
Charge-transfer dynamics in van der Waals heterojunctions formed by thiophene-based semiconductor polymers and exfoliated franckeite investigated from resonantly core-excited electrons†
Yunier
Garcia-Basabe
 *a,
David
Steinberg
b,
Lara M.
Daminelli
a,
Cesar D.
Mendoza
c,
E. A. Thoroh
de Souza
b,
Flavio C.
Vicentin
d and
Dunieskys
G. Larrudé
b
*a,
David
Steinberg
b,
Lara M.
Daminelli
a,
Cesar D.
Mendoza
c,
E. A. Thoroh
de Souza
b,
Flavio C.
Vicentin
d and
Dunieskys
G. Larrudé
b
aUniversidade Federal da Integração Latino-Americana, UNILA, 85867-970, Foz do Iguaçu, Brazil
bMackGraphe-Graphene and Nanomaterial Research Center, Mackenzie Presbyterian University, 01302-907, São Paulo, Brazil
cDepartamento de Física, Pontifícia Universidade Católica do Rio de Janeiro, 22451-900, Rio de Janeiro, Brazil
dBrazilian Synchrotron Light Laboratory (LNLS), Brazilian Center for Research in Energy and Materials (CNPEM), Campinas, 13083-970, Sao Paulo, Brazil
First published on 6th July 2021
Abstract
Organic/inorganic van der Waals heterojunctions formed by a combination of 2D materials with semiconductor polymer films enable the fabrication of new device architectures that are interesting for electronic and optoelectronic applications. Here, we investigated the charge-transfer dynamics at the interface between 2D layered franckeite (Fr) and two thiophene-based conjugated polymers (PFO-DBT and P3HT) from the resonantly core-excited electron. The unoccupied electronic states of PFO-DBT/Fr and P3HT/Fr heterojunctions were studied using near-edge X-ray absorption fine structure (NEXAFS) and resonant Auger (RAS) synchrotron-based spectroscopies. We found evidence of ultrafast (subfemtosecond charge-transfer times) interfacial electron delocalization pathways from specific electronic states. For the interface between the PFO-DBT polymer and exfoliated franckeite, the most efficient interfacial electron delocalization pathways were found through π*(S–N) and π*(S–C) electronic states corresponding to the benzothiadiazole and thiophene units. On the other hand, for the P3HT polymer, we found that electrons excited to π–π* and S1s–π*(C–C) electronic states of the P3HT polymer are the most affected by the presence of exfoliated franckeite and consequently are the main interfacial electron-transfer pathways in this heterojunction. Our results have important implications in understanding how ultrafast electron delocalization is taking place in organic/inorganic van der Waals heterojunctions, which is relevant information in designing new devices involving these systems.
1 Introduction
Recently, hybrid organic/inorganic van der Waals heterojunctions composed of organic semiconductor polymers and layered two-dimensional (2D) inorganic materials have become attractive options for designing and manufacturing the next generation of novel devices.1–4 Organic semiconductor polymers are an important component for fabricating organic/inorganic van der Waals heterojunctions due to their chemical versatility, mechanical flexibility, low-cost processing, and simple manufacturing process.5–8 Specifically, thiophene-based conjugated polymers have been extensively investigated due to their potential applications such as in organic photovoltaics (OPVs), organic field-effect transistors (OFETs) and organic light-emitting diodes (OLEDs).9–13On the other hand, layered two-dimensional (2D) inorganic materials, such as graphene, hexagonal boron nitride (hBN) and transition metal dichalcogenides (TMDs), have been the most studied materials owing to their novel and unique physical properties.14–16 Recently, a naturally occurring sulfosalt franckeite has emerged as an attractive 2D layered material. Structurally, the franckeite mineral is formed by the alternated stacking of tin disulfide-based (SnS2) and lead sulfide-based (PbS) layers.17 Franckeite is an air-stable p-type doping material and has a narrow band gap of below 0.7 eV.18,19 Although there have been few studies on franckeite, it is considered to have great potential in optoelectronic devices.20–22
Here, we investigated the van der Waals heterojunctions formed by thiophene-based conjugated polymers and exfoliated layered franckeite. The main advantage of using layered franckeite in organic–inorganic van der Waals heterojunctions is for its p-type doping characteristics. By contrast, 2D layered materials like MoS2 are generally n-type semiconductors.23 Therefore, van der Waals heterojunctions using thiophene-based conjugated polymers and exfoliated layered franckeite can be an alternative in building a hybrid p–n heterojunction, the essential component in modern electronic devices.24–28 For this study, we selected two thiophene-based conjugated polymers to construct the organic/inorganic van der Waals heterojunctions with exfoliated layered franckeite: (1) donor–acceptor copolymer organic films of PFO-DBT (poly[2,7-(9,9-dioctylfluorene)-alt-4,7-bis(thiophen-2-yl)benzo-2,1,3-thiadiazole]) composed of electron-rich thiophene and electron-deficient benzothiadiazole units, and (2) regioregular poly[3-hexylthiophene-2,5-diyl] (P3HT). These two polymers are electron-donor materials extensively studied for organic optoelectronic devices such as OPVs and OLEDs.29–34 Hence, they could be considered as a promising n-type part of the hybrid p–n heterojunction. The chemical structures of PFO-DBT and P3HT polymers and franckeite are displayed in Scheme 1. Two heterojunctions were achieved by stacking PFO-DBT and P3HT on top of exfoliated layered franckeite and were labeled as PFO-DBT/Fr and P3HT/Fr, respectively.
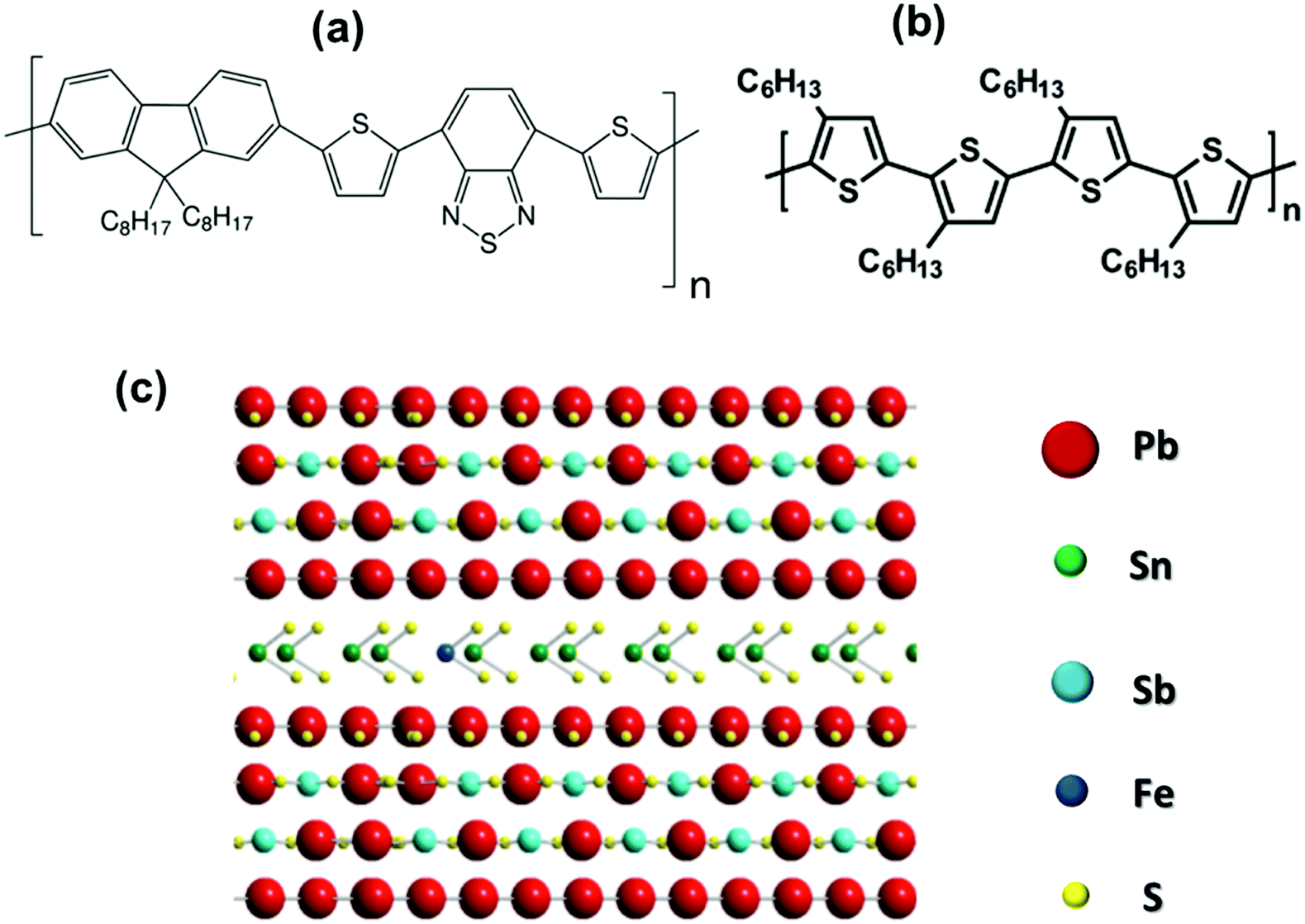 | ||
| Scheme 1 Representation of the chemical structures of (a) PFO-DBT, (b) P3HT, and (c) franckeite. | ||
In this regard, understanding how the charge-transfer process occurs at the interface between the organic polymer and the exfoliated layered franckeite in the p–n heterojunction is essential information for the design of more efficient optoelectronic devices. For this purpose, we used the core-hole clock (CHC) approach, using the core-hole lifetime as an internal reference clock for dynamic processes.35,36 The CHC approach is an elementally sensitive and orbital-specific synchrotron-based spectroscopy method that is capable of reaching the charge-transfer dynamics on the attosecond (10−18 s) time scale.37,38
In this paper, the charge-transfer time (τCT), calculated from the branching of competing core-hole decay channels, was used as a quantitative parameter to evaluate the electron-delocalization dynamics of excited electrons over the unoccupied electronic states of the PFO-DBT/Fr and P3HT/Fr heterojunctions. We found evidence of ultrafast interfacial electron-delocalization pathways from specific electronic states. For the PFO-DBT/Fr heterojunction, delocalization on a sub-femtosecond time scale was found for electrons excited in the π*(S–N) and π*(S–C) electronic states, corresponding to the benzothiadiazole and thiophene units, respectively. This result is strong evidence that both electronic states are the most efficient interfacial electron-delocalization pathways in the PFO-DBT/Fr heterojunction. On the other hand, for the P3HT/Fr heterojunction, we found electrons excited to the π–π* and S1s–π*(C–C) electronic states of the P3HT polymer as the main interfacial electron-transfer pathways in this heterojunction. These findings provide valuable insight in understanding how ultrafast electron delocalization is taking place in organic/inorganic van der Waals heterojunctions, which can motivate the design of new devices involving these systems.
2 Results and discussion
The elemental chemical compositions of the PFO-DBT/Fr and P3HT/Fr heterojunctions were estimated from the survey XPS spectra, as shown in Fig. 1a. The core-level Pb4f, Sn3d, Sb4s, C1s, N1s (for PFO-DBT), and S2s peaks found in the spectra for PFO-DBT/Fr and P3HT/Fr confirm the presence of the constituent elements in these heterojunctions. Additionally, Si2p and O1s core-level peaks are observed due to the SiO2/Si substrate.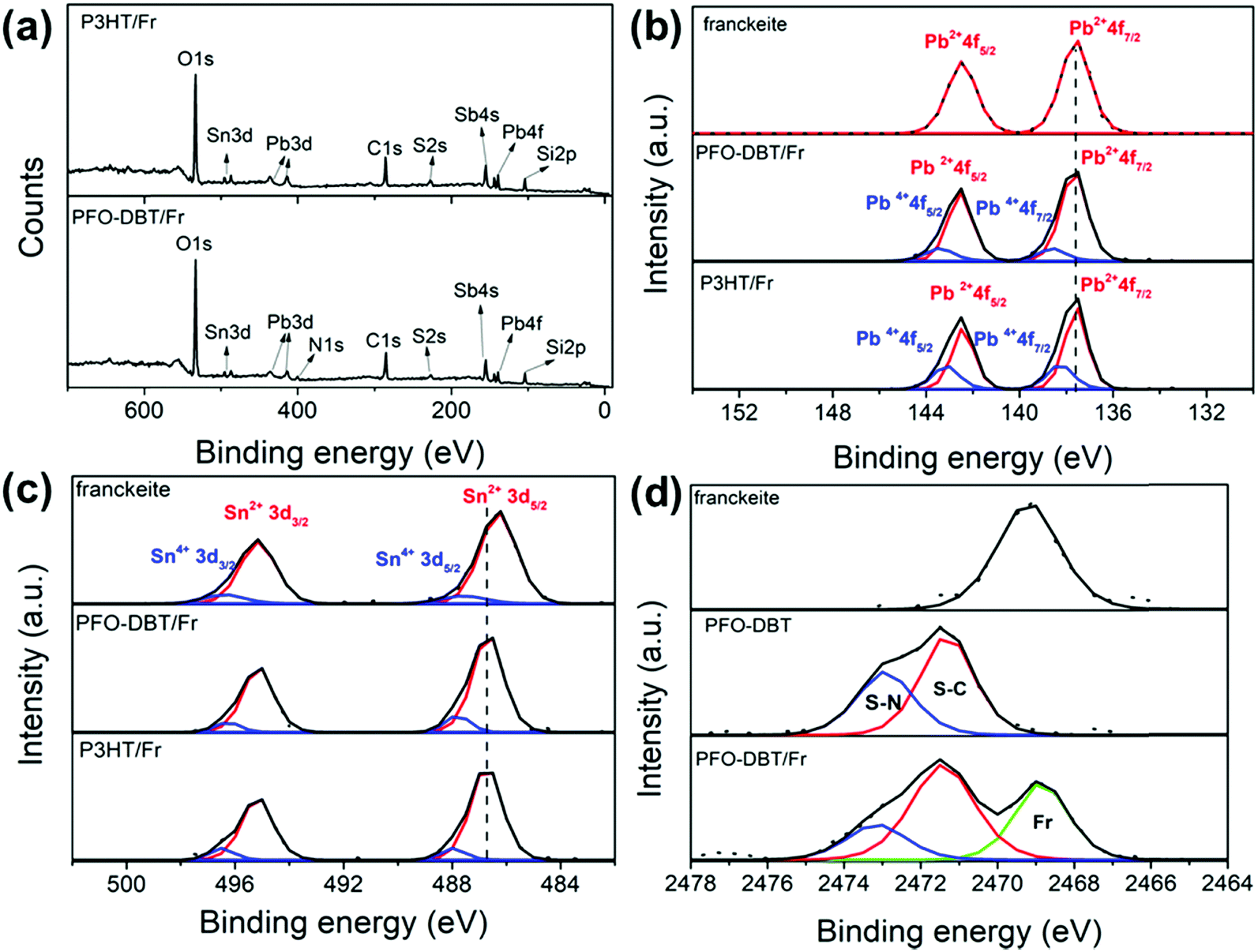 | ||
| Fig. 1 (a) XPS survey spectra of the PFO-DBT/Fr and P3HT/Fr heterojunctions. (b) and (c) Pb4f and Sn3d high-resolution XPS spectra of exfoliated franckeite and the PFO-DBT/Fr and P3HT/Fr heterojunctions, respectively. (d) S1s high-resolution XPS spectra of exfoliated franckeite and the PFO-DBT/SiO2 and PFO-DBT/Fr thin films. | ||
Fig. 1b and c shows the Pb4f and Sn3d high-resolution XPS spectra of both heterojunctions and pristine exfoliated franckeite. Two spin–orbit doublets are observed in the Pb4f and Sn3d XPS spectra of both heterojunctions, which are associated with the Pb2+, Pb4+, Sn2+ and Sn4+ oxidation states.19Fig. 1d shows the S1s XPS spectra of exfoliated franckeite, PFO-DBT/SiO2 and PFO-DBT/Fr. The S1s XPS spectrum of PFO-DBT/Fr is composed of three features. The feature at the binding energy of 2469.5 eV (green curve) is attributed to sulfur species in franckeite. The other two features at binding energies of 2471.5 eV and 2473.0 eV are associated with S–C (red curve) and S–N (blue curve) bonding in the benzothiadiazole and thiophene units, respectively. S1s XPS spectra corresponding to P3HT/SiO2 and P3HT/Fr thin films are shown in Fig. S2 in the ESI.† More representative differences between the electronic structures of exfoliated franckeite and the PFO-DBT/Fr and P3HT/Fr heterojunctions are observed in the high-resolution Pb4f and Sn3d XPS spectra. From a comparison of the Pb4f XPS spectra, we notice an increase of the Pb4+ oxidation species for the PFO-DBT/Fr and P3HT/Fr heterojunctions. These results show that there is an electronic coupling between the polymer and franckeite.
On the other hand, from analysis of the Sn3d XPS spectra, it is possible to observe a blue shift (an increase in binding energy) in the PFO-DBT/Fr and P3HT/Fr heterojunctions compared with the exfoliated franckeite thin film. Such a blue shift can be considered as evidence that confirms the electron transfer from the P3HT and PFO-DBT polymers to franckeite.39
The unoccupied electronic structures of exfoliated franckeite/SiO2, the PFO-DBT/SiO2 copolymer thin film and the PFO-DBT/Fr heterojunction were investigated using sulfur K-edge NEXAFS spectra as displayed in Fig. 2. The franckeite structure is formed by alternating phase segregation of pseudo-hexagonal Sn-rich (SnS2) and pseudo-tetragonal Pb-rich (PbS) layers, separated by a van der Waals gap. The S K-edge NEXAFS spectrum of exfoliated franckeite is described as a combination of the SnS2 and PbS phases. The S K-edge NEXAFS spectrum of the PbS layer is characterized by transitions from S1s to mixed S3p and Pb6p unoccupied electronic states in the conduction band.40,41 On the other hand, the S K-edge NEXAFS spectrum of the SnS2 layer is formed by transitions from core-level S1s to antibonding states made up of S3p states mixed with Sn5s and Sn5p states.42,43
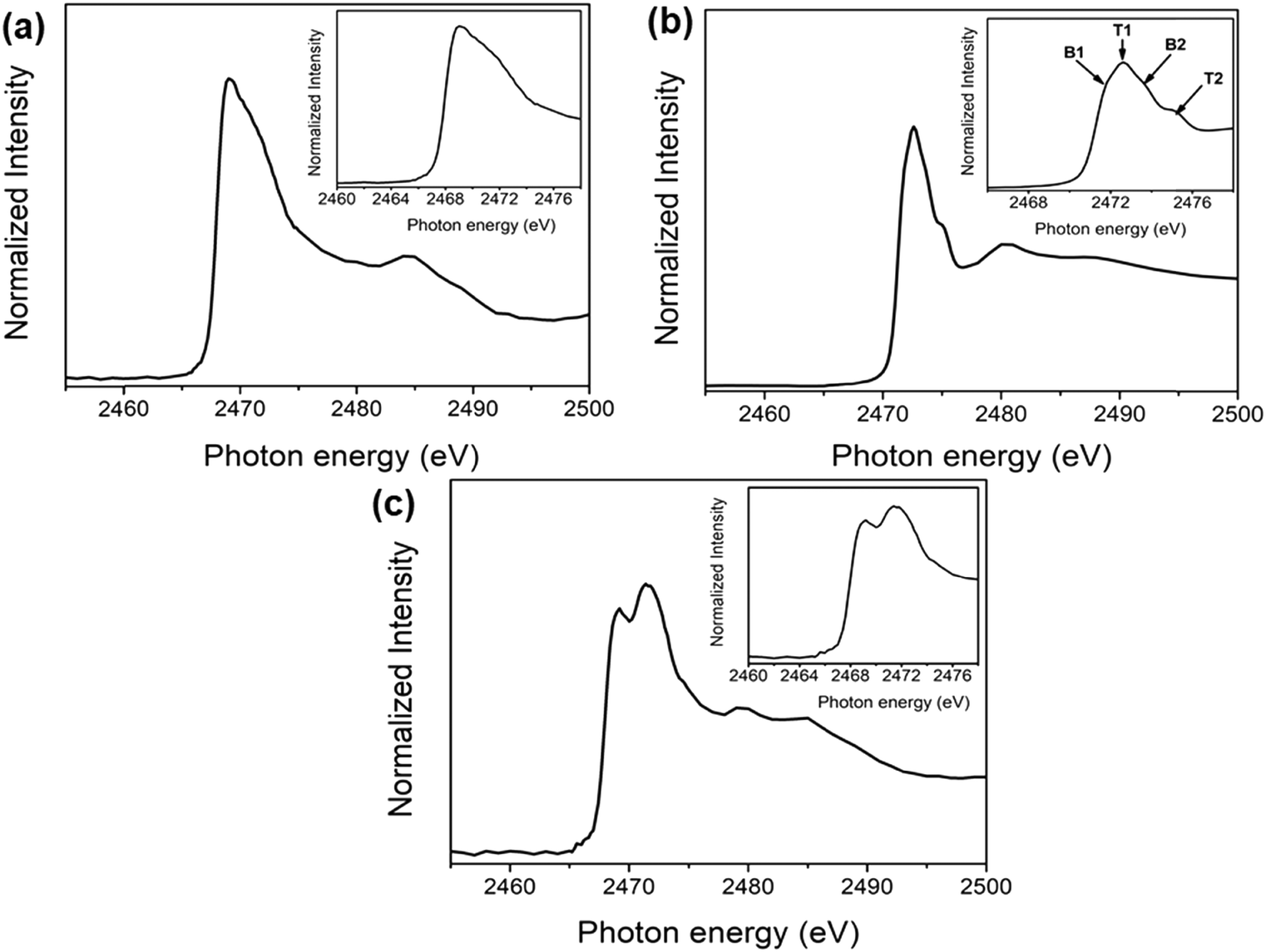 | ||
| Fig. 2 Sulfur K-edge NEXAFS thin-film spectra of (a) exfoliated franckeite/SiO2, (b) PFO-DBT/SiO2, and (c) PFO-DBT/Fr. | ||
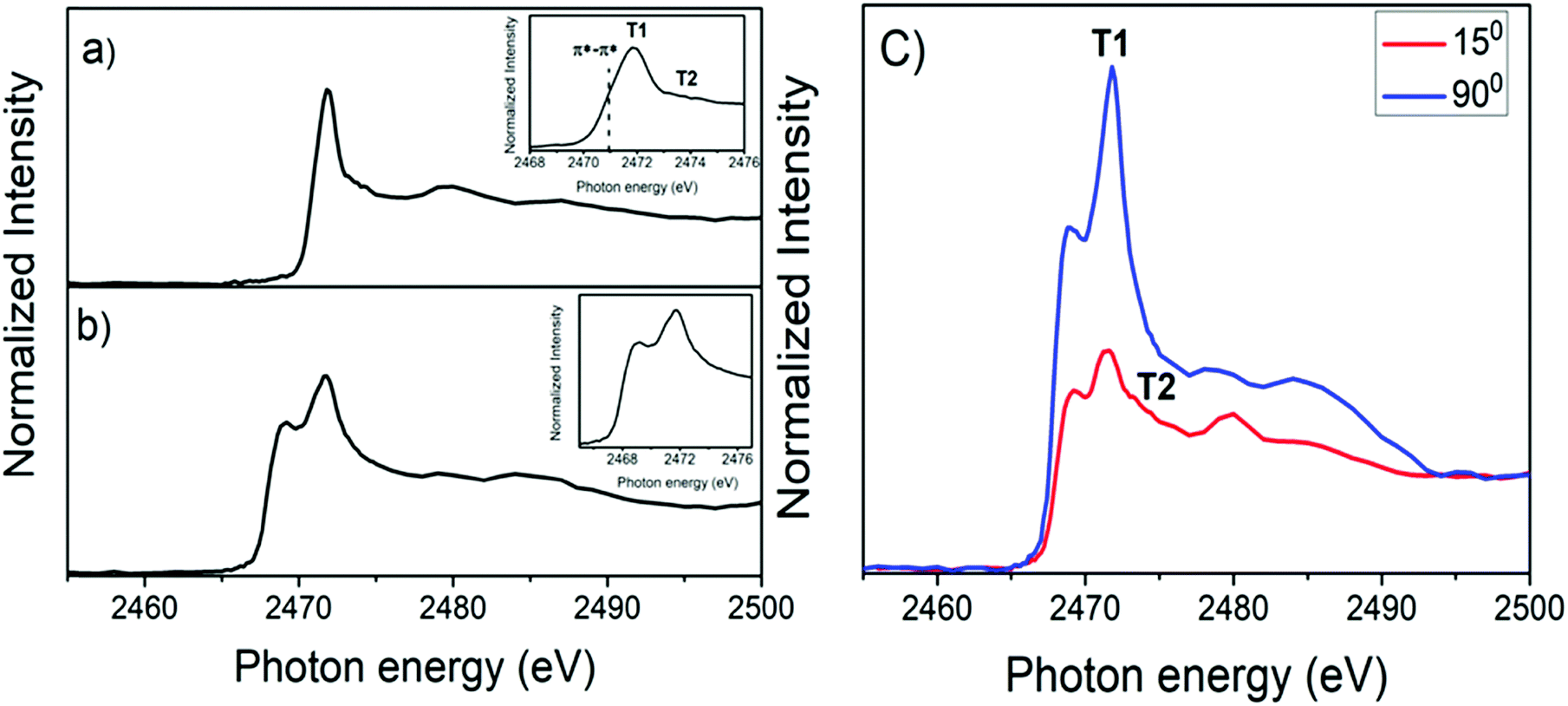
Four features characterized the sulfur K-edge NEXAFS spectrum of the PFO-DBT/SiO2 donor–acceptor copolymer (see the inset of Fig. 2b): the B1 (2471.8 eV) and B2 (2473.5 eV) signals are attributed to the S 1s–π*(S–N) and σ*(S–N) transitions, respectively, of the benzothiadiazole unit,44,45 while the T1 (2472.5 eV) and T2 (2474.5 eV) signals correspond to the S 1s–π* and σ* transitions, respectively, of the thiophene unit.44,45 For the PFO-DBT/Fr sample, the sulfur K-edge NEXAFS spectrum is formed by a convolution of the PFO-DBT copolymer and franckeite unoccupied electronic states.
The angular dependence of S K-edge NEXAFS spectra for PFO-DBT/Fr is shown in Fig. 3. This figure shows the spectra collected at normal (90°) and grazing (15°) incidence angles. According to this analysis, we can see that the intensities corresponding to S 1s–π*(S–N) of the benzothiadiazole unit (B1) and S 1s–π*(S–C) of the thiophene unit (T1) electronic transitions increase in the spectrum collected at a normal incident angle and decrease when compared with the spectrum collected at a grazing angle. An opposite tendency is observed for the S 1s–σ*(S–C, T2) and S 1s–σ*(S–N, B2) transitions. This tendency suggests that the thiophene and benzothiadiazole units in the PFO-DBT copolymer are almost coplanar, presenting a nearly edge-on average molecular orientation in the PFO-DBT/Fr heterojunction.46,47
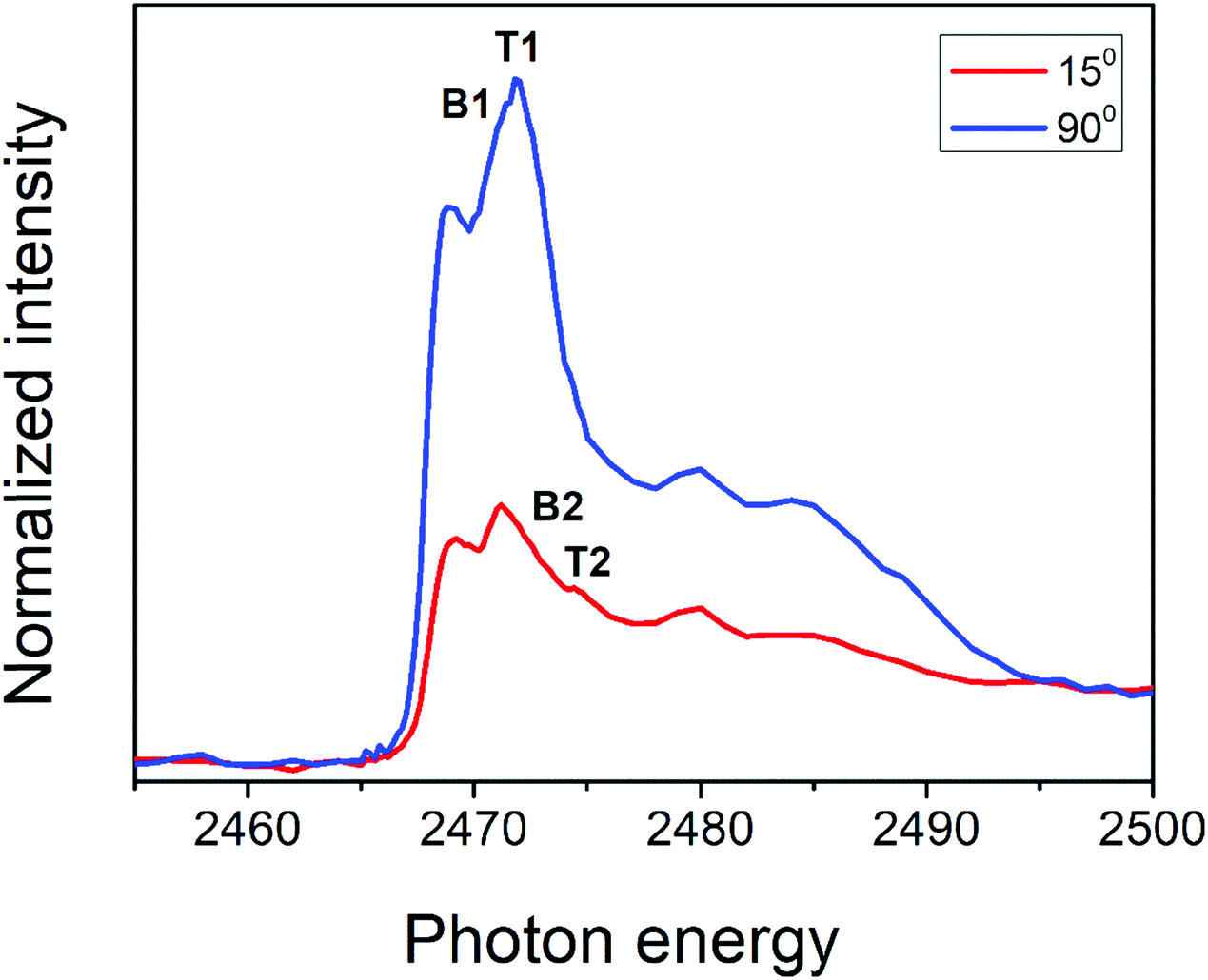 | ||
| Fig. 3 Polarized S K-edge NEXAFS dependence for the PFO-DBT/Fr thin film. The incident angle with respect to the film surface is also displayed. | ||
The electron delocalization and interfacial charge-transfer (CT) dynamics processes in the PFO-DBT/Fr heterojunction were investigated using the core-hole clock approach from the S K-L2,3L2,3 resonant Auger decay spectra. A detailed description of the core-hole clock approach and its application to thiophene-based polymers can be found elsewhere.39,44,48–50 Typically, the decay of a core excitation yields a spectral signature that is characteristic for a given system. The different decay Auger processes due to a core-level excitation are summarized in Scheme 2. They can be divided in two categories: resonant and non-resonant decay processes. Inside the resonant category, we can find two different decay processes: participator and spectator. In the participator decay channel, the excited electron is involved in the core-hole decay process, leaving the system with one hole (1h) in the valence band, while in the spectator process (Scheme 2B), the excited electron stays in the unoccupied valence states and does not participate in the decay process, with two-hole and one-electron final states (2h1e). The focus in this analysis is the spectator (SP) Auger decay process (Scheme 2B), which is the main resonant decay channel due to S1s core-level excitation.39,44,46,48–51 The other possible Auger decay channel is when the electron is transferred out of the atom during the core-hole lifetime (Scheme 2C). This case is considered to be a non-resonant decay process, where two holes (2h) in the valence band final state are reached by the system. It is energetically equivalent to normal Auger (NA) decay owing to a direct core-level photoionization process. The spectator and normal Auger decay channels are independent and competitive processes, and the branching among them is used to estimate the charge-transfer time (via the equation τCT = (Ispectator/Inormal) × τCH) using the core-hole lifetime (τCH) as the internal reference clock. In this approach depending on the data quality, the accessible τCT range is 0.1 × τCH ≤ τCT ≤ 10 × τCH. Assignment of the spectator and normal Auger decay channels is based on their overall dependence when the incident photon energy is tuned across the S1s excitation resonance. For the spectator decay channel, the Auger electron's kinetic energy is proportional to the incident photon energy. By contrast, for the normal Auger decay channel, the electron's kinetic energy is independent of the incident photon energy.
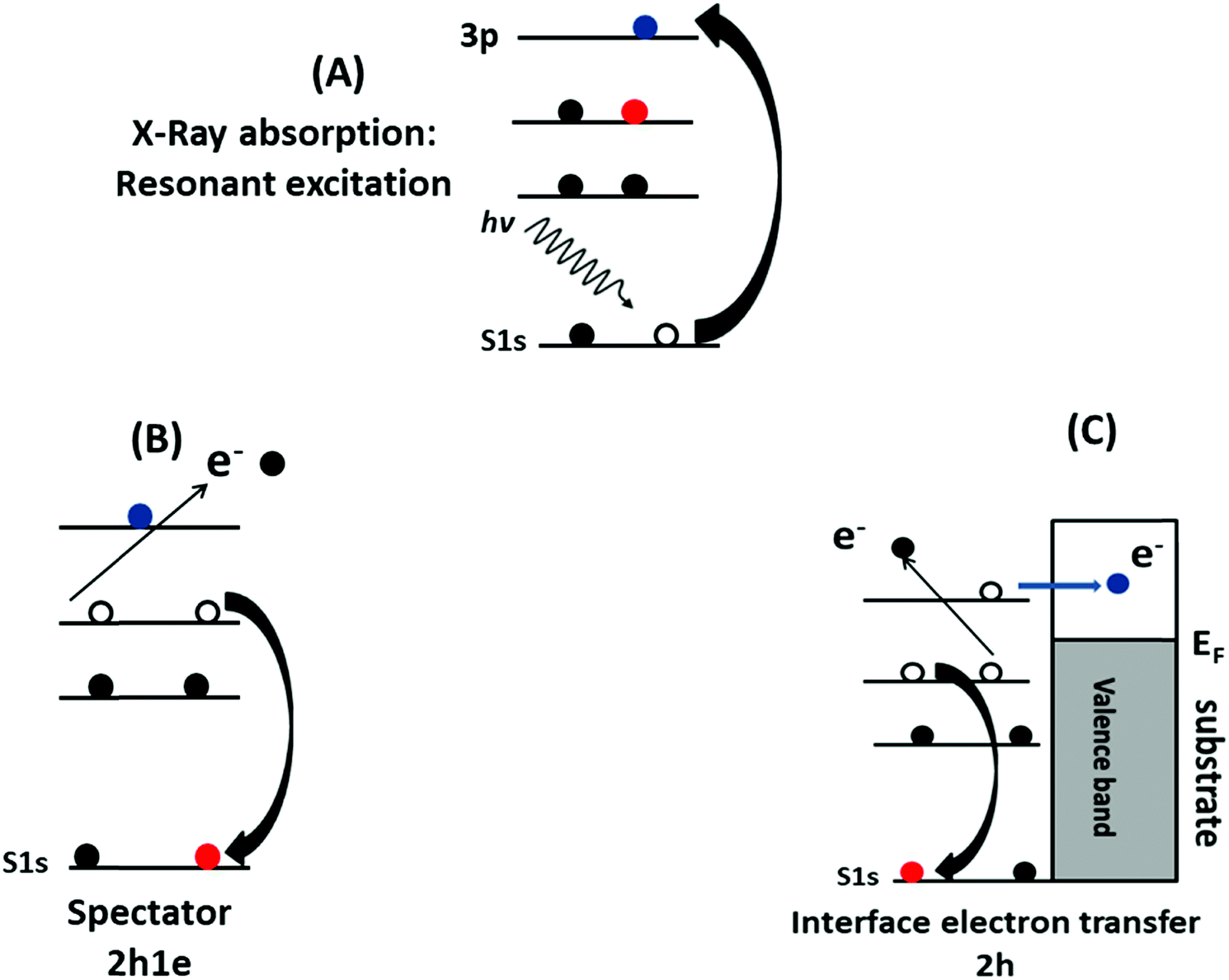 | ||
| Scheme 2 Schematic representation of the core-hole clock method. (A) Core-level resonant excitation into the unoccupied electronic state. (B) The spectator decay process with the 2h1e final state. (C) The electron is transferred to the substrate (or molecular environment) and core-hole decay proceeds via normal Auger decay with the 2h final state. | ||
Fig. 4 shows the deconvolution procedures of the S K-L2,3L2,3 RAS spectra of PFO-DBT, exfoliated franckeite and PFO-DBT/Fr thin films collected at excitation energies of 2471.8 eV, 2472.5 eV, 2473.5 eV, and 2474.5 eV, corresponding to the B1 (S 1s–π*(S–N)), T1 (S 1s–π* S–C), B2 (S 1s–σ*(S–N)) and T2 (S 1s–σ* (S–C)) transitions in the PFO-DBT copolymer. Some physical restrictions were imposed in the deconvolution procedures: the kinetic energy of the NA contribution is constant, and the line width of the spectator signal is smaller than that of the normal Auger contribution. The PFO-DBT and exfoliated franckeite S K-L2,3L2,3 Auger decay spectra consist of 1S and 1D Auger multiplets of the S3p states. The non-resonant Auger, or normal Auger, spectrum of the PFO-DBT copolymer, measured at an X-ray energy of hν = 2500 eV (above the ionization potential), is shown in Fig. S3 (ESI†); it is principally characterized by the two-hole (2h) final state appearing at the kinetic energy of 2112.0 eV. The overall evolution of intensity and peak width of the decay channels when the incident photon energy is tuned across the S 1s excitation resonance for the PFO-DBT/SiO2 thin film is shown in Fig. 5. Spectator decay features reach an intensity maximum and line sharpening for specific photon energies.44,51,52 The SP1, SP2, SP3 and SP4 decay channels achieved a maximum intensity and a minimum peak width for photon energies corresponding to B1, T1, B2 and T2 excitations. This behavior is associated with electron localization in these states.51 Then, we confirmed that the SP1, SP2, SP3 and SP4 decay channels in the RAS spectra are attributed to spectator decay contributions (the 2h1e final state) with the electron localized in the electronic state of π*(S–N), π*(S–C), s–σ*(S–N) and σ*(S–C), respectively. The NA (2h) Auger decay contribution appears at a constant kinetic energy of 2112.0 eV and with an almost constant peak width that is independent of the excitation energy. The NA decay channel in the RAS spectra is associated with the charge-transfer process during the core-hole lifetime. A new decay contribution was found in the RAS spectrum collected at an excitation energy of 2474.5 eV, associated with Rydberg states appearing close to the sulfur 1s ionization potential.44,51
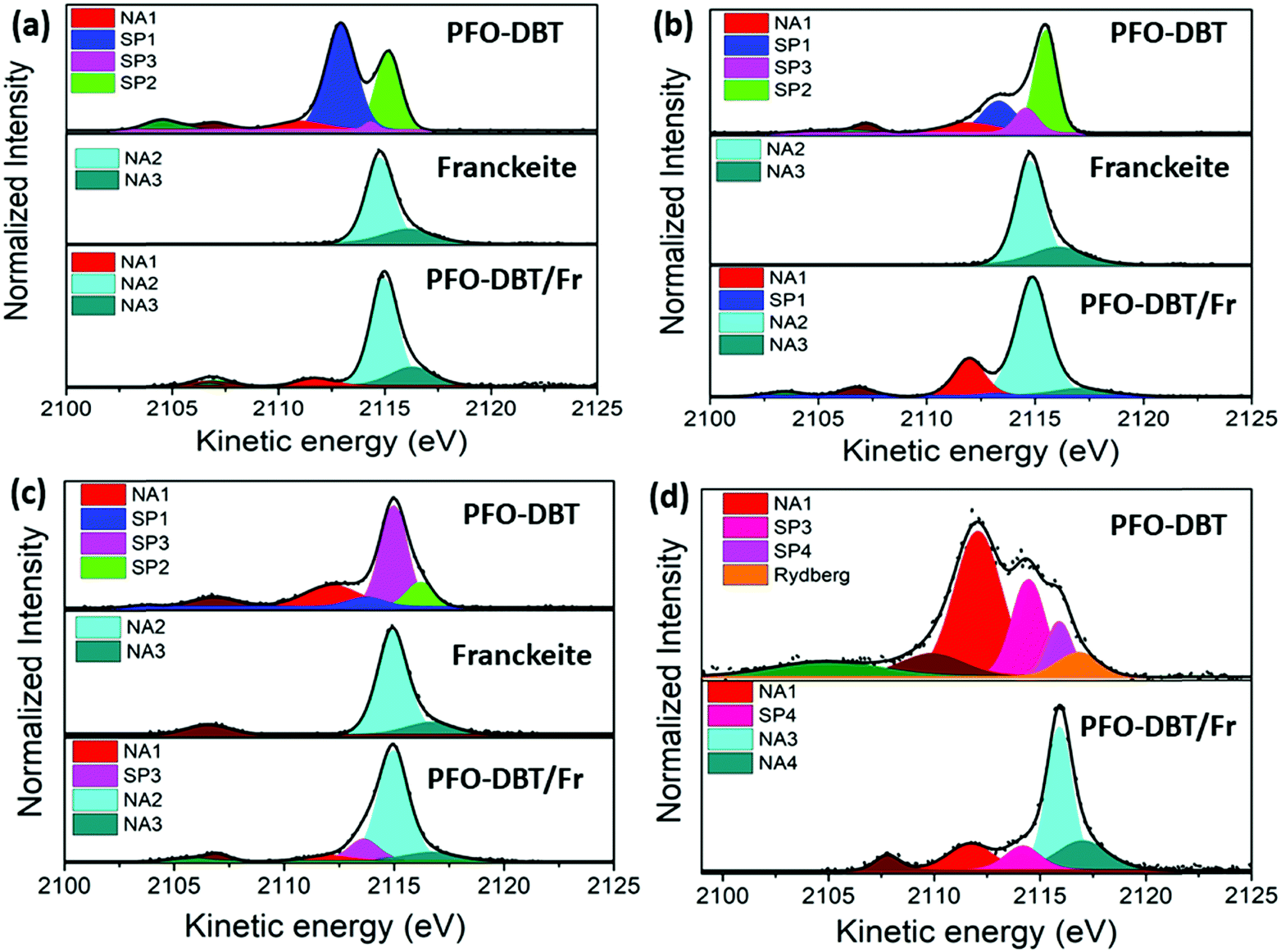 | ||
| Fig. 4 S K-L2,3L2,3 RAS spectra of PFO-DBT, exfoliated franckeite and PFO-DBT/Fr thin films collected at the photon energies of (a) 2471.8 eV (B1), (b) 2472.5 eV (T1), (c) 2473.5 eV (B2), and (d) 2474.5 eV (T2). | ||
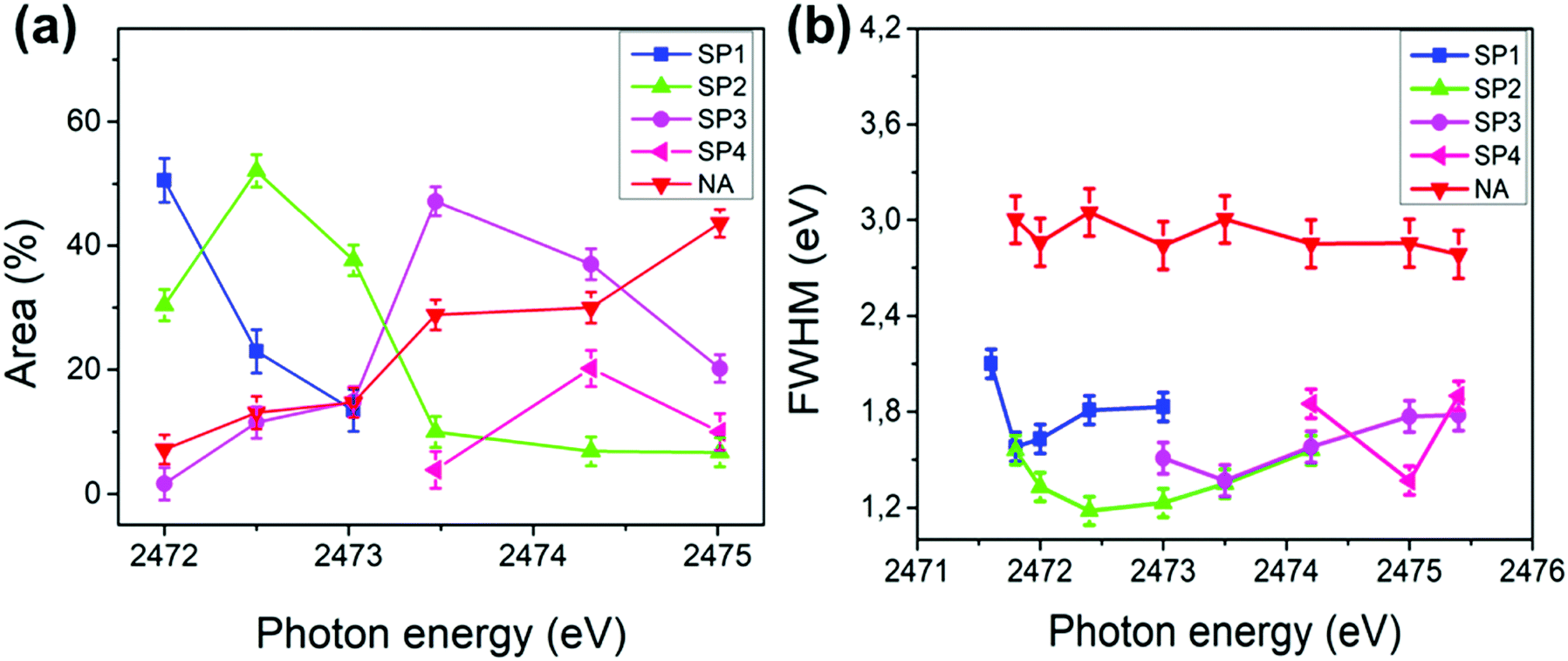 | ||
| Fig. 5 Area % (a) and full width at half maximum (FWHM) (b) plotted against the incident photon energy of the decay channels (SP1, SP2, SP3, SP4 and NA) in the S K-L2,3L2,3 RAS spectra of the PFO-DBT/SiO2 thin film. The error bar of 3% is also represented. | ||
The S K-L2,3L2,3 RAS spectrum of exfoliated franckeite is composed of two decay contributions appearing at constant kinetic energies of ∼2115 eV and ∼2116 eV, independent of the incoming photon energy. These two contributions at the same kinetic energies (∼2115 eV and ∼2116 eV) also appear in the non-resonant Auger spectrum of franckeite (measured at hν = 2500 eV) presented in Fig. S3 (ESI†). Therefore, these RAS spectra contributions are attributed to the normal Auger decay channels NA2 and NA3 due to the charge-transfer process in the core-hole lifetime. On the other hand, the S K-L2,3L2,3 RAS spectra of the PFO-DBT/Fr heterojunction are a convolution of the PFO-DBT and franckeite decay channels.
The electron-delocalization dynamics at the PFO-DBT/Fr heterojunction interface were analyzed quantitatively through the charge-transfer time (τCT) values, determined from the ratio between spectators and normal Auger signals using the equation τCT = (Ispectator/INA)τCH and the S1s core-hole lifetime τCH of 1.27 fs as the internal reference clock.53
We concentrated this analysis investigating the effect of franckeite in the electron delocalization degree of the unoccupied states of the PFO-DBT copolymer. The τCT of the PFO-DBT/SiO2 thin film was calculated using the integral intensities of their corresponding spectators (SP1, SP2, SP3 and SP4) and the normal Auger (NA1) contributions identified in each spectrum. In the PFO-DBT/Fr heterojunction case, the normal Auger (NA2 and NA3) contributions corresponding to the franckeite species were not considered for calculating the τCT values. The values of τCT obtained for the electrons excited to the B1 and B2 states of the benzothiadiazole unit and for the electrons excited to the T1 and T2 electronic states of the thiophene unit are summarized in Table 1.
| Electronic transition | τ CT (fs) | |
|---|---|---|
| PFO-DBT/SiO2 | PFO-DBT/Fr | |
| The τCT standard deviation values are shown in parentheses. | ||
| B1 π*(S–N) | >12.7 | >0.127 |
| T1 π*(S–C) | 8.33 (3) | 0.158 (4) |
| B2 σ*(S–N) | 2.69 (5) | 2.96 (4) |
| T2 σ*(S–C) | 1.04 (4) | 1.81 (6) |
According to the results presented in Table 1, we can see that the electron-delocalization process for the electrons excited to π*(S–N) and π*(S–C) in the benzothiadiazole and thiophene units is faster than those in the PFO-DBT/Fr heterojunction compared with the PFO-DBT/SiO2 copolymer. The opposite behavior is observed for electrons excited to the s–σ*(S–N) and T2 σ*(S–C) unoccupied electronic states. Therefore, we can conclude that PFO-DBT interacts with franckeite through the benzothiadiazole and thiophene π* electronic states. This result could be associated with the almost planar configuration of the PFO-DBT copolymer observed from analysis of the angular dependence of the S K-edge NEXAFS spectra. According to previous reports, a more planar structure in the copolymer improves the π-electron delocalization.
A similar analysis was performed with a van der Waals heterojunction constructed from the poly-3-hexylthiophene (P3HT) semiconductor polymer and exfoliated franckeite. Fig. 6a and b shows the S K-edge NEXAFS spectra for P3HT deposited on the SiO2 substrate (P3HT/SiO2) and the P3HT/Fr heterojunction thin film, respectively. According to previous reports, the S K-edge NEXAFS spectrum for the P3HT polymer is characterized by two features inside the resonance.39,52,54 These features were assigned to the overlapping of the S 1s → π*(C–C) and S 1s → σ*(S–C) transitions, which will be labeled here as T1 and T2, respectively. In addition, a small shoulder was identified at 2471.8 eV at the low photon energy side, corresponding to the π* symmetry state due to a strong π–π interchain interaction in the P3HT polymer.52 The angular dependence of the P3HT/Fr S K-edge NEXAFS spectrum is presented in Fig. 6c. At the normal (90°) incident angle, T1 transitions increase and T2 transitions decrease, while at the grazing (15°) incident angle, the opposite behavior is observed. This latter observation suggests that the P3HT polymer is edge-on oriented with respect to the franckeite/SiO2 substrate.39 Similar to the case of PFO-DBT/Fr, the S K-edge NEXAFS spectrum of the P3HT/Fr heterojunction is formed by the convolution of P3HT and franckeite unoccupied electronic states.
 | ||
| Fig. 6 Sulfur K-edge NEXAFS spectra of (a) P3HT/SiO2 and (b) P3HT/Fr, and the angular dependence of the S K-edge NEXAFS spectrum for the P3HT/Fr thin film (c). | ||
Fig. 7 shows the results of the deconvolution process for the S K-L2,3L2,3 RAS decay spectra collected at three excitation energies corresponding to the π–π interchain (a) and T1 (b) and T2 (c) electronic transitions. According to previous reports,39,52 the P3HT RAS spectra are composed of two spectator decay channels S1s–π*(C–C) and S1s–σ*(S–C), labeled here as SP1 and SP2, and the normal Auger (NA) contribution appearing at a constant kinetic energy of ∼2112.0 eV. A new decay contribution associated with Rydberg states was found in the RAS spectrum collected at the excitation energy of the T2 transition. The S K-L2,3L2,3 RAS spectrum of P3HT/Fr is a convolution of P3HT and the franckeite Auger decay channel.
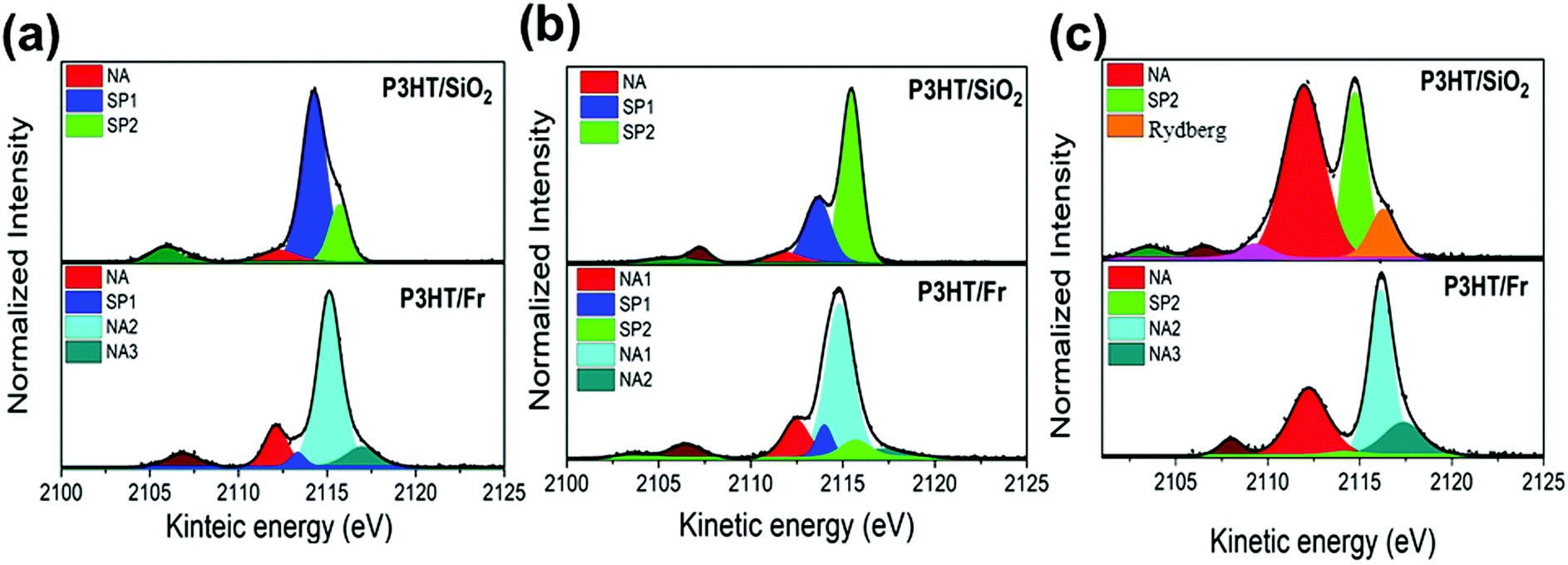 | ||
| Fig. 7 S K-L2,3L2,3 RAS spectra of the P3HT/SiO2 and P3HT/Fr thin films collected at photon energies of (a) 2471.8 eV, (b) 2472.6 eV, and (c) 2474.5 eV. | ||
Table 2 summarize the values of τCT for P3HT/SiO2, and P3HT/Fr obtained for electrons excited to the π–π*, S1s–π*(C–C) and S1s–σ*(S–C) states. The τCT values for P3HT/Fr were calculated using only the spectator (SP1 and/or SP2) and the normal Auger (NA1) decay channels of the P3HT polymer.
| Electronic transition | τ CT (fs) | |
|---|---|---|
| P3HT/SiO2 | P3HT/Fr | |
| The τCT standard deviation values are shown in parentheses. | ||
| S1s-π–π | >12.7 | 0.27 (4) |
| T1 S1s–π*(C–C) | 7.30 (7) | 1.26 (5) |
| T2 S1s σ*(S–C) | 1.02 (4) | 0.24 (2) |
Comparing the results presented in Table 2 for P3HT/SiO2 and P3HT/Fr/SiO2, we can observe that the presence of franckeite decreases the τCT values in the P3HT species. This can be interpreted as a strong electronic coupling between P3HT and franckeite unoccupied electronic states. However, the τCT values for electrons excited to π–π and S1s–π*(C–C) are the most affected by the presence of franckeite. Then, we can conclude that these two states are the main interfacial electron-transfer pathways in the P3HT/Fr heterojunction. Similar results were reported by Garcia-Basabe et al., investigating the electronic coupling at the interface between the P3HT polymer and 2D layered materials (exfoliated black phosphorus54 and MoS239). Furthermore, a previous study on the P3HT-MoS2 heterojunction,39 using time-resolved fluorescence lifetime measurements, showed similar results for the CT investigated using the CHC technique, from a qualitative point of view (i.e., a reduction in CT in the heterojunction compared with the isolated film), which reinforces the potential of the CHC method for investigating the interfacial charge transfer in organic–inorganic heterojunctions.
3 Experimental
3.1 Sample preparation
PFO-DBT/SiO2 and P3HT/SiO2 polymer thin films were prepared following the procedure described by Hao et al.55 The films were deposited on a silicon dioxide wafer by a spin-coating method at 1200 rpm for 60 s and using a solution of PFO-DBT and P3HT polymers dissolved in chloroform (0.5 mg mL−1). Later, the films were placed in a UHV chamber and annealed at 457 K for 2 h to remove the residual solvent and improve the crystallinity of the polymer.Exfoliated franckeite films were prepared using micromechanical exfoliation via a scotch tape method56 and transferred directly onto a silicon dioxide wafer. The PFO-DBT/Fr and P3HT/Fr heterojunctions on top of exfoliated franckeite were prepared using the spin-coating method under the same conditions used for preparing the PFO-DBT/SiO2 and P3HT/SiO2 polymer thin films.
3.2 Characterization
Raman spectra were collected using a WITec Alpha 300R confocal Raman imaging microscope. The spectra were measured using an excitation line of 488 nm and a 100× objective lens at room temperature under a nitrogen flow and an average laser power of 0.5 mW. All the measurements were made using a high-resolution 2400 g mm−1 Blaze 500 nm grating. These analysis results are presented in Fig. S1A and B in the ESI.† The morphology and thickness of exfoliated franckeite were studied by AFM microscopy (Bruker-Dimension Icon) operated in tapping mode. The results of this analysis are also presented in Fig. S1C and D in the ESI.†The S K-edge X-ray near-edge absorption (NEXAFS) and S K-L2,3L2,3 resonant Auger spectroscopy (RAS) measurements were carried out in the (SXS) beamline at the Brazilian Synchrotron Light Source (LNLS).57 A Si(111) double-crystal monochromator with an energy bandwidth of 0.48 eV was used to cover the sulfur K-edge. Experiments were performed under the so-called Auger resonant-Raman conditions, where the total experimental resolution is better than the natural-lifetime broadening of the core-excited states. It is a necessary condition to measure the dispersion of the spectral features related to the final states reached after the decay process. Near-edge X-ray absorption fine structure spectra were recorded in the total electron yield (TEY) mode normalized by the spectrum simultaneously obtained with a photon flux monitor (Au grid) to correct the fluctuation intensity of beam. Spectra are averages from at least three scans, and the background is corrected by linear pre-edge subtraction and linear regression beyond the edge.
S K-L2,3L2,3 RAS and X-ray photoelectron spectra (XPS) were collected inside an ultra-high vacuum (UHV) chamber with a base pressure of 10−8 mbar using a hemispherical electron energy analyzer (Specs model PHOIBOS 150) with a 45° take-off direction of Auger electrons and 25 eV of pass energy. The photon energy was calibrated using the Au 4f7/2 core level at 84.0 eV of a gold foil. The total energy resolution was 0.76 eV. The fitting analysis of the RAS spectra was performed using the CASA XPS software package (version 2.3.2) using two Pseudo-Voigt profile functions: (1) the Sum Form (SGL), which is a linear combination of Gaussian (G) and Lorentzian (L) functions, where the mixing is determined by m = p/100, GL(100) is a pure Lorentzian and GL(0) is pure Gaussian; and (2) the product form, which is the product of Gaussian (G) and Lorentzian (L) functions where the mixing is determined by m = p/100, GL(100) is a pure Lorentzian and GL(0) is pure Gaussian. For both cases, the background was corrected using a Shirley function. The result of this analysis is shown in Tables S1 and S2 in the ESI† showing that the different peak-fitting methods will not greatly affect the CT values. The possible surface charge effect (shift in electron energy) was monitored using the C (1s) (C–C) photoemission line localized at a binding energy of 285 eV. The sample degradation was monitored by measuring the NEXAFS spectra after each RAS experiment.
Conclusions
In summary, the electron-delocalization dynamics at the interface between 2D layered franckeite and two thiophene-based conjugated polymers (PFO-DBT and P3HT) from the resonantly core-excited electron have been investigated. We showed the ultrafast interfacial electron-delocalization pathways from specific electronic states for the PFO-DBT/Fr and P3HT/Fr heterojunctions. For the PFO-DBT/Fr heterojunction, the most efficient interfacial electron-delocalization pathways were in the π*(S–N) and π*(S–C) electronic states and through its benzothiadiazole and thiophene units. On the other hand, for the P3HT/Fr heterojunction, we found that electrons excited to the π–π* and S1s–π*(C–C) electronic states of the P3HT polymer are the most affected by the presence of franckeite and, consequently, are the main interfacial electron-transfer pathways in this heterojunction.Author contributions
Yunier Garcia-Basabe and Dunieskys G. Larrudé: conceptualization, methodology, writing – original draft, writing – review and editing, supervision, visualization, validation, project administration and funding acquisition. David Steinberg, Lara M. Daminelli and Cesar D. Mendoza: investigation and writing – review and editing. E. A. Thoroh de Souza and Flavio C. Vicentin: methodology, validation, writing – review and editing, writing – original draft, supervision and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the CNPq (grant numbers 408265/2016-7 and 314406/2018-2) and the São Paulo Research Foundation (FAPESP), grant 18/08988-9, CAPES and MackPesquisa. The research was partially supported by the LNLS – National Synchrotron Light Laboratory (SXS- 20180128) and CNPEM-LNNano (XPS-21999), Brazil. We are also grateful to the Araucaria Foundation for the productivity scholarship grant 068/2019 (Garcia-Basabe, Y.). We would also like to thank FAPERJ for a PDR10 scholarship (Cesar D. Mendoza, Processo E-26/202.357/2019).References
- J. Azadmanjiri, J. Wang, C. C. Berndt and A. Yu, J. Mater. Chem. A, 2018, 6, 3824–3849 RSC.
- M. Liras, M. Barawi and V. A. De La Peña O’Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- M. Gobbi, E. Orgiu and P. Samorì, Adv. Mater., 2018, 30, 1706103 CrossRef.
- Y. Wang, L. Wang, F. Liu, Z. Peng, Y. Zhang and C. Jiang, Org. Electron., 2020, 83, 105778 CrossRef CAS.
- M. Xu, M. Nakamura, M. Sakai and K. Kudo, Adv. Mater., 2007, 19, 371–375 CrossRef CAS.
- A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8475–8491 CrossRef CAS.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- X. Zhang, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C. J. Liu, S. C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- F. Zhang, D. Wu, Y. Xu and X. Feng, J. Mater. Chem., 2011, 21, 17590–17600 RSC.
- F. A. Larik, M. Faisal, A. Saeed, Q. Abbas, M. A. Kazi, N. Abbas, A. A. Thebo, D. M. Khan and P. A. Channar, J. Mater. Sci.: Mater. Electron., 2018, 29, 17975–18010 CrossRef CAS.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Top. Curr. Chem., 2017, 375, 84 CrossRef.
- K. Zhang, Y. Feng, F. Wang, Z. Yang and J. Wang, J. Mater. Chem. C, 2017, 5, 11992–12022 RSC.
- P. Avouris, Nano Lett., 2010, 10, 4285–4294 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS.
- E. Makovicky, V. Petrícek, M. Dušek and D. Topa, Am. Mineral., 2011, 96, 1686–1702 CrossRef CAS.
- A. J. Molina-Mendoza, E. Giovanelli, W. S. Paz, M. A. Ninõ, J. O. Island, C. Evangeli, L. Aballe, M. Foerster, H. S. J. Van Der Zant, G. Rubio-Bollinger, N. Agraït, J. J. Palacios, E. M. Pérez and A. Castellanos-Gomez, Nat. Commun., 2017, 8, 1–9 CrossRef.
- M. Velický, P. S. Toth, A. M. Rakowski, A. P. Rooney, A. Kozikov, C. R. Woods, A. Mishchenko, L. Fumagalli, J. Yin, V. Zólyomi, T. Georgiou, S. J. Haigh, K. S. Novoselov and R. A. W. Dryfe, Nat. Commun., 2017, 8, 1–11 CrossRef.
- S. Gan, Y. Zhao, X. Dai and Y. Xiang, Results Phys., 2019, 13, 102320 CrossRef.
- J. Li, L. Du, J. Huang, Y. He, J. Yi, L. Miao, C. Zhao and S. Wen, Nanophotonics, 2020, 10, 927–935 CrossRef.
- K. Ray, A. E. Yore, T. Mou, S. Jha, K. K. H. Smithe, B. Wang, E. Pop and A. K. M. Newaz, ACS Nano, 2017, 11, 6024–6030 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- A. S. Sarkar and S. K. Pal, J. Phys. Chem. C, 2017, 121, 21945–21954 CrossRef CAS.
- J. Dong, F. Liu, F. Wang, J. Wang, M. Li, Y. Wen, L. Wang, G. Wang, J. He and C. Jiang, Nanoscale, 2017, 9, 7519–7525 RSC.
- D. He, Y. Pan, H. Nan, S. Gu, Z. Yang, B. Wu, X. Luo, B. Xu, Y. Zhang, Y. Li, Z. Ni, B. Wang, J. Zhu, Y. Chai, Y. Shi and X. Wang, Appl. Phys. Lett., 2015, 107, 183103 CrossRef.
- X. Yu, J. Zhang, Z. Zhao, W. Guo, J. Qiu, X. Mou, A. Li, J. P. Claverie and H. Liu, Nano Energy, 2015, 16, 207–217 CrossRef CAS.
- R. Cheng, D. Li, H. Zhou, C. Wang, A. Yin, S. Jiang, Y. Liu, Y. Chen, Y. Huang and X. Duan, Nano Lett., 2014, 14, 5590–5597 CrossRef CAS PubMed.
- E. Wang, L. Wang, L. Lan, C. Luo, W. Zhuang, E. Wang, L. Wang, L. Lan, C. Luo and W. Zhuang, Appl. Phys. Lett., 2008, 92, 23 Search PubMed.
- P. Vanlaeke, A. Swinnen, I. Haeldermans, G. Vanhoyland, T. Aernouts, D. Cheyns, C. Deibel, J. D’Haen, P. Heremans, J. Poortmans and J. V. Manca, Sol. Energy Mater. Sol. Cells, 2006, 90, 2150–2158 CrossRef CAS.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- Q. Zhou, Q. Hou, L. Zheng, X. Deng, G. Yu and Y. Cao, Appl. Phys. Lett., 2004, 84, 1653–1655 CrossRef CAS.
- J. Luo, X. Li, Q. Hou, J. Peng, W. Yang and Y. Cao, Adv. Mater., 2007, 19, 1113–1117 CrossRef CAS.
- D. C. Watters, H. Yi, A. J. Pearson, J. Kingsley, A. Iraqi and D. Lidzey, Macromol. Rapid Commun., 2013, 34, 1157–1162 CrossRef CAS PubMed.
- A. Föhlisch, S. Vijayalakshmi, F. Hennies, W. Wurth, V. R. R. Medicherla and W. Drube, Chem. Phys. Lett., 2007, 434, 214–217 CrossRef.
- D. Menzel, Chem. Soc. Rev., 2008, 37, 2212–2223 RSC.
- A. Föhlisch, P. Feulner, F. Hennies, A. Fink, D. Menzel, D. Sanchez-Portal, P. M. Echenique and W. Wurth, Nature, 2005, 436, 373–376 CrossRef.
- C. N. Eads, D. Bandak, M. R. Neupane, D. Nordlund and O. L. A. Monti, Nat. Commun., 2017, 8, 1–7 CrossRef CAS PubMed.
- Y. Garcia-Basabe, G. G. Parra, M. B. Barioni, C. D. Mendoza, F. C. Vicentin and D. Larrudé, Phys. Chem. Chem. Phys., 2019, 21, 23521–23532 RSC.
- I. N. Demchenko, M. Chernyshova, X. He, R. Minikayev, Y. Syryanyy, A. Derkachova, G. Derkachov, W. C. Stolte, E. Piskorska-Hommel, A. Reszka and H. Liang, J. Phys.: Conf. Ser., 2013, 430, 012030 CrossRef CAS.
- O. Trejo, K. E. Roelofs, S. Xu, M. Logar, R. Sarangi, D. Nordlund, A. L. Dadlani, R. Kravec, N. P. Dasgupta, S. F. Bent and F. B. Prinz, Nano Lett., 2015, 15, 7829–7836 CrossRef CAS PubMed.
- I. Lefebvre-Devos, J. Olivier-Fourcade, J. C. Jumas and P. Lavela, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3110 CrossRef CAS.
- L. Meng, X. Zhou, S. Wang, Y. Zhou, W. Tian, P. Kidkhunthod, S. Tunmee, Y. Tang, R. Long, Y. Xin and L. Li, Angew. Chem., 2019, 131, 16821–16828 CrossRef.
- Y. Garcia-Basabe, C. F. N. Marchiori, C. E. V. De Moura, A. B. Rocha, L. S. Roman and M. L. M. Rocco, J. Phys. Chem. C, 2014, 118, 23863–23873 CrossRef CAS.
- A. P. Hitchcock, R. S. DeWitte, J. M. Van Esbroeck, P. Aebi, C. L. Frenc, R. T. Oakley and N. P. C. Westwood, J. Electron Spectrosc. Relat. Phenom., 1991, 57, 165–187 CrossRef CAS.
- Y. Garcia-Basabe, C. F. N. Marchiori, B. G. A. L. Borges, N. A. D. Yamamoto, A. G. Macedo, M. Koehler, L. S. Roman and M. L. M. Rocco, J. Appl. Phys., 2014, 115, 134901 CrossRef.
- A. S. Anselmo, A. Dzwilewski, K. Svensson and E. Moons, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 176–182 CrossRef CAS.
- C. Arantes, B. G. A. L. Borges, B. Beck, G. Araújo, L. S. Roman and M. L. M. Rocco, J. Phys. Chem. C, 2013, 117, 8208–8213 CrossRef CAS.
- Y. Garcia-Basabe, B. G. A. L. Borges, D. C. Silva, A. G. Macedo, L. Micaroni, L. S. Roman and M. L. M. Rocco, Org. Electron., 2013, 14, 2980–2986 CrossRef CAS.
- F. O. L. Johansson, M. Ivanović, S. Svanström, U. B. Cappel, H. Peisert, T. Chassé and A. Lindblad, J. Phys. Chem. C, 2018, 122, 12605–12614 CrossRef CAS.
- K. Yoshii, Y. Baba and T. A. Sasaki, Phys. Status Solidi, 1998, 206, 811–822 CrossRef CAS.
- Y. Garcia-Basabe, D. Ceolin, A. J. G. Zarbin, L. S. Roman and M. L. M. Rocco, RSC Adv., 2018, 8, 26416–26422 RSC.
- J. L. Campbell and T. Papp, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- Y. Garcia-Basabe, V. O. Gordo, L. M. Daminelli, C. D. Mendoza, F. C. Vicentin, F. Matusalem, A. R. Rocha, C. J. S. de Matos and D. G. Larrudé, Appl. Surf. Sci., 2021, 541, 148455 CrossRef CAS.
- X. T. Hao, T. Hosokai, N. Mitsuo, S. Kera, K. K. Okudaira, K. Mase and N. Ueno, J. Phys. Chem. B, 2007, 111, 10365–10372 CrossRef CAS PubMed.
- B. Wang, J. Park, D. R. Dreyer, S. Park, W. Bielawski, R. S. Ruoff, Z. Liu, J. T. Robinson, X. Sun, H. Dai, B. Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts, R. S. Ruoff, P. Avouris, C. Dimitrakopoulos, Y. Si, E. T. Samulski, C. Hill, N. Carolina, A. K. Geim, A. H. Macdonald, A. K. Geim, A. H. Macdonald, K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang and P. Article, Mater. Today, 2004, 306, 666–669 Search PubMed.
- M. Abbate, F. C. Vicentin, V. Compagnon-Cailhol, M. C. Rocha and H. Tolentino, J. Synchrotron Radiat., 1999, 6, 964–972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01694f |
| This journal is © the Owner Societies 2021 |