of alkynes using a (PhS)2/(Ph2P)2 binary system, Tetrahedron Lett., 2008, 49, 4043–4046 CrossRef CAS.
- D. Kaur, P. Sharma and P. V. Bharatam, A comparative study on the nature and strength of O-O, S-S, and Se-Se bond, J. Mol. Struct.: THEOCHEM, 2007, 810, 31–37 CrossRef CAS.
- S. Kawaguchi and A. Ogawa, Applications of Diphosphines in Radical Reactions, Asian J. Org. Chem., 2019, 8, 1164–1173 CrossRef CAS.
- V. T. Hung, C. C. Tran, Y. Yamamoto, S. Kodama, A. Nomoto and A. Ogawa, Clarification on the reactivity of diaryl diselenides toward hexacyclohexyldilead under light, Molecules, 2021, 26, 6265 CrossRef CAS PubMed.
- C. H. W. Jones and R. D. Sharma, 125Te NMR And Mössbauer Spectroscopy of Tellurium-phosphine Complexes and the Tellurocyanates, Organometallics, 1987, 6, 1419–1423 CrossRef CAS.
- M. S. Hill, M. F. Mahon and T. P. Robinson, Calcium-centred phosphine oxide reactivity: P-C metathesis, reduction and P-P coupling, Chem. Commun., 2010, 46, 2498–2500 RSC.
- Y. Shinro, S. Kosei, Y. Masakuni, M. Toshihisa and O. Atsuyoshi, The Arbuzov Reaction of Alkyl Diphenylphosphinites with 10-Methylacridinium Ion. Kinetic Study on the Formation and the Decomposition of Phosphonium Intermediates, Bull. Chem. Soc. Jpn., 1993, 66, 2077–2083 CrossRef.
- A. Kinbara, M. Ito, T. Abe and T. Yamagishi, Nickel-catalyzed C-P cross-coupling reactions of aryl iodides with H-phosphinates, Tetrahedron, 2015, 71, 7614–7619 CrossRef CAS.
- C. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education, 5th edn, 2018 Search PubMed.
- We previously proposed related electrostatic and steric contributions to rationalise the thermodynamics of homometathesis P–P equilibria, see ref. 12.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2162103. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01093c |
| This journal is © The Royal Society of Chemistry 2022 |