
Building ordered nanoparticle assemblies inspired by atomic epitaxy
Jiaming
Liu
,
Jingjing
Wei
 and
Zhijie
Yang
*
and
Zhijie
Yang
*
Key Laboratory of Colloid and Interface Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: zyangchem@sdu.edu.cn
First published on 18th August 2021
Abstract
Self-assembly of inorganic nanoparticles into mesoscopic or macroscopic nanoparticle assemblies is an efficient strategy to fabricate advanced devices with emergent nanoscale functionalities. Furthermore, assembly of nanoparticles onto substrates may enable the fabrication of substrate-integrated devices, akin to atomic crystal growth on a substrate. Recent progress in nanoparticle assembly suggests that ordered nanoparticle assemblies could be well produced on a selected substrate, referred to as soft epitaxial growth. Herein, recent advances in soft epitaxial growth of a nanoparticle assembly are presented, including the assembly strategies, the choice of substrate and the epitaxial modes. Perspectives are also discussed for the material design based on substrate-integrated soft epitaxial growth.
 Zhijie Yang | Zhijie Yang received his MSc in Chemistry from Shandong University in 2010 and earned his PhD in 2014 from Université Pierre et Marie Curie (now Sorbonne Université) under the supervision of Marie-Paule Pileni. From 2014–2015 he held a postdoctoral position in Pileni's research group at Sorbonne Université. From 2016 to 2018 he held a postdoctoral position at the Center for Soft and Living Matter of IBS in South Korea. He was appointed Full Professor in Physical Chemistry at Shandong University in 2018. His current research interest focuses on the surface chemistry of nanoparticles and hybrid materials from chemical self-assembly. |
1. Introduction
Crystallization is ubiquitous in nature arranging atoms, ions, molecules or macromolecules into three-dimensional periodic structures. Similarly, assembly of nanoparticles is usually analogous to the crystallization of atoms, which represents a practical approach for the fabrication of mesoscopic as well as macroscopic materials with emergent nanoscale functionalities.1–5 The last two decades have witnessed tremendous progress in the assembly of nanoparticles into highly ordered nanoparticle superlattices.6–11 Different from individual nanoparticles or the disordered counterparts, ordered nanoparticle superlattices show collective optical, electrical and magnetic properties arising from the electronic coupling effect between neighbouring nanoparticles with engineered interparticle distances.12–16Nanoparticles synthesized in a non-polar phase are commonly coated with monolayer organic ligands.17,18 Therefore, the forces that direct the nanoparticle assembly are also determined by the surface organic coating layer other than the entropy, including hydrogen bonding, electrostatic forces, steric repulsion, van der Waals attraction, hydrophobic interactions, magnetic dipolar interactions, and depletion forces.9,19–22 To understand the forces between nanoparticles, manipulation of the nanoparticles as “artificial atoms” assembled into a designed structure and realization of their functions are the ultimate goals for nanoparticle assemblies. Hence, determining the crystal growth principles from atomic crystals is required to design and fabricate nanoparticle superlattices with increasing complexities.23,24
At the atomic and molecular scales, epitaxial growth (EG) is an efficient strategy for producing new materials for specific device fabrication.25–27 The chemical vapour deposition (CVD) method is commonly used to grow an epitaxial layer on a single crystal substrate, and the as-grown layer is usually single-crystalline.28,29 Furthermore, liquid-phase epitaxial growth (LPG) methods have been widely used in the preparation of high-quality semiconductor single crystal thin films since it is a growth process at or close to thermodynamic equilibrium.30 Three typical epitaxial growth modes at the atomic scale have been prepared, including the Frank–van der Merve (F–vdM) mode, Stranski–Krastanow (S–K) mode and Volmer–Weber (V–W) mode, as illustrated in Fig. 1.31,32 The F–vdM mode demonstrates the layer-by-layer epitaxial growth; the S–K model is layered growth followed by island growth; the V–W mode is dominantly island growth. The growth mode is determined by the competition of the interaction between an epitaxy atom and the substrate and the interactions between epitaxy atoms. Besides, the lattices misfit degree of substrate and adlayer is also an important factor for epitaxial growth.23
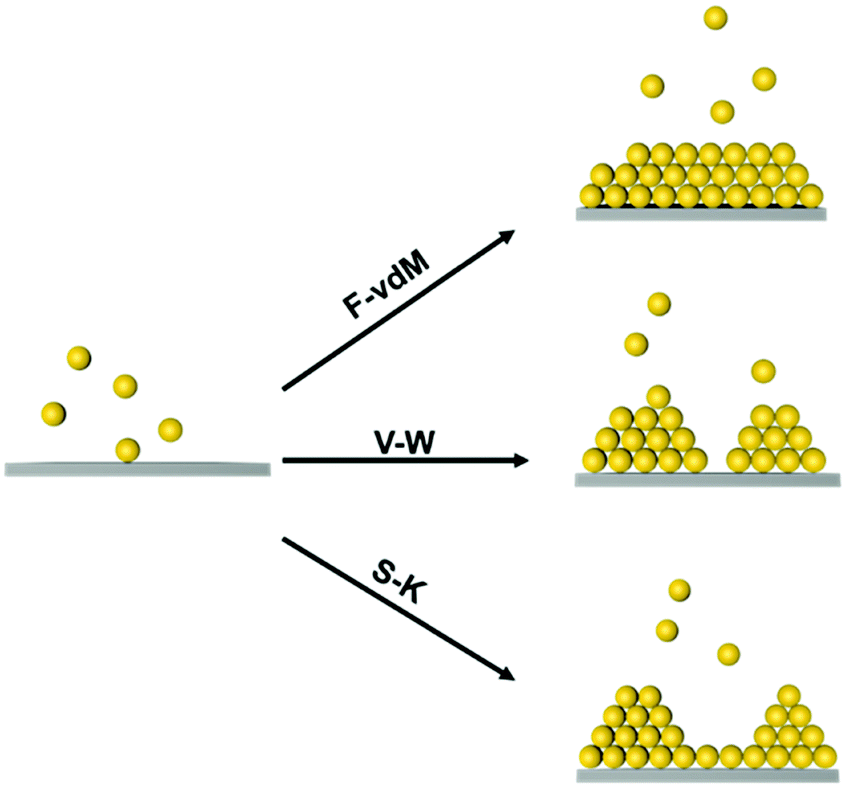 | ||
| Fig. 1 Schematic illustration of three different modes in atomic epitaxial growth. | ||
Inspired by the atomic epitaxial growth, the nanoparticles could also be used as the epitaxial “artificial atoms” for the creation of desired nanoparticle superlattices, which is referred to as soft epitaxial growth, SEG.33 From the perspective of self-assembly, the study of nanoparticle superlattice SEG will promote the understanding of interactions at the nanoscale. The weak interaction of multiple objects including the interaction between neighbouring nanoparticles, and between nanoparticles and a substrate compete in the process of soft epitaxy growth, generating different epitaxy modes. From the perspective of material design, SEG directed nanoparticle assemblies as well as the new interface created between the nanoparticle superlattice material and the substrate material will bring new properties and functions. From the perspective of physical properties, understanding the electron (photoelectron) and energy transfer between the nanoparticle superlattice and the substrate material will be helpful for the design of new photoelectric devices, including photocatalysts, photodetectors, etc.
Similar to atomic epitaxial growth, the misfit degree of the substrate and epitaxial adlayer is the key factor that determines the epitaxial growth efficiency and structures.27 The lattice misfit degree is commonly modulated by adjusting the interactions between epitaxial nanoparticle and substrate, including the van der Waals interactions, the electrostatic interactions, the hydrogen bond and coordination bond, etc. When the attractive force between the nanoparticles and substrate is overwhelming, the nanoparticles would be kinetically trapped onto the substrate irreversibly, generating a loose-ordered grown layer of nanoparticles. This always happens in electrostatic force or coordinating interaction directed layer-by-layer nanoparticle assembly.34,35 When the interaction the between nanoparticles and substrate is much weaker than that of neighbouring nanoparticles, self-sorted assembly is dominant. Therefore, a well-ordered epitaxial nanoparticle assembly can be initiated only when the attraction between the nanoparticles and substrate matches the interaction between nanoparticles. Unlike atomic epitaxial growth, SEG directed nanoparticle assemblies to some extent tolerate the misfit because of the softness of nanoparticle surface coating agents. The nanoparticle could squeeze their coating layer for the fitting of an adlayer to the substrate.
In this perspective, we start with a brief introduction to nanoparticle assemblies, emphasizing the role of soft coating agents in controlling the self-assembly of nanoparticles. We then discuss self-assembly of nanoparticles on diverse substrates, highlighting recent advances in building nanoparticle superlattices from SEG strategies.
2. Nanoparticle self-assemblies
Organic surface coating agents tethered on inorganic nanoparticle cores promote the colloidal stability of these nanoparticles and make them good candidates for self-assembly, forming long range ordered superlattices.36 From a thermodynamic point of view, the self-assembly of a large number of particles in a closed system at equilibrium can be regarded as an optimized process to reduce the Helmholtz free energy (F). The free energy F of the system is determined mainly by the internal energy (U) and the entropy (S).37 Different from micro-particles that are assembled driven by entropy, the internal energy of nanoparticles is remarkably influenced by their surface coating agents, which makes the self-assembly of nanoparticles more sophisticated. For example, when Au and Ag nanoparticles were passivated by HS(CH2)10COO−Na+ and HS(CH2)11NMe3+Cl− ligands, respectively, they had negative and positive charges in polar solvent, promoting the attractive electrostatic forces between nanoparticles during assembly.9 Moreover, the controllable assembly of nanoparticles can be accurately realized to construct a variety of superlattice structures through the grafting of single stranded DNA molecules on the nanoparticles and complementary base pairs between DNA molecules.38–41When the nanoparticles are coated with monolayer alkyl coatings, the self-assembly behaviour will be modulated by these coatings, which could be demonstrated through the optimal packing model, OPM:42
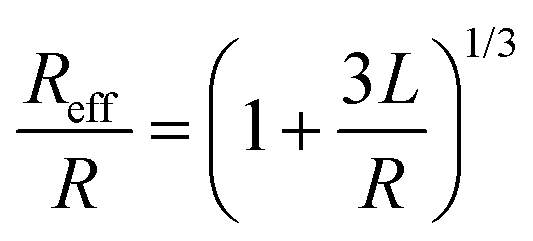 | (1) |
Based on the above discussion on the self-assembly of nanoparticles and its driving force, interaction between nanoparticles is mainly regulated in two ways: (1) van der Waals interactions between alkyl chains tethered on the surface of nanoparticles; (2) building the “donor–acceptor” relation of nanoparticles, including electrostatic forces and hydrogen bonding, etc. Thereby, linking the substrate materials to the nanoparticles through the above mentioned two strategies, can be applied to realize nanoparticle soft epitaxial growth, leading to nanoparticle superlattices with increasing complexities that cannot be engineered under conventional conditions.
3. SEG directed nanoparticle superlattices by van der Waals interactions
The self-assembled alkyl chains on nanoparticles synthesized from nonpolar solvents provide the steric repulsion for their stabilization in nonpolar solvent. As the nanoparticle colloidal solution is concentrated and the volume is decreased, the nanoparticles interact with their neighbours and the ligands interdigitate, producing ordered structures on desired substrates. When the arbitrary substrates are functionalized with a similar alkyl chain, the alkyl chains tethered on nanoparticles would interact with those tethered on substrates, providing attractive van der Waals forces that direct the assembly of nanoparticles on the substrates. For the first assumption, the interaction energy can be estimated by two paralleled C12 chains, which are mostly studied in the literature.46 The van der Waals attraction (UC12) between two nearest parallel alkyl chains of length L from N identical basic units (L = Nλ) and separated by a distance D has been given by Salem:47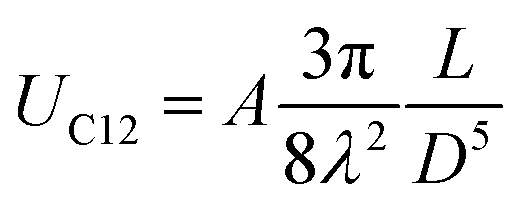 | (2) |
| Uattr ≈ (−4.8kBT) × (2L − d) | (3) |
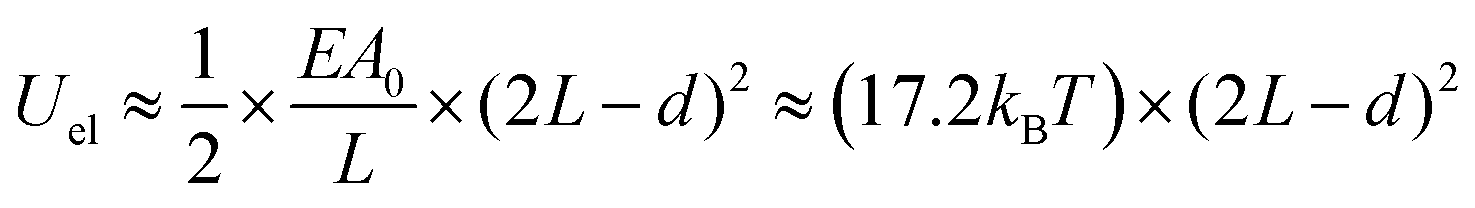 | (4) |
Hence, it is crucial to functionalize the substrate with a monolayer of alkyl chains, and three main strategies are applied including: (1) self-assembled nanoparticle superlattices as substrates; (2) inorganic materials functionalized with alkyl chains; (3) supramolecular structures assembled from small organic molecules bearing lateral alkyl chains.
3.1 Nanoparticle superlattices as substrates
The nanoparticle superlattices themselves are excellent candidates for soft epitaxial growth since long range ordered nanoparticle superlattices provide a coherent alkyl chain layer, directing the epitaxial nanoparticle assemblies (Fig. 2a). Moreover, the property of generated coherent alkyl chain, such as their density, thickness and topology could be precisely modulated by the nanoparticle diameter, their coatings and the superlattice structures, which would enrich the epitaxial growth features.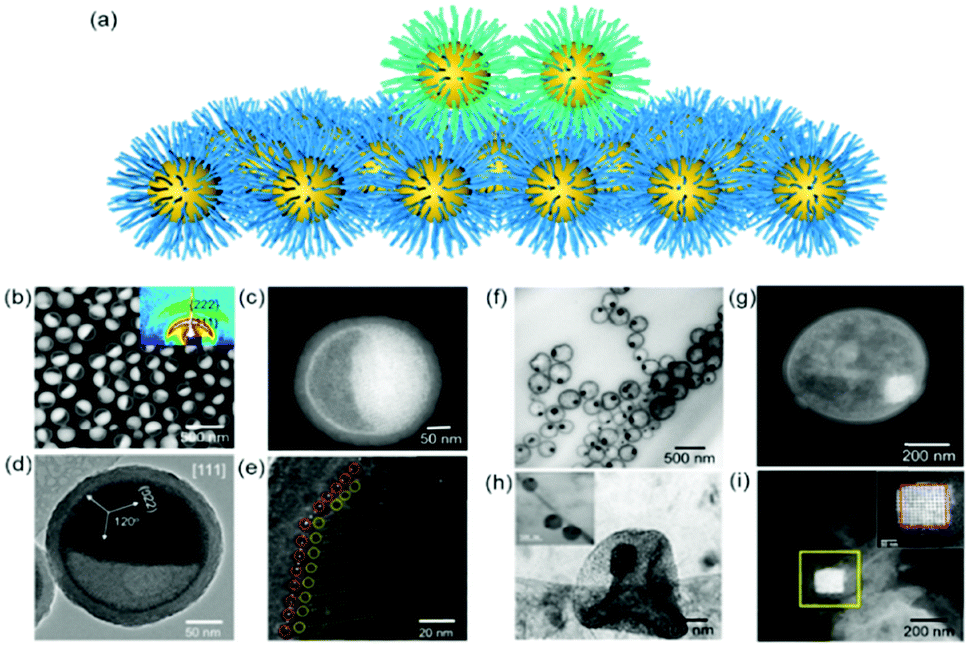 | ||
| Fig. 2 (a) Schematic illustration of nanoparticle epitaxial assembly on a nanoparticle superlattice substrate. (b and c) HAADF-STEM images and (d and e) TEM images of half-filled colloidosomes. (f) TEM, (h) cryo-TEM, and (g and i) HADDF-STEM images of binary nanoparticle superlattices grown in colloidosomes.48 | ||
Yang et al. achieved soft epitaxial growth in a confined space.48 In this work, oleic acid coated Fe3O4 nanoparticles were dispersed in the chloroform phase of emulsions and then assembled by evaporating the carrier solvent. These nanoparticles firstly stabilized at the water/chloroform interface, forming close packed monolayer hollow assemblies, named colloidosomes. Then the extra dispersed nanoparticles were found assembled on the formed monolayer assemblies until all the nanoparticles were consumed, producing half-filled colloidosome structures (as shown in Fig. 2b and c). High resolution TEM images in Fig. 2d and e show that the lattices of the epitaxial part are well fitted with the pre-formed nanoparticle monolayer at the interface, which is similar to the traditional F–vdM epitaxial growth mode (layer-by-layer). To further expand the possibility of epitaxial growth, nanoparticles of two different sizes (Fe3O4 of 6.5 nm and Au of 3.5 nm) instead of only Fe3O4 nanoparticles were used for the assembly in emulsions. The epitaxial assembly behaviour is largely distinct from the single-component ones. As shown in Fig. 2f–i, discrete NaZn13 type binary superlattices assembled from Fe3O4 and Au nanoparticles were formed and localized inside the Fe3O4 nanoparticle pre-assembled monolayer shell. This is mainly caused by the lattice misfit between NaZn13 type superlattices and the Fe3O4 monolayer shell. This is well fitted to the V–W epitaxy mode (island growth). It is worth noting here that in this work, the SEG growth takes place in a confined space, generating uniform colloids dispersed well in water.
Two dimensional nanoparticle superlattices are also efficient substrates for soft epitaxial growth. For example, Talapin's group systematically studied the influence of lattice mismatch coefficients between the substrate superlattice and adlayer nanoparticles on the SEG (Fig. 3).33 A PbS nanoparticle superlattice monolayer was first prepared on a Si substrate, which was applied as the soft substrate for the deposition of Au nanoparticles by immersing the substrate directly into an Au nanoparticle colloidal solution. Fast deposition of nanoparticles induced the adlayer lattice misfit due to kinetic trapping of nanoparticles. To further decrease the lattice misfit, the samples were subjected to annealing in solvent vapour towards a thermodynamically favoured configuration. The more ordered structures were obtained by efficient strain relaxation. When the substrate of PbS nanoparticle superlattices was replaced by binary nanoparticle superlattices (assembled from Fe3O4 and Au nanoparticles), the Au nanoparticles were assembled in the interstitial sites between the Fe3O4 nanoparticles.
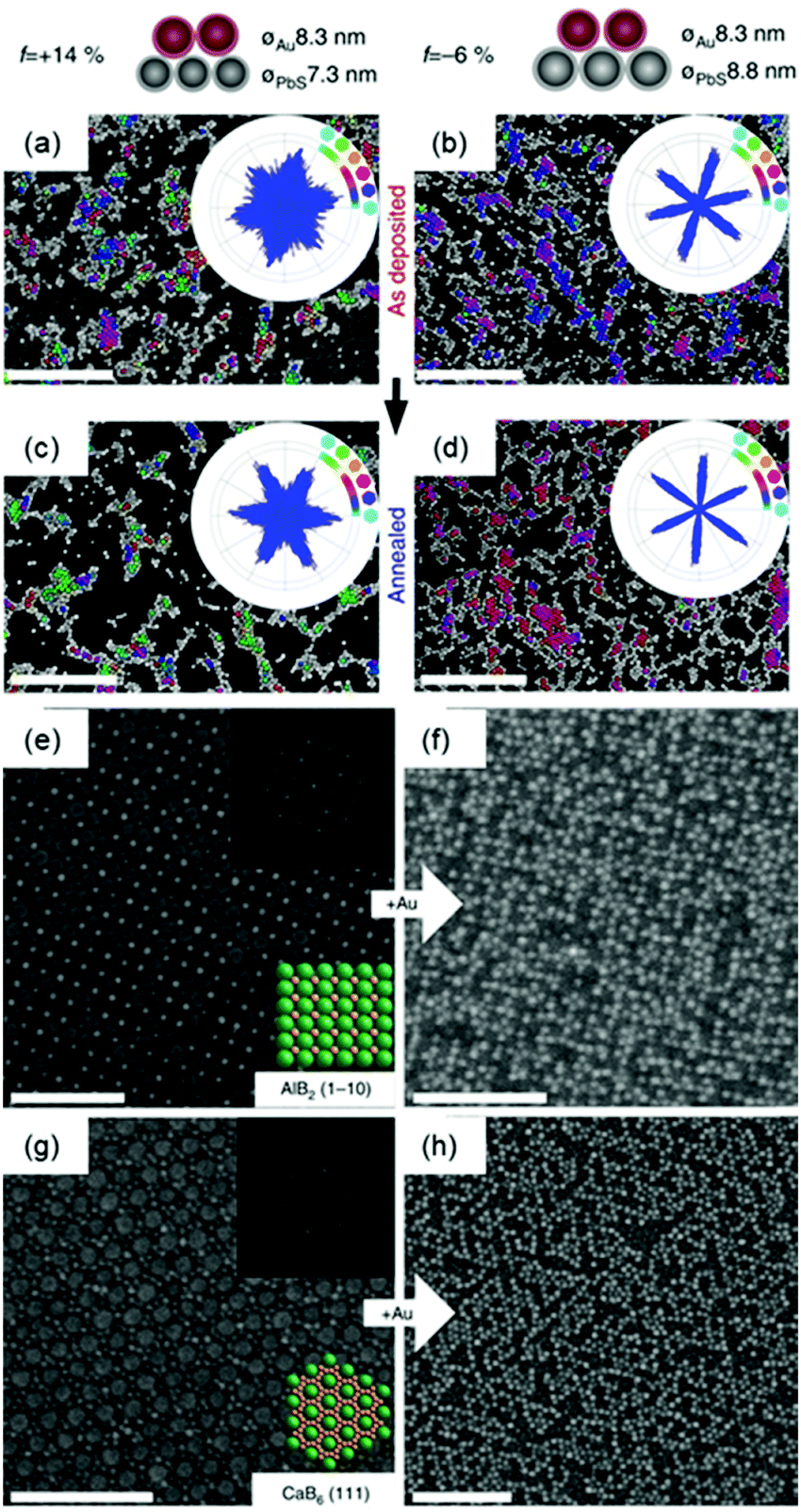 | ||
| Fig. 3 (a–d) SEM images and bond angle plots of Au nanoparticles assembled on PbS nanoparticle monolayers before (a and b) and after (c and d) solvent annealing at different lattice misfits. (e) SEM image of the AlB2 structure assembled from 15.5 nm Fe3O4 and 8.3 nm Au nanoparticles. (f) SEM image of the epitaxial assembly formed by the deposition of 8.3 nm Au nanoparticles on the structure in (e). (g) SEM image of the 111 projection of the CaB6 structure assembled from 15.5 nm Fe3O4 and 6.0 nm Au nanoparticles. (h) SEM image of the epitaxial assembly formed by the deposition of 8.3 nm Au nanoparticles on the structure in (g). Upper insets in (e and g) are fast Fourier transform (FFT) patterns of the images. Lower insets in (e and g) are models of the lattice projections. All scale bars are 100 nm.33 | ||
3.2 Alkyl chain functionalized materials as a substrate
Functionalizing a substrate with a layer of alkyl chains (Fig. 4a), is well-known as a self-assembled monolayer (SAM) created by the chemisorption of head groups of surfactant molecules onto a substrate followed by the slow organization of tail groups. Selecting the type of head group depends on the chemical composition of the underlying substrate. Typically, alkanethiols are applied as noble metal substrates, exemplified as gold and silver in seminal reviews in this field. Other than thiol groups, other polar head groups, such as amine, hydroxyl, and carboxylic acid, are applied for SAM generation.49,50 In comparison with nanoparticle superlattice substrates, SAM functionalized substrates are more generalized and molecularly uniform. Assembly of nanoparticles onto a flat substrate functionalized with SAM could be predictable with an hexagonal close-packed structure, which would be much more interesting when the substrate is curved. For instance, Dong et al. reported an epitaxial assembly of metal oxide nanoparticles onto anodized aluminum oxide (AAO) nanotubes (Fig. 4b–d).51 These AAO nanotubes were tethered with a layer of oleic acid, where covalent bonding between aluminum ions and carboxylic acid of oleic acid takes place. This surface coating enables the assembly of MnO nanoparticles onto the inner wall of AAO nanotubes, resulting in hexagonal close-packed structures. Furthermore, these MnO nanoparticle arrays can be transformed into Mn3O4 nanoparticle superlattices after a post-assembly oxidation-etching treatment. Furthermore, this assembly strategy could be applied for the assembly of CdSe nanorods within the CdSe nanotubular structure, as illustrated in Fig. 4f.52 As a consequence, 1D–2D CdSe mixed dimensional heterostructures from the same materials could be fabricated.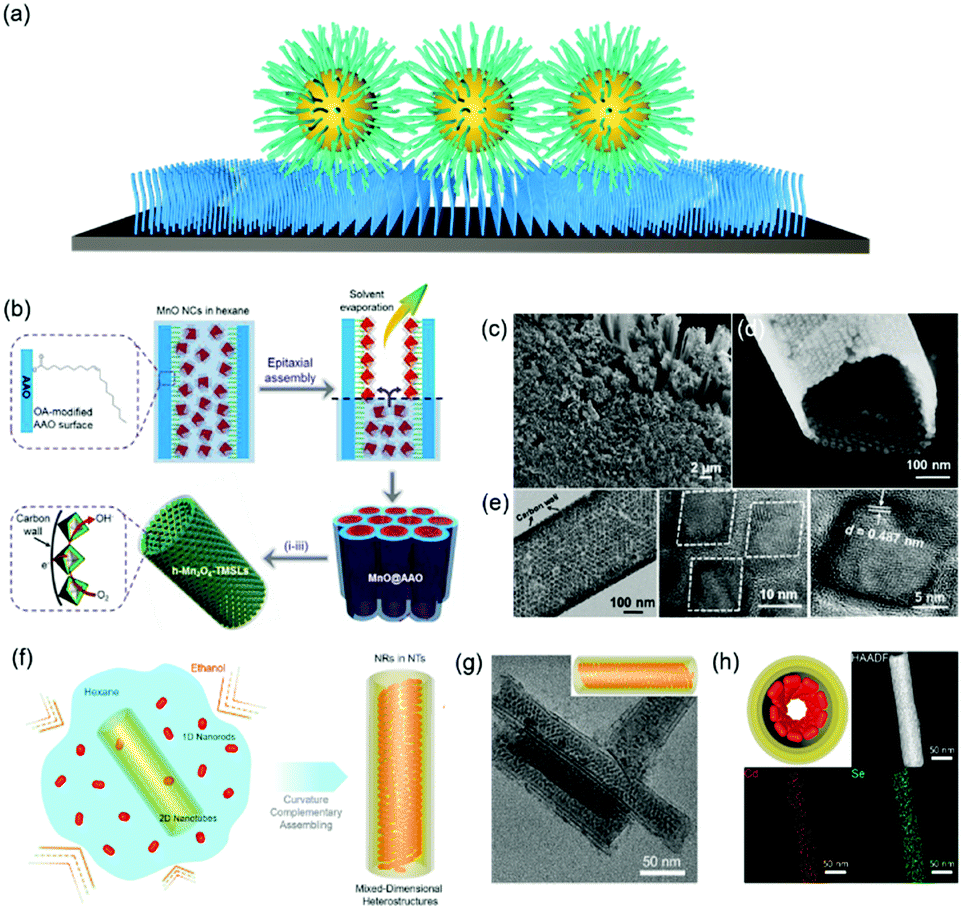 | ||
| Fig. 4 (a) Schematic illustration of alkyl chain functionalized materials as substrates. (b) Schematic illustration of tubular monolayer superlattices of Mn3O4 nanocrystals. (c and d) SEM images and (e) TEM images of tubular nanocrystal superlattices. (f) Illustration of the assembly process of CdSe 1D/2D MDHs. (g) TEM images and 3D illustrations and (h) cross-section illustrations, HAADF-STEM, and EDS-mapping images of MDHs-AR4.51,52 | ||
The development of nanolithography enables the selective functionalization of a substrate with SAM as desired. For example, when the patterned substrates are selectively functionalized with alkyl chains, epitaxial assembly of nanoparticles would selectively take place at the site of interest, leading to nanoparticle superlattices with desired morphologies, such as one-dimensional chains, two-dimensional rings as well as three-dimensional clusters with well-defined geometry. We note that the surface functionalized strategy usually results in a layered assembly of nanoparticles onto the desired substrate, analogous to the F–vdM SEG mode. Because the surface of the substrate is flat, there are larger van der Waals interactions than those between two nanoparticles.
3.3 Self-assembled supramolecular structures as a substrate
A common feature of the above two strategies is that these substrates are premade prior to the epitaxial assembly of nanoparticles, hence the tenability of the substrate is very limited, which constrains the further development of the SEG of nanoparticles into diverse nanoparticle superlattices, including dynamic and reconfigurable ones. Besides, the substrates in the above two strategies are merely the substrate. In other words, the synergistic properties between the substrates and the as-formed nanoparticle superlattices are very poor and limited, which fall behind the increasing demand of the functional materials. In this regard, self-assembled supramolecular structures from small organic molecules could be an alternative strategy to achieve hybrid functional materials with a combination of both inorganic and organic elements.The delicate design of functional organic molecules enables the specific supramolecular interaction between molecular building blocks, including the hydrogen bonding, coordinating bonding, π–π interactions and steric repulsions, which leads to nanostructures with desired dimensions and/or morphologies (Fig. 5a).53–55 The simplest model of supramolecular assembly is the assembly of amphiphilic surfactants in water, resulting in supramolecular structures dispersed in water. For example, Lin et al. co-assembled cetyltrimethylammonium bromide (CTA+Br−) coated gold nanoparticles with two-dimensional supramolecular materials with a similar structure to CTA+, and found that the gold nanoparticles epitaxially assembled on the surface of the two-dimensional supramolecular materials, forming two-dimensional nanoparticle superlattice materials with densely packed structures.56 This method can be applied to diverse nanoparticles coated with CTAB surfactants, leading to two-dimensional nanoparticle superlattices with either spherical or non-spherical nanoparticle building blocks. Nevertheless, the surface coatings also restrict the application scope of this method, because most of the nanoparticles are synthesized in non-polar solvent and covalently coated with a layer of alkyl chains that are difficult to replace with a weaker ligand, i.e. CTAB.
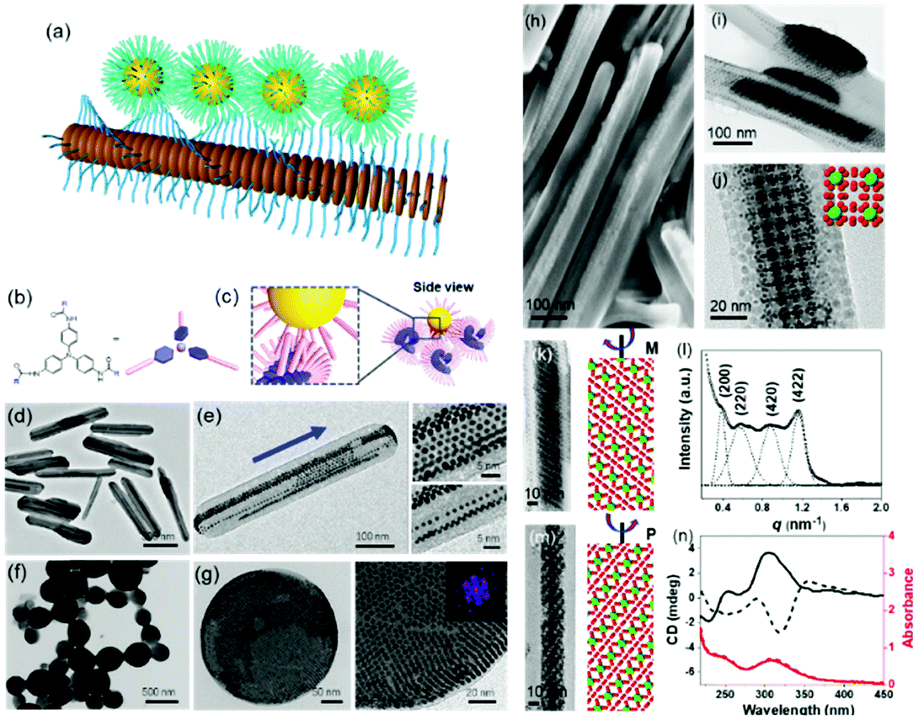 | ||
| Fig. 5 (a) Schematic illustration of nanoparticle epitaxial growth on supramolecular structures. (b and c) Scheme of a soft epitaxial self-assembly system based on TATA supramolecular assembly. TEM images of the directed assembly of Au nanocrystals by TATA-C7-4 (d and e) and TATA-C7-1′ (f and g) supramolecular structures. SEM (h) and TEM images (i–m) of binary nanocrystal superlattices constructed with supramolecular assemblies as the substrate; (l) SAXS profile and (n) CD spectra of binary nanocrystal superlattices.57 | ||
Considering the surface coating feature of nanoparticles, our group proposed that supramolecular materials can be modified by alkylation and then interact with nanoparticles to initiate soft epitaxy assembly of nanoparticles.57 In this way, the additional surface ligand exchange and recodification are not necessary. Fig. 5b shows the molecular structure of tris-amide triarylamines derivatives (TATA) with three alkyl chains at the peripheral side. The π–π interactions coupled with the hydrogen bonding interactions give rise to one-dimensional supramolecular fibrils. Furthermore, an important feature of supramolecular fibrils is that they are functionalized with alkyl chains. Hence, these TATA supramolecular fibrils can be regarded as nanowires functionalized with a layer of alkyl chains. First, bundling between fibrils takes place, leading to supramolecular nanorods. Second, these supramolecular nanorods could be used as a substrate for the SEG of nanoparticles, producing one-dimensional nanoparticle chains or ribbons, depending on the amount of supplying nanoparticles. Furthermore, by changing the alkyl chain structure of TATA molecules, the assembled supramolecular structures varied from one dimensional nanorods to nanospheres. The nanoparticles assembled along the coherent alkyl chain outside the supramolecular assemblies, as shown in Fig. 5d–g. Here, the interactions between TATA supramolecular structures and nanoparticles are much stronger than those between two nanoparticles, thereby giving rise to the epitaxial self-assembly of nanoparticles over TATA assemblies. When the alkyl chain of TATA molecules is a straight chain or branched chain with α-substitutions, the TATA molecules form H-type aggregates. When the alkyl chain is a γ-substituted branched chain, TATA molecules assemble into J-type aggregates. When these H-type aggregates are used as substrates, the nanoparticles form a one-dimensional structure. When J-type aggregates are used as substrates, the nanoparticles are assembled on the surface of supramolecular aggregates in a planar epitaxial mode (F–vdM mode) to form a two-dimensional superlattice structure of nanoparticles with a hexagonal close-packed arrangement.
These TATA supramolecular structures could also be applied for the assembly of nanoparticles with two distinct sizes. When Au and Fe3O4 nanoparticles with two distinct sizes were applied for the SEG on TATA supramolecular structures, NaZn13 type binary nanoparticle superlattices were formed and exhibited left-handed or right-handed helicity from batch to batch (Fig. 5h–n). Different from the nanoparticle superlattice substrate, these supramolecular structures show strong lattice misfit tolerance, giving rise to nanoparticle superlattices with registered morphology from the substrate.
When the substituted alkyl chain is homochiral, it could be expected that chiral nanoparticle superlattices can be produced. Very recently, our group reported the assembly of Au nanoparticles into chiral nanoparticle superlattices in the presence of chiral porphyrin molecules. Chiral porphyrin molecules are self-assembled into diverse supramolecular structures, depending on the specific metalation and metal-coordination reactions (Fig. 6).58 Then Au nanoparticles are able to deposit on the as-formed porphyrin supramolecular structures through hetero chain–chain van der Waals interactions. We note that these chiral nanoparticle assemblies are the first example on the basis of chirality transfer through weak van der Waals forces across three orders of magnitude. Although the chiral nanoparticle assemblies have been engineered, the affinity between chiral moieties and nanoparticles is built through specific ionic pairs, hydrogen bonding pairs, metal coordination pairs or covalent bonding, with interaction energies much larger than the present chiral nanoparticle assemblies from SEG processes.59
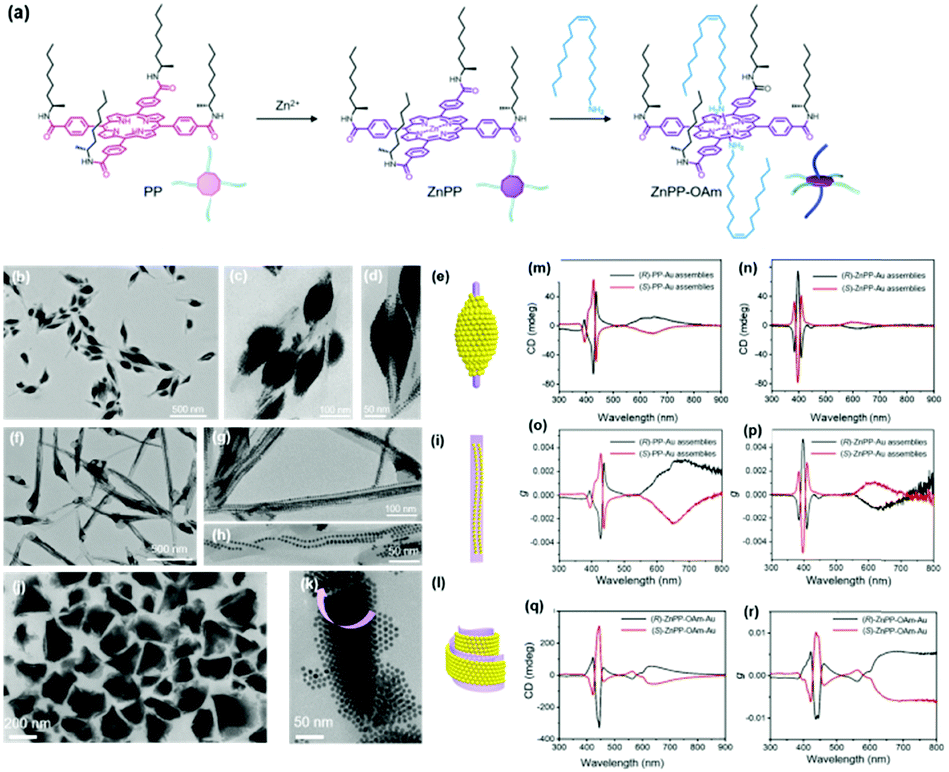 | ||
| Fig. 6 (a) Molecular structures of three porphyrin derivatives. (b–d) TEM images of (R)-PP-Au assemblies. (e) Model of the 3D nanoparticle assemblies grown on (R)-PP structures. (f–h) TEM images of (R)-ZnPP-Au assemblies. (i) Model of the 3D nanoparticle assemblies grown on (R)-ZnPP structures. (g and k) TEM images of (S)-ZnPP-OAm-Au assemblies. (l) Model of the 3D nanoparticle assemblies grown on (S)-ZnPP-OAm structures. The circular dichroism spectra and g factor spectra of supramolecular structure colloidal suspensions of (S/R)-PP-Au assemblies (m and o), (S/R)-ZnPP-Au assemblies (n and p), and (S/R)-ZnPP-OAm-Au assemblies (q and r).58 | ||
4. SEG based on complementary interactions
The construction of complementary interactions is ubiquitous in biology, exemplified as base pairs in double stranded nucleic acids consisting of two nucleobases bound to each other by hydrogen bonds, and lock-key configurations in enzyme catalysis. In artificial systems, host–guest complementary interactions lead to complexes comprised of two or more molecules that are held together in specific conformation.60 Hence, it is possible to build nanoparticle superlattices on the basis of complementary interactions between nanoparticles and the arbitrary substrate. In this part, we will discuss two main complementary pairs that have been applied to build nanoparticle superlattices, including host–guest complexation and DNA base pairing interactions.4.1 Construction of host–guest interactions for epitaxial growth
Host guest complexation has shown its potential for applications in supramolecular catalysis, sensing, conformational switching and environmental remediation. Here, we show that this host–guest complexation can be implanted into nanoparticle assemblies, which consequently regulates their assembly behaviour. An earlier report showed that a host–guest pair between β-cyclodextrin (β-CD) and an adamantly functionalized molecule could be applied for the assembly of β-CD functionalized silica nanoparticles onto the β-CD functionalized substrate through adamantly functionalized dendrimers.61 This method could result in the layered assembly of silica nanoparticles onto the substrate. Another example showed that the host–guest pair could be a cucurbit[n]uril (CB) based system.62 First, the CB molecule interacted with a substrate grafted with a layer of viologendecanethiol, which could further interact with naphthalene-functionalized nanoparticles, leading to the assembly of nanoparticles onto the substrate. In the end, neither of the two strategies produced nanoparticle assemblies with periodic ordering, which is probably due to the strong attractive forces between the host–guest pairs.Recently, we showed that porous organic cage (POC) molecules with open windows could be applied as molecular hosts.63 A significant feature of POCs is that they possess a rigid cavity and can interact with other molecules through their windows.64–66 Therefore, the POCs are used as hosts for the construction of host–guest interactions with other molecules for assembly. In this work, we found that POCs synthesized from a [4+6] amine-aldehyde condensation reaction is able to interact with a long alkyl chain through hydrophobic interactions, akin to the host–guest complexation. When the nanoparticles are tethered with alkyl chains, they are able to interact with POCs crystals through the inclusion of alkyl chains into the cavity of POCs. Consequently, nanoparticles are able to self-assemble at the surface of POCs crystals. Furthermore, the results show that spherical nanoparticles assembled on a POC crystal adopt the F–vdM mode, sitting on eight (111) crystal facets with a closed packing structure (Fig. 7c–h). However, cubic or octahedral nanoparticles assembled onto the POC crystal following the V–W mode. All the nanocubes or nano-octahedron assembled on one crystal facet of the POC crystal (Fig. 7). This is due to the competition of forces between nanoparticles and those between nanoparticle and substrate. When the interaction between nanoparticles is stronger than that of the nanoparticle and POC crystal, the V–W epitaxial growth mode is dominant, otherwise the F–vdM mode is dominent.
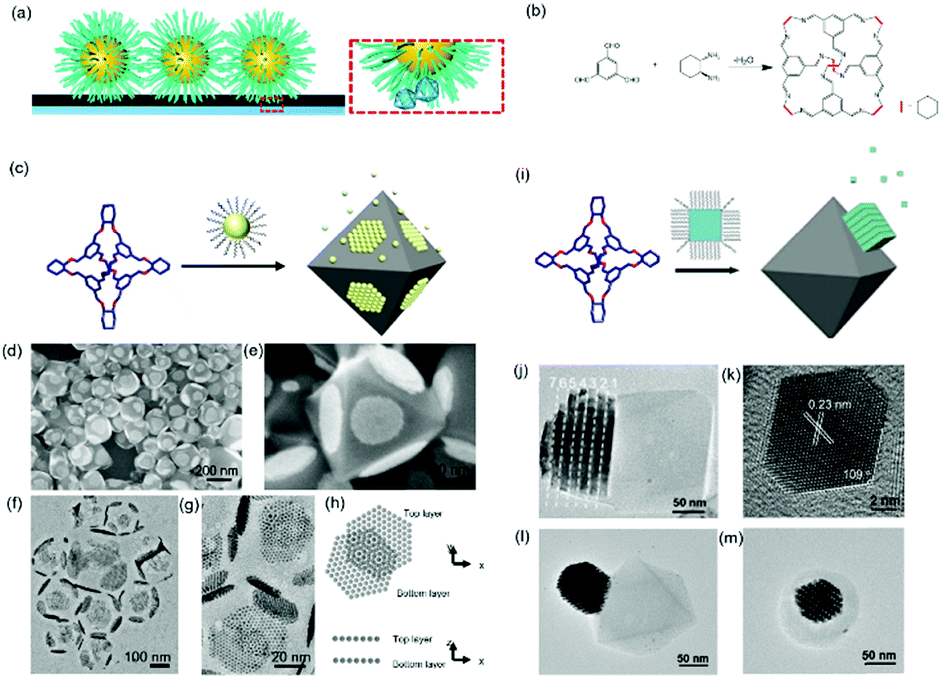 | ||
| Fig. 7 (a) Schematic illustration of host–guest interaction pairs between nanoparticles and POCs. (b) The chemical reaction of POC synthesis. Schematic illustration of spherical (c) and cubic (i) nanoparticle assembly onto the POC assembled crystals. SEM images (d and e) and TEM images (f and g) of spherical nanoparticle-POC assemblies. (h) The carton image of the top and bottom layer of nanoparticles on two facets of POC crystal. (j) TEM image of nanocube-POC assemblies. (k) TEM images of one octahedral nanoparticle. (l and m) TEM images of octahedral nanoparticle-POC assemblies.63 | ||
4.2 Base pair complementation of nucleic acid molecules
DNA hybridization has been applied to assemble nanoparticles into superlattices with crystalline structures that are surprisingly rich.39 It was found that DNA's three-dimensional double helix structure (fixed pitch, fixed diameter) has more advantages than other materials in guiding nanoparticles to three-dimensional ordered assembly. The specific recognition between base pairs and the ability to control of DNA chain length and base sequence make it a powerful weapon for assembly at the nanoscale. The programmability of DNA makes it an extremely attractive structure-oriented ligand.This strategy can also be applied to study the SEG assembly of nanoparticle superlattices. Macfarlane and co-workers used this technique and a combination of DNA functionalized nanoparticles and a substrate functionalized by DNA strands to engineer an epitaxial assembly process.67 They found that single-crystal Winterbottom shapes of nanoparticle crystals are formed by controlling the interfacial energies between the crystals and fluid, the substrate and the crystal, and the substrate and the fluid (Fig. 8). Other examples show that DNA-grafted nanoparticles self-assembled into two-dimensional colloidal films can be applied as a substrate for soft epitaxial assembly.68,69 For instance, Mirkin and coworkers used DNA-coated nanoparticles as more elastic and malleable building elements to better adapt to the lattice misfit, as shown in Fig. 9.69 Subsequent studies showed that superlattice thin films assembled by DNA-functionalized nanoparticles can store elastic strains by deforming and rearranging, with lattice mismatches up to ±7.7%, significantly exceeding the ±1% lattice mismatches allowed by atomic thin films. Importantly, these DNA-coated nanoparticles undergo a progressive and coherent relaxation, dissipating the strain elastically and irretrievably through the formation of dislocations or vacancies. It is therefore possible to grow heteroepitaxial colloidal films by controlling “soft” programmable atomic equivalents of nanometers and microstructures using rigid nanocrystals coated with soft compressible polymeric materials.
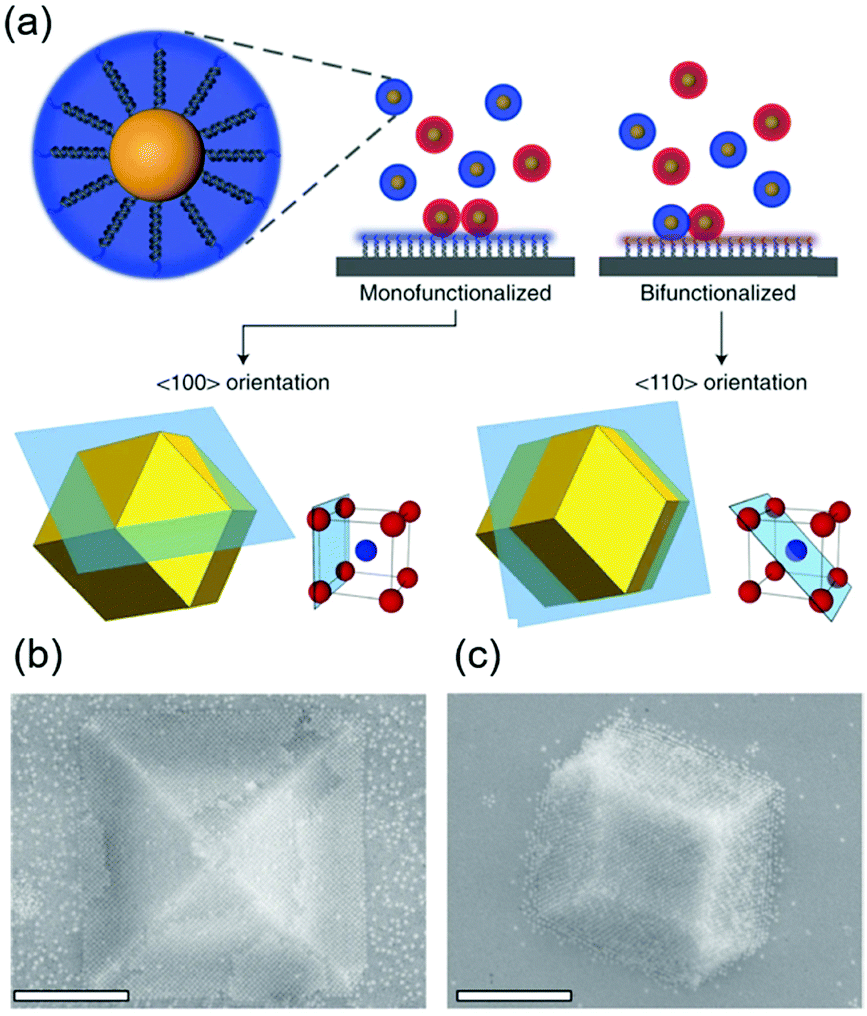 | ||
| Fig. 8 (a) Scheme of DNA-functionalized nanoparticles assembled onto a DNA-functionalized substrate. (b and c) SEM images of 〈100〉 or 〈110〉 oriented crystals depending on the substrate functionalization. Scale bars, 1 μm.67 | ||
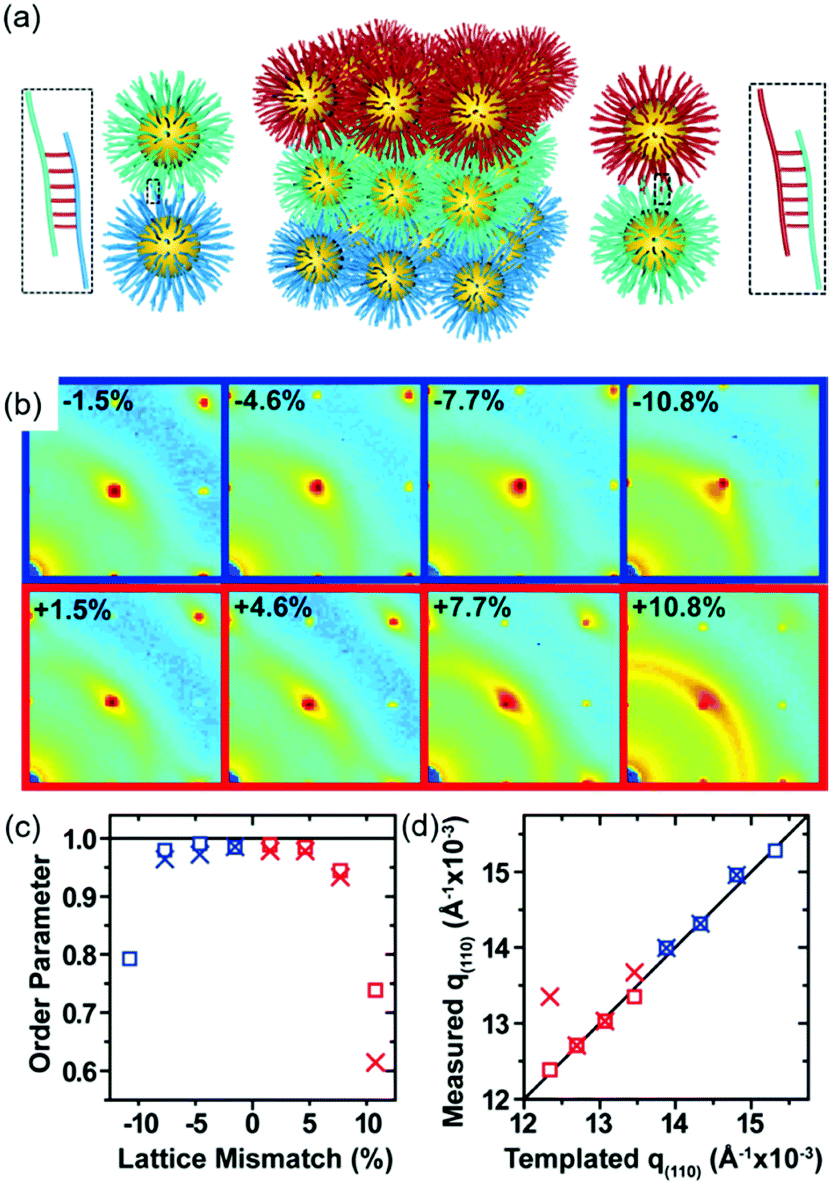 | ||
| Fig. 9 (a) Schematic illustration of epitaxial assembly directed by DNA strains. Nanoparticle assembled thin films maintain coherency with the patterned crystallography up to ±7.7% lattice mismatch. (b) The 2D transmission SAXS data centered on the (110) reciprocal spot, (c) order parameter, calculated by comparing the integrated intensity of the (110) spot to the intensity of the amorphous ring, as a function of lattice mismatch, and (d) the maximum q(110) value from the measured SAXS data compared to the templated q(110) position.69 | ||
5. Interaction energy in SEG processes
The attractive interaction energy between nanoparticles and a substrate is the main driving force for SEG processes. Generally, when the attractive interaction between nanoparticles and the substrates is stronger than that between two nanoparticles, the layered assembly F–vdM mode dominates. In fact, the layered assembly of nanoparticles onto the substrate is the most probable case, even when the interaction between nanoparticles and the substrates is not directional, because a repulsive interaction is present when the nanoparticles are dispersed in solvents. However, it would be of particular interest when the substrates are also dispersed in the same solvent, which consequently induces the competitive self-assembly upon triggering the self-assembly. Here, we summarized the attractive forces in the cases we discussed above (Table 1), including the hydrogen bonding, van der Waals, and diverse host guest systems. It can be seen that the attractive van der Waals energy between unit alkyl chains is ∼1kBT, which is much weaker than the hydrogen bonding, CB-based host–guest pairs or CD-based host–guest pairs. This clearly indicates that the hydrogen bonding or host–guest complexation is more directional. On the other hand, strong attractive interactions could also lead to irreversible attachment of nanoparticles onto the substrate, resulting in disordered nanoparticle assemblies. In fact, this layered assembly of nanoparticles onto the substrate with disordered structures could also be observed in conventional layer-by-layer self-assembly, triggered by the attractive electrostatic energy.70 Since the energy strength in the electrostatics could be regulated through tuning the density of charges and ionic strength, it is possible that the SEG process with ordered nanoparticle assemblies could be achieved through building the electrostatic pairs.71| Type of interaction | Interaction energy | Energy strength |
|---|---|---|
| Covalent, metallic | Short range, complicated | ∼100kBT |
| Electrostatic |
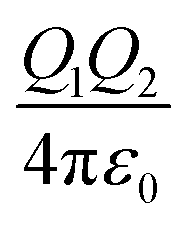
|
Variable |
| Hydrogen bonding | Short range, complicated | ∼5–10kBT |
| van der Waals (alkyl chains) |
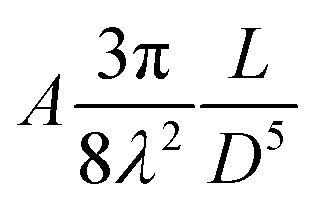
|
∼1kBT |
| CB based host–guest system | Hydrophobic, complicated | ∼20–40kBT |
| CD based host–guest system | Hydrophobic, complicated | ∼1–10kBT |
| POC based host–guest system | Hydrophobic, complicated | ∼2kBT |
6. Summary and outlook
Traditional atomic epitaxy on substrates has pushed the development of theories of crystallization on a substrate. When translating these ideas to epitaxial assembly of nanoparticles on a substrate, the traditional epitaxy theories work well in most cases. However, epitaxial assembly of nanoparticles can tolerate much larger lattice misfit between the nanoparticle superlattice and the substrate. The soft organic shell coated on nanoparticles which undergoes deformation during the assembly process may account for this difference. Importantly, organic ligands impart the multiple supramolecular interactions between nanoparticles into the nanoparticles, which in turn regulate the competitive interactions between nanoparticle–nanoparticle and nanoparticle–substrate, giving rise to varied structures of nanoparticle superlattices. Additionally, the interactions between nanoparticle and substrate enable the construction of a phase boundary, which allow for the design and fabrication of hybrid materials with tailor-made properties. Uncovering the charge carrier transport, photoelectron transfer and/or energy transport between the nanoparticle superlattice and the underlying substrate will provide a productive avenue to design new photoelectric devices, including photocatalysis systems and photodetectors. For example, when quantum dots are self-assembled on 2D quantum sheets, it is possible to engineer 0D/2D mixed-dimensional heterojunctions. Such mixed dimensional heterojunctions combine the merits from the large optical absorbance coefficients of 0D quantum dots and the high charge carrier mobility of 2D materials, which leads to nanostructured photocatalysts with mixed-dimensional heterojunctions.Although several studies have shown that epitaxial assembly of nanoparticles can be achieved through spherical nanoparticles, it is still challenging to engineer the epitaxial assembly of non-spherical nanoparticles.72–74 Considering that shape anisotropy at the nanoscale would provide exotic anisotropic physical properties, it would be interesting to explore the synergistic physical properties of anisotropic nanoparticle superlattices and the substrate of interest.75,76
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21972076). Z. Y. thanks Taishan Scholars Program of Shandong Province (tsqn201812011).Notes and references
- S. H. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed.
- M. P. Pileni, J. Phys. Chem. B, 2001, 105, 3358–3371 CrossRef CAS.
- J. Wei, N. Schaeffer and M. P. Pileni, J. Am. Chem. Soc., 2015, 137, 14773–14784 CrossRef CAS PubMed.
- J. Hao, Y. Yang, F. Zhang, Z. Yang and J. Wei, J. Phys. Chem. C, 2020, 124, 14775–14786 CrossRef CAS.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- E. Rabani, D. R. Reichman, P. L. Geissler and L. E. Brus, Nature, 2003, 426, 271–274 CrossRef CAS PubMed.
- M. P. Pileni, Acc. Chem. Res., 2007, 40, 685–693 CrossRef CAS PubMed.
- N. Goubet, H. Portales, C. Yan, I. Arfaoui, P. A. Albouy, A. Mermet and M. P. Pileni, J. Am. Chem. Soc., 2012, 134, 3714–3719 CrossRef CAS PubMed.
- A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CrossRef CAS.
- F. X. Redl, K. S. Cho, C. B. Murray and S. O'Brien, Nature, 2003, 423, 968–971 CrossRef CAS PubMed.
- E. V. Shevchenko, M. Ringler, A. Schwemer, D. V. Talapin, T. A. Klar, A. L. Rogach, J. Feldmann and A. P. Alivisatos, J. Am. Chem. Soc., 2008, 130, 3274–3275 CrossRef CAS PubMed.
- R. Y. Wang, J. P. Feser, J. S. Lee, D. V. Talapin, R. Segalman and A. Majumdar, Nano Lett., 2008, 8, 2283–2288 CrossRef CAS PubMed.
- M. Gauvin, Y. Wan, I. Arfaoui and M.-P. Pileni, J. Phys. Chem. C, 2014, 118, 5005–5012 CrossRef CAS.
- N. Goubet, C. Yan, D. Polli, H. Portales, I. Arfaoui, G. Cerullo and M. P. Pileni, Nano Lett., 2013, 13, 504–508 CrossRef CAS PubMed.
- P. Yang, I. Arfaoui, T. Cren, N. Goubet and M.-P. Pileni, Nano Lett., 2012, 12, 2051–2055 CrossRef CAS PubMed.
- D. Anselmetti, A. Baratoff, H. J. Güntherodt, E. Delamarche, B. Michel, C. Gerber, H. Kang, H. Wolf and H. Ringsdorf, Europhys. Lett., 1994, 27, 365–370 CrossRef CAS.
- D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12, 4723–4730 CrossRef CAS.
- A. M. Jackson, J. W. Myerson and F. Stellacci, Nat. Mater., 2004, 3, 330–336 CrossRef CAS PubMed.
- R. Klajn, K. J. M. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261–264 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- J. W. Matthews and A. E. Blakeslee, J. Cryst. Growth, 1974, 27, 118–125 CAS.
- Z. Zhang and M. G. Lagally, Science, 1997, 276, 377–383 CrossRef CAS PubMed.
- O. Ambacher, J. Phys. D: Appl. Phys., 1998, 31, 2653–2710 CrossRef CAS.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- K. L. Choy, Prog. Mater. Sci., 2003, 48, 57–170 CrossRef CAS.
- G. Fiori, F. Bonaccorso, G. Iannaccone, T. Palacios, D. Neumaier, A. Seabaugh, S. K. Banerjee and L. Colombo, Nat. Nanotechnol., 2014, 9, 768–779 CrossRef CAS PubMed.
- F. F. Lange, Science, 1996, 273, 903–909 CrossRef CAS PubMed.
- I. V. Markov, Fundamentals of Nucleation, Crystal Growth and Epitaxy, World Scientific, Singapore, 1995 Search PubMed.
- E. Bauer and J. H. van der Merwe, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 3657–3671 CrossRef CAS PubMed.
- S. M. Rupich, F. C. Castro, W. T. M. Irvine and D. V. Talapin, Nat. Commun., 2014, 5, 1–10 Search PubMed.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831–1841 CrossRef CAS PubMed.
- F. X. Xiao, M. Pagliaro, Y. J. Xu and B. Liu, Chem. Soc. Rev., 2016, 45, 3088–3121 RSC.
- H. Heinz, C. Pramanik, O. Heinz, Y. Ding, R. K. Mishra, D. Marchon, R. J. Flatt, I. Estrela-Lopis, J. Llop, S. Moya and R. F. Ziolo, Surf. Sci. Rep., 2017, 72, 1–58 CrossRef CAS.
- M. I. Bodnarchuk, M. V. Kovalenko, W. Heiss and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 11967–11977 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- S. J. Tan, M. J. Campolongo, D. Luo and W. Cheng, Nat. Nanotechnol., 2011, 6, 268–276 CrossRef CAS PubMed.
- U. Landman and W. D. Luedtke, Faraday Discuss., 2004, 125, 1–22 RSC.
- A. I. Henry, A. Courty, M. P. Pileni, P. A. Albouy and J. Israelachvili, Nano Lett., 2008, 8, 2000–2005 CrossRef CAS PubMed.
- B. A. Korgel, S. Fullam, S. Connolly and D. Fitzmaurice, J. Phys. Chem. B, 1998, 102, 8379–8388 CrossRef CAS.
- Y. Nagaoka, R. Tan, R. Li, H. Zhu, D. Eggert, Y. A. Wu, Y. Liu, Z. Wang and O. Chen, Nature, 2018, 561, 378–382 CrossRef CAS PubMed.
- F. W. DelRio, C. Jaye, D. A. Fischer and R. F. Cook, Appl. Phys. Lett., 2009, 94, 131909 CrossRef.
- L. Salem, J. Chem. Phys., 1962, 37, 2100–2113 CrossRef CAS.
- Z. Yang, T. Altantzis, D. Zanaga, S. Bals, G. V. Tendeloo and M. P. Pileni, J. Am. Chem. Soc., 2016, 138, 3493–3500 CrossRef CAS PubMed.
- J. C. Love, D. B. Wolfe, R. Haasch, M. L. Chabinyc, K. E. Paul, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 2003, 125, 2597–2609 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- T. Li, B. Xue, B. Wang, G. Guo, D. Han, Y. Yan and A. Dong, J. Am. Chem. Soc., 2017, 139, 12133–12136 CrossRef CAS PubMed.
- Y. Wei, F. Zhang, J. Wei and Z. Yang, Small, 2021, e2102047 CrossRef PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- E. Moulin and J. J. Armao, Acc. Chem. Res., 2019, 52, 975–983 CrossRef CAS PubMed.
- H.-X. Lin, L. Chen, D.-Y. Liu, Z. C. Lei, Y. Wang, X.-S. Zheng, B. Ren, Z.-X. Xie, G. D. Stucky and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 2828–2831 CrossRef CAS PubMed.
- F. Zhang, F. Yang, Y. Gong, Y. Wei, Y. Yang, J. Wei, Z. Yang and M. P. Pileni, Small, 2020, 16, 2005701 CrossRef CAS PubMed.
- F. Yang, X. Liu and Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 14671–14678 CrossRef CAS PubMed.
- S. Mokashi, Y. Zhou, S. C. Brooks and N. L. Rosi, Adv. Mater., 2020, 32, 1905975 CrossRef PubMed.
- M. Cheng, F. Shi, J. Li, Z. Lin, C. Jiang, M. Xiao, L. Zhang, W. Yang and T. Nishi, Adv. Mater., 2014, 26, 3009–3013 CrossRef CAS PubMed.
- V. Mahalingam, S. Onclin, M. Péter, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Langmuir, 2004, 20, 11756–11762 CrossRef CAS PubMed.
- F. Tian, N. Cheng, N. Nouvel, J. Geng and O. A. Scherman, Langmuir, 2010, 26, 5323–5328 CrossRef CAS PubMed.
- M. Hua, J. Hao, Y. Gong, F. Zhang, J. Wei, Z. Yang and M.-P. Pileni, ACS Nano, 2020, 14, 5517–5528 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- D. J. Lewis, L. Z. Zornberg, D. J. D. Carter and R. J. Macfarlane, Nat. Mater., 2020, 19, 719–724 CrossRef CAS PubMed.
- M. X. Wang, S. E. Seo, P. A. Gabrys, D. Fleischman, B. Lee, Y. Kim, H. A. Atwater, R. J. Macfarlane and C. A. Mirkin, ACS Nano, 2017, 11, 180–185 CrossRef CAS PubMed.
- P. A. Gabrys, S. E. Seo, M. X. Wang, E. Oh, R. J. Macfarlane and C. A. Mirkin, Nano Lett., 2018, 18, 579–585 CrossRef CAS PubMed.
- V. Mohanta and S. Patil, Langmuir, 2013, 29, 13123–13128 CrossRef CAS PubMed.
- V. Liljestrom, A. Ora, J. Hassinen, H. T. Rekola, Nonappa, M. Heilala, V. Hynninen, J. J. Joensuu, R. H. A. Ras, P. Torma, O. Ikkala and M. A. Kostiainen, Nat. Commun., 2017, 8, 671 CrossRef PubMed.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- R. Guo, J. Mao, X. M. Xie and L. T. Yan, Sci. Rep., 2014, 4, 7021 CrossRef PubMed.
- G. Zhu, Z. Huang, Z. Xu and L. T. Yan, Acc. Chem. Res., 2018, 51, 900–909 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–55 CrossRef CAS PubMed.
- M. Jones and C. Mirkin, Nature, 2012, 491, 42–43 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2021 |