
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtom-efficient transition-metal-free arylation of N,O-acetals using diarylzinc reagents through Zn/Zn cooperativity†
Andryj M.
Borys‡
 ,
Jose M.
Gil-Negrete‡
and
Eva
Hevia
*
,
Jose M.
Gil-Negrete‡
and
Eva
Hevia
*
Departement für Chemie, Biochemie und Pharmazie, Universität Bern, Switzerland. E-mail: eva.hevia@dcb.unibe.ch
First published on 11th August 2021
Abstract
Exploiting the cooperative action of Lewis acid Zn(C6F5)2 with diarylzinc reagents, the efficient arylation of N,O-acetals to access diarylmethylamines is reported. Reactions take place under mild reaction conditions without the need for transtion-metal catalysis. Mechanistic investigations have revealed that Zn(C6F5)2 not only acts as a Lewis acid activator, but also enables the regeneration of nucleophilic ZnAr2 species, allowing a limiting 50 mol% to be employed.
Capable of exceptional functional group tolerance, arylzinc compounds are some of the most widely used organometallic reagents in synthesis for C–C bond forming processes.1 Frequently the use of transition-metal catalysis is required to maximise yields and selectivities.2 However, recent advances have shown that in certain cases organozinc reagents can react effectively with organic electrophiles in the absence of catalysts. Thus, direct cross-coupling with aryl halides has been reported by Uchiyama and Wang although harsh reaction conditions are required (90–130 °C, 24 h).3 Ingleson has also shown that coupling of benzyl and alkyl halides with ZnAr2 can take place efficiently at room temperature using non-ethereal solvents.4 More recently, Knochel has used ArZnX reagents to prepare triarylmethanes via sequential cross-couplings with benzal diacetates.5 These reactions are proposed to operate via a two-step SN1-type mechanism with the initial formation of a reactive ketone oxonium intermediate. Related to Knochel's work, we have reported the stereoselective cross-coupling between glycosyl bromides and ZnAr2 reagents facilitated by Lewis acidic bis(pentafluorophenyl)zinc, Zn(C6F5)2.6 These processes are underpinned by the special cooperation between the two different types of arylzinc reagents, with Zn(C6F5)2 facilitating bromine abstraction of the substrate, forming a highly electrophilic oxocarbenium species that, in turn, reacts with ZnAr2 to give the desired arylated product. Extending the scope of this Zn/Zn′ cooperative partnership beyond glycosylation reactions, here we report a new transition-metal free method to access synthetically relevant functionalised diarylmethylamines, assessing the role played by each organozinc component. Diarylmethylamines are important organic scaffolds present in many pharmaceuticals and biologically active molecules.7 Previous studies have shown that they are accessible via one-step three component reactions by coupling of arylzinc reagents with secondary amines and aldehydes.8 On the downside, these processes typically require cobalt or copper catalysis, as well as the use of excess arylzinc reagent and/or high reaction temperatures. Therefore, this study asked, could we develop a method to circumvent these additional requirements?
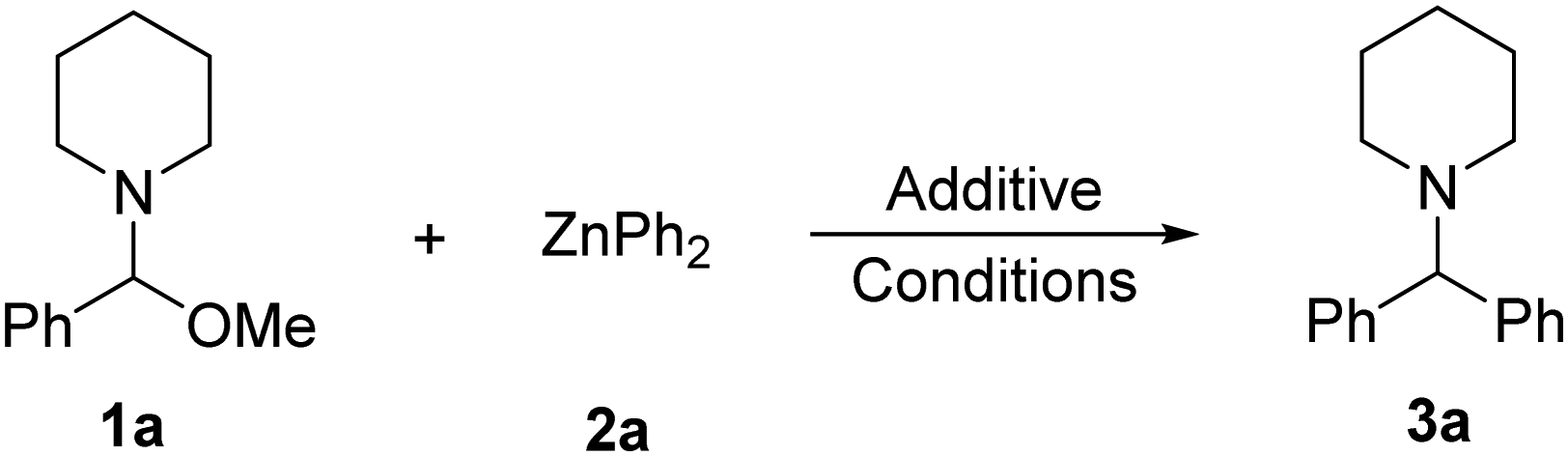
We first attempted the direct synthesis of diarylmethylamines from N,O-acetals using diarylzinc reagents. However, no reaction is seen on treating N,O-acetal (1a) with 0.5 equivalents of ZnPh2 (2a) in THF at room temperature (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | ZnPh2 (mol%) | Additive (mol%) | Yield of 3a (%) |
| 1 | 1 h, THF, rt | 50 | — | 0 |
| 2 | 1 h, toluene, rt | 50 | — | 47 |
| 3 | 1 h, toluene, rt | 100 | — | 93 |
| 4 | 1 h, toluene, rt | 50 | Zn(C6F5)2 (10) | 63 |
| 5 | 1 h, toluene, rt | 50 | B(C6F5)3 (10) | 43 |
| 6 | 1 h, toluene, rt | 50 | GaCl3 (10) | 52 |
| 7 | 1 h, toluene, rt | 50 | ZnBr2 (10) | 47 |
| 8 | 1 h, toluene, rt | 50 | Zn(C 6 F 5 ) 2 (50) | 94 |
Contrastingly, switching to toluene as the reaction solvent, a 47% yield of the corresponding diarylmethanamine product (3a) was obtained after 1 hour (entry 2), although approximately half of the N,O-acetal did not react suggesting that only one phenyl from ZnPh2 is active in the arylation process. Indeed, using 1 equivalent of ZnPh2, a 93% yield of 3a was now obtained (entry 3). Striving for a more atom-economical process, the reaction was attempted with PhZnBr, but this only gave a 21% yield of 3a, likely due to the poor solubility of the zinc reagent in non-donor solvents (see ESI† for further screening and optimisation). Considering the marked solvent effect observed, we pondered if the use of Lewis acids as additives could facilitate the atom-efficient arylation process. Pleasingly, adding 10 mol% Zn(C6F5)2 in combination with 0.5 equivalents of ZnPh2 increased the yield of 3a up to 63% (entry 4), whilst B(C6F5)3, GaCl3 or ZnBr2 surprisingly had much less impact (entries 5–7). This suggests that Zn(C6F5)2 plays a more intimate role beyond simple Lewis acid substrate activation (vide infra). On increasing the quantity of Zn(C6F5)2 up to 50 mol%, complete transfer of both Ph groups in ZnPh2 was observed to give diarylmethanamine 3a in 94% yield (entry 8). Notably, no C6F5-substitution was detected, showing that Zn(C6F5)2 is inert towards the direct arylation reaction.
A series of spectroscopic and structural mechanistic studies were carried out to understand how Zn(C6F5)2 enables the substitution of both phenyl groups from ZnPh2 to the N,O-acetal. When 1-methoxy-(4-fluorophenyl)methyl-piperidine (1f) is treated with 1 equivalent of ZnPh2 (2a) in toluene-d8, complete consumption is observed within 20 minutes; this is marked by the almost complete disappearance of the benzylic singlet at δ 4.51 in the 1H NMR spectrum, and appearance of a new benzylic singlet at δ 4.07 for the product (3i) (see Fig. S1 in ESI†). An additional singlet is observed at δ 3.63, which is attributed to PhZnOMe, the expected by-product of the reaction. This species does not appear to react further with an excess of 1f, which is consistent with only one phenyl group from ZnPh2 being active towards the arylation process.
A rational synthesis of the proposed phenylzinc methoxide was performed by treating ZnPh2 with an equimolar amount of MeOH in toluene, leading to a mixture of tetrameric [PhZnOMe]4 (4) and heptanuclear [{PhZnOMe}6ZnOMe2] (5) in approximately a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 1). Heteroleptic (5) is methoxy-rich and is postulated to form via redistribution9 of 4 since it was present in similar ratios with respect to 4 for repeated syntheses and recrystallisations. Isolated in low yields by adjusting the stoichiometry and crystallisation conditions (see ESI†), 5 is not observed in the reaction between 1f and ZnPh2, and there is no evidence from variable temperature NMR studies to suggest that 4 and 5 can interconvert. The OMe signal of 4 in the 1H NMR spectrum matches the PhZnOMe species that is formed in the reaction mixture (Fig. S1, ESI†). Consistent with the reactivity studies described above, no further formation of arylation product 3a was observed when treating N,O-acetal 1a with isolated crystals of phenylzinc methoxide 4.
:1 ratio (Fig. 1). Heteroleptic (5) is methoxy-rich and is postulated to form via redistribution9 of 4 since it was present in similar ratios with respect to 4 for repeated syntheses and recrystallisations. Isolated in low yields by adjusting the stoichiometry and crystallisation conditions (see ESI†), 5 is not observed in the reaction between 1f and ZnPh2, and there is no evidence from variable temperature NMR studies to suggest that 4 and 5 can interconvert. The OMe signal of 4 in the 1H NMR spectrum matches the PhZnOMe species that is formed in the reaction mixture (Fig. S1, ESI†). Consistent with the reactivity studies described above, no further formation of arylation product 3a was observed when treating N,O-acetal 1a with isolated crystals of phenylzinc methoxide 4.
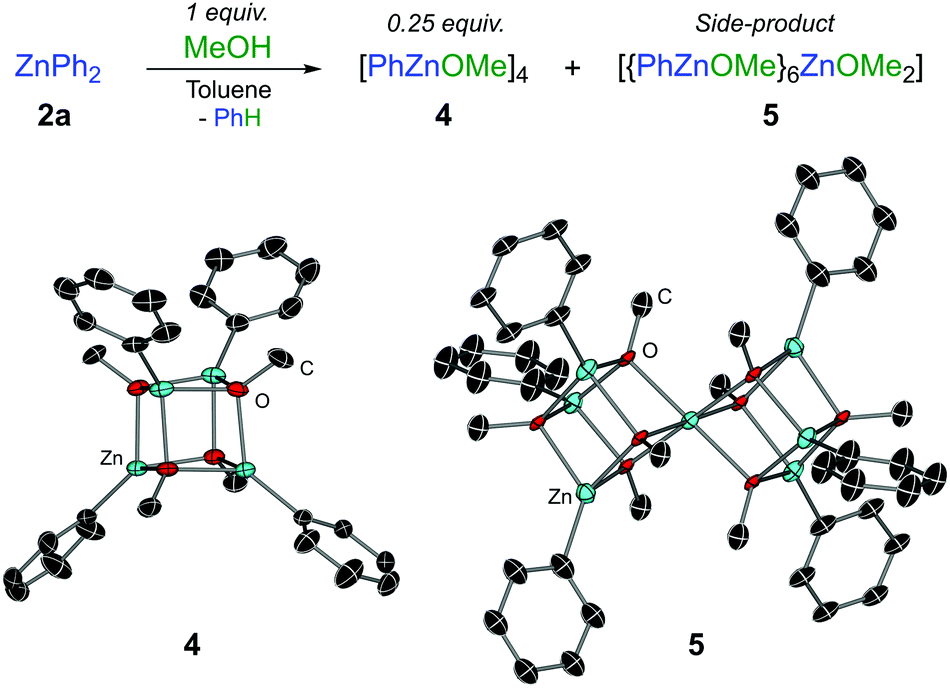 | ||
| Fig. 1 Synthesis of stoichiometric and OMe-rich phenyl zinc-methoxides, 4 and 5. Molecular structures of 4 and 5. Thermal ellipsoids shown at 30% probability. Hydrogen atoms and solvents of crystallisation have been omitted for clarity. | ||
The next step was to investigate the role of Zn(C6F5)2 in the transformation and explain how it enables the reaction to occur efficiently with a limiting 50 mol% of the diarylzinc reagent. Combining 1f with one equivalent of Zn(C6F5)2 in toluene-d8 suggests coordination and adduct formation of I (Scheme 1a) as evidenced by the broadening and shifting of the 1H and 19F NMR signals (see Fig. S2–S3 in ESI†).
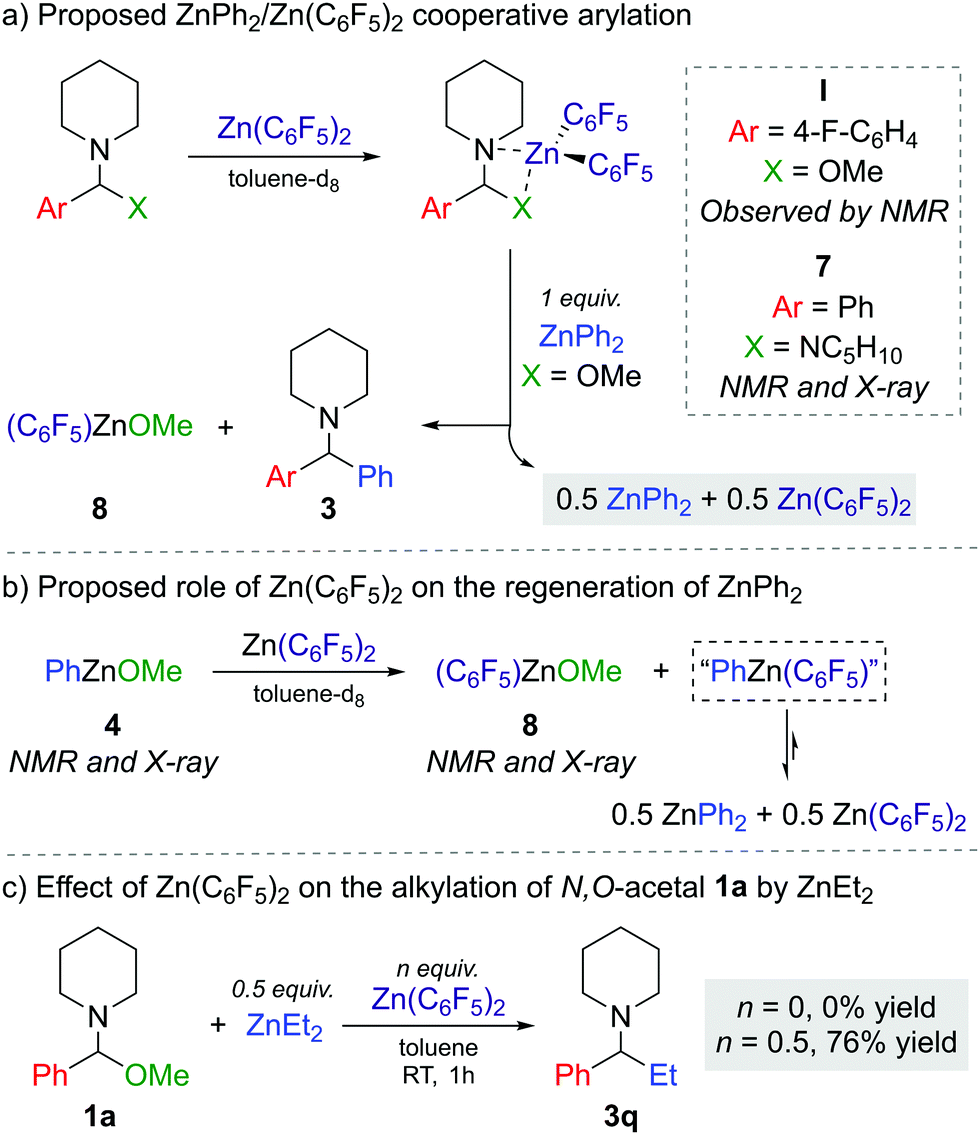 | ||
| Scheme 1 Mechanistic studies on the arylation of N,O-acetals by ZnAr2/Zn(C6F5)2 combinations. | ||
While I could not be isolated as a solid and it partially decomposes over time in solution, coordination adduct [{PhCH(NC5H10)2}Zn(C6F5)2] (7) was obtained and structurally authenticated by treating Zn(C6F5)2 with the related N,N′-aminal 1,1 (phenylmethylene)dipiperidine, 6 (Scheme 1a). In 7, the N,N′-aminal coordinates to the Zn centre in a chelating fashion via its two N atoms (Fig. 2a). Interestingly, 7 is significantly more robust that I in solution and it does not undergo arylation with ZnPh2 even under forcing refluxing conditions. This can be attributed to the greater strength of the C–N bonds in the N,N′-aminal versus the C–O bond in 1f.10 Related to these findings it can be hypothesised that the coordination of N,O-acetal to the Lewis acid would increase the electrophilicity of the substrate and enable the less nucleophilic PhZnOMe intermediate to transfer its remaining Ph group. However, no other Lewis acids appeared to promote the reaction and 50 mol% of Zn(C6F5)2 is needed, implying that its role is not catalytic in nature. Instead, we observed that Zn(C6F5)2 reacts directly with the PhZnOMe by-product to regenerate the more nucleophilic diarylzinc reagent together with the formation of (C6F5)ZnOMe (Scheme 1a and b). This would justify the necessity for requiring 50 mol% Zn(C6F5)2 to enable complete transfer of both aryl groups from the zinc reagent. Similarly to 4, treating a toluene solution of freshly sublimed Zn(C6F5)2 with equimolar MeOH yields the tetrameric species [(C6F5)ZnOMe]4 (8) (Fig. 2b). Interestingly, the 1H and 19F NMR signals for 8 appear to differ slightly when compared to in situ ‘(C6F5)ZnOMe’, prepared from isolated [PhZnOMe]4 (4) with regeneration of the nucleophilic diarylzinc reagent (Fig. S4–S5, ESI†). We attribute these differences to the formation of different aggregates and/or solvates in solution, but the possibility of heteroleptic/mixed aryl zinc alkoxides cannot be fully discarded. However, on spiking each of the three (C6F5)ZnOMe solutions with THF, almost identical 19F NMR spectra were obtained, consistent with the hypothesis that different aggregates/solvates coexist (Fig. S6, ESI†). Furthermore, ZnPh2 (now as the THF adduct) can be identified by 1H NMR spectroscopy (Fig. S7, ESI†), illustrating the ability of Zn(C6F5)2 to regenerate the more nucleophilic diarylzinc reagent. Overall, we propose that Zn(C6F5)2 first coordinates to the N,O-acetal 1, forming adduct I, activating the substrate towards arylation by nucleophilic ZnPh2 to give the corresponding diarylmethanamine 3 and PhZnOMe (Scheme 1a and b). This step may occur via the in situ formation of an iminium intermediate or in a concerted manner.5,11 The marked solvent effect observed in these transformations, with Lewis donor THF completely inhibiting formation of 3, supports the key role of the coordination of Zn(C6F5)2 to the substrate to facilitate the arylation process.6 By-product PhZnOMe reacts in turn with Zn(C6F5)2 to form (C6F5)ZnOMe (the ultimate by-product of the reaction, Scheme 1a and b), along with the regeneration of 0.5 equivalents of ZnPh2 and Zn(C6F5)2, resulting from the redistribution of PhZn(C6F5) (Scheme 1b).6,12 The regenerated ZnPh2 can react further with N,O-acetal, whilst the unreacted 0.5 equivalents of Zn(C6F5)2 enable the continued recycling of PhZnOMe that is formed in the reaction. In this regard almost identical 19F NMR spectra are obtained when combining isolated [PhZnOMe]4 (4) with Zn(C6F5)2 (0.25:1 ratio), or a mixture of isolated [(C6F5)ZnOMe]4 (6), ZnPh2 and Zn(C6F5)2 (0.25:0.5:0.5 ratio) (Fig. S8, ESI†).
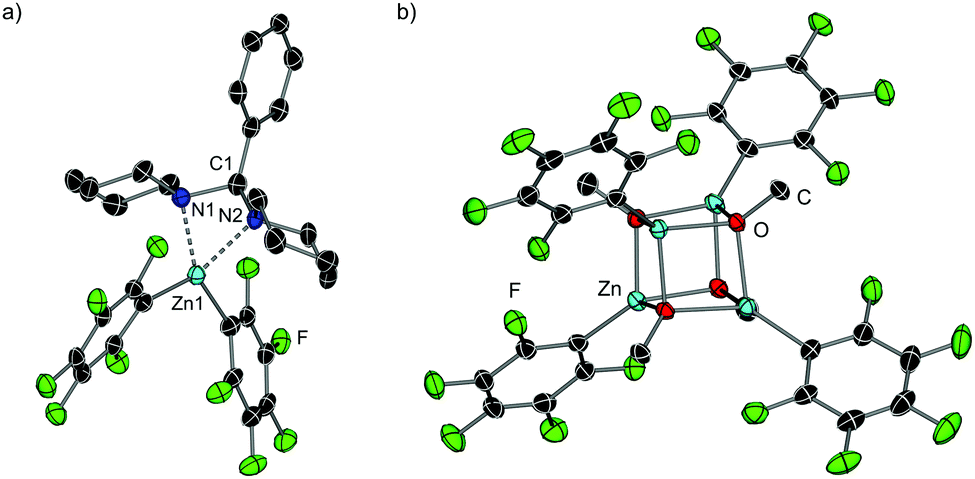 | ||
| Fig. 2 Molecular structures of (a) [{PhCH(NC5H10)2}Zn(C6F5)2] (7) and (b) [(C6F5)ZnOMe]4, (8). Thermal ellipsoids shown at 30% probability. Hydrogen atoms and solvent molecules of crystallisation omitted for clarity. | ||
The participation of Zn(C6F5)2 enables the use of 50 mol% of ZnPh2, which can be particularly useful when employing more complex aryl scaffolds on zinc, as only limiting amounts are required to achieve high yields, in contrast to other methods where an excess of the organozinc reagent is typically needed.8
We also found that when 1a is reacted with 0.5 equiv. ZnEt2 no alkylation is observed, whereas introducing Zn(C6F5)2 (0.5 equiv.) furnishes 3q in a 76% yield (Scheme 1c). This can be attributed to the reduced Lewis acidity of ZnEt2 (in comparison to ZnPh2), so on its own it cannot activate 1a towards C–O bond cleavage, requiring the initial formation of coordination adduct akin to I (Scheme 1a) which can then react with ZnEt2.
Having gained some mechanistic insights, we then went on to explore the scope of the reaction (Fig. 3). Diarylmethanamine products (3b–3g) were realised in high isolated yields (84–92%) using a range of diarylzinc reagents (2b–g) furnished with electron-donating or electron-withdrawing groups, ortho-substituents and even heteroaryls. For the synthesis of 3d and 3g, the reaction was performed at 90 °C due to the poor solubility of the ZnAr2 species in toluene. In all cases, although the reactions were complete within 1 hour, it was critical that the diarylzinc reagents were free of residual Et2O to enable the transformation. Next, the scope of the N,O-acetals was probed (Fig. 3). The reaction was successfully carried out with a variety of substrates bearing different functional groups in the aromatic ring. Product yields and reaction times remained consistent in the case of both electron-donating and electron-withdrawing groups. Compounds 3b, 3c and 3e could be prepared in similar yields when compared to varying the ZnAr2 reagent, offering a second route to mixed-diarylmethanamine species. Additionally, products 3k and 3l, containing sensitive cyano and nitro-functional groups, could be obtained in good yields (81% and 58% respectively) with no significant signs of substrate decomposition or side-products. N,O-acetals prepared from amines other than piperidine also reacted smoothly with ZnPh2 under the optimised conditions. Compound 3m, derived from morpholine, was obtained in 86% yield, while compound 3n containing N-methylpiperazine, was isolated in 72% yield. Compound 3n is commonly known as Cyclizine, and is a widely employed anticholinergic drug, which highlights the potential of the reaction to access relevant bioactive molecules. The reaction could also be performed with the acyclic N,O-acetal, resulting in product 3o in 76% yield. Finally, the reaction was also possible with an N-substituted oxazolidine, which resulted in smooth ring opening to afford amino-alcohol 3p in a 74% yield.
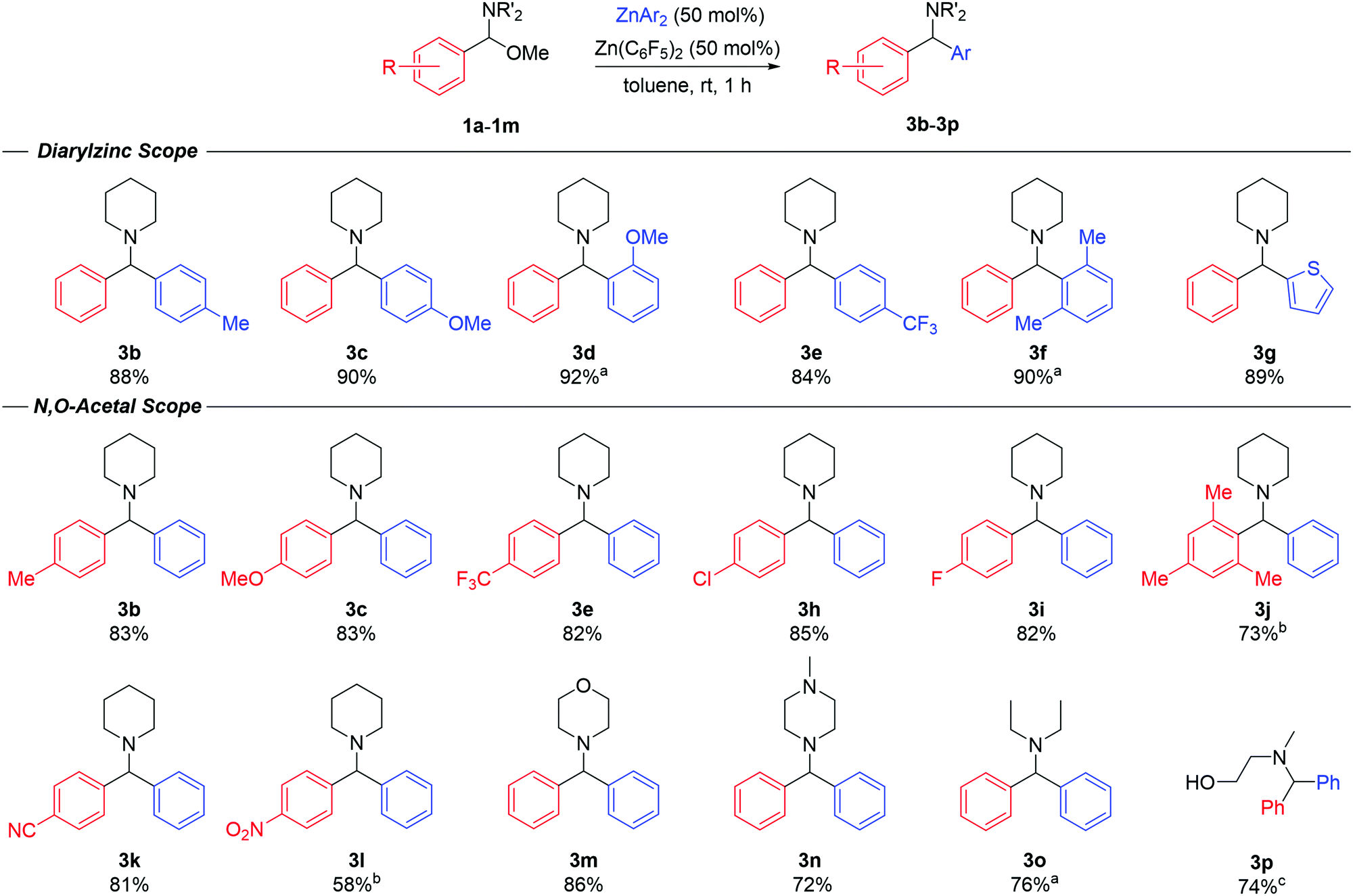 | ||
| Fig. 3 Substrate scope for the arylation of N,O-acetals with diarylzinc reagents. Yields refer to isolated compounds after column chromatography. aReaction performed at 90 °C. bThe N,O-acetal was employed without prior purification. cYield determined by 1H NMR spectroscopy using hexamethylcyclotrisiloxane as an internal standard. | ||
To conclude, we have demonstrated how Zn/Zn′ cooperativity through the use of nucleophilic diarylzinc reagents and Lewis acidic Zn(C6F5)2, enables the atom-efficient and transition-metal free arylation of N,O-acetals to afford a range of diarylmethanamines. Mechanistic studies show the double role of Zn(C6F5)2 which not only activates the substrate towards the arylation process but also effectively regenerates ZnAr2 from the inactive ArZnOMe by-product, allowing a limiting 50 mol% of ZnAr2 to be employed.
We thank the SNSF (188573) and the University of Bern for their generous sponsorship of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Rappoport and I. Marek, ed., The Chemistry of Organozinc Compounds, Wiley, 2006 Search PubMed.
- A. de Meijere, S. Bräse and M. Oestreich, ed., Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014 Search PubMed.
- H. Minami, X. Wang, C. Wang and M. Uchiyama, Eur. J. Org. Chem., 2013, 7891–7894 CrossRef CAS.
- J. J. Dunsford, E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 5688–5692 CrossRef CAS PubMed.
- B. Wei, Q. Ren, T. Bein and P. Knochel, Angew. Chem., Int. Ed., 2021, 60, 10409–10414 CrossRef CAS PubMed.
- A. Hernán-Gómez, S. A. Orr, M. Uzelac, A. R. Kennedy, S. Barroso, X. Jusseau, S. Lemaire, V. Farina and E. Hevia, Angew. Chem., Int. Ed., 2018, 57, 10630–10634 CrossRef.
- (a) D. Ameen and T. J. Snape, Med. Chem. Commun., 2013, 4, 893–907 RSC; (b) D. Roy and G. Panda, ACS Omega, 2020, 5, 19–30 CrossRef CAS PubMed.
- (a) E. Le Gall, M. Troupel and J. Y. Nédélec, Tetrahedron, 2006, 62, 9953–9965 CrossRef CAS; (b) S. Sengmany, E. Le Gall, C. Le Jean, M. Troupel and J. Y. Nédélec, Tetrahedron, 2007, 63, 3672–3681 CrossRef CAS; (c) E. Le Gall, C. Haurena, S. Sengmany, T. Martens and M. Troupel, J. Org. Chem., 2009, 74, 7971–7973 CrossRef.
- A. Charette, A. Beauchemin, S. Francoeur, F. Bélanger-Gariépy and G. D. Enright, Chem. Commun., 2002, 466–467 RSC.
- (a) N. Sakai, H. Hori, Y. Yoshida, T. Konakahara and Y. Ogiwara, Tetrahedron, 2015, 71, 4722–4729 CrossRef CAS; (b) H. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2012, 134, 4011–4014 CrossRef CAS PubMed.
- E. Le Gall, S. Sengmany, C. Hauréna, E. Leonel and T. Martens, J. Organomet. Chem., 2013, 736, 27–35 CrossRef CAS.
- M. Fontes, X. Verdaguer, L. Solà, M. A. Pericàs and A. Riera, J. Org. Chem., 2004, 69, 2532–2543 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2099646–2099649. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04137a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |