
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic, porous hydrogels templated by lyotropic chromonic liquid crystals†
Suitu
Wang
 ,
Daniel P.
Maruri
,
Jennifer M.
Boothby‡
,
Xili
Lu
,
Laura K.
Rivera-Tarazona
,
Victor D.
Varner
and
Taylor H.
Ware
*
,
Daniel P.
Maruri
,
Jennifer M.
Boothby‡
,
Xili
Lu
,
Laura K.
Rivera-Tarazona
,
Victor D.
Varner
and
Taylor H.
Ware
*
Department of Bioengineering, The University of Texas at Dallas, Richardson, Texas, USA. E-mail: taylor.ware@utdallas.edu
First published on 24th June 2020
Abstract
Approaches to control the microstructure of hydrogels enable the control of cell–material interactions and the design of stimuli-responsive materials. We report a versatile approach for the synthesis of anisotropic polyacrylamide hydrogels using lyotropic chromonic liquid crystal (LCLC) templating. The orientational order of LCLCs in a mold can be patterned by controlling surface anchoring conditions, which in turn patterns the polymer network. The resulting hydrogels have tunable pore size and mechanical anisotropy. For example, the elastic moduli measured parallel and perpendicular to the LCLC order are 124.9 ± 6.4 kPa and 17.4 ± 1.1 kPa for a single composition. The resulting anisotropic hydrogels also have 30% larger swelling normal to the LCLC orientation than along the LCLC orientation. By patterning the LCLC order, this anisotropic swelling can be used to create 3D hydrogel structures. These anisotropic gels can also be functionalized with extracellular matrix (ECM) proteins and used as compliant substrata for cell culture. As an illustrative example, we show that the patterned hydrogel microstructure can be used to direct the orientation of cultured human corneal fibroblasts. This strategy to make anisotropic hydrogels has potential for enabling patternable tissue scaffolds, soft robotics, or microfluidic devices.
1. Introduction
Devices with user-defined mechanical properties, stimulus response, and cell–material interactions can be fabricated by programming the microstructure of soft materials. Traditionally, soft materials with programmed microstructures are generated via top-down patterning technologies, such as 3D printing or photolithography.1–3 These programmed materials exhibit controllable structural anisotropy and stimulus response, enabling their potential applications as controllable actuators,4,5 patterned tissue scaffolds,6 and microfluidic devices.7 Control of the microstructure is typically constrained by the spatial resolution of the patterning technique, and the treatment conditions intrinsic to each technique can limit the types of materials that can be successfully patterned. By comparison, bottom-up approaches, where conditions during material synthesis are controlled to dictate the microstructure, can be harnessed to provide control at length scales and in materials that are difficult to pattern using top-down approaches.8 Bottom-up approaches, such as the directed self-assembly of either liquid crystals or nanoparticles, can result in anisotropic glassy polymer networks, elastomers, and hydrogels.9–14Anisotropic hydrogels with controlled microstructures are well-suited to interact with biological systems because they can simulate the anisotropic environment of living systems. Therefore, a number of strategies have been explored to synthesize these anisotropic hydrogels.15,16 For example, mechanically stretching a hydrogel network and fixing the oriented polymer chains with additional crosslinking can result in structural anisotropy.17,18 By orienting nanofillers in the monomer solution, anisotropic mechanical properties can be obtained after polymerization.19,20 Heterogeneous hydrogels with controlled microstructures and microscale mechanics can be fabricated via the liquid–liquid phase separation of polypeptide and poly(ethylene glycol) solutions.21,22 Anisotropic hydrogels with oriented voids can also be fabricated by polymerization after directional freezing or in the presence of lyotropic liquid crystals to serve as templates.14,23–26 However, most of these methods cannot produce user-defined, arbitrary orientation, limiting potential applications. A new and versatile approach to in plane and through thickness control the microstructure of hydrogels could enable the synthesis of new materials with high-resolution control of the microstructure.
Lyotropic chromonic liquid crystals (LCLCs) are potentially useful templates to generate anisotropic hydrogels.27 LCLCs, which are quite different from typical amphiphiles, usually have plank-like, aromatic cores with hydrophilic groups on the periphery.28 These molecules aggregate into stacks when dissolved in water, and these stacks form mesophases when in specific temperature and composition ranges. As the concentration is increased or temperature is lowered, isotropic (I) solutions can order into nematic (N), columnar (M), and rectangular (O) phases.29–34 A variety of strategies can be used to pattern the molecular orientation of LCLCs, such as surface anchoring controlled by grooved surfaces or photoaligned dyes,35–38 3D confinement,39,40 and magnetic fields.41 These methods have enabled spatial control over the alignment of LCLCs. The resulting anisotropic, aqueous solution can be used to provide structure to biological systems or for chemical reactions.42,43
Here, we report a versatile approach to synthesize anisotropic polyacrylamide hydrogels with tunable microstructures via polymerization in different LCLC phases. By only changing the temperature of polymerization, hydrogels with either a homogeneous, macroporous, fibrous, or cellular microstructure can be fabricated. Importantly, if the monomer solution is oriented, an aligned fibrous or cellular microstructure results. The resulting hydrogels have tunable and anisotropic pore size, swelling, and mechanical properties. The orientation of the microstructure can be controlled both within the plane and through the thickness of the sample. Spatial patterns can lead to programmable shape change, morphing a flat material into a 3D structure. Finally, we show that these anisotropic materials can be functionalized with extracellular matrix (ECM) proteins and used as substrata for cell culture. Human corneal fibroblasts cultured on patterned, anisotropic polyacrylamide gels adopt defined cellular patterns.
2. Experimental
2.1 Materials
Disodium cromoglycate (DSCG, 98% purity) was purchased from Alfa Aesar (Haverhill, USA). Acrylamide (AAm, 99% purity), N,N′-methylenebis(acrylamide) (MBAAm), N,N,N′,N′-tetramethylethylenediamine (TEMED), ammonium persulfate (APS), 3-(trimethoxysilyl) propyl methacrylate, Sulfo-SANPAH, DMEM, Triton X-100, and DAPI were purchased from Sigma-Aldrich (St. Louis, USA). Methacryloxyethyl thiocarbonyl rhodamine B (PolyFluor 570) was purchased from Polysciences. Alconox, dimethylformamide (DMF), acetone, isopropanol, and toluene were purchased from Fisher Chemicals. RM257 was purchased from Wilshire Technologies. Irgacure 651 was donated by Ciba. Type I Collagen was purchased from PureCol (Advanced Biomatrix, San Diego, USA). Fetal Bovine Serum was purchased from R&D Systems (Minneapolis, MN). Pen/Strep/Amp B was purchased from Lonza (Walkersville, MD). Paraformaldehyde was purchased from Electron Microscopy Sciences (Hatfield, PA). Goat Serum was purchased from Gibco (Gaithersburg, MD). Phalloidin was purchased from Invitrogen (Carlsbad, CA). Brilliant yellow (50% purity, Sigma) dissolved in DMF at a concentration of 10 wt% was filtered three times before use. All chemicals were used as received without further purification.2.2 Preparation
Rubbed glass molds. Clean glass slides were rubbed with P400 3M sandpaper. A mold was then assembled. The rubbed sides were then made to face inward and parallel to each other, and 30 μm thick paper spacers were used between the glass slides. A UV-curable thiol-acrylate glue was used to hold the molds together.
Photo-patterned glass molds. Clean glass slides were exposed to O2 plasma for 60 seconds. A solution of 10 wt% brilliant yellow dissolved in DMF was spincoated on one surface of each glass slide at 3000 rpm for 30 seconds. Brilliant yellow coated glass slides were photo-patterned using polarized light. Each orientation was exposed for 3 minutes with a previously described system.44 Brilliant yellow coated glass slides were then spincoated with a 10 wt% RM257 solution dissolved in toluene at 3000 rpm for 30 seconds. This coating was then dried at 50 °C for 5 minutes, and the coating was then exposed to 365 nm UV light for 10 minutes at an intensity of 4 mW cm−2 to polymerize RM257.
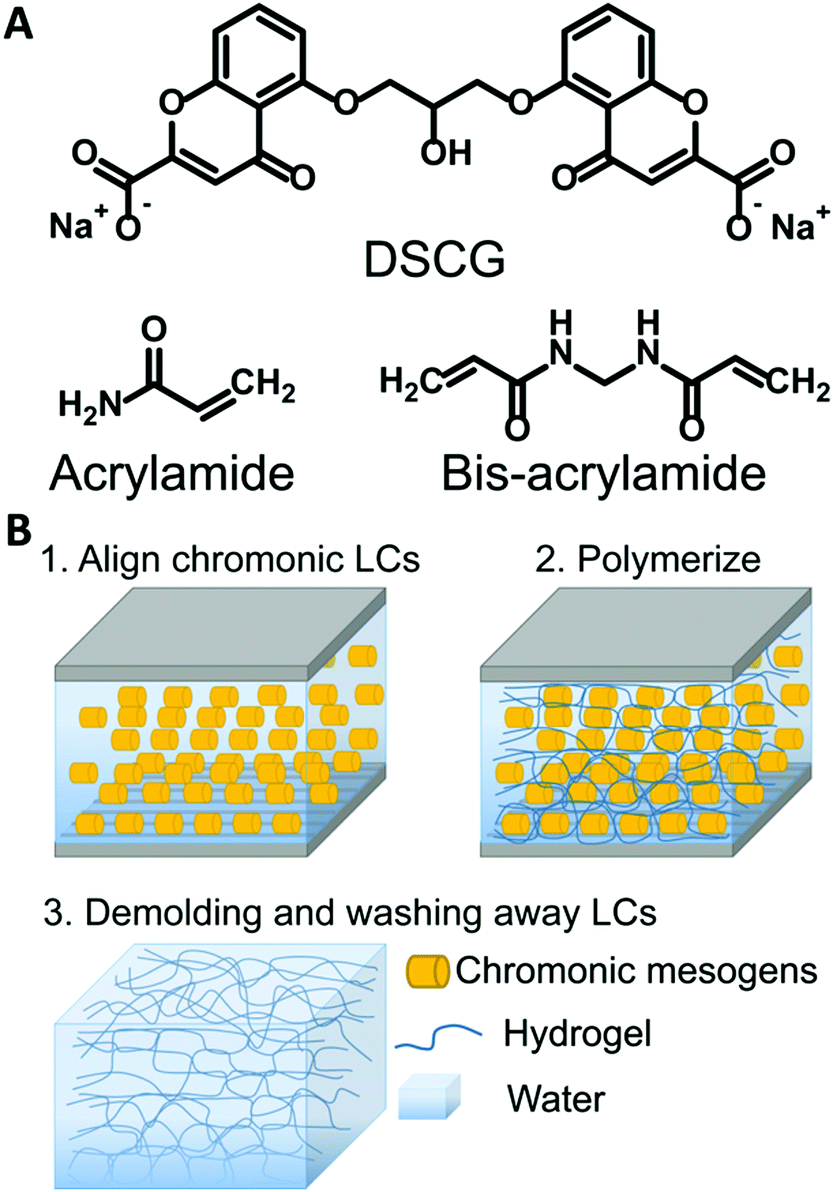 | ||
| Fig. 1 Schematic of the templating process. (A) Chemical structure of DSCG and monomers. (B) Illustration of the synthesis of anisotropic hydrogels templated by chromonic liquid crystals. | ||
2.3 Characterization
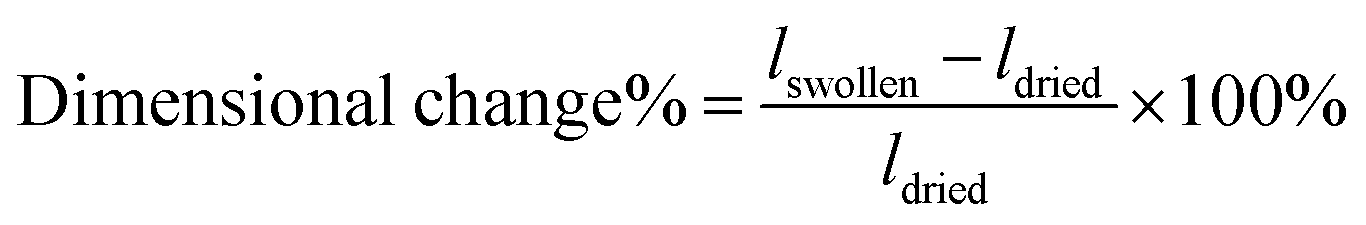
Glass slide functionalization. Rubbed glass slides were serially washed by sonication with 5% Alconox water solution, acetone, and isopropanol, each for 10 minutes. After cleaning, the glass slides were kept in deionized water overnight to keep the surface hydrated. Next, the clean glass slides were treated with a 5% (v/v) solution of 3-(trimethoxysilyl)propyl methacrylate (Sigma-Aldrich) in toluene for 30 minutes at 60 °C, washed with toluene, dried with nitrogen gas, and baked at 120 °C for 5 minutes.
Gel preparation. Uniaxially oriented hydrogels were synthesized with LCLCs in the O-phase in rubbed molds, as described above. Polydomain O-phase templated hydrogels were prepared in non-rubbed glass molds. In the absence of rubbing, the LCLCs adopt a polydomain LCLC texture. This polydomain texture is comprised of randomly oriented domains on the order of 100 μm in dimension. At the millimeter scale and larger, the samples are expected to be isotropic in nature (Fig. S1D, ESI†). Isotropic hydrogels were synthesized in the isotropic phase at 60 °C in rubbed cells. PAAm hydrogels were prepared at room temperature in rubbed cells, but in these monomer solutions, DSCG was replaced by additional water. Spatially patterned aligned hydrogels were prepared by first creating a mold with spatially patterned alignment. Clean glass slides were partially covered by masking tape. P400 3M sandpaper was used to rub the uncovered part along the long axis of the glass slide. This tape was then removed and used to cover the rubbed portion. P400 3M sandpaper was used to rub the remaining part along the short axis of the glass slide. In all molds used to synthesize hydrogels for cell culture, one side of the rubbed glass mold was methacrylate functionalized to facilitate hydrogel attachment while the other side facilitated release.
Gel functionalization. To enable cell attachment, the different polyacrylamide substrates were functionalized with unpolymerized type I collagen via the heterobifunctional crosslinker sulfosuccinimidyl 6-(4′-azido-2′-nitrophenylamino)hexanoate (sulfo-SANPAH; Sigma-Aldrich, St. Louis, MO). Briefly, the gels were rinsed with a 50 mM solution of HEPES, treated with a 1 mg ml−1 solution of sulfo-SANPAH in sterile water, and exposed to UV light for 10 min. This process was repeated, and the gels were rinsed with 50 mM HEPES to remove any excess sulfo-SANPAH. The gels were then incubated with a neutralized solution of 50 μg ml−1 unpolymerized type I collagen (PureCol; Advanced Biomatrix, San Diego, CA) at 37 °C for 30 minutes and rinsed with Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO), as described previously.45 Prior to plating cells, the gels were incubated with DMEM at 37 °C for an additional 30 minutes.
Cell culture. Cell culture studies were performed using the previously characterized human corneal fibroblast cell line, HTK.46,47 Prior to plating on the collagen-functionalized polyacrylamide substrates, HTK cells were cultured at 37 °C in tissue culture flasks containing DMEM supplemented with 10% fetal bovine serum (FBS; R&D Systems, Minneapolis, MN) and 1% penicillin/streptomycin/amphotericin B (Lonza, Walkersville, MD). The cells were then plated onto the polyacrylamide gels at a density of 30
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per ml and incubated at 37 °C for 48 hours.
000 cells per ml and incubated at 37 °C for 48 hours.
Fluorescence imaging. The samples were fixed in 3% paraformaldehyde in PBS for 20 minutes at room temperature, then washed three times with PBS and permeabilized in 0.5% Triton X-100 in PBS for 20 minutes. The cells were then blocked with 10% goat serum (Gibco, Gaithersburg, MD) in PBS for 1 hour at room temperature, incubated with Alexa Fluor 594 phalloidin (1
:200 dilution) (Invitrogen, Carlsbad, CA) for 60 minutes at 37 °C, washed three times with PBS, and incubated with DAPI (1:1000 dilution) (Invitrogen, Carlsbad, CA) for 20 minutes at room temperature. Confocal microscopy of fixed samples was performed with a Zeiss LSM 800 microscope using a computer-controlled, motorized stage and a 20×, NA 0.8, Plan-Apochromat air objective (Zeiss).
Cell nuclei angle measurements. Quantitative image analysis was performed in ImageJ. As a first approximation, we used the angle of the major axis of a best-fit ellipse of the cell nucleus to quantify the orientation of the cultured fibroblasts. This angle was measured with respect to the horizontal axis in each image, which corresponded to the direction of sandpaper rubbing along the glass slide. The number of cell nuclei was counted by ImageJ. Each substrate was imaged in 3 representative locations with each image representing an area of 0.4 mm2. We analyzed >500 nuclei for each treatment condition for n = 6 substrates from 3 different experimental replicates.
Cell nuclei orientation index. To quantify the degree of the cell alignment, an orientation index (OInuclei) was calculated using the following equation,
| OInuclei = {2〈cos2(θ)〉 − 1} × 100% |
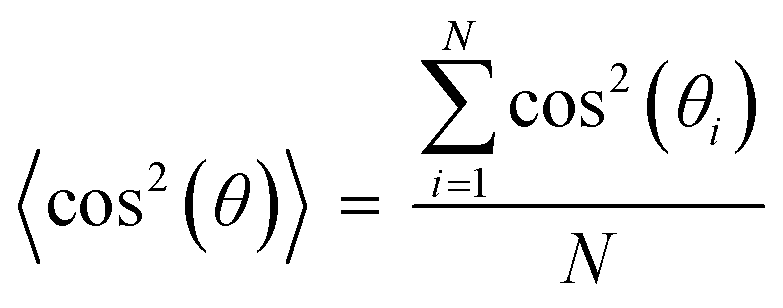
In this equation, θi represents the angle of the cell nucleus, and N is the total number of nuclei analyzed on a given substrate. An OI of 100% corresponds to perfect alignment to the horizontal axis, an OI of 0 corresponds to random alignment to the horizontal axis, and an OI of −100% corresponds to perpendicular alignment to the horizontal axis.
Statistical analysis. Analysis of the normality and variance showed non-normal data with equal variance. Statistical comparisons were made using the non-parametric Kruskal–Wallis test followed by Dunn's post-hoc test. For all tests, a significance level of 0.05 was used.
Cell aspect ratio and area. The cell morphology was measured by ImageJ. The diameters of the major and the minor axes of the cell were recorded. The cell aspect ratio (r) was defined by the ratio of the diameters of the major axis to the minor axis. The cell area was obtained by ImageJ directly. We analyzed 300 cells for each treatment condition for n = 6 substrates from 3 different experimental replicates.
Statistical analysis. Analysis of the normality and variance showed normal data with equal variance. Statistical comparisons were made using the 1-way ANOVA test followed by a post-hoc Tukey test. For all tests, a significance level of 0.05 was used.
3. Results and discussion
In this work, we describe the microstructure of hydrogels polymerized in different phases of an LCLC solution. The morphologies of the resulting anisotropic hydrogels are characterized. The effect of the morphology on the mechanical properties is described. Finally, aligned hydrogels are used to orient human corneal fibroblasts.3.1 Effect of the polymerization phase on the PAAm hydrogel structures
In order to template a hydrogel with an LCLC phase, the phase behavior of the monomer-LCLC solutions must be controlled. Typically, it is expected that by adding water-soluble compounds that do not form LCLC phases to LCLC solutions, the phase transition temperatures will be reduced.48 Solutions of 18 wt% DSCG in water serve as our LCLC, as this solution typically is nematic at room temperature.29 By adding 9 wt% AAm to this DSCG solution, the LCLC phases remain, but the phase transition temperatures decreased as compared to the neat DSCG solution, as determined by POM and DSC. On cooling from the isotropic phase, a broad exothermic event is observed from 44 °C to 8 °C, and a narrow exothermic event is observed from 1 °C to −6 °C (Fig. 2A). The latent heat of these transformations is 4.31 J g−1 and 4.77 J g−1, respectively. Based on prior work on DSCG solutions,32 we assign the higher temperature transition as isotropic to nematic + isotropic to nematic and the second transition as the nematic to O-phase. The same phase transitions were observed in the heating cycle. By further cooling to −25 °C, the aqueous solution froze (Fig. S2, ESI†).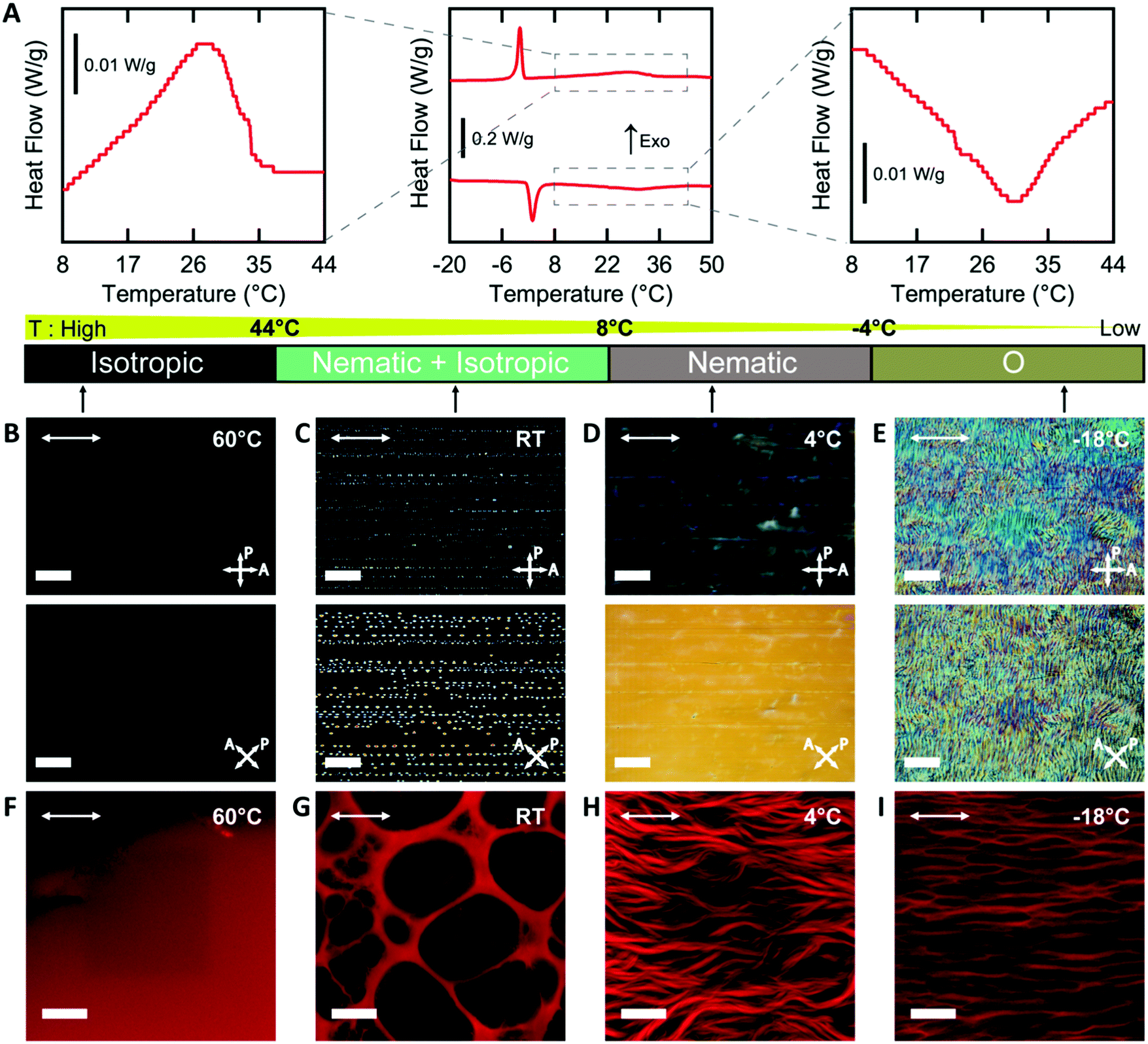 | ||
| Fig. 2 Tunable hydrogel morphology by polymerizing in different chromonic liquid crystal phases. (A) DSC thermogram of the DSCG/AAm solution with insets showing the transition peaks in detail on heating and cooling. (B–E) Polarized Optical Microscopy images of the DSCG/AAm solution in isotropic (60 °C), nematic + isotropic (RT), nematic (4 °C), and O- (−18 °C) phases (from left to right, scale bars 200 μm). (F–I) Confocal images of hydrogel microstructures after polymerization in the corresponding phase (hydrogels fluoresce red, scale bars 20 μm). Arrows in the top left corner of the images depict the direction of rubbing. | ||
Variable temperature POM of AAm-DSCG solutions in a rubbed cell at different temperatures supports the presence of these transitions. At 60 °C, the solution is dark between crossed polarizers, indicating that it is an isotropic liquid (Fig. 2B). At room temperature, birefringent nematic tactoids and isotropic regions are observed (Fig. 2C). At 4 °C, the entire sample is a nematic (Fig. 2D). Importantly, the nematic phase is oriented by the presence of scratches on the interior surface of the mold and shows a typical dark-bright pattern on rotation between crossed polarizers, as has been observed for neat DSCG solutions. At −18 °C, the homogeneous nematic texture is lost, and a texture associated with the O-phase is observed.30,31,34,49 This O-phase texture also exhibits changes in brightness when rotated between crossed polarizers, indicating potential alignment of this phase (Fig. 2E).
The temperature of polymerization can be used to change the phase of the monomer solution, which in turn controls the microstructure of the hydrogel. All hydrogels were observed after washing away the DSCG and allowing the gel to reach equilibrium swelling in water. When polymerized in the isotropic phase, a homogenous PAAm hydrogel is formed (Fig. 2F). In the biphasic nematic + isotropic phase region, the resulting hydrogels form a macroporous structure (Fig. 2G). As has been previously observed with other additives in LCLCs,48 the acrylamide monomers are expected to be preferentially excluded to the isotropic regions. In this system, this exclusion leads to gelation only in isotropic regions. These pores are neither uniformly spherical nor elliptical, and the pores have similar dimensions to the nematic tactoids in the monomer solution. In the nematic phase, fibrous structures of PAAm hydrogels form with the fibrils oriented along the nematic director (Fig. 2H). When polymerization occurs in the O-phase, anisotropic cellular structures are observed (Fig. 2I). These fibrous and cellular microstructures likely result from phase separation between LCLCs and polymer chains. Based on prior work on polymer-stabilized liquid crystals, similar fibrous and cellular structures are observed when polymerization occurs in the thermotropic nematic phase.9,50 However, the mechanisms of the formation of the distinct network morphologies in nematic and O-phase are still unclear. In the following sections, only the cellular structures resulting from polymerization in the O-phase are discussed.
3.2 Effect of crosslinker concentration on the pore morphology and mechanical and swelling properties of aligned cellular PAAm hydrogels
To further understand the aligned cellular PAAm hydrogels, the influence of crosslink density on the porous structure and corresponding mechanical and swelling properties was investigated. All samples were prepared in the O-phase, and the crosslinker concentration (0.1, 0.3, and 0.5 wt% MBAAm) was varied. Aligned cellular structures with different pore sizes can be observed by confocal microscopy for each composition (Fig. S3, ESI†). Pore size decreases with increasing crosslink density, both parallel and perpendicular to the alignment direction. The average pore sizes along the rubbing direction are 25.8 ± 7.8, 19.5 ± 9.2, and 7.2 ± 3.0 μm, while the average pore sizes perpendicular to the director are 6.5 ± 2.1, 4.6 ± 1.9, and 1.6 ± 0.6 μm with 0.1, 0.3 and 0.5 wt% crosslinker respectively (Fig. 3A). One reasonable explanation of the decreasing pore size with increased crosslink density is that higher content of MBAAm forms a crosslinked network at lower monomer conversion, which prevents further phase separation.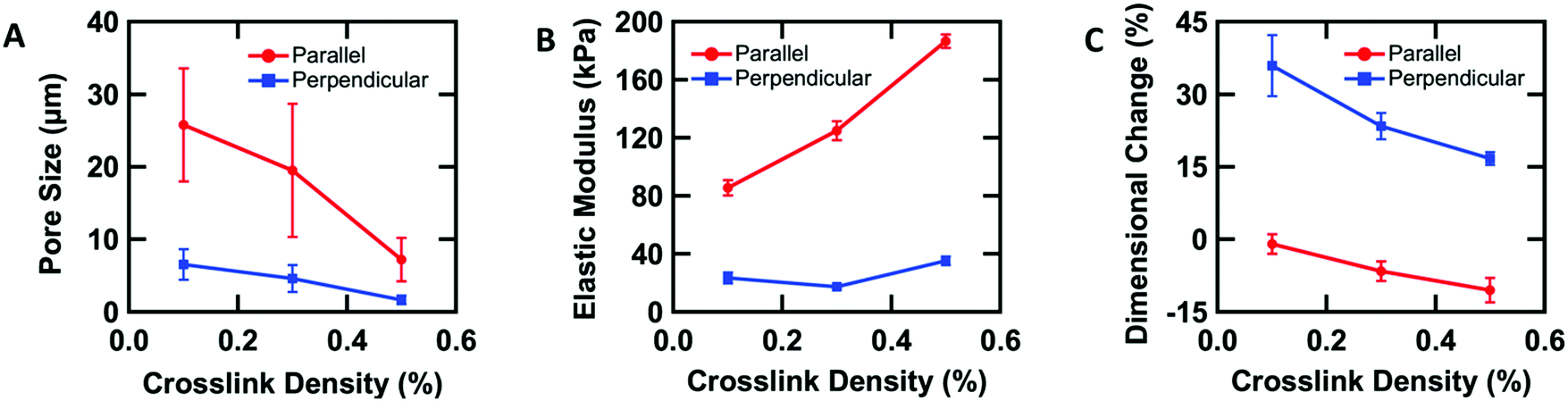 | ||
| Fig. 3 Mechanical anisotropy induced by the anisotropic structure. (A) Pore size along and perpendicular to the nematic director as a function of crosslink density. (B) Elastic modulus of the hydrogels along and perpendicular to the nematic director as a function of crosslink density. (C) Swelling along and perpendicular to the nematic director as a function of crosslink density. | ||
In addition to changes in morphology, the amount of crosslinker can be used to control the anisotropic mechanical properties of the aligned cellular hydrogels. Young's modulus was measured both parallel and perpendicular to the alignment by the micro-tensile test in water (Fig. S4, ESI†). Upon increasing the crosslink density from 0.1, 0.3, to 0.5 wt%, the elastic moduli increase (85.4 ± 5.4, 124.9 ± 6.4, and 186.4 ± 4.6 kPa respectively) along the rubbing direction (Fig. 3B). However, the elastic modulus perpendicular to the alignment shows little difference with increasing crosslink density. We note that for 0.3 wt% crosslinker, the elastic modulus parallel to the director is 6 times higher than the elastic modulus perpendicular to the alignment, which indicates that the aligned cellular structures induce sizeable mechanical anisotropy. This mechanical anisotropy is also confirmed by comparing with a 0.3 wt% isotropic PAAm hydrogel which has an isotropic elastic modulus of 40.4 ± 6.8 kPa. This anisotropy is substantially larger than the anisotropy observed in hydrogels with mechanically elongated polymer chains or oriented cylindrical clay nanofibers, which ranges from 2 to 3.16,51
The anisotropic porous structures generated by polymerization in the oriented O-phase swell anisotropically. To measure swelling anisotropy, the gel was allowed to dry within the glass mold, and the sample was then immersed in water to allow the DSCG to be removed from the gel and for the gel to reach its equilibrium shape. The hydrogels swell 30% more perpendicular to the alignment than along the alignment (Fig. 3C). By comparison, a 0.3 wt% isotropic PAAm hydrogel swells isotropically (Fig. S5A and B, ESI†), which also confirms that the swelling anisotropy of anisotropic hydrogels is induced by the anisotropic porous structure. As crosslink density increases, the swelling ratio decreases, as expected, but the anisotropy of the material remains apparent. The negative dimensional change of hydrogels at higher crosslink densities parallel to the alignment is affected by three factors (Fig. S5C and D, ESI†). The first factor is that the whole hydrogel will swell when absorbing water. The second factor is that the DSCG within the network will be removed. The third is that the porosity enables larger expansion in the perpendicular direction. These three competing factors cause the very small or negative dimensional change for the parallel direction.
3.3 Patterned alignment in PAAm hydrogels
By controlling the orientation of the LCLC monomer solution, hydrogels with through-thickness patterned microstructures can be synthesized. By building molds where the rubbing directions of the two sides of the glass mold are perpendicular to one another, the monomer solution can be induced to undergo a 90° twist in molecular orientation. At each surface, the LCLC aligns according to the rubbing direction. Confocal images (Fig. 4A) indicate the twisted pores of the resulting hydrogels through the thickness. As the orientation of the pores within the hydrogel varies through the thickness, the anisotropy of swelling should also vary through the thickness. When rectangular strips are cut from the twisted porous gel, the change in swelling anisotropy results in out of plane bending or twisting. By varying the cutting angle with respect to the rubbing direction of the top layer, the initially flat hydrogel films morph into 3D bent or helical structures (Fig. 4B). In the 0° and 90° samples, surface alignment is along the long and short axes of the gel strip. The resulting hydrogels bend either along the short axis or long axis of the strip; both shapes are observed with each alignment. The presence of metastable shapes in these hydrogels is reminiscent of the shape transformations of the tape spring.52 Samples with surface alignments between 0° and 90° form helical ribbons. By increasing the angle from 0° to 90°, the helical angle of hydrogels also increases accordingly while the helical pitch decreases (Fig. 4C). Similar behavior has been observed for twisted nematic liquid crystal elastomers.53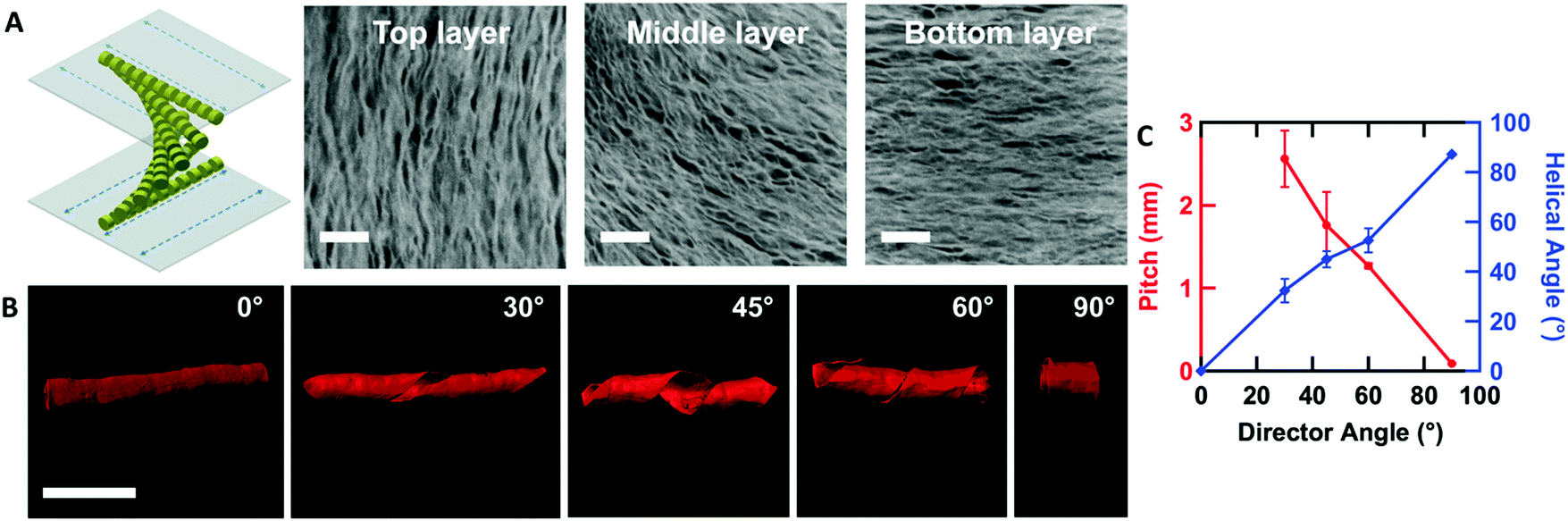 | ||
| Fig. 4 Twisted aligned PAAm hydrogels. (A) Twisted aligned hydrogels induced by twisted oriented LCLCs (confocal images indicate the Z-stacking structure of hydrogels from top layer to bottom layer, scale bar 10 μm). (B) Confocal images of 2D hydrogel films forming 3D shapes by varying the cutting angle from 0° to 90° (scale bars 2 mm). (C) Pitch and helical angle of the 3D shapes measured as a function of cutting angle (the pitch of the 0° sample is infinite). | ||
The orientation of the LCLC monomer solution can also be controlled spatially. We adopted a strategy for LCLC photo-patterning previously described by Peng et al.37 Briefly, a layer of photosensitive dye can be oriented with spatial control using linearly polarized light. An additional layer of a polymerized hydrophobic RM257 layer, a thermotropic reactive mesogen, is then coated on top of this dye. This crosslinked RM257 layer, in turn, orients the LCLC monomer solution and hydrogel microstructure. We first patterned a film with two domains oriented at 90° to one another. POM images with a waveplate indicate the LCLC pattern and the corresponding hydrogel alignment after polymerization (Fig. 5A). We note that the observed birefringence is a product of both the DSCG and the RM257 alignment layers. However, confocal images (Fig. 5B) taken from three spots of this sample after washing away the DSCG also show a clear microstructural change according to the photoalignment pattern. More complex patterns can also be designed, such as the letters “CLC” and the director field associated with an azimuthal +1 topological defect. To visualize these mm-scale patterns, the hydrogel with the DSCG still present is heated to isotropic and then cooled to the nematic–isotropic region while being viewed with POM. In the isotropic phase, the birefringence is only due to the alignment layers. During the cooling process, the hydrogel serves as a template to guide the orientation of the DSCG tactoids, which form elongated structures as dictated by the photoalignment process (Fig. 5C and D). Notably, due to the interaction between the RM257 layers and hydrogels during polymerization, RM257 layers attach to both sides of the resulting hydrogels, which limit their applications (Fig. S6, ESI†). Soaking in 1 M NaOH solution for 24 hours can be used to induce degradation of RM257 layers. However, this method results in the hydrogel also becoming partially degraded, which changes the mechanical properties of the original hydrogels. In order to use these hydrogels without a hydrophobic coating, we adopted spatially selective rubbing of the surfaces of the glass mold to control hydrogel microstructures.
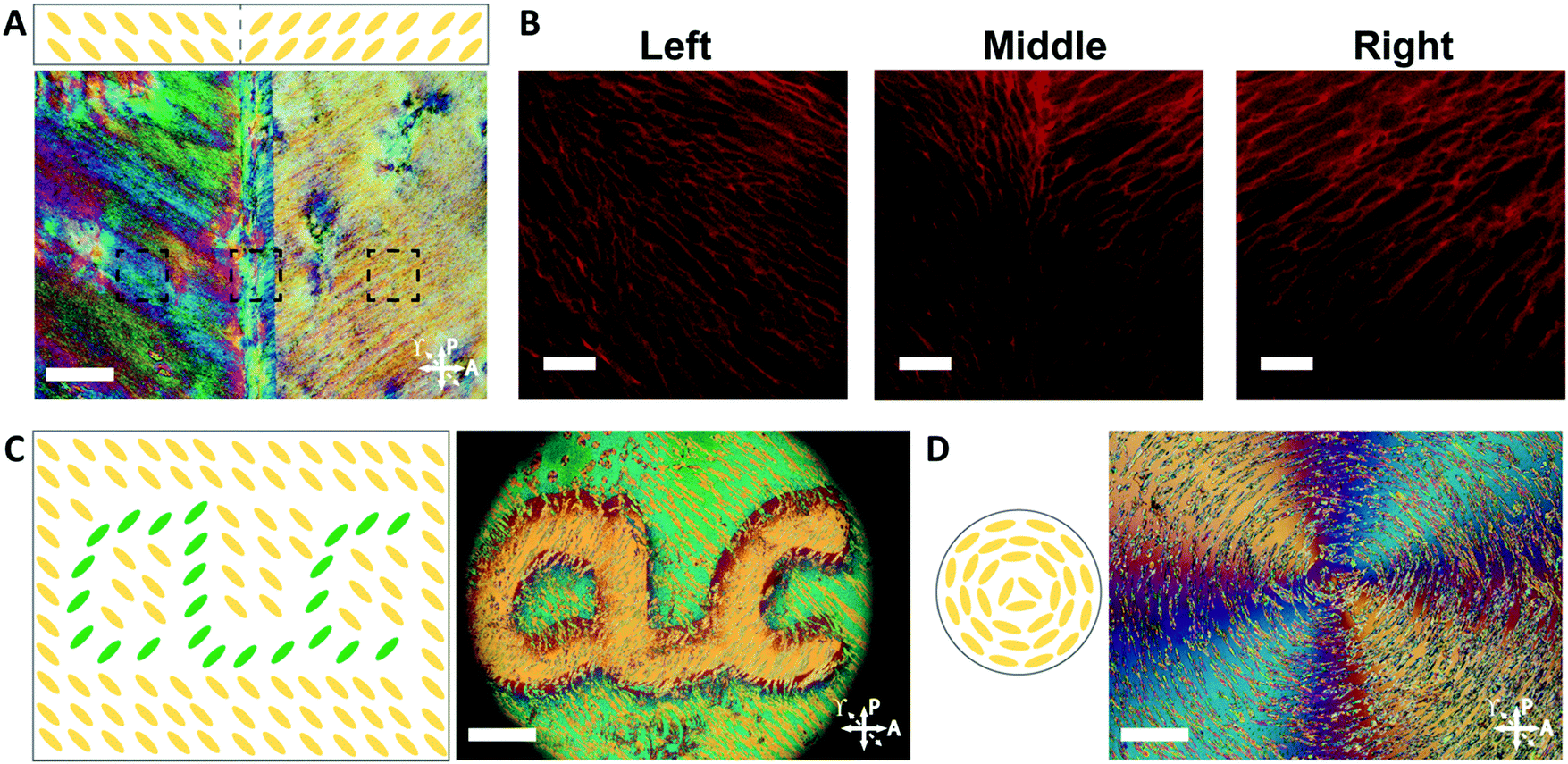 | ||
| Fig. 5 Spatially patterned PAAm hydrogels. (A) POM image at room temperature of the patterned hydrogel after polymerization and before washing the DSCG solution (scale bar 200 μm). (B) Confocal images indicating the patterned hydrogel microstructure after removal of the DSCG (scale bars 30 μm). (C and D) Schematic illustration of chromonic liquid crystal pattern and the corresponding POM image of the “CLC” and “+1 defect” patterned hydrogel/DSCG solution after polymerization when heated and cooled from the isotropic phase, the resulting hydrogel structures can be shown by the liquid crystal tactoids which follow the hydrogel network (scale bars 400 μm). Dotted arrows indicate the direction of an inserted quarter waveplate to distinguish perpendicular states. | ||
3.4 Anisotropic properties of LCLC-templated hydrogels used to create defined patterns of cultured corneal fibroblasts
The oriented porous structures of the LCLC-templated hydrogels are reminiscent of the aligned, fibrous networks of ECM proteins present within many biological tissues. For example, in the human cornea, which is situated at the anterior aspect of the eye, the stromal compartment contains lamellae of aligned type I collagen fibrils that endow the tissue with its transparent optical properties.54,55 A population of cells called corneal keratocytes maintain this highly organized ECM and mediate the wound healing response in the event of an injury.56 Previous experiments have suggested that the aligned ECM provides an important topographical cue that can modulate the differentiation and motility of these cells.57,58 To demonstrate the potential application of these anisotropic hydrogels for cell and tissue engineering applications, we cultured human corneal fibroblasts (HTKs) on polyacrylamide substrates functionalized with unpolymerized type I collagen and determined their effect on cell alignment.The microstructure of the anisotropic hydrogel, as dictated by the orientation of the LCLC monomer solution during gel polymerization, can be used to control the alignment of cultured HTKs. Cells were cultured on four different types of substrates: (1) hydrogels that are uniaxially oriented by polymerization in the aligned O-phase, (2) hydrogels polymerized in a polydomain O-phase, (3) hydrogels polymerized in the isotropic phase within rubbed molds, and (4) hydrogels polymerized in rubbed molds without DSCG. These gels were selected to elucidate the effects of orientation and porosity on cell attachment and alignment. On aligned polyacrylamide substrates, the HTKs strongly orient in the direction of the aligned porous microstructure after 48 hours of culture (Fig. 6A and B). By contrast, a much smaller orientation preference is exhibited on the other three substrates. HTK alignment is quantified by analyzing the orientation of cell nuclei with respect to the alignment of the glass rubbing direction (Fig. 6C). We computed a running average for each substrate to ensure that a sufficient number of nuclei were analyzed and obtained an orientation index for the cells on each substrate (Fig. S7, ESI†). For aligned porous gels, greater than 70% of nuclei are aligned within 20° of the glass rubbing direction. The orientation index is 79.8% ± 8.3% for cells cultured on these substrates (Fig. 6D). In contrast, cell nuclei are less aligned on the other three substrates. The orientation indices are 40.6% ± 10.1% for polydomain PAAm, 27.9% ± 14.1% for isotropic PAAm, and 22.8% ± 14.1% for pure PAAm. Statistical analysis reveals a significant difference between aligned PAAm vs. isotropic PAAm and aligned PAAm vs. pure PAAm (P < 0.05), suggesting that cell orientation is determined by the alignment of the porosity of the hydrogel and not by the submicrometer-scale grooves used to orient the liquid crystals. Although the difference of orientation index between aligned PAAm and polydomain PAAm is substantial, we do not observe a significant difference. The partial alignment of the HTKs on the polydomain samples may indicate that the polydomain sample has some residual alignment resulting from flow during synthesis.
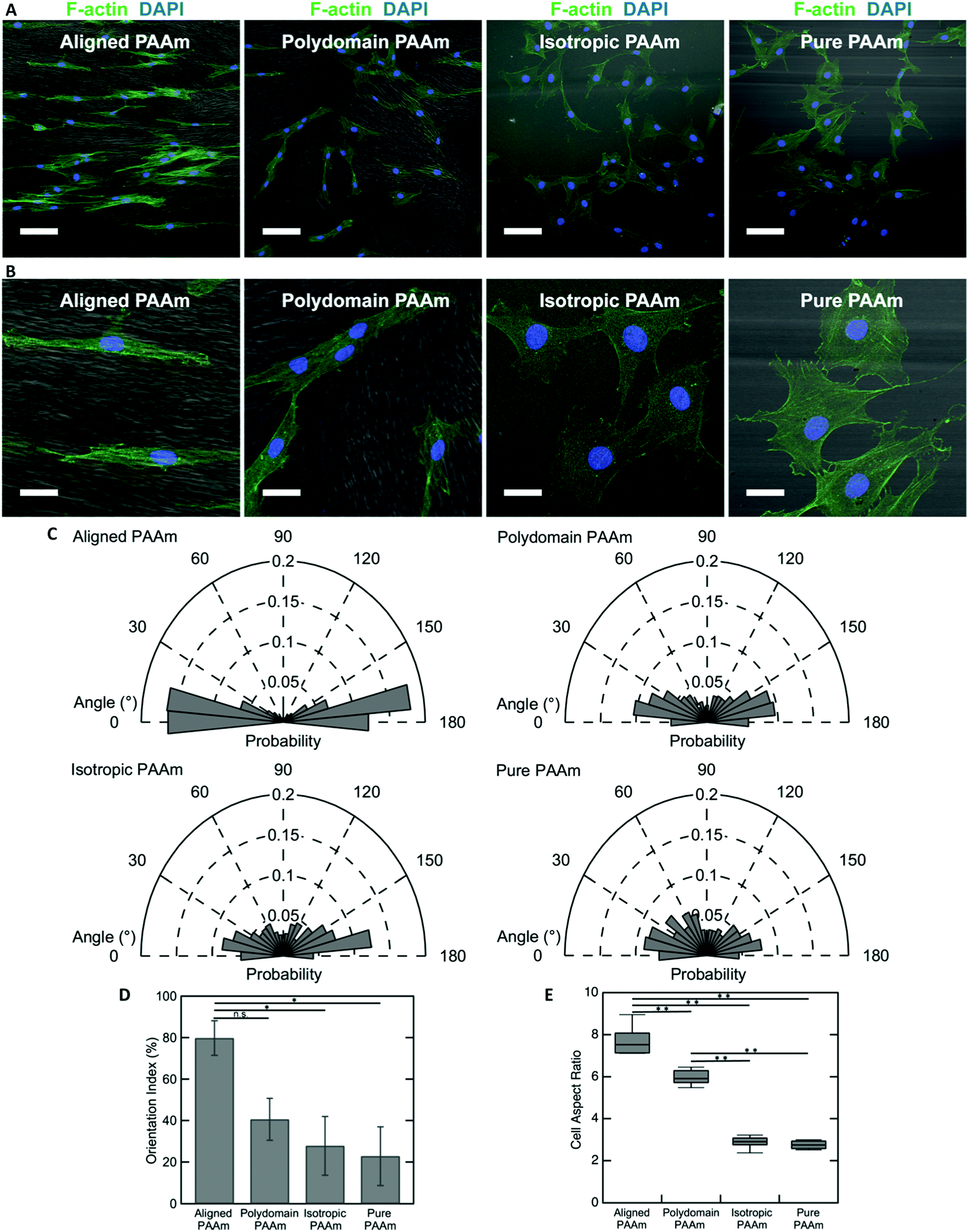 | ||
| Fig. 6 Human corneal fibroblast orientation induced by different PAAm substrates. Confocal images of human corneal fibroblast cell line (A) (scale bars 100 μm) and (B) (scale bars 30 μm) on different patterned PAAm substrates after incubating for 2 days. Cells were stained using phalloidin (green) and DAPI (blue) to visualize F-actin and nuclei, respectively. The gel microstructure (grayscale) was imaged using DIC. (C) Rose plot of cell nucleus orientation angle with respect to the rubbing direction (horizontal axis). (D) Cell orientation index on different PAAm substrates towards the horizontal axis (*, P < 0.05). (E) Cell aspect ratio cultured on different PAAm substrates (**, P < 0.001). | ||
The cell area and aspect ratio are also modulated by the microstructure of the substrate. HTKs cultured on either aligned or polydomain PAAm gels exhibit geometries that are elongated along the local pore wall direction. These cells have a significantly increased aspect ratio, with the HTKs on the aligned PAAm gels exhibiting the largest values (Fig. 6B and E) (P < 0.001), as compared to cells cultured on either isotropic or pure PAAm substrates, which have more homogeneous microstructures (P < 0.001). The projected cell area is also weakly modulated by changes in the gel microstructure. The area of cultured HTKs is somewhat lower on gels that have been polymerized with DSCG, as compared to pure PAAm substrates (Fig. S8A, ESI†), with cells on pure PAAm substrates being significantly larger in area as compared to cells on the aligned PAAm and isotropic PAAm (p < 0.05) substrates. It is unclear if the presence of DSCG alters the stiffness of the gel, which might regulate the observed differences in cell area. Still, despite these differences, the cells attach to all hydrogel substrates with a similar affinity, as the average number of cells per unit area is not affected by the gel microstructure (Fig. S8B, ESI†).
Different spatial patterns of cellular alignment can also be created on individual substrates, using a spatially patterned gel microstructure. An illustration of the alignment pattern generated using spatially patterned rubbing is shown in Fig. 7A. After 48 hours of culture, HTKs align with the local direction of the hydrogel microstructure, generating clear spatial patterns of cellular alignment (Fig. 7B). At the interface between the perpendicular regions of the gel, HTK orientation changes dramatically (Fig. 7C). Therefore, spatially patterned hydrogel microstructures templated by directed self-assembly of liquid crystals can in turn spatially control cell orientation. While surface topography is a well-known strategy to induce cell alignment,59 the formation of topography directed by self-assembly and not top-down methods such as lithography may enable new strategies to generate high fidelity patterns of cell alignment in non-planar scaffolds. We note that within chemically identical gels, the substratum microstructure can be widely varied between homogeneous, macroporous, fibrous, and cellular. We believe that this templating technique may be applicable to 3D scaffolds. However, two limiting factors are that the surface anchoring techniques shown here are only active ∼100 μm from an aligning surface and that the pore size is currently too small to enable cell penetration. In combination with well-known strategies to functionalize polyacrylamide hydrogels with extracellular proteins, the programmable microstructure of these templated hydrogels may enable new strategies to elucidate the effects of topography, stiffness, and other environmental cues on cellular behavior.
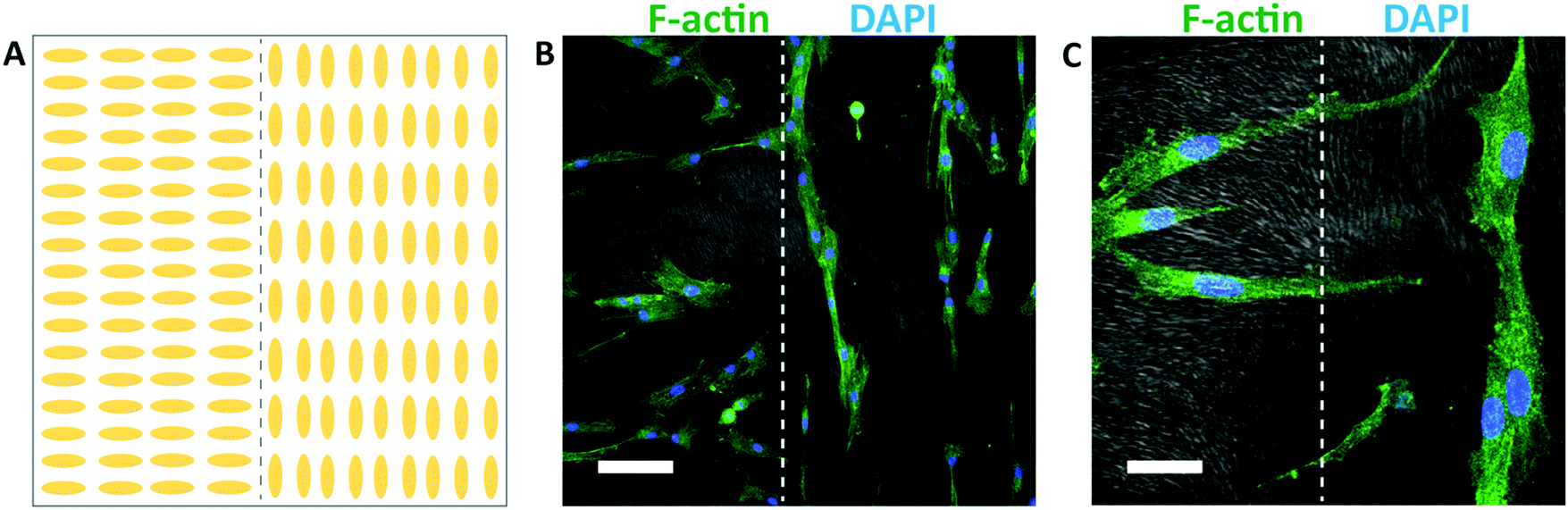 | ||
| Fig. 7 Patterned human corneal fibroblast orientation. (A) Illustration of the alignment of the substrate microstructure. (B) Composite confocal fluorescence image depicting fibroblast alignment on patterned hydrogel substrates (scale bar 100 μm). Cells were stained using phalloidin (green) and DAPI (blue) to visualize F-actin and nuclei, respectively. The gel microstructure (grayscale) was imaged using DIC. (C) A higher magnification confocal image showing the interface of the alignment domains (scale bar 30 μm). | ||
4. Conclusions
In summary, we put forward a new strategy to make anisotropic polyacrylamide hydrogels. By utilizing the nature of LCLC phases, substantially different hydrogel structures were formed by simply varying the polymerization temperature. Polymerization in the O-phase of the monomer mixture leads to a highly oriented cellular microstructure. This O-phase can be aligned uniaxially, with a through-thickness twist, or in a spatially varied pattern to produce porous polyacrylamide hydrogels templated by this structure. The anisotropic porosity induces mechanical anisotropy, which results in anisotropic elastic modulus and swelling ratio. Cultured corneal fibroblasts follow this orientation. It is anticipated that this new strategy to synthesize anisotropic hydrogels has potential applications in patterned tissue scaffolds and in the synthesis of programmable stimuli-responsive materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is in part based on work supported by the National Science Foundation under Grant No. 1663367. This work was supported in part by the NIH grant R01 EY030190.References
- A. Sydney Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef CAS PubMed.
- H. W. Kang, S. J. Lee, I. K. Ko, C. Kengla, J. J. Yoo and A. Atala, Nat. Biotechnol., 2016, 34, 312–319 CrossRef CAS PubMed.
- S. Ma, B. Yu, X. Pei and F. Zhou, Polymer, 2016, 98, 516–535 CrossRef CAS.
- M. Schaffner, J. A. Faber, L. Pianegonda, P. A. Rühs, F. Coulter and A. R. Studart, Nat. Commun., 2018, 9, 878 CrossRef PubMed.
- C. P. Ambulo, J. J. Burroughs, J. M. Boothby, H. Kim, M. R. Shankar and T. H. Ware, ACS Appl. Mater. Interfaces, 2017, 9, 37332–37339 CrossRef CAS PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- R. M. Erb, J. S. Sander, R. Grisch and A. R. Studart, Nat. Commun., 2013, 4, 1712 CrossRef PubMed.
- I. Dierking, Adv. Mater., 2000, 12, 167–181 CrossRef CAS.
- I. Dierking, L. L. Kosbar, A. Afzali-Ardakani, A. C. Lowe and G. A. Held, Appl. Phys. Lett., 1997, 71, 2454 CrossRef CAS.
- T. H. Ware, M. E. McConney, J. J. Wie, V. P. Tondiglia and T. J. White, Science, 2015, 347, 982–984 CrossRef CAS PubMed.
- R. S. Kularatne, H. Kim, M. Ammanamanchi, H. N. Hayenga and T. H. Ware, Chem. Mater., 2016, 28, 8489–8492 CrossRef CAS.
- J. M. Boothby, J. Samuel and T. H. Ware, Soft Matter, 2019, 15, 4508–4517 RSC.
- M. A. DePierro, K. G. Carpenter and C. A. Guymon, Chem. Mater., 2006, 18, 5609–5617 CrossRef CAS.
- K. Sano, Y. Ishida and T. Aida, Angew. Chem., Int. Ed., 2018, 57, 2532–2543 CrossRef CAS PubMed.
- L. E. Millon, H. Mohammadi and W. K. Wan, J. Biomed. Mater. Res., Part B, 2006, 79, 305–311 CrossRef CAS PubMed.
- S. H. Kim, S. K. Im, S. J. Oh, S. Jeong, E. S. Yoon, C. J. Lee, N. Choi and E. M. Hur, Nat. Commun., 2017, 8, 14346 CrossRef CAS PubMed.
- P. Lin, T. Zhang, X. Wang, B. Yu and F. Zhou, Small, 2016, 12, 4386–4392 CrossRef CAS PubMed.
- J. Ramón-Azcón, S. Ahadian, M. Estili, X. Liang, S. Ostrovidov, H. Kaji, H. Shiku, M. Ramalingam, K. Nakajima, Y. Sakka, A. Khademhosseini and T. Matsue, Adv. Mater., 2013, 25, 4028–4034 CrossRef PubMed.
- Q. Lu, S. Bai, Z. Ding, H. Guo, Z. Shao, H. Zhu and D. L. Kaplan, Adv. Mater. Interfaces, 2016, 3, 1500687 CrossRef.
- H. K. Lau, L. Li, A. K. Jurusik, C. R. Sabanayagam and K. L. Kiick, ACS Biomater. Sci. Eng., 2017, 3, 757–766 CrossRef CAS.
- H. K. Lau, A. Paul, I. Sidhu, L. Li, C. R. Sabanayagam, S. H. Parekh and K. L. Kiick, Adv. Sci., 2018, 5, 1701010 CrossRef PubMed.
- J. Wu, Q. Zhao, J. Sun and Q. Zhou, Soft Matter, 2012, 8, 3620–3626 RSC.
- M. Barrow and H. Zhang, Soft Matter, 2013, 9, 2723–2729 RSC.
- H. Bai, A. Polini, B. Delattre and A. P. Tomsia, Chem. Mater., 2013, 25, 4551–4556 CrossRef CAS PubMed.
- C. L. Lester, S. M. Smith, C. D. Colson and C. A. Guymon, Chem. Mater., 2003, 15, 3376–3384 CrossRef CAS.
- S. W. Tam-Chang and L. Huang, Chem. Commun., 2008, 1957–1967 RSC.
- P. J. Collings, J. N. Goldstein, E. J. Hamilton, B. R. Mercado, K. J. Nieser and M. H. Regan, Liq. Cryst. Rev., 2015, 3, 1–27 CrossRef CAS.
- N. Zimmermann, G. Jünnemann-Held, P. J. Collings and H. S. Kitzerow, Soft Matter, 2015, 11, 1547–1553 RSC.
- J. Lydon, J. Mater. Chem., 2010, 20, 10071–10099 RSC.
- D. Goldfarb, Z. Luz, N. Spielberg and H. Zimmermann, Mol. Cryst. Liq. Cryst., 1985, 126, 225–246 CrossRef CAS.
- H. Lee and M. M. Labes, Mol. Cryst. Liq. Cryst., 1983, 91, 53–58 CrossRef CAS.
- D. Goldfarb, M. M. Labes and R. Poupko, Mol. Cryst. Liq. Cryst., 1982, 87, 259–279 CrossRef CAS.
- D. Perahia, D. Goldfarb and Z. Luz, Mol. Cryst. Liq. Cryst., 1984, 108, 107–123 CrossRef CAS.
- P. van der Asdonk, P. J. Collings and P. H. J. Kouwer, Adv. Funct. Mater., 2017, 27, 1701209 CrossRef.
- Y. Guo, H. Shahsavan, Z. S. Davidson and M. Sitti, ACS Appl. Mater. Interfaces, 2019, 11, 36110–36117 CrossRef CAS PubMed.
- C. Peng, Y. Guo, T. Turiv, M. Jiang, Q. H. Wei and O. D. Lavrentovich, Adv. Mater., 2017, 29, 1606112 CrossRef PubMed.
- Y. Guo, M. Jiang, C. Peng, K. Sun, O. Yaroshchuk, O. D. Lavrentovich and Q. H. Wei, Crystals, 2017, 7, 8 CrossRef.
- J. Fu, K. Nayani, J. O. Park and M. Srinivasarao, NPG Asia Mater., 2017, 9, e393 CrossRef CAS.
- C. F. DIetrich, P. Rudquist, K. Lorenz and F. Giesselmann, Langmuir, 2017, 33, 5852–5862 CrossRef CAS PubMed.
- H. Lee and M. M. Labes, Mol. Cryst. Liq. Cryst., 1982, 84, 137–157 CrossRef CAS.
- C. Peng, T. Turiv, Y. Guo, Q. H. Wei and O. D. Lavrentovich, Science, 2016, 354, 882–885 CrossRef CAS PubMed.
- S. Zhou, A. Sokolov, O. D. Lavrentovich and I. S. Aranson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1265–1270 CrossRef CAS PubMed.
- J. M. Boothby and T. H. Ware, Soft Matter, 2017, 13, 4349–4356 RSC.
- M. Miron-Mendoza, E. Graham, P. Kivanany, J. Quiring and W. M. Petroll, Invest. Ophthalmol. Vis. Sci., 2015, 56, 2079–2090 CrossRef CAS PubMed.
- J. V. Jester, J. Huang, S. Fisher, J. Spiekerman, J. H. Chang, W. E. Wright and J. W. Shay, Investig. Ophthalmol. Vis. Sci., 2003, 44, 1850–1858 CrossRef PubMed.
- W. M. Petroll, L. Ma, A. Kim, L. Ly and M. Vishwanath, J. Cell. Physiol., 2008, 217, 162–171 CrossRef CAS PubMed.
- L. Tortora, H. S. Park, S. W. Kang, V. Savaryn, S. H. Hong, K. Kaznatcheev, D. Finotello, S. Sprunt, S. Kumar and O. D. Lavrentovich, Soft Matter, 2010, 6, 4157–4167 RSC.
- J. R. Magana, M. Homs, C. Solans, M. Obiols-Rabasa, L. M. Salonen and C. Rodríguez-Abreu, J. Phys. Chem. B, 2016, 120, 250–258 CrossRef CAS PubMed.
- G. A. Held, L. L. Kosbar, I. Dierking, A. C. Lowe, G. Grinstein, V. Lee and R. D. Miller, Phys. Rev. Lett., 1997, 79, 3443–3446 CrossRef CAS.
- K. Kaneda, K. Uematsu, H. Masunaga, Y. Tominaga, K. Shigehara and K. Shikinaka, J. Fiber Sci. Technol., 2014, 70, 137–144 Search PubMed.
- M. R. Schultz, M. J. Hulse, P. N. Keller and D. Turse, Composites, Part A, 2008, 39, 1012–1017 CrossRef.
- Y. Sawa, F. Ye, K. Urayama, T. Takigawa, V. Gimenez-Pinto, R. L. B. Selinger and J. V. Selinger, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6364–6368 CrossRef CAS PubMed.
- M. J. Hogan, J. A. Alvarado and J. E. Weddell, Histology of the human eye: an atlas and textbook, 1971 Search PubMed.
- K. M. Meek, Biophys. Rev., 2009, 1, 83–93 CrossRef CAS PubMed.
- M. E. Fini, Prog. Retin. Eye Res., 1999, 18, 529–551 CrossRef CAS.
- K. E. Myrna, R. Mendonsa, P. Russell, S. A. Pot, S. J. Liliensiek, J. V. Jester, P. F. Nealey, D. Brown and C. J. Murphy, Investig. Ophthalmol. Vis. Sci., 2012, 53, 811–816 CrossRef CAS PubMed.
- P. B. Kivanany, K. C. Grose, N. Yonet-Tanyeri, S. Manohar, Y. Sunkara, K. H. Lam, D. W. Schmidtke, V. D. Varner and W. M. Petroll, J. Funct. Biomater., 2018, 9, 54 CrossRef CAS PubMed.
- A. Curtis and C. Wilkinson, Biomaterials, 1997, 18, 1573–1583 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00904k |
| ‡ Current address: Johns Hopkins University Applied Physics Laboratory, Laurel, Maryland, USA. |
| This journal is © The Royal Society of Chemistry 2020 |