
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A hydrogen-driven biocatalytic approach to recycling synthetic analogues of NAD(P)H†
Holly A.
Reeve‡
 *a,
Jake
Nicholson
a,
Farieha
Altaf
a,
Thomas H.
Lonsdale
a,
Janina
Preissler
b,
Lars
Lauterbach
bc,
Oliver
Lenz
b,
Silke
Leimkühler
d,
Frank
Hollmann
e,
Caroline E.
Paul
*e and
Kylie A.
Vincent
*a
*a,
Jake
Nicholson
a,
Farieha
Altaf
a,
Thomas H.
Lonsdale
a,
Janina
Preissler
b,
Lars
Lauterbach
bc,
Oliver
Lenz
b,
Silke
Leimkühler
d,
Frank
Hollmann
e,
Caroline E.
Paul
*e and
Kylie A.
Vincent
*a
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: holly@hydregenoxford.com; kylie.vincent@chem.ox.ac.uk
bDepartment of Chemistry, Technische Universität Berlin, Strasse des 17. Juni 135, 10623 Berlin, Germany
cRWTH Aachen University iAMB – Institute of Applied Microbiology Worringer Weg 1, 52074 Aachen, Germany
dDepartment of Molecular Enzymology, Institute of Biochemistry and Biology, University of Potsdam, Germany
eDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629HZ Delft, The Netherlands. E-mail: c.e.paul@tudelft.nl
First published on 18th August 2022
Abstract
We demonstrate a recycling system for synthetic nicotinamide cofactor analogues using a soluble hydrogenase with turnover number of >1000 for reduction of the cofactor analogues by H2. Coupling this system to an ene reductase, we show quantitative conversion of N-ethylmaleimide to N-ethylsuccinimide. The biocatalyst system retained >50% activity after 7 h.
Oxidoreductases catalyse an impressive array of redox reactions with exquisite selectivity, however an estimated 50% are dependent on the nicotinamide adenine dinucleotide cofactors (NADH or NADPH, Fig. 1a(i)). There is increasing interest in replacing these expensive biological cofactors by synthetic, artificial cofactors, Fig. 1a(ii).1–4 Although these artificial cofactors can be produced cost-effectively, it is still undesirable to use them at stoichiometric levels due to poor atom economy imposed on the reaction, the cost of product clean-up and possible inhibition of enzymes at high cofactor concentrations. For these reasons, methods for recycling the artificial cofactors are required.1,2
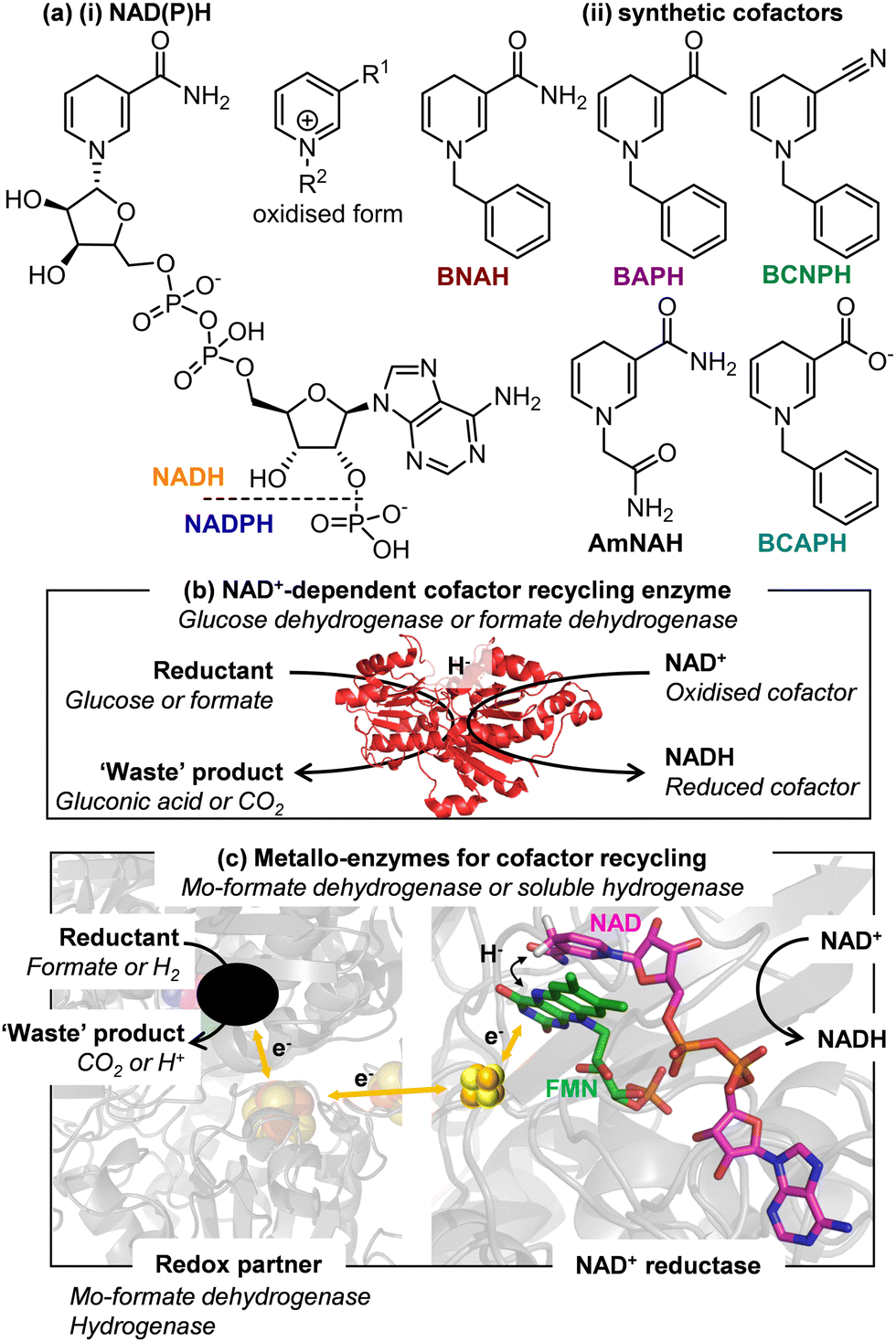 | ||
| Fig. 1 (a) Structure of (i) native and (ii) artificial nicotinamide cofactors. (b) Industry standard cofactor recycling enzymes operate via direct hydride transfer between a substrate and a cofactor. (c) Cofactor recycling using a metallo-enzyme, where electrons from a redox partner (Mo-formate dehydrogenase, or hydrogenase unit) transfer via a FeS cluster chain for reduction of an enzyme bound flavin able to reduce NAD+ to NADH, thus coupling an oxidative half reaction at one catalytic moiety, to cofactor recycling in an NAD+ reductase moiety. | ||
Methods for recycling NADH and NADPH, are well-established and employ a second enzyme (most commonly glucose dehydrogenase, GDH, or formate dehydrogenase, FDH) acting on a sacrificial substrate (glucose or formate, respectively), Fig. 1b.5,6 These enzymes tend to be highly cofactor-specific. At present, there are conflicting reports in the literature as to whether these enzymes are able to recycle artificial cofactor analogues. Here we investigate the activity of a GDH and a FDH for artificial cofactor recycling and show that neither of the enzymes is able to act on the set of artificial cofactors trialled in this study. Non-enzymatic routes to recycling the reduced artificial cofactors have been reported, but are limited to non-selective chemical methods and use of iridium-based homogeneous catalysts and none have been able to convincingly show multiple turnovers.7 GDH from Sulfolobus sulfataricus (SsGDH) showed promise for acting on artificial cofactors, however a double variant I192T/V306I only slightly improved the catalytic efficiency to 1.64 × 10−3 mM−1 s−1 (turnover frequency TOF of 0.54 min−1) for generating the reduced artificial cofactor 1-benzyl-1,4-dihydronicotinamide (BNAH) or 5.17 × 10−3 mM−1 s−1 (TOF of 2.52 min−1) for generating 1-phenethyl-1,4-dihydronicotinamide (P2NAH), compared to the wild type enzyme forming NADH with catalytic efficiency of 15.94 mM−1 s−1 (TOF of 407 min−1).8 An engineered F420-NADPH oxidoreductase from Thermobifida fusca (TfuFNO, G29W variant) provided a TOF of 252 min−1 for reducing the cofactor mimic 1-benzyl-3-acetylpyridine BAP+), yet also requires glucose-driven recycling of the F420 cofactor.9
Here, we establish a H2-driven biocatalytic system for reducing nicotinamide cofactor analogues, and demonstrate it as a catalytic recycling system by coupling it to an ene reductase from the Old Yellow Enzyme (OYE) family. OYEs are becoming established as selective catalysts for the reduction of activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-double bonds.10–12 Catalysis by these enzymes occurs in two steps; a hydride transfer from NAD(P)H to reduce the flavin prosthetic group, followed by hydride transfer from the prosthetic flavin to the substrate, generating the product with potentially two stereogenic centres, in a trans-addition fashion.13
C-double bonds.10–12 Catalysis by these enzymes occurs in two steps; a hydride transfer from NAD(P)H to reduce the flavin prosthetic group, followed by hydride transfer from the prosthetic flavin to the substrate, generating the product with potentially two stereogenic centres, in a trans-addition fashion.13
Many OYEs show cofactor promiscuity,14–17 with reduced flavins also shown to be able to act as reductants.18–21 OYEs are well-known to accept reducing equivalents from artificial cofactors, and in some cases show preference for them over the native nicotinamide cofactors.14,16 Tolerance of OYEs for a range of reductants can be explained by their stepwise mechanism which involves no direct contact between the cofactor/reductant and the alkene substrate.14–16 In contrast, GDH and FDH rely on direct hydride transfer between the oxidised cofactor and the sacrificial substrate meaning that favourable orientation of the two molecules within the enzyme active site is critical for hydride transfer to take place, Fig. 1b, and hence they are not easily adapted for recycling artificial cofactor analogues.
With these observations in mind, we sought to investigate three NAD+-linked metallo-enzymes that contain a conserved flavin (FMN) active site for NAD+/NADH cycling, hypothesising that reduction of the artificial cofactor analogues might be possible at the bound flavin active site. In these enzymes, oxidation of a reductant (H2 or formate) at one metallo-catalytic moiety releases electrons which are transferred, via an internal iron-sulfur cluster electron-transport chain, to the enzyme-bound flavin, the site of cofactor reduction, Fig. 1c. We demonstrate here that these flavin-containing enzymes are able to accept a number of artificial cofactors and form a viable recycling system for them. We have previously demonstrated use of soluble hydrogenases (SHs) in H2-driven NAD(P)H recycling.22,23 When coupled to an NADH-dependent enzyme, this facilitates atom-efficient biocatalysis with both protons and electrons from H2 being incorporated into the final product. An SH has also been employed in H2-driven recycling of reduced flavin.21 Here, we find that the soluble hydrogenases can be re-purposed to supply reduced artificial cofactor to the OYE from Thermus scotoductus (TsOYE) for carvone or N-ethylmaleimide reduction, with catalytic turnover of the artificial cofactor by the SH.
Initially, the activity of five enzymes for reduction of each of the artificial cofactors was investigated. The enzymes chosen are: a standard FDH(Evocatal) and GDH (Johnson Matthey), the NAD+-linked soluble hydrogenase (SH) enzymes from Hydrogenophilus thermoluteolus (HtSH) and Ralstonia eutropha (also known as Cupriavidus necator) (ReSH) and the NAD+-linked Mo-formate dehydrogenase from Rhodobacter capsulatus (Mo-FDH) (see ESI,† General information for details of enzyme sources.) The activities of FDH and GDH were first investigated using standard solution assays monitored with UV-visible spectroscopy using a plate reader. The activities towards NAD+ were 0.25 and 8.0 U mg−1 for FDH and GDH, respectively. However, no activity was observed for either of these enzymes with the synthetic cofactors, BNA+, BAP+, BCNP+, BCAP+ or BuNA+ (see structures in Fig. 1(a)(ii) for the corresponding reduced forms, BNAH, BAPH, BCNPH, and BCAPH or Fig. S1 (ESI†) for BuNAH). Results are shown in the ESI,† Fig. S1. These results are consistent with several earlier reports that alcohol dehydrogenases are generally not able to accept synthetic cofactors.24
To analyse the activity of HtSH and ReSH towards the native and artificial cofactors, reaction mixtures containing oxidised cofactors (2 mM) were prepared in H2-saturated buffer (50 mM MOPS-NaOH pH 7, 25 °C for ReSH and 50 mM Tris–HCl pH 8, 50 °C for HtSH) containing DMSO (2% v/v). These conditions were chosen based on conditions found to favour NAD+ reduction by each SH. The reactions were initiated by addition of SH (40 μg) to the sealed reaction mixtures, with H2 continually bubbling through the headspace, according to previously published procedures.25 The reactions were monitored using in situ UV-Vis spectroscopy (ESI,† Fig. S2).
The time course data from these experiments was used to determine the activity of the soluble hydrogenase, calculated using literature extinction coefficients at λmax for each cofactor.26 The results are summarised in Table 1. The highest activities were observed for HtSH with BAP+ and BCNP+, so these combinations were taken forward for further work. The SH enzymes act via direct hydride transfer from a bound flavin which is re-reduced by electrons from H2 oxidation at a separate catalytic hydrogenase moiety, Fig. 1(b). This supports the hypothesis that flavin active sites facilitate use of artificial cofactors.
| Enzyme | Cofactor | Temperature (°C) | pH | Activity (mU mg−1) |
|---|---|---|---|---|
| Reactions were performed in 50 mM MOPS-NaOH buffer, pH 7.0 or 50 mM Tris–HCl buffer pH 8.0 according to the pH indicated.a Cofactor degradation occurred above 60 °C. See Fig. 1 for the structures of the reduced forms. | ||||
| ReSH | NAD+ | 32 | 8 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| BCNP+ | 32 | 8 | 5.1 | |
| AmNA+ | 32 | 8 | 3.8 | |
| BCAP+ | 32 | 8 | 3.0 | |
| BAP+ | 20 | 8 | 3.1 | |
| BAP+ | 32 | 8 | 9.6 | |
| BAP+ | 32 | 7 | 5.2 | |
| HtSH | NAD+ | 50 | 7 | 9400 |
| BAP+ | 50 | 7 | 92.6 | |
| BAP+ | 60a | 7 | 102.9 | |
| BCNP+ | 50 | 7 | 206.8 | |
| AmNA+ | 50 | 7 | 56.7 | |
To further probe this, a molybdenum-containing formate dehydrogenase which operates with a similar mechanism was chosen. R. capsulatus Mo-FDH is shown to be able to catalyse formate-driven reduction of BCNP+, BNA+ and BAP+. However, in each case the reactions stop after about 3 min, showing that the enzyme has a short half-life under these conditions (see ESI,† Fig. S3). Therefore, although this Mo-FDH is able to act on the artificial cofactors, it is not suitable for application in cofactor recycling. Due to this limitation, further characterisation was not carried out.
To demonstrate that the reduced artificial cofactor produced by SH is suitable for biocatalysis, H2-driven cofactor generation was coupled with the thermostable ene reductase, TsOYE, for the chemoselective reduction of carvone. Use of the thermostable HtSH and TsOYE required experiments to be performed at elevated temperatures (50 °C).
Initially, HtSH was supplied with oxidised cofactor (BCNP+, 2 mM) in the presence of H2. In situ UV-vis spectroscopy was used to monitor the generation of reduced cofactor (Fig. 2a). At ca. 50 min, after an initial lag phase, the concentration of reduced BCNPH approached 1 mM. Aliquots of TsOYE (2 μM) and carvone (1 mM) were injected into the cuvette. An immediate drop in the concentration of BCNPH was observed, suggesting that the reduced cofactor had been completely consumed by the ene reductase for the reduction of carvone over 15 min. A sample was taken for analysis by GC, confirming complete conversion of carvone to (+)-dihydrocarvone (Fig. 2b, red line). This demonstrates that BCNPH produced by HtSH is compatible with the ene reductase.
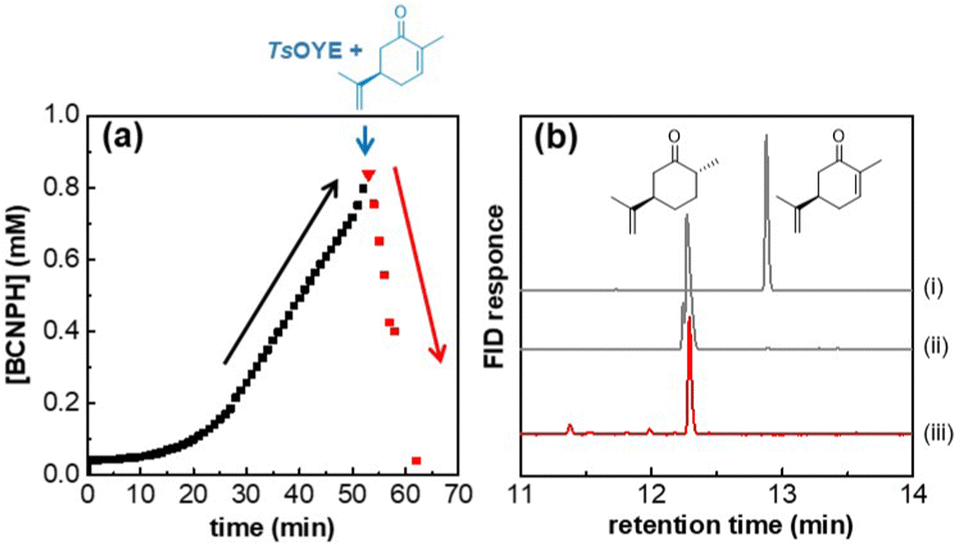 | ||
| Fig. 2 (a) Time course for H2-driven generation of BCNPH by HtSH (0.2 mg mL−1) before and after addition of TsOYE (2 μM) and carvone (1 mM), indicated by the red triangle (▼). Reaction performed in 50 mM MOPS-NaOH buffer, pH 7.0 and 50 °C, and BCNP+ (2 mM). H2 was flowed through the headspace throughout the experiment. (b) Gas chromatograms of (i) (R)-carvone and (ii) (+)-dihydrocarvone standards and of (iii) reaction mixture after 62 min, corresponding to the last time point in (a), red square at 62 min. | ||
Experiments were designed to achieve cofactor turnover by increasing the substrate concentration and lowering the cofactor loading. Due to the relatively low activity of native HtSH towards the artificial cofactor, significant evaporation of carvone occurred on the time scale required for H2-driven batch reactions, complicating analysis. An alternative substrate, N-ethylmaleimide, was chosen and shown to be relatively stable with respect to evaporation and hydrolysis over an hour at 50 °C, although some loss was observed by 2 h (see ESI,†). The product was determined to be stable under the same conditions over 2 h. To avoid hydrolysis and/or evaporation of N-ethylmaleimide over longer timeframes, the reaction was carried out as a fed-batch reaction. The fed-batch reaction was monitored using UV-Vis spectroscopy to ensure a build-up of reduced cofactor to a concentration of ca 0.2 mM, before addition of an aliquot of N-ethylmaleimide (0.22 mM injections, red arrows, Fig. 3a). After the first injection of substrate, the concentration of reduced artificial cofactor dropped nearly to zero, suggesting that it had been consumed by TsOYE for conversion of N-ethylmaleimide to N-ethylsuccinimide. The concentration of reduced cofactor then immediately increased, demonstrating that the N-ethylmaleimide had been completely consumed, such that a build-up of reduced cofactor could occur again. Further injections of N-ethylmaleimide were made, in each case the concentration of reduced cofactor drops to around zero, before increasing again. This result suggests that the cofactor can be efficiently cycled between the two enzymes.
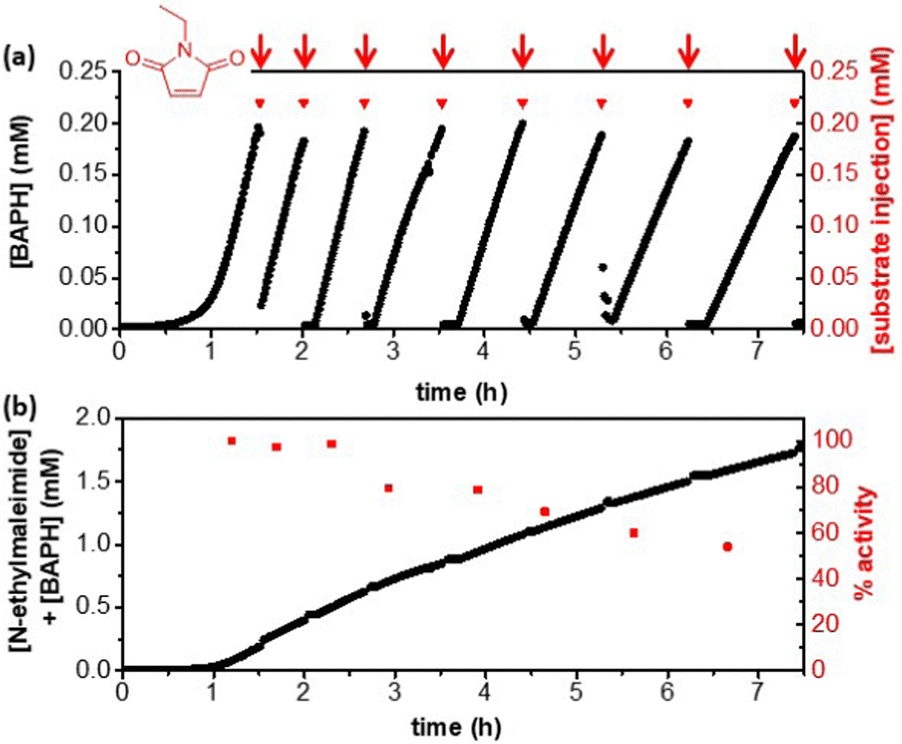 | ||
| Fig. 3 Fed-batch reaction for H2-driven artificial cofactor recycling using HtSH (208 μg) coupled to N-ethylmaleimide reduction by TsOYE (2 μM). (a) Time course for concentration of BAPH, monitored in situ. Injections of substrate (0.22 mM) denoted by red triangles and arrows. (b) The sum of BAPH recorded concentration and N-ethylmaleimide injected over time giving the total concentration of reduced products (black dots). Percent activity of HtSH remaining for each cycle compared to the first (red squares). Reaction performed in 50 mM MOPS-NaOH buffer pH 7.0 and 50 °C in the presence of BAP+ (0.5 mM), with H2 flowing through the headspace. | ||
The sum of the concentrations of N-ethylmaleimide injected and BAPH recorded gives a predicted product concentration over time (Fig. 3b, black circles). After 8 injections the N-ethylsuccinimide concentration was determined to be 1.9 mM (see ESI,†), confirming full conversion of substrate to product and complete compatibility of the artificial cofactor in this two-enzyme system. Since the concentration of artificial cofactor used was 0.5 mM, this demonstrates a TONBAP of >3. The HtSH activity can be calculated from the build-up of BAPH, and is calculated for each cycle compared to the first (Fig. 3b, red squares). The HtSH activity is approximately constant at 30 mU mg−1 for the first 2 h, this is a third of the maximum activity observed in Table 1, likely due to the lower cofactor concentration used (0.5 mM compared with 2 mM). This activity corresponds to a HtSH turnover frequency of >1.6 s−1 and leads to a total turnover of >1400 mol product mol−1HtSH after ca. 8 h. There is a modest drop in activity over time, with more than 50% activity retained after 7 h.
Overall, these results demonstrate a H2-driven enzymatic recycling system for artificial cofactors, that is highly atom-efficient when coupled to CC-bond reductions, and operates at relatively low cofactor concentrations. This system therefore overcomes key limitations with implementing artificial cofactors for biocatalysis.
The GDH and FDH tested in this study showed no activity for the set of artificial cofactors. However the fact that activity for the artificial cofactors was observed with Mo-FDH and two hydrogenases, each of which has a flavin active site for NAD+/NADH cycling, lends support to the idea that the artificial cofactors are best tolerated by enzymes with an exposed flavin prosthetic group. The Mo-FDH showed poor longevity during catalytic reduction of the artificial cofactors, but the hydrogenases, in particular HtSH, showed promising stability, even at 50 °C. It is likely that the activity of these enzymes for the artificial cofactors could be improved further by enzyme engineering to boost affinity for the artificial cofactors. To conclude, we showcase a proof-of-concept system for H2-driven recycling of synthetic analogues of NAD(P)H.
HAR, KAV, FH and CEP conceptualised the study, acquired funding and wrote the draft manuscript. Experimental investigation was carried out by HAR, JN, FA and TH; JP, LL, OL and SL provided enzymes; CEP and FH provided artificial cofactors and enzymes. HAR and JN analysed data. All authors reviewed and edited the manuscript and approved the final version.
This research was financially supported by Engineering and Physical Sciences Research Council (EPSRC) IB Catalyst Award EP/N013514/1 (to HAR and KAV), and NWO VENI grant number 722.015.011 (to CEP). Research by JP, LL, OL and SL was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany′s Excellence Strategy – EXC 2008 – 390540038 – UniSysCat.
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Zachos, C. Nowak and V. Sieber, Curr. Opin. Chem. Biol., 2018, 49, 59–66 CrossRef PubMed.
- C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2014, 4, 788–797 CrossRef CAS.
- A. Guarneri, W. J. H. van Berkel and C. E. Paul, Curr. Opin. Biotechnol, 2019, 60, 63–71 CrossRef CAS PubMed.
- E. King, S. Maxel and H. Li, Curr. Opin. Biotechnol, 2020, 66, 217–226 CrossRef CAS PubMed.
- W. A. van der Donk and H. M. Zhao, Curr. Opin. Biotechnol, 2003, 14, 421–426 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Appl. Biochem. Biotechnol., 1987, 14, 147–197 CrossRef CAS PubMed.
- Y. Okamoto, V. Köhler, C. E. Paul, F. Hollmann and T. R. Ward, ACS Catal., 2016, 6, 3553–3557 CrossRef CAS.
- C. Nowak, A. Pick, P. Lommes and V. Sieber, ACS Catal., 2017, 7, 5202–5208 CrossRef CAS.
- J. Drenth, G. Yang, C. E. Paul and M. W. Fraaije, ACS Catal., 2021, 11, 11561–11569 CrossRef CAS PubMed.
- H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 CrossRef CAS PubMed.
- C. K. Winkler, K. Faber and M. Hall, Curr. Opin. Chem. Biol., 2018, 43, 97–105 CrossRef CAS PubMed.
- T. Ress, W. Hummel, S. P. Hanlon, H. Iding and H. Gröger, ChemCatChem, 2015, 7, 1302–1311 CrossRef CAS.
- R. M. Kohli and V. Massey, J. Biol. Chem., 1998, 273, 32763–32770 CrossRef CAS PubMed.
- C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernández, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180–183 CrossRef CAS PubMed.
- T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033–1039 CrossRef CAS PubMed.
- S. A. Löw, I. M. Löw, M. J. Weissenborn and B. Hauer, ChemCatChem, 2016, 8, 911–915 CrossRef.
- W. B. Black, L. Y. Zhang, W. S. Mak, S. Maxel, Y. T. Cui, E. King, B. Fong, A. S. Martinez, J. B. Siegel and H. Li, Nat. Chem. Biol., 2020, 16, 87–94 CrossRef CAS PubMed.
- H. S. Toogood, T. Knaus and N. S. Scrutton, ChemCatChem, 2014, 6, 951–954 CrossRef CAS.
- J. Kim, S. H. Lee, F. Tieves, D. S. Choi, F. Hollmann, C. E. Paul and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 13825–13828 CrossRef CAS PubMed.
- S. J. Srinivasan, S. E. Cleary, M. A. Ramirez, H. A. Reeve, C. E. Paul and K. A. Vincent, Angew. Chem., Int. Ed., 2021, 60, 13824–13828 CrossRef PubMed.
- A. Al-Shameri, S. J. P. Willot, C. E. Paul, F. Hollmann and L. Lauterbach, Chem. Commun., 2020, 56, 9667–9670 RSC.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2013, 280, 3058–3068 CrossRef CAS PubMed.
- A. K. Holzer, K. Hiebler, F. G. Mutti, R. C. Simon, L. Lauterbach, O. Lenz and W. Kroutil, Org. Lett., 2015, 17, 2431–2433 CrossRef CAS PubMed.
- K. E. Taylor and J. B. Jones, J. Am. Chem. Soc., 1976, 98, 5689–5694 CrossRef CAS PubMed.
- H. A. Reeve, L. Lauterbach, O. Lenz and K. A. Vincent, ChemCatChem, 2015, 7, 3480–3487 CrossRef CAS PubMed.
- A. Guarneri, A. H. Westphal, J. Leertouwer, J. Lunsonga, M. C. R. Franssen, D. J. Opperman, F. Hollmann, W. J. H. van Berkel and C. E. Paul, ChemCatChem, 2020, 12, 1368–1375 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Enzyme activities, experimental conditions and methods. See DOI: https://doi.org/10.1039/d2cc02411j |
| ‡ Current address: HydRegen Limited, Centre for Innovation and Enterprise, Begbroke Science Park, Oxford, OX5 1PF, UK. |
| This journal is © The Royal Society of Chemistry 2022 |