
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase-selective active sites on ordered/disordered titanium dioxide enable exceptional photocatalytic ammonia synthesis†
Jinsun
Lee‡
ab,
Xinghui
Liu‡
ab,
Ashwani
Kumar‡
ab,
Yosep
Hwang
ab,
Eunji
Lee
c,
Jianmin
Yu
ab,
Young Dok
Kim
b and
Hyoyoung
Lee
 *abde
*abde
aCenter for Integrated Nanostructure Physics (CINAP), Institute for Basic Science (IBS), 2066 Seoburo, Jangan-gu, Suwon 16419, Republic of Korea. E-mail: hyoyoung@skku.edu
bDepartment of Chemistry, Sungkyunkwan University, 2066 Seoburo, Jangan-gu, Suwon 16419, Republic of Korea
cDepartment of Energy Science, Sungkyunkwan University, 2066 Seoburo, Jangangu, Suwon 16419, Republic of Korea
dDepartment of Biophysics, Sungkyunkwan University, 2066 Seoburo, Jangan-gu, Suwon 16419, Republic of Korea
eCreative Research Institute, Sungkyunkwan University, 2066 Seoburo, Jangan-gu, Suwon 16419, Republic of Korea
First published on 9th July 2021
Abstract
Photocatalytic N2 fixation to NH3via defect creation on TiO2 to activate ultra-stable N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N has drawn enormous scientific attention, but poor selectivity and low yield rate are the major bottlenecks. Additionally, whether N2 preferentially adsorbs on phase-selective defect sites on TiO2 in correlation with appropriate band alignment has yet to be explored. Herein, theoretical predictions reveal that the defect sites on disordered anatase (Ad) preferentially exhibit higher N2 adsorption ability with a reduced energy barrier for a potential-determining-step (*N2 to NNH*) than the disordered rutile (Rd) phase of TiO2. Motivated by theoretical simulations, we synthesize a phase-selective disordered-anatase/ordered-rutile TiO2 photocatalyst (Na-Ad/Ro) by sodium-amine treatment of P25-TiO2 under ambient conditions, which exhibits an efficient NH3 formation rate of 432 μmol g−1 h−1, which is superior to that of any other defect-rich disordered TiO2 under solar illumination with a high apparent quantum efficiency of 13.6% at 340 nm. The multi-synergistic effects including selective N2 chemisorption on the defect sites of Na-Ad with enhanced visible-light absorption, suitable band alignment, and rapid interfacial charge separation with Ro enable substantially enhanced N2 fixation.
N has drawn enormous scientific attention, but poor selectivity and low yield rate are the major bottlenecks. Additionally, whether N2 preferentially adsorbs on phase-selective defect sites on TiO2 in correlation with appropriate band alignment has yet to be explored. Herein, theoretical predictions reveal that the defect sites on disordered anatase (Ad) preferentially exhibit higher N2 adsorption ability with a reduced energy barrier for a potential-determining-step (*N2 to NNH*) than the disordered rutile (Rd) phase of TiO2. Motivated by theoretical simulations, we synthesize a phase-selective disordered-anatase/ordered-rutile TiO2 photocatalyst (Na-Ad/Ro) by sodium-amine treatment of P25-TiO2 under ambient conditions, which exhibits an efficient NH3 formation rate of 432 μmol g−1 h−1, which is superior to that of any other defect-rich disordered TiO2 under solar illumination with a high apparent quantum efficiency of 13.6% at 340 nm. The multi-synergistic effects including selective N2 chemisorption on the defect sites of Na-Ad with enhanced visible-light absorption, suitable band alignment, and rapid interfacial charge separation with Ro enable substantially enhanced N2 fixation.
1. Introduction
Ammonia (NH3) is not only the most important industrial chemical but also the most promising carbon-free energy alternative due to its high hydrogen density (17.8 wt%).1 Industrial NH3 production has heavily relied on the traditional Haber–Bosch process (150–350 atm, 350–550 °C), which consumes ∼2% of the global energy supply and accounts for 1.6% of total global CO2 emissions.2,3 To circumvent these issues, an alternative N2 fixation method operating under milder reaction conditions is necessary, and in response to this, electrocatalytic and photocatalytic N2 reduction reactions (NRRs) have emerged as promising choices as they can be performed under green and sustainable conditions.4–9 However, the reduction of N2 to NH3 under ambient conditions is extremely difficult due to the inherent nature of the NN (bond dissociation energy: 940.95 kJ mol−1) along with additional thermodynamically challenging multistep processes. Although recent progress in the photocatalytic NRR demonstrated that N2 fixation can be promoted using various semiconductors including two-dimensional materials such as Mxenes,10 TiO2 nanosheets,11 Au/end-CeO2,12 Bi5O7X (X = Cl, Br, I) nanotubes,13,14 and single atoms anchored on various supports,15,16 the selectivity and NH3 yield rate for most of them are far from satisfactory. The two main reasons for the poor photocatalytic NRR are (i) poor interfacial charge transfer (CT) due to the high recombination rate of photogenerated holes and electrons and (ii) weak N2 adsorption ability.9 Therefore, a novel strategy to design photocatalysts with efficient charge separation and high N2 adsorption ability is urgently needed to improve the performance of the photocatalytic NRR.
More recently, defect engineering of photocatalysts has received immense attention as it offers unsaturated sites for N2 chemisorption, further weakening the NN and lowering the activation energy barrier for the NRR.17 For instance, Hirakawa et al. reported that introducing oxygen vacancies (Ov) on commercial JRC-TIO-6 (rutile phase, R) can boost the photocatalytic NRR with a solar-to-chemical conversion (SCC) efficiency of 0.02%.18 Zhao et al. elucidated that Ov along with lattice strain rich ultrathin anatase (A) nanosheets obtained via a facile copper-doping strategy exhibited strong chemisorption and activation of N2 and water, resulting in high rates of NH3 evolution under visible-light irradiation.11 However, the defect sites introduced additionally act as the recombination centers of the photogenerated carriers and limit the efficiency of the reported photocatalysts. Therefore, despite such significant efforts on the defect engineering of the TiO2 surface, the lack of fundamental understanding of the nature of phase-selective defect sites, high recombination rate, poor visible-light harvesting ability, harsh defect-engineering procedure, and insufficient chemisorption/activation of NN severely hamper the photocatalytic NRR kinetics. In addition, one important issue remained unanswered “Whether N2 chemisorption happens at all the Ov sites, or it is specific to a particular phase of P25-TiO2 (A: 75% and R: 25%) in correlation with appropriate band alignment for activating N2 and subsequent conversion to NH3” deserves to be further investigated.
Herein, we first investigated the N2 fixation activity of single-phase ordered (Ro, Ao) and disordered (Rd, Ad) TiO2 by theoretical calculations, which suggested that creating Ov on the anatase phase of TiO2 (Ad) is conducive for N2 adsorption, significantly reducing the energy barrier of the potential-determining step (PDS: *N2 to NNH*), boosting the intrinsic NRR over the rutile phase (Rd). Encouraged by theoretical predictions, we developed a facile strategy to synthesize a phase-selective Ad/Ro heterostructure by alkali metal (Na) amine treatment of P25-TiO2 (Na-Ad/Ro) under ambient conditions, which exhibits an efficient adsorption/desorption kinetics of N2/NH3 specifically on the Na-Ad phase and faster hole transport through the heterointerface region of the Ro phase hindering charge recombination, significantly facilitating water oxidation to produce necessary protons for efficient N2 fixation. The novel Na-Ad/Ro without any co-catalyst possesses a superior NH3 formation rate (432 μmol g−1 h−1), which is ∼125 and 5.5 times higher than that of bare TiO2 and Na-Ad, respectively under solar illumination, exhibiting one of the superior NRR performances with a high selectivity of 97.6%. Theoretical simulations clearly revealed that high intrinsic N2 chemisorption ability with the reduced energy barrier for PDS selectively originated from defect sites on Na-Ad rather than Ro and Li-Rd (Li-EDA treated Ro) along with a suitable band alignment promoting the NRR over hydrogen (H2) generation on Na-Ad/Ro. The multi-synergistic effects induced by phase-selective defect sites on Na-Ad/Ro, increased visible light absorption, and enhanced interfacial charge separation are found to be the key factors for higher photocatalytic activity.
2. Results and discussion
2.1 Theoretical calculation predictions
To understand the nature of phase-selective defect sites, density functional theory (DFT) calculations were first conducted to study the intrinsic N2 adsorption ability on the specific phase of ordered (Ro, Ao) and defect-rich disordered (Li-Rd, Na-Ad) TiO2 samples. The optimized atomic models with adsorbed N2 molecules on Li-Rd, Na-Ad, Ro, and Ao are presented in Fig. 1a–d. The computed N2 adsorption energy (Eads) values on Ro and Ao are close to 0 eV, indicating that the adsorption/activation of N2 in the absence of defect-sites on Ao and Ro hardly takes place at room temperature (Fig. 1e). In contrast, N2 can readily adsorb on Na-Ad with an Eads of −0.21 eV (thermodynamically favorable), while N2 adsorption on Li-Rd is an endothermic process (Eads: +0.61 eV), suggesting that the defect sites on the Ad phase of TiO2 selectively prefer N2 chemisorption over the Rd phase. Furthermore, we examined the Gibbs free energy diagrams of the NRR along the alternating pathway on Na-Ad and Li-Rd (Fig. 1f). Similar to the Eads trend, the first hydrogenation step (PDS: *N2 to NNH*) on Na-Ad is energetically more favorable than that on Li-Rd, boosting NH3 synthesis for the former. In a nutshell, DFT calculations certify that phase-selective defect sites on the Ad phase of TiO2 (Na-Ad) show higher N2 adsorption ability with superior intrinsic NRR activity over defect sites on the Rd phases of TiO2 (Li-Rd).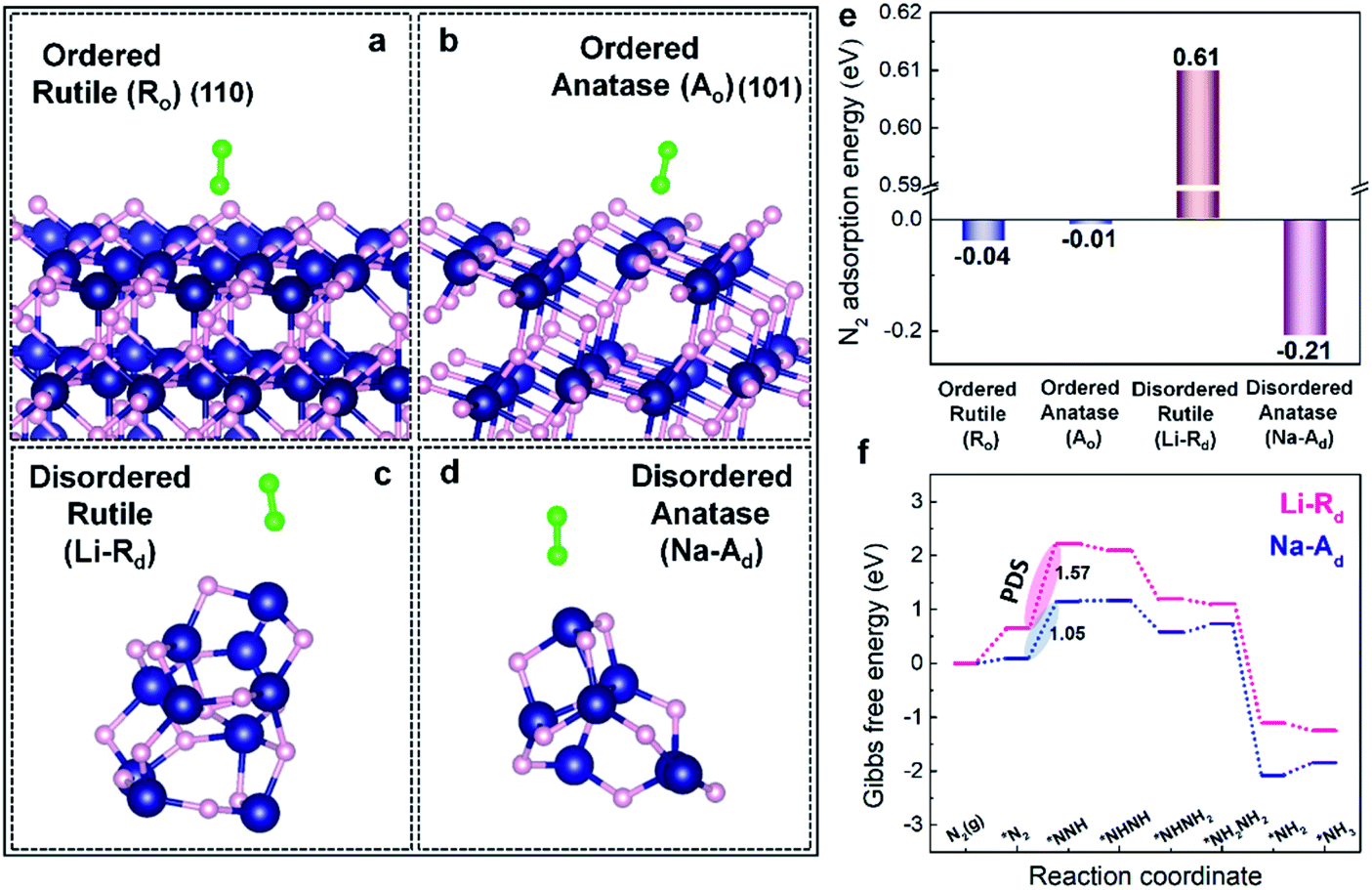 | ||
| Fig. 1 DFT calculations of N2 adsorption on ordered/disordered TiO2 and the reaction pathway. (a–d) Optimized atomic models with N2 molecules on Ro, Ao, Li-Rd, and Na-Ad. (e) N2 adsorption energy on Ro, Ao, Li-Rd, and Na-Ad atomic models. (f) Gibbs free energy (ΔG) profiles of N2 fixation over Li-Rd and Na-Ad along the alternating pathway. Navy, pink, light green balls represent titanium, oxygen, and nitrogen, respectively. | ||
2.2 Synthesis and structural analysis of ordered/disordered TiO2 photocatalysts
Motivated by the theoretical predictions, a series of selectively ordered/disordered phases of P25-TiO2 were synthesized by alkali-metal amine treatment also known as “dissolving-metal reduction” following our previous reports with slight modifications,19,20 as it is an effective strategy to create Ov on the specific phase of TiO2. Using lithium and the sodium ethylenediamine complex (Li, Na-EDA) with different reduction abilities of the solvated electrons can selectively create defect sites (Ti3+ adjacent to Ov) on Ro and Ao phases, respectively, to generate Li-Rd and Na-Ad, under mild conditions. The X-ray diffraction (XRD) (Fig. S1†) peaks indicate a gradual decrease in the specific phase of P25-TiO2 with the reaction time (1, 3, and 7 days) by Li/Na-EDA treatments. Fig. 2a reveals that either the Ro or Ao phase in mixed-phase P25-TiO2 nearly disappeared after 7 days of treatment, denoted as Li-Ao/Rd and Na-Ad/Ro, respectively, which is additionally supported by Raman spectra (Fig. S2†). The high-resolution transmission electron microscopy (HRTEM) image with the selected area electron diffraction (SAED) pattern of Na-Ad/Ro revealed the formation of a distinct heterointerface between the crystalline phase of Ro and disordered phase of Na-Ad along with uniform distribution of Ti and O as confirmed by EDX mapping (Fig. 2b and S3†). The ordered lattice fringes with a spacing of 3.6 Å and 2.1 Å can be assigned to the Ao(101) and Ro(111) plane (Fig. S4a and b†), respectively, along with each disordered phase starting from the surface to the core after 5 days of treatment, further confirming the random distribution of atoms in the disordered phase by HADDF-STEM (Fig. S4c–f†). Thus, our phase-selective disordered method successfully introduces defect sites on the specific phase of the TiO2 surface, which could selectively capture inert N2 and boost the NRR.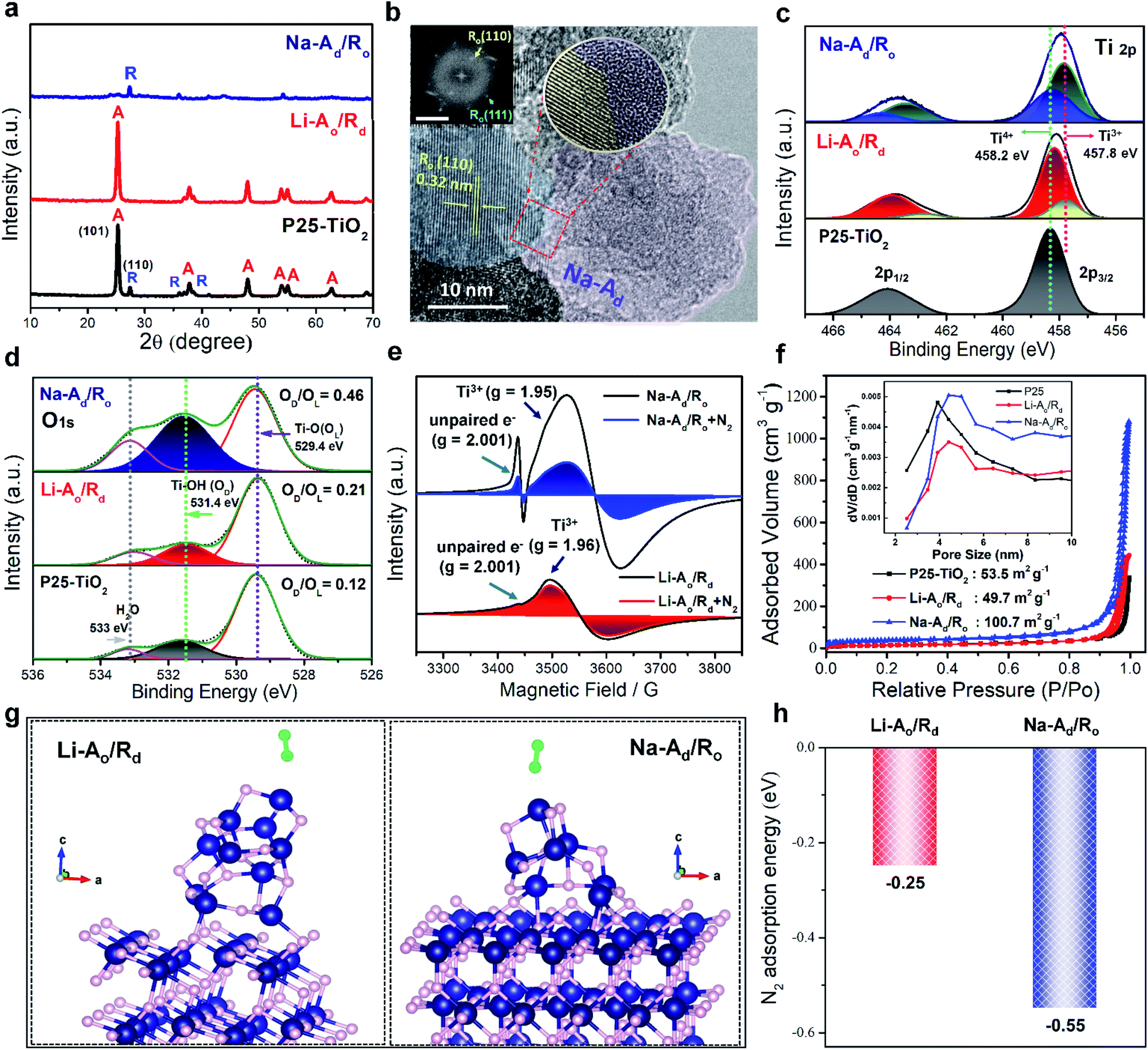 | ||
| Fig. 2 Structural characterization and simulation for N2 adsorption on P25-TiO2, Li-Ao/Rd, and Na-Ad/Ro. (a) The powder X-ray diffraction spectra (XRD). (b) HR-TEM image of Na-Ad/Ro (inset scale bar: 5 nm−1). XPS spectra of (c) Ti 2p and (d) O 1s. (e) Electron paramagnetic resonance (EPR) spectra of Li-Ao/Rd and Na-Ad/Ro before and after N2 saturation at 77 K. (f) N2 adsorption–desorption isotherms of P25-TiO2, Li-Ao/Rd and Na-Ad/Ro (inset, corresponding pore-size distribution). (g) Optimized atomic models with N2 molecules on Li-Ao/Rd and Na-Ad/Ro. (h) N2 adsorption energy of Li-Ao/Rd and Na-Ad/Ro. Navy, pink, light green balls represent titanium, oxygen, and nitrogen, respectively. | ||
To investigate the chemical composition and valence states of the elements over the selectively reduced TiO2 catalysts, high-resolution X-ray photoelectron spectroscopy (XPS) was employed. Compared to P25-TiO2, the binding energy (BE) of the Ti 2p doublet peaks at 458.2 and 463.9 eV consistent with the typical characteristic peaks of Ti 2p1/2 and Ti 2p3/2 for Ti4+ showed a negative shift to lower BE for Li-Ao/Rd and Na-Ad/Ro, attributed to the appearance of a low-valence Ti3+ at 457.8 and 462.8 eV on the surface (Fig. 2c).21 The removal of lattice oxygen left excessive electrons trapped by the adjacent Ti4+ species, generating Ti3+ sites or unpaired electrons, which could behave as an active center for surface adsorption of key N2 adsorbates. The O 1s XPS spectra exhibit three peaks at 529.4, 531.4, and 533 eV, attributed to the TiO2 lattice oxygen (Ti–O, OL), surface hydroxyl group (Ti–OH, defective oxygen: OD) originating from the reaction between chemisorbed water and the oxygen vacancy of disordered TiO2, and adsorbed H2O molecule, respectively (Fig. 2d). In Fig. S5a–d,† there is a gradual increase of Ti3+ sites with a simultaneous increase of OD/OL ratio for Li-Ao/Rd (0.21) and Na-Ad/Ro (0.46) as a function of reaction time, confirming the generation of Ti3+ with oxygen vacancies during alkali-metal amine treatment.22 Furthermore, the absence of all possible impurities like EDA (ethylenediamine), alkali ions (Na+), and chloride (Cl−) in the samples which may cause exaggerated and erroneous NH3 yield was confirmed by N 1s, Na 1s, and Cl 2p XPS spectra, respectively, (Fig. S5e–h†).23 We further quantified the amount of Ti3+ on Li-Ao/Rd, Na-Ad/Ro, Li-Rd, and Na-Ad by XPS peak deconvolution and CO2 temperature-programmed desorption (CO2-TPD) measurements, as summarized in Table 1. Atomic percentages of Ti3+/Ti4+ over the samples in the Ti 2p orbitals of XPS were calculated (Fig. S6†). CO2-TPD was also investigated to monitor surface defects (Ti3+) existing in disordered structures (Fig. S7 and Table S1†). CO2-TPD analysis between 150 and 500 °C resulted in two ranges: one range between 170 and 204 °C attributed to CO2 molecules bound to a regular five-coordinate Ti4+ site in the ordered TiO2 and the other over 280 °C corresponds to CO2 molecules bound to defective Ti3+ species in the disordered structure, indicating Ti3+ species bonded CO2 more strongly than the regular five-coordinate Ti4+ sites.24 From the above results, it was found that the surface defects (Ti3+) with oxygen vacancies on phase-selective disordered TiO2 were successfully created by Li/Na-EDA treatment while P25-TiO2 had a few defect sites.
| Sample | Surface areaa (m2 g−1) | E g (eV) | Quantity of Ti3+c (%/mmol g−1) | k H2 (μmol g−1 h−1) | k NH3 (μmol g−1 h−1) | Selectivity of N2 reductionf (%) | Selectivity of H2 evolutiong (%) | NH3 yield per BETh (μmol h−1 m−2 g) |
|---|---|---|---|---|---|---|---|---|
| a Determined by Brunauer–Emmett–Teller (BET) for N2 adsorption/desorption isotherms. b Determined by a plot of the Kubelka–Munk function versus the energy of light absorbed. c Quantification of Ti3+ concentration over all samples measured by XPS (%) and CO2-TPD (mmol g−1). d The H2 production rate (kH2) during the N2 reduction reaction under solar light under closed conditions. All the H2 gas was detected by syringe using GC-TCD. e The NH3 production rate (kNH3) during the N2 reduction reaction under solar light determined by cation exchange chromatography (IC). f Selectivity for N2 reduction (%) = 3kNH3/Re × 100%. Electron consumption rate for the reduced products (μmol g−1 h−1), Re = 2kH2 + 3kNH3. g Selectivity for H2 evolution (%) = 2kH2/Re × 100%. h NH3 yield per BET surface area = kNH3 × 50 mg/BET. | ||||||||
| P25-TiO2 | 53.5 | 3.12 | 2.6/0.1 | 18.3 | 3.4 | 21.8 | 78.2 | 3.2 |
| Li-Ao/Rd | 49.7 | 2.84 | 21.8/0.4 | 213 | 23 | 14.0 | 86.0 | 23.1 |
| Na-Ad/Ro | 100.7 | 2.65 | 62.1/0.6 | 15.7 | 432 | 97.6 | 2.4 | 214.5 |
| Na-Ad | 143.7 | 2.66 | 73.4/NA | Trace | 77.1 | NA | NA | 26.8 |
| Li-Rd | 14.5 | 2.75 | 74.8/NA | Trace | 4.2 | NA | NA | 14.5 |
The N2 adsorption capacity is one of the key factors impacting NH3 synthesis. The N2 physisorption ability of defect sites on Na-Ad/Ro and Li-Ao/Rd compared to P25-TiO2 was firstly investigated by electron paramagnetic resonance (EPR) analysis. Each catalyst (20 mg) was separately placed in a quartz EPR tube and analyzed at 77 K before and after N2 (99.999%) gas saturation (20 torr) (Fig. 2e). Commercial TiO2 with Ti4+ sites are diamagnetic (3d0, S = 0) in nature whereas defective TiO2 contains paramagnetic Ti3+ centers (3d1, S = 1/2) also known as “spin-active defects” generated due to oxygen vacancies.25 Both Na-Ad/Ro and Li-Ao/Rd show distinctive EPR signals of paramagnetic Ti3+ (g = 1.96) and unpaired electrons (g = 2.001) trapped in the oxygen vacancies while P25-TiO2 showed a negligible EPR signal (Fig. S8†). The Na-Ad/Ro exhibits a much higher number and signature of spin active Ti3+ sites compared to Li-Ao/Rd, suggesting a high density of electron spin polarization arising from the spin active Ti3+ centers on Na-Ad/Ro. Especially, these spin active Ti3+ centers containing unpaired electrons in Na-Ad can effectively interact with the N2 adsorbates expected to be effective for N2 fixation, which is corroborated by the significantly quenched EPR signal of Na-Ad/Ro after N2 saturation (Fig. 2e). Additionally, the BET specific surface area of all samples was examined by N2 adsorption–desorption measurements in Fig. 2f, which revealed that the P25-TiO2, Li-Ao/Rd, and Na-Ad/Ro are mesoporous in nature with an average pore distribution in the range of 3.5 to 4.5 nm. The specific surface area for Na-Ad/Ro (100.67 m2 g−1) is much higher than that of Li-Ao/Rd (49.7 m2 g−1) with a slightly larger average pore size compared to P25-TiO2, implying higher N2 adsorption with better mass transport, suitable for enhancing N2 fixation. Interestingly, the Na-Ad (143.74 m2 g−1) exhibited a much higher surface area than Li-Rd (14.508 m2 g−1) even with abundant Ti3+ species in both cases, indicating high intrinsic N2 adsorption ability of the defect sites on Na-Ad, consistent with the calculated N2 adsorption energy (Fig. 1e and S9†). To further understand the chemisorption ability of surface defects on Na/Li–TiO2 samples compared to P25-TiO2, N2/NH3-TPD measurements were then employed (Fig. S10 and Table S2†).26,27 The area of N2 desorption follows the order of Na-Ad > Na-Ad/Ro > Li-Ao/Rd ∼ Li-Rd > P25-TiO2, indicating that Na-Ad and Na-Ad/Ro have higher and stronger N2 adsorption capacities than Li-Rd and Li-Ao/Rd. Meanwhile, NH3-TPD was evaluated to monitor the NH3 desorption from the catalysts.27 Most of the NH3 desorbed on Na-Ad/Ro and Na-Ad relatively at a low temperature (100–194 °C), which indicates its weak adsorption on defect sites compared to Li-Ao/Rd and Li-Rd, implying much faster reaction kinetics and mass diffusion for the former. Likewise, the optimized N2-Ead of Na-Ad on Na-Ad/Ro (−0.55 eV) exhibits more negative values than that of Li-Rd on Li-Ao/Rd (−0.25 eV) as calculated by DFT (Fig. 2g and h) and AIMD simulations (Movies S1–S6†),28–32 suggesting that N2 adsorption is favorable on the defect sites of Na-Ad over Li-Rd in the heterostructure, further verifying our above-mentioned claims. Note that we also considered the N2-Ead at the interface between Na-Ad and Ro, showing that N2 hardly adsorbs during the relaxation process (Fig. S11†). These results concluded that N2 molecules selectively adsorb on the surface defects of the Na-Ad phase, neither on the Li-Rd phase nor at the heterointerface.
2.3 Photocatalytic N2 reduction activity and durability over ordered/disordered TiO2 samples
To confirm the effect of phase-selective defect sites on N2 fixation, we assessed the efficacy of Na-Ad/Ro and other catalysts for the photocatalytic NRR, as schematically illustrated in Fig. S12†. Generally, the catalyst needs to be well-dispersed in a mixture containing 50 mL of N2-saturated water and 7 mL of isopropyl alcohol (IPA) as the hole and hydroxyl radical scavenger under 1 sun illumination with continuous N2 gas flow (99.999%, flow rate: 0.3 L min−1, cc). One can see that the 7 day treated Na-Ad/Ro exhibits the highest rate of NH4+ production (432 μmol g−1 h−1) with a high NRR selectivity of 97.6%, followed by Na-Ad (77.1 μmol g−1 h−1), Li-Ao/Rd (23 μmol g−1 h−1), Li-Rd (4.2 μmol g−1 h−1) and P25-TiO2 (3.4 μmol g−1 h−1), benefitted by excellent N2 adsorption ability of the Na-Ad phase, as measured by ion chromatography (IC) (Fig. 3a and Table S3†). The Na-Ad/Ro showed the highest NH3 yield at an optimum amount of 50 mg, while the NH3 yield rate gradually decreased at a higher amount due to the poor penetration of photons and hindered mass diffusion (Fig. 3b).33 It is interesting to note that in the absence of IPA, Na-Ad/Ro also showed a reasonable NH3 production rate of 327 μmol g−1 h−1 along with oxygen (O2) generation as confirmed by GC-TCD (Fig. S13a and b†), with a stoichiometric ratio of NH3 to O2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5), N2 + 3H2O → 2NH3 + 3/2O2, suggesting that the photogenerated holes are efficiently consumed via the water oxidation process. The NH3 yield rate and apparent quantum efficiency (AQE) for Na-Ad/Ro can be comparable and superior to most of the reported photocatalysts with/without hole scavengers under solar light irradiation (Table S4†). Additionally, photocatalytic nitrate (NO3−) reduction activity was evaluated; since N
:1.5), N2 + 3H2O → 2NH3 + 3/2O2, suggesting that the photogenerated holes are efficiently consumed via the water oxidation process. The NH3 yield rate and apparent quantum efficiency (AQE) for Na-Ad/Ro can be comparable and superior to most of the reported photocatalysts with/without hole scavengers under solar light irradiation (Table S4†). Additionally, photocatalytic nitrate (NO3−) reduction activity was evaluated; since N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds have relatively lower dissociation energy (204 kJ mol−1) than NN bonds (945 kJ mol−1),34 Na-Ad/Ro exhibits the highest NO3− reduction activity under sunlight and 100 W LED light compared to Li-Ao/Rd and P25-TiO2, confirming the superior intrinsic activity for the former (Fig. S13c†). Furthermore, high NRR activity for Na-Ad/Ro over other catalysts was supported by the indophenol-blue method as an alternative analytical method, which can be used to ensure the produced NH4+, as our solution is neutral or slightly basic before and the after reaction, as detected with a pH meter (Fig. S14a–c†).35 To evaluate the effect of different ratios of Na-Ad and Ro phases on the NRR performance, we synthesized two different ratios of Na-Ad/Ro (90:10 and 50:50) and compared their NRR performance with main Na-Ad/Ro (75:25) (Fig. S14d†). As revealed in Fig. S14d,† Na-Ad/Ro (75:25) exhibited the highest NH3 yield compared to other control ratios (90:10 and 50:50). Additionally, we conducted a blank experiment in the absence of any catalysts under the same conditions, which resulted in a non-traceable amount of NH3 as confirmed by IC and the indophenol blue method, revealing that the photocatalytic NRR is the sole reason for a high rate of NH3 production rather than any possible contamination from air or other impurities (Fig. S14d†). For the control experiment, we synthesized a heterostructure containing 75% of Li-Rd and 25% of Ao and performed the NRR under the same conditions. However, its NH3 yield (4.8 μmol g−1 h−1) quantified by IC was negligible because of poor N2 adsorption ability on the abundant Li-Rd phase. The superior intrinsic N2 adsorption ability of Na-Ad over Li-Rd was further corroborated by the BET specific surface area-normalized NH3 yield (Table 1), which strongly reinforces the significance of phase-selective active sites for N2 fixation.
O bonds have relatively lower dissociation energy (204 kJ mol−1) than NN bonds (945 kJ mol−1),34 Na-Ad/Ro exhibits the highest NO3− reduction activity under sunlight and 100 W LED light compared to Li-Ao/Rd and P25-TiO2, confirming the superior intrinsic activity for the former (Fig. S13c†). Furthermore, high NRR activity for Na-Ad/Ro over other catalysts was supported by the indophenol-blue method as an alternative analytical method, which can be used to ensure the produced NH4+, as our solution is neutral or slightly basic before and the after reaction, as detected with a pH meter (Fig. S14a–c†).35 To evaluate the effect of different ratios of Na-Ad and Ro phases on the NRR performance, we synthesized two different ratios of Na-Ad/Ro (90:10 and 50:50) and compared their NRR performance with main Na-Ad/Ro (75:25) (Fig. S14d†). As revealed in Fig. S14d,† Na-Ad/Ro (75:25) exhibited the highest NH3 yield compared to other control ratios (90:10 and 50:50). Additionally, we conducted a blank experiment in the absence of any catalysts under the same conditions, which resulted in a non-traceable amount of NH3 as confirmed by IC and the indophenol blue method, revealing that the photocatalytic NRR is the sole reason for a high rate of NH3 production rather than any possible contamination from air or other impurities (Fig. S14d†). For the control experiment, we synthesized a heterostructure containing 75% of Li-Rd and 25% of Ao and performed the NRR under the same conditions. However, its NH3 yield (4.8 μmol g−1 h−1) quantified by IC was negligible because of poor N2 adsorption ability on the abundant Li-Rd phase. The superior intrinsic N2 adsorption ability of Na-Ad over Li-Rd was further corroborated by the BET specific surface area-normalized NH3 yield (Table 1), which strongly reinforces the significance of phase-selective active sites for N2 fixation.
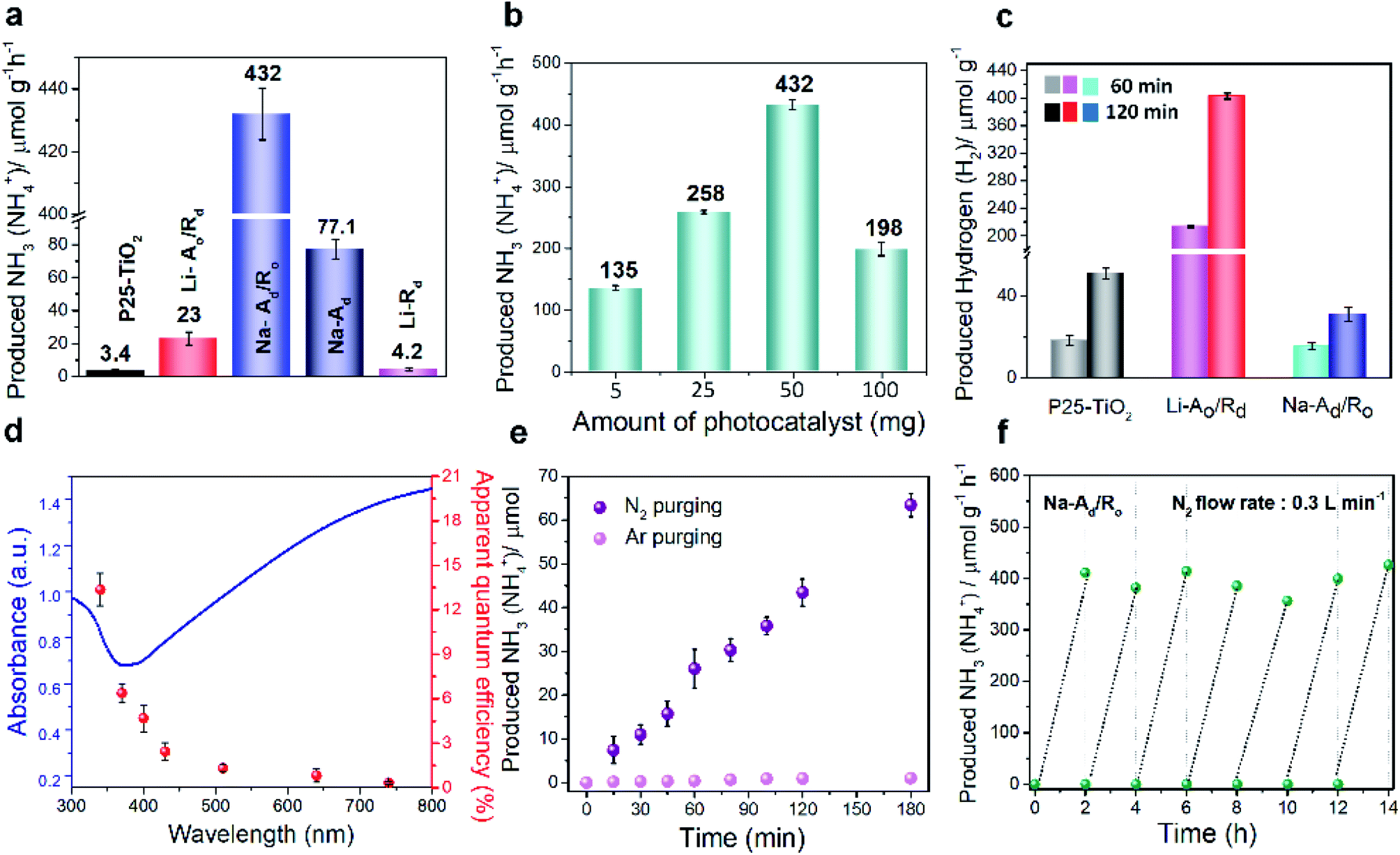 | ||
| Fig. 3 Photocatalytic N2 fixation activities of various ordered/disordered TiO2 samples. (a) The rate of produced NH3 (NH4+) over P25-TiO2, Li-Ao/Rd, Na-Ad/Ro, Li-Rd and Na-Ad in N2 saturated water with IPA (14%) as the hole/hydroxyl radical scavenger under solar illumination. (b) The rate of produced NH3 (NH4+) for Na-Ad/Ro at various amounts of the catalyst for photocatalytic N2 reduction. (c) The rate of produced H2 over P25-TiO2, Li-Ao/Rd and Na-Ad/Ro in N2 saturated-water with IPA (14%) as the hole scavenger under solar illumination. (d) UV-vis diffuse reflectance spectrum (UV-DRS, left axis) and calculated apparent quantum efficiency (AQE, right axis) for N2 fixation over Na-Ad/Ro under monochromatic light irradiation. (e) Time-dependent rates of produced NH3 (NH4+) for Na-Ad/Ro under N2 and Ar gas purging. (f) Cycling test of photocatalytic N2 fixation over Na-Ad/Ro. | ||
To confirm the origin of the NH3 generated from the supplied N2 gas, 15N2 (98 atom%, 15N) isotope labeling experiments were carefully performed for 50 min at 8 sccm controlled by the mass flow control (MFC) system (Fig. S15†). Double coupling of 15NH4+ and triple coupling of 14NH4+ are identified by 1H nuclear magnetic resonance (1H NMR), signifying that the N2 gas is the only source of NH3, further supported by mass spectra,18 EDS analysis of Na-Ad/Ro and additional control NRR experiments under Ar-saturated solution (Fig. S15c and S16†).23,36 Importantly, some reported papers claimed that oxygen vacancies introduced on catalysts could behave as active sites for water dissociation and boost HER activity.22,37–39 Hence, it is necessary to examine the HER activity and selectivity over disordered catalysts in a closed system under the same conditions for the NRR (Fig. 3c and Table 1). Interestingly, the Na-Ad/Ro (H2 yield rate: 15.7 μmol g−1 h−1) exhibits poor HER activity compared to Li-Ao/Rd (H2 yield rate: 213 μmol g−1 h−1), which may be attributed to the high surface coverage of N2 molecules on the defect sites of Na-Ad rather than the proton (H+), but an opposite trend to OV defective sites on Li-Rd, as revealed by experimental analysis and theoretical calculations. Additionally, hydrazine (N2H4) and nitrate acid (HNO3) are possible by-products that can be detected by the Watt and Chrisps method and IC, respectively, and negligible N2H4 and a trace amount of NO3− (0.17 μmol g−1 h−1) were detected during the NRR, suggesting high selectivity of NH3 synthesis for Na-Ad/Ro (Fig. S17, S18 and Table S5†).40 To confirm the effect of trace NO3− produced during the reaction on the total yield of NH3 for Na-Ad/Ro, twice the amount of trace NO3− (0.34 μmol g−1 h−1) was added into the catalyst dispersed solution and examined under solar light for 2 h under the same NRR conditions. The NH3 yield of only 0.357 ppm (1.05 μmol h−1 g−1) detected by IC was well within the error range. Therefore, the influence of trace NO3− on the total yield of NH3 was negligible. In Fig. 3d, the wavelength-dependent quantum efficiency (QE) experiment for N2 fixation on Na-Ad/Ro under monochromatic light irradiation closely fits the absorption spectrum, showing approximately 13.6%, 6.4%, 4.7%, 2.4%, 1.3%, 0.8% and 0.3% at 340, 370, 400, 430, 510, 640 and 740 nm, respectively. However, the apparent QE of Na-Ad/Ro over 500 nm is inconsistent with the visible light absorption distribution in the ultraviolet-visible spectrum. Creating oxygen defects on disordered TiO2 can harvest visible light, however, the excited electrons which are located at deep trap states are unable to be utilized for the NRR because they cannot be excited and migrated thermally to the CB as well as their states are located well below the N2/NH3 redox potential, which makes them inactive for N2 fixation.41 Although these trap sites are inactive for N2 fixation, they are the main sites for enhancing the N2 adsorption on the surface of the Na-Ad accelerating NRR process.
Besides superior activity, the durability of the catalyst is another crucial parameter for practical applications. The cycling tests for Na-Ad/Ro were conducted for 3 h under 1 sun illumination with continuous N2 gas supply showing linearly produced NH4+, quantified by IC (Fig. 3e) and the indophenol-blue method (Fig. S19†). Moreover, the rate of NH3 yield for Na-Ad/Ro remained constant for 7 consecutive cycles (Fig. 3f), suggesting its excellent stability towards the NRR. After stability tests, careful examination of XPS, XRD, and TEM characterization on Na-Ad/Ro phases shows no distinct changes over surface Ti3+ and the amorphous structure of the Na-Ad junction with the crystalline Ro phase, indicating the robustness of Na-Ad/Ro towards the photocatalytic NRR, however, a slight decrease of the Ov sites was observed probably due to water molecules or reactants/intermediates after stability tests (Fig. S20†). From the results, we found that the photocatalytic NRR showed a close relationship with a specific type of phase-selective absorption site for N2 fixation rather than all possible surface defects sites. Although sufficient defect sites on Li-Rd and Li-Ao/Rd were present, poor intrinsic N2 adsorption ability and favorable HER kinetics significantly hamper the formation of NH3 and its selectivity (Table 1).16
2.4 Optical and charge transfer (CT) properties and band alignment over ordered/disordered TiO2 samples
To develop an in-depth mechanistic understanding of the outstanding NRR performance of Na-Ad/Ro over the other catalysts, the electronic band position was determined based on the Tauc plots, UPS, and valence-band XPS spectra (VB-XPS) (Fig. 4a–e and S21†).42 It is known that a heterointerface between two different phases is an important factor leading to rapid interfacial CT and modulation of the energy band alignment.19 The secondary-electron cut-off energy for Na-Ad and Ro was measured by UPS as 16.40 and 16.3 eV, respectively (Fig. 4a), and the Fermi level (Ef) was then estimated to be −0.04 and 0.06 eV (vs. NHE, pH = 7), respectively. The VB positions with respect to Ef were determined by VB-XPS as 2.10 and 1.86 eV, respectively (Fig. 4b). To further obtain the conduction-band position (ECB) of Na-Ad and Ro, the energy bandgap (Eg) was also obtained by linear extrapolation of the Tauc plots (αhν)1/2vs. hν, obtaining indirect bandgap energies of 2.49 and 3.04 eV, respectively (Fig. 4c). Consequently, the ECB of Na-Ad and Ro can be further determined as −0.39 and −1.18 eV, respectively (vs. NHE, pH = 7) (Fig. 4d). In general, the energy band at the interface of two different semiconductors would be rearranged by a band alignment through contacts until Ef is aligned at the same level. The possible electronic band structure of Na-Ad/Ro (Fig. 4d) is finally depicted after rearrangement. For Na-Ad, both CB and VB potentials are downshifted while those for Ro are upshifted, which is a type-II band structure, indicating that the CB potential of Na-Ad (−0.39 eV) is suitable for N2/NH3 and the VB potential of Ro (1.81 eV) is much closer for the water oxidation reaction.43 Combined with a suitable band-alignment and enhanced visible-light absorption, possible interfacial charge separation at the heterointerface where electrons on Na-Ad are consumed to activate the adsorbed N2 molecules and holes on Ro oxidize the water molecules in the meantime (Fig. 4d) corroborates the superior NRR selectivity of Na-Ad/Ro. Additionally, we also delineated the possible electronic band structure of Li-Ao/Rd and P25-TiO2 (Fig. S21†). Interestingly, we found that the CB potential of Li-Ao/Rd and P25-TiO2 is placed nearby HER potentials with low N2 adsorption, resulting in poor NRR selectivity (14%) and high HER kinetics (86%) for Li-Ao/Rd. | ||
| Fig. 4 Spectroscopic analysis for possible charge carrier dynamics and the corresponding energy level diagram depicting the photochemical reaction. (a) UPS spectra and (b) valence band XPS edge spectra of Ro and Na-Ad. (c) Kubelka–Munk function versus the photon energy graphs and calculated bandgap of Ro, Li-Rd, and Na-Ad. (d) Estimation of energy band structures of Na-Ad, Ro, and Na-Ad/Ro. (e) Photoluminescence (PL) spectra (λex = 375 nm: λem = 506 nm) and (f) time-resolved photoluminescence (TRPL) of P25-TiO2, Na-Ad and Na-Ad/Ro. The scatter points were fitted using a biexponential decay. (g) Transient photocurrent response of Na-Ad/Ro and Na-Ad. | ||
In order to further understand the interfacial charge carrier dynamics, photoluminescence (PL) spectra of P25-TiO2, Na-Ad, and Na-Ad/Ro were examined (Fig. 4e). All catalysts exhibit a distinct PL peak centered approximately at 506 nm upon excitation by a 375 nm laser. After defect formation, the PL intensity of Na-Ad is quenched at 550 nm corresponding to defect states attributed to Ti3+ on oxygen vacancies and further quenched for Na-Ad/Ro. This PL quenching behavior corresponds to facile CT between Na-Ad and Ro at the interface. We then used TRPL spectroscopy to probe charge carrier dynamics over Na-Ad and Na-Ad/Ro films by electrophoretic deposition (10 V, 15 s each on the ITO substrate, inset) in Fig. 4f. The light intensity decay curves through two relaxation pathways can be fitted with an exponential equation as shown below:44
| f(t) = A0 + A1(exp(−t/τ1) + A2(exp(−t/τ2)), | (1) |
2.5 Proposed mechanistic pathway for N2 fixation
To identify the time-dependent change of N-related functional groups in the early stages of the photocatalytic NRR, in situ diffuse reflectance infrared Fourier transform (in situ DRIFT) measurements were conducted (Fig. S23†).11,14,45 To exclude the background spectra of adsorbed gas and impurities, the sample was fully dried at 333 K in a vacuum oven overnight, and then was placed into a DRIFT specimen chamber. Several absorption bands gradually increase under 1 sun irradiation from 0 to 60 min in the range of 1100–1700 cm−1 (Fig. 5a). The FT-IR spectra of N-related functional groups represent six bands: 1123, 1289, 1329, 1425, 1551, and 1660 cm−1. The interference from CO2 and H2O from the air was removed by a background calibration. The bands at 1123 and 1660 cm−1 can be assigned to N–N stretching in chemisorbed N2Hy species on catalysts. Two weak bands at 1329 and 1551 cm−1 are characteristic of adsorbed NH3 and sharp bands at 1289 and 1425 cm−1 can be assigned to adsorbed NH4+ (–NH2 wagging and H–N–H bending of adsorbed N2Hy species, respectively), which are intensified with the increase of reaction time. Absorption bands in the range of 3100–3600 cm−1 for Na-Ad/Ro and Na-Ad are shown in Fig. 5b and S24.† The intensity of peaks at 3150 and 3334 cm−1 attributed to an N–H stretching mode (νN–H) gradually increases, followed by the increased peaks at 3527 cm−1 assigned to the O–H stretching (νTiO–H) mode after water supply.46 As a result, in situ measurements elucidate that inert NN is efficiently activated on phase-selective defect sites on Na-Ad/Ro under 1 sun irradiation, leading to the subsequent NN cleavage to NH3 or NH4+ at the last step.
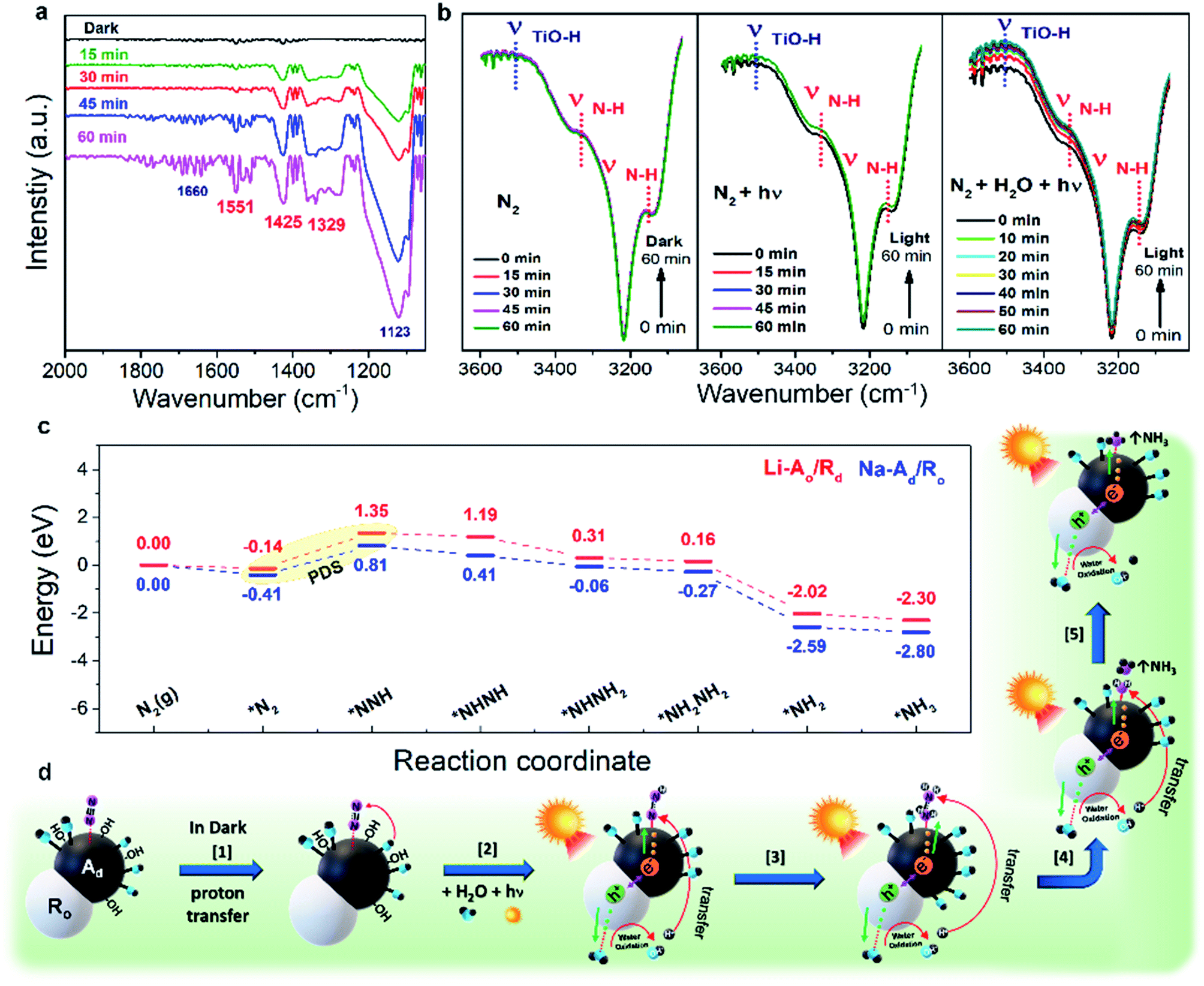 | ||
| Fig. 5 Proposed mechanistic pathway for N2 fixation. (a) Fourier Transform Infrared (FT-IR) spectra during the N2 reduction reaction in the presence of water and N2 under solar illumination for 60 min. (b) Time-dependent change of in situ Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectra of Na-Ad/Ro after N2 adsorption in the dark for 60 min, sunlight irradiation for 60 min and then water vapor supply for 60 min simultaneously (from left to right) under ambient conditions. (c) Gibbs free energy diagram of the N2 reduction reaction on Na-Ad/Ro and Li-Ao/Rd. (d) Schematic illustration of the alternating pathway during the photocatalytic N2 fixation on Na-Ad/Ro. | ||
To gain further insight into the NRR mechanism of phase-selective defect sites over Li-Ao/Rd(110) and Na-Ad/Ro(101), the DFT calculations were carried out as shown in Fig. 5c. After successful *N2 adsorption, the Gibbs free energy for the first hydrogenation step is significant as a potential determining step (PDS). One can see that the PDS (*N2 to N–NH*) on Na-Ad/Ro(101) was indeed favored by a lower energy barrier (1.22 eV), in comparison to 1.49 eV for Li-Ao/Rd, indicating that Na-Ad/Ro possesses better NRR intrinsic activity than Li-Ao/Rd. Note that the alternating pathway is the thermodynamically favorable process compared to the distal pathway after the PDS (Fig. S25†). Subsequent N–NH* hydrogenation to NH–NH* requires a low energy barrier for Na-Ad/Ro compared to Li-Ao/Rd, which is thermodynamically more feasible during the NRR. Corresponding visual images of different steps involved in the DFT calculation for both Na-Ad/Ro and Li-Ao/Rd are presented in Fig. S26.† Based on in situ DRIFT and theoretical calculations, we deduced a possible NRR pathway on Na-Ad/Ro in Fig. 5d and additionally, and listed the chemical equations (Discussion S1†). The photo-driven NH3 synthesis proceeds through the activation of N2 by the supply of multiple exciton transfer using photons as a driving force. At first, there is a proton-coupled electron transfer step (Step 1) where the N2 adsorbed on Ti3+ species in Na-Ad (*NN) interact with H atoms of the adjacent Ti3+–OH, resulting in the appearance of an N–H stretching vibration (νN–H) band. After 1 sun irradiation and water supply (Step 2), protons (H+) from the oxidized water via photogenerated holes (h+) at the VB of Ro and electrons (e−) at the CB of Na-Ad generate the adsorbed chemical species (*NNH) as follows: *NN + H+ + e− → *NNH˙. Consequently, transferring four e− and H+ to *NH2–NH2 (Step 3) on Na-Ad produces free-NH3 (Step 4), and the remaining *NH2 is hydrogenated and regenerates the Ti3+ sites after NH3 desorbed from the catalysts for the next N2 adsorbates (Step 5). This ideal alternating pathway on Na-Ad/Ro which involves the one-by-one addition of H atoms and electrons to simultaneously break down the NN bond can efficiently produce NH3.47
3. Conclusions
In summary, superior NRR activity with high selectivity was achieved by phase-selective defect sites on mixed-phase commercial P25-TiO2. Our DFT simulations revealed that defect sites on the disordered Na-Ad phase exhibit higher N2 adsorption ability and better intrinsic NRR activity than those on the disordered Li-Rd phase. Consistently, the experimental findings confirmed highly selective N2 affinity on defect sites on Na-Ad over Li-Rd. Moreover, Na-Ad can harvest the visible-range spectrum ascribed to the bandgap reduced by Ti3+ sites on oxygen vacancies. Upon integration with the Ro phase, a suitable CB band position for the NRR over the HER and rapid interfacial charge separation at the heterointerface for Na-Ad/Ro facilitate the desired intermediates (*NNH) via subsequent hydrogenation steps along with a thermodynamically favorable process verified by charge dynamics and band structure analysis combined with DFT simulations. As a result, noble-metal free Na-Ad/Ro achieved a superior NH3 formation rate of 432 μmol g−1 h−1, which is ∼125 times higher than that of bare TiO2 (3.4 μmol g−1 h−1) under simulated AM 1.5G light irradiation at room temperature with a high AQE of 13.6%, suppressing H2 generation. This finding highlights a promising approach to demonstrate the importance of phase-selective active sites with a suitable band position to develop a novel photocatalyst for future energy-related applications.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
J. S., X. L. and A. K. equally contributed to this work. J. S. and Y. H. conceived and designed the experiments. J. S. and Y. H. carried out the synthesis of the materials and photocatalytic NRR. X. L. conducted the DFT calculation. A. K., J. Y. and Y. D. K. supported the data analysis and supported in situ measurements. E. L. conducted the PL measurements. H. L. supervised the project. J. S., A. K., and H. L. wrote the paper. All authors discussed the results and commented on the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R011-D1) and was partially supported by the Korea Medical Device Development Fund grant funded by the Korea government (the Ministry of Science and ICT, the Ministry of Trade, Industry and Energy, the Ministry of Health & Welfare, the Ministry of Food and Drug Safety) (Project Number: KMDF_PR_20200901_0004) and Scale-up Support Program for Environmental Small-Medium Enterprise (00005002700).References
- R. F. Service, Science, 2014, 345, 610 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636 CrossRef CAS.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183 RSC.
- S. Li, Y. Wang, J. Liang, T. Xu, D. Ma, Q. Liu, T. Li, S. Xu, G. Chen, A. M. Asiri, Y. Luo, Q. Wu and X. Sun, Mater. Today Phys., 2021, 18, 100396 CrossRef CAS.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- T. Xu, B. Ma, J. Liang, L. Yue, Q. Liu, T. Li, H. Zhao, Y. Luo, S. Lu and X. Sun, Acta Phys.-Chim. Sin., 2021, 37(7), 2009043 Search PubMed.
- T. Wang, S. Li, B. He, X. Zhu, Y. Luo, Q. Liu, T. Li, S. Lu, C. Ye, A. M. Asiri and X. Sun, Chin. J. Catal., 2021, 42, 1024–1029 CrossRef CAS.
- T. Wang, Q. Liu, T. Li, S. Lu, G. Chen, X. Shi, A. M. Asiri, Y. Luo, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 884–888 RSC.
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- Q. Liu, L. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102–4110 RSC.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- H. Jia, A. Du, H. Zhang, J. Yang, R. Jiang, J. Wang and C.-Y. Zang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS PubMed.
- Y. Shiraishi, M. Hashimoto, K. Chishiro, K. Moriyama, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2020, 142, 7574–7583 CrossRef CAS PubMed.
- P. Li, Z. Zhou, Q. Wang, M. Guo, S. Chen, J. Low, R. Long, W. Liu, P. Ding, Y. Wu and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- C. Liu, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- C. Zhang, D. Wang, Y. Wan, R. Lv, S. Li, B. Li, X. Zou and S. Yang, Mater. Today, 2020, 40, 18–25 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929 CrossRef CAS PubMed.
- K. Zhang, L. Wang, J. K. Kim, M. Ma, G. Veerappan, C.-L. Lee, K.-j. Kong, H. Lee and J. H. Park, Energy Environ. Sci., 2016, 9, 499–503 RSC.
- H. Hwang, S. Oh, J. Shim, Y. Kim, A. Kim, D. Kim, J. Kim, S. Bak, Y. Cho, V. Q. Bui, T. A. Le and H. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 35693 CrossRef CAS PubMed.
- H. Choi, J. Lee, D. Kim, A. Kumar, B. Jeong, K.-J. Kim, H. Lee and J. Y. Park, Catal. Sci. Technol., 2021, 11, 1698–1708 RSC.
- J. Lee, A. Kumar, T. Yang, X. Liu, A. R. Jadhav, G. H. Park, Y. Hwang, J. Yu, C. T. K. Nguyen, Y. Liu, S. Ajmal, M. G. Kim and H. Lee, Energy Environ. Sci., 2020, 13, 5152–5164 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nie-lander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- L.-B. Xiong, J.-L. Li, B. Yang and Y. Yu, Nanomater, 2012, 2012, 831524 Search PubMed.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, Š. Kment, R. Zbořil and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS PubMed.
- Y. Liu, Q. Xu, X. Fan, X. Quan, Y. Su, S. Chen, H. Yu and Z. Cai, J. Mater. Chem. A, 2019, 7, 26358–26363 RSC.
- P. Kuśtrowski, L. Chmielarz, E. Bożek, M. Sawalha and F. Roessner, Mater. Res. Bull., 2004, 39, 263–281 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Park, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- A. Kumar, V. Q. Bui, J. Lee, A. R. Jadhav, Y. Hwang, M. G. Kim, Y. Kawazoe and H. Lee, ACS Energy Lett., 2021, 6, 354–363 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, ACS Catal., 2017, 7, 3713–3720 CrossRef CAS.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- C. Guo, J. Ran, A. Vasileff and S. Qiao, Energy Environ. Sci., 2018, 11, 45 RSC.
- A. Kumar and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2017, 9, 41906–41915 CrossRef CAS PubMed.
- S. Parvin, A. Kumar, A. Ghosh and S. Bhattacharyya, Chem. Sci., 2020, 11, 3893–3902 RSC.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- S. Kohtani, A. Kawashima and H. Miyabe, Catalysts, 2017, 7(10), 303 CrossRef.
- Y.-H. Chen, J. K. Ye, Y. J. Chang, T. W. Liu, Y. H. Chuang, W. R. Liu, S. H. Liu and Y. C. Pu, Appl. Catal., B, 2021, 284, 119751 CrossRef CAS.
- M.-H. Vu, M. Sakar and T.-O. Do, Catalysts, 2018, 8(12), 621 CrossRef.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126–4136 CrossRef CAS.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen, J. Liu, J. Zhao and L. Zhang, Chem, 2019, 5, 2702–2717 CAS.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03223b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |