
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering of polyethylene-like polyphosphoesters: solid-state NMR characterization and surface chemistry of anisotropic polymer nanoplatelets†
Jens C.
Markwart
a,
Oksana
Suraeva
a,
Tobias
Haider
a,
Ingo
Lieberwirth
 a,
Robert
Graf
*a and
Frederik R.
Wurm
*ab
a,
Robert
Graf
*a and
Frederik R.
Wurm
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: graf@mpip-mainz.mpg.de
b“Sustainable Polymer Chemistry”, MESA+ Institute for Nanotechnology, Faculty of Science and Technology, Universiteit Twente, PO Box 217, 7500 AE Enschede, The Netherlands. E-mail: frederik.wurm@utwente.nl
First published on 29th October 2020
Abstract
Anisotropic materials with very high aspect ratios, such as nanoplatelets, are an exciting class of materials due to their unique properties based on their unilamellar geometry. Controlling their size and surface-functionality is challenging and has only be achieved in some cases for synthetic polymers. We present a general approach to prepare polymer-nanoplatelets with control over the lateral size and basal functionality, by simple polycondensation of precisely spaced and functional phosphate groups in polyethylene-like polymers. Because of the relatively large size of the phosphate groups, they are expelled from the bulk crystal to the basal surface. This allows to control the chain-folding during crystallization, which we analyzed via solid-state NMR and TEM. Furthermore, we present chemistry “on the surface” of the platelets: the pendant ester group at the phosphate offers the possibility to introduce functional groups accessible for further chemical modification on the crystal surface. This is shown by introducing a 2-acetylthioethyl ester group and subsequently cleaving this 2-acetylthioethyl ester group to the free P–OH. Together, these results render polyethylene-like polyphosphoesters a versatile platform for soft-matter nanoplatelets as functional colloids.
Introduction
Due to the pioneering work of Karl Ziegler and Giulio Natta, the synthesis of polyethylene and propylene at mild temperatures and pressure became possible, which has led to more than hundred million tons of polyethylene (PE) and polypropylene (PP) produced per year.1–3 The application of these polymers is determined by their bulk properties like melting or glass transition temperature. Today, PE and PP are known as commodity material with various applications.4–7In contrast to these bulk materials, planar materials with very high aspect ratios are an exciting material class due to their unique properties based on their unilamellar geometry, allowing anisotropic material properties. Examples for such materials are graphene or inorganic platelets, which are often used to reinforce bulk polymers.8 To date, only very few reports on chemically functional platelet-shaped nanomaterials based on commodity plastics like polyethylene were reported.9–11
We utilized phosphorus chemistry and simple polycondensation reactions to introduce precisely spaced phosphoester defects in a polymer chain to achieve polyethylene-like polyphosphoesters (PPEs).12 PPEs have been a focus of our research in recent years ranging from biomedical to material science applications.13,14 For the PE-like PPEs, the phosphates can introduce additional functionality but act as defects for the crystallization, as they are too large to be incorporated into the lamellar crystal. This led to a layered morphology of crystallites in the bulk.12,15 Besides, the phosphate units also confine the thickness of the polymer lamellae, which resulted in the formation of anisotropic polymer platelets, when crystallized from dilute solution15 or at the air–water interface.16 In addition, the pendant ester group at the phosphate gives the possibility for the introduction of functional groups which are accessible for further chemical modification on the crystal surface after crystallization from solution. Ramakrishnan's group followed a related concept for the preparation of liquid crystals from bulk by preparing polyethylene-like polyesters, carrying long-chain alkyl segments in the backbone and either poly(ethylene glycol) (PEG) or fluorinated pendant groups at precisely placed positions.17
The crystallization of PE from dilute solution was used already more than 50 years ago to prepare PE-single crystals.18,19 The conditions for crystallization from solution of polymers influence the crystal properties and can be controlled for example by the rate of cooling,20–22 concentration,23 or techniques like self-seeding.19,24 However, with all techniques it is challenging to adjust the lateral size and thickness of the materials at the same time. Precisely control over the chain folding by defect engineering allows eliminating the variable of varying thickness and focusing on lateral size control during crystallization from dilute solution.
Taken together, long-chain aliphatic polyphosphoesters are a versatile platform to prepare nanoplatelets with very high aspect ratio and distinct chemical functionality on their surface. The pendant ester group was used for the first time to tailor a multi-step reaction “on surface” of the nanoplatelets. The versatility of the chemistry in the pendant group renders the nanoplatelets as highly functional colloidal platform which can be utilized in various future applications.
Results and discussion
Acyclic diene metathesis polymerization (ADMET) allows the preparation of PE-like materials; in our group, it was extensively used for the preparation of phosphate-containing polymers.15,25–30 Here, we synthesized two different monomers equipped with two undecenyl chains for the olefin metathesis starting from POCl3. M1, a phosphodiester with an ethoxy side group were prepared according to literature.31,32 In addition, a new monomer, M2, was prepared by the reaction of 10 equivalent phosphorus oxychloride with 1 equivalent of 2-bromoethanol and in a second step with 2 equivalents of 10-undecen-1-ol, in order to install the alkyl-bromide functionality as a side chain. The excessive 10-undecen-1-ol was removed by flash chromatography over alumina. The resulting monomer is a liquid at room temperature and is soluble in aromatic and halogenated solvents (e.g. toluene, dichloromethane, chloroform) and insoluble in water. The alkyl-bromide in the pendant chain acts as versatile electrophilic functionality, as it can be functionalized by various nucleophilic substitutions after the synthesis or after polymerization (Scheme 1 shows all chemical structures and reactions).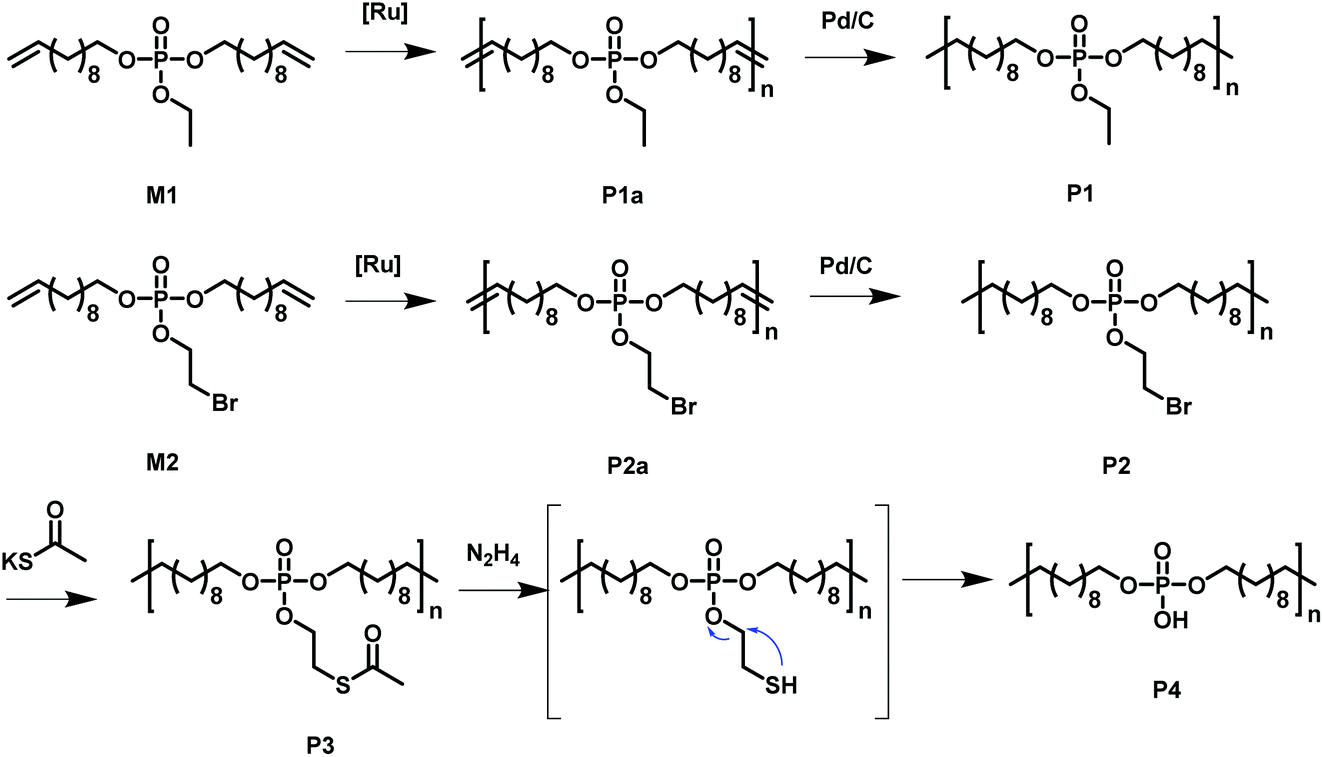 | ||
| Scheme 1 Overview of the monomers used to synthesize the functional PE-like polymers by ADMET polycondensation with subsequent hydrogenation and functionalization. | ||
The phosphate groups in the polymer chain are precisely spaced after the ADMET polymerization. The aliphatic methylene spacer between them was adjusted to 20 units (–(CH2)20–) to guarantee their ability to crystallize. If crystallized from dilute solution, this resulted in lamellar crystals with the phosphate groups on two opposing sides. With the possibility of installing chemical functionality in the phosphate unit, this strategy gives access to polymer platelets bearing functional groups on their surface which are accessible for further surface chemistry (Scheme 2).
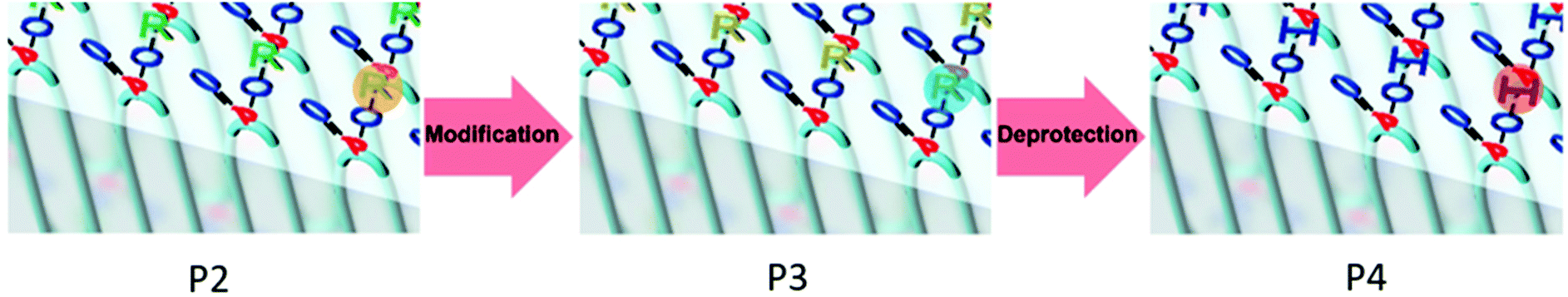 | ||
| Scheme 2 Schematic representation of the multi-step reaction, which was conducted “on surface” of the polymer platelets: 1. step: modification of the Br-group to a thioacetate-group. 2. step: cleavage of the 2-acetylthioethyl protective group. | ||
To prove that the phosphate groups are expelled from the crystal and arranged in a single plain, P1 was synthesized from M1 according to literature32 and analyzed by transmission electron microscopy (TEM). Previous studies on the crystallization of PPEs with varying lenghts of methylene pacers between the phosphate units revealed a transition from pseudo hexagonal an to a orthorhombic phase with increasing chain-length.33 From the electron diffraction pattern in Fig. 1a the only observed lattice distance dhkl amounts to 4.1 Å. When assuming an orthorhombic unit cell, as it is common for polyethylenes, the (110) and the (200) (110) ≈ (200). The lattice constants extracted from this assumption amount to a = 8.2 Å and b = 4.7 Å, where a becomes nearly equal to 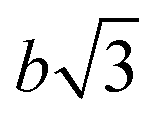 and hence the unit cell becomes pseudo-hexagonal.34 Using the unit cell parameters obtained by transmission electron microscopy studies, different models for the local planar arrangement of the 31P sites relative to the orthorhombic/pseudo-hexagonal lattice of the aliphatic chains can be derived (Fig. 2 top) and compared to structural information gathered by solid-state NMR.
and hence the unit cell becomes pseudo-hexagonal.34 Using the unit cell parameters obtained by transmission electron microscopy studies, different models for the local planar arrangement of the 31P sites relative to the orthorhombic/pseudo-hexagonal lattice of the aliphatic chains can be derived (Fig. 2 top) and compared to structural information gathered by solid-state NMR.
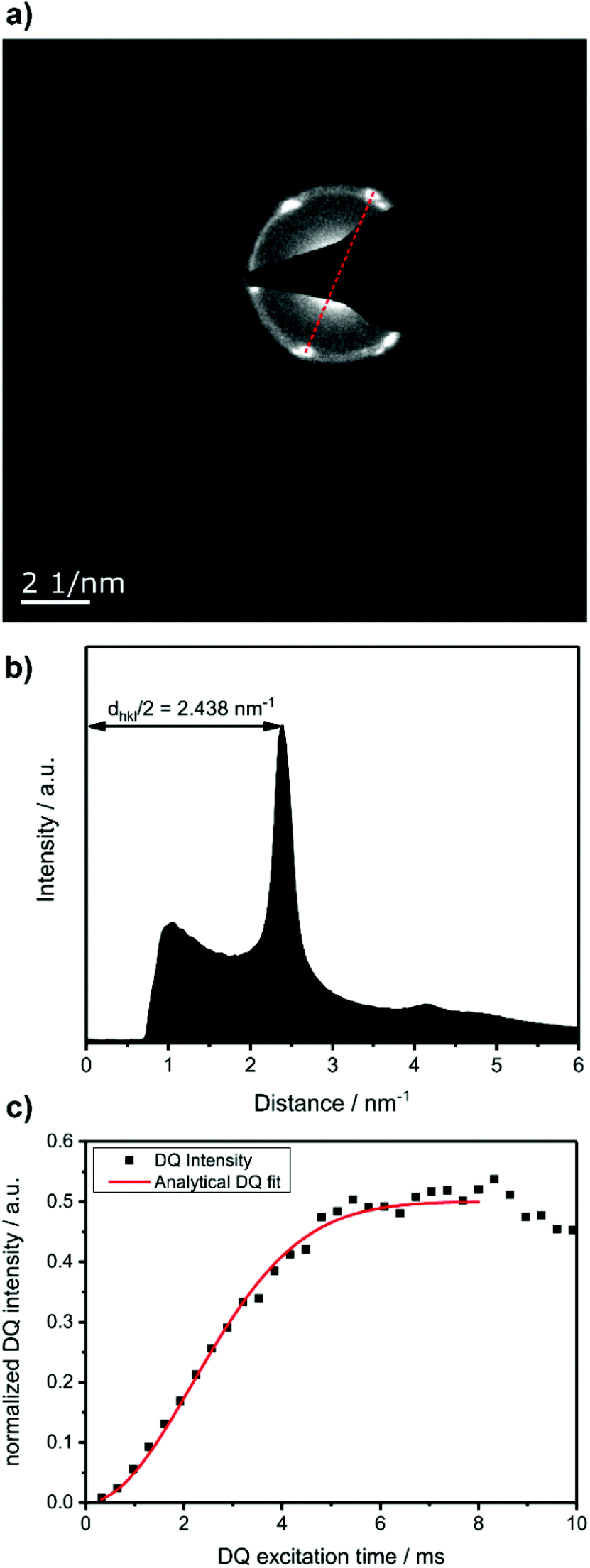 | ||
| Fig. 1 (a) Diffraction pattern of a single crystal P1 showing the (110) and (200) plane. (b) Radial intensity profile of the diffraction pattern. (c) Double quantum build-up curve of solution crystallized P1, recorded at 25 kHz MAS, T = 25 °C, and 202 MHz 31P Larmor frequency. | ||
 | ||
| Fig. 2 Arrangement of the P atoms on the crystal surface depending on the direction of the chain folding along the crystal lattice, indicated by the black arrow (a) polymer chain propagating along the (110) direction of the crystal lattice (Model 1). (b) Polymer chain propagating along the b axis of the crystal shifted by b/2 (Model 2). (c) Polymer chain propagating along the b axis of the crystal shifted by b/2 alternating (Model 3). (bottom) Plane on which M2 equals the experimentally obtained value of 0.197 kHz2. | ||
Commonly used solid-state NMR methods like 31P magic angle spinning (MAS) NMR or 31P{1H} CP-MAS NMR may probe the local chemical environment of the 31P sites in the solid-state, however, they will not provide any information on spatial proximities of the chemically isolated 31P sites in P1. Utilizing double-quantum (DQ) MAS-NMR methods, it is possible to probe molecular proximities in the range of 0.5 nm and thus gaining information regarding conformations and packing.35 In addition to 31P–31P double-quantum single-quantum (DQ-SQ) correlation experiments, probing directly spatial proximities between chemically distinced 31P sites, this method can probe as well 31P–31P DQ dipolar coupling between chemically equivalent 31P sites based on measurements of build-up curves. These build-up curves (Fig. 1c) are sensitive to P–P inter-nuclear distances, as the nuclear dipolar coupling constant 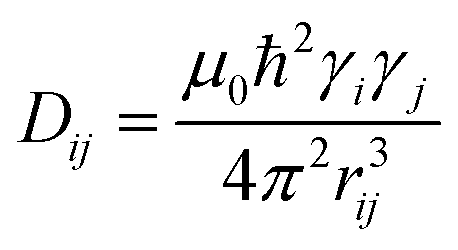 between two spins i and j is proportional to the inverse cube of the inter-nuclear distance rij, and the strongest 31P dipolar coupling, thus the closest spatial proximity, determines predominantly the initial t2-proportional rising of the double quantum build-up curve (with μ0 the vacuum permittivity; ħ the Planck constant; γi and γj are the nuclear gyro-magnetic ratios of the two spins involved in the dipolar coupling). Moreover, the long term behavior of DQ build up curves provides qualitative information on the distribution of the 31P dipolar couplings and spatial proximities. In crystalline arrangements with well-defined distinct distances between neighboring 31P sites, oscillatory behavior of the DQ build-up curves is observed (see Fig. 6b, in the publication by Saalwächter et al.36). For very long excitation times, these oscillations vanish in powder samples due to the orientation dependence of the DQ excitation efficiency. In less controlled assemblies with broad distributions of distances and thus dipolar couplings, the oscillatory behavior of the DQ build-up curve vanishes due to destructive interferences of the different oscillation frequencies of discrete dipolar coupling values completely. In these cases, a second moment description of the dipolar coupling assuming a dipolar interaction of a single site with continuously distributed surrounding 31P sites is more appropriate and provides a better description of the DQ build-up behavior. The smooth DQ build-up curve with its quadratic initial behavior obtained for the local packing of the 31P sites in crystallized P1 samples shown in Fig. 1 indicates that the second moment analysis of the local packing should be appropriate (i.e. indicating a crystalline, planar arrangement of P on the crystal surface). Please note, that the fluctuations occurring in the DQ build-up curve for longer DQ excitation times result from increasing relative experimental uncertainties with increasing DQ excitation times and thus do not indicate the presence of discrete dipolar coupling values resulting from a crystalline lattice (which should vanish for long DQ excitation times) like the oscillations mentioned above.
between two spins i and j is proportional to the inverse cube of the inter-nuclear distance rij, and the strongest 31P dipolar coupling, thus the closest spatial proximity, determines predominantly the initial t2-proportional rising of the double quantum build-up curve (with μ0 the vacuum permittivity; ħ the Planck constant; γi and γj are the nuclear gyro-magnetic ratios of the two spins involved in the dipolar coupling). Moreover, the long term behavior of DQ build up curves provides qualitative information on the distribution of the 31P dipolar couplings and spatial proximities. In crystalline arrangements with well-defined distinct distances between neighboring 31P sites, oscillatory behavior of the DQ build-up curves is observed (see Fig. 6b, in the publication by Saalwächter et al.36). For very long excitation times, these oscillations vanish in powder samples due to the orientation dependence of the DQ excitation efficiency. In less controlled assemblies with broad distributions of distances and thus dipolar couplings, the oscillatory behavior of the DQ build-up curve vanishes due to destructive interferences of the different oscillation frequencies of discrete dipolar coupling values completely. In these cases, a second moment description of the dipolar coupling assuming a dipolar interaction of a single site with continuously distributed surrounding 31P sites is more appropriate and provides a better description of the DQ build-up behavior. The smooth DQ build-up curve with its quadratic initial behavior obtained for the local packing of the 31P sites in crystallized P1 samples shown in Fig. 1 indicates that the second moment analysis of the local packing should be appropriate (i.e. indicating a crystalline, planar arrangement of P on the crystal surface). Please note, that the fluctuations occurring in the DQ build-up curve for longer DQ excitation times result from increasing relative experimental uncertainties with increasing DQ excitation times and thus do not indicate the presence of discrete dipolar coupling values resulting from a crystalline lattice (which should vanish for long DQ excitation times) like the oscillations mentioned above.
In Fig. 2, we have drawn three possibilities, how the polymer chains may propagate along with the unit cell of the crystallites, with the phosphate groups positioned between the lattice positions of the alkyl chain in an alternating pattern as illustrated in Fig. 2. For clarity, only the phosphorus atoms of the top surface are shown. In Model 1 (Fig. 2a) the polymer chain propagates diagonal to the unit cell along the (110) direction of the crystal. In Model 2 (Fig. 2b), the polymer chain propagates along the b axis of the unit cell, but the neighboring polymer chain is shifted by b/2. In Model 3 (Fig. 2c), the polymer chains propagate along the b axis, but the shift alternates between + and −b/2.
From the different models, the distance rij between two phosphate groups in close spatial proximity is known and thus can be used to calculate the 31P dipolar second moment (M2) 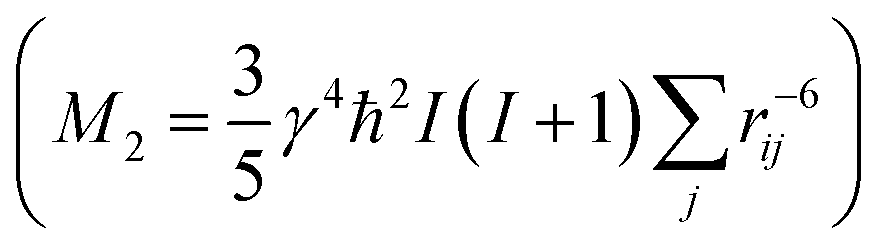 37 and compare it to the M2 obtained by the experimental DQ build-up curves of 0.197 kHz2 (for details on data acquisition, processing and analysis see Solid-state NMR Experimental section). For the calculation of M2, five unit cells in each direction were taken into account. Note that the integral contribution of more remote 31P sites to the value of M2 is less than 0.1%, as their contribution to the second moment decreases with the 6th power of the distance. For all 3 models, the calculation yields a M2 of ∼0.14 kHz2 with only minor variations, which is significantly lower than the M2 value obtained from the experimental DQ build-up curves. However, the crystals were measured in a dry state, so that the stacking of multiple crystalline layers is possible. Considering the adjacent-reentry model, the phosphate groups are located on the crystal surface and the interactions of phosphate groups in the c direction with neighboring crystalline layers have also to be taken into account. It should be noted that the interaction between the two phosphate planes within a single platelet can be neglected because the distance between them is by far too large. Shifting the parallel surfaces of the two single crystals in x, y, and z direction relative to each other the spatial distribution of the second moment M2 is computed for the different models (for illustration see Fig. S16 in the ESI†). From the computed three dimensional arrays of M2 values model dependent bended iso-surface on which M2 equals the experimentally obtained value 0.197 kHz2 (Fig. 2 bottom). These iso-surfaces predict distances of 4.0–5.2 Å between the phosphorus atoms in the z-direction, which is in a reasonable range.
37 and compare it to the M2 obtained by the experimental DQ build-up curves of 0.197 kHz2 (for details on data acquisition, processing and analysis see Solid-state NMR Experimental section). For the calculation of M2, five unit cells in each direction were taken into account. Note that the integral contribution of more remote 31P sites to the value of M2 is less than 0.1%, as their contribution to the second moment decreases with the 6th power of the distance. For all 3 models, the calculation yields a M2 of ∼0.14 kHz2 with only minor variations, which is significantly lower than the M2 value obtained from the experimental DQ build-up curves. However, the crystals were measured in a dry state, so that the stacking of multiple crystalline layers is possible. Considering the adjacent-reentry model, the phosphate groups are located on the crystal surface and the interactions of phosphate groups in the c direction with neighboring crystalline layers have also to be taken into account. It should be noted that the interaction between the two phosphate planes within a single platelet can be neglected because the distance between them is by far too large. Shifting the parallel surfaces of the two single crystals in x, y, and z direction relative to each other the spatial distribution of the second moment M2 is computed for the different models (for illustration see Fig. S16 in the ESI†). From the computed three dimensional arrays of M2 values model dependent bended iso-surface on which M2 equals the experimentally obtained value 0.197 kHz2 (Fig. 2 bottom). These iso-surfaces predict distances of 4.0–5.2 Å between the phosphorus atoms in the z-direction, which is in a reasonable range.
For these predicted distances, the phosphorous attached ethoxy chains between the crystal surfaces have a density between 1012 and 1320 kg m−3, which compares very well to the density of PEG (∼1128 kg m−3). It should be pointed out, that the lowest density value of 1012 kg m−3 results from an unfavorable geometry, where the 31P atoms of neighboring layers would sit directly on top of each other causing maximal steric interaction between the attached ethoxy groups.
In contrast to pure polyethylene, the situation of local densities in the crystalline and the non-crystalline part of the material in the precise phosphorylated PE chains is significantly different. The phosphate groups, expelled from the solution crystallized PE, is difficult to compare with a non-crystalline PE chain. The local structure is not necessarily non-crystalline with a lower density as the phosphate group has completely different possibilities to organize locally. An overall tilting of the chains would lead to a slightly increased in plane distance of the 31P sites. The resulting lower M2 value for a single layer would still require the presence of the double layer in order to explain the results, however, with a slightly reduced inter layer distance. Keeping the density of the attached ethoxy groups in mind, the distance between two adjacent “31P layers” do not vary significantly.
Surface modification of polymer platelets
The chemical accessibility of the pendant chains in the phosphates at the surface of the polymer platelets was studied using a multi-step reaction, which was conducted “on surface” (Scheme 2). The polymer platelets were prepared by solution crystallization of P2, which was prepared as followed: M2 was polymerized via ADMET (P2a, Mn = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1, Mw/Mn = 2.07), the purified polymer was then hydrogenated using a Pd/C catalyst to give P2 (Mn = 15900, Mw/Mn = 1.67). To prepare the dispersion of the polymer platelets of P2, the polymer was dissolved in ethyl acetate at 60 °C at a concentration of 1 mg mL−1. Then the solution was cooled in a temperature bath, which was set to 20 °C. A part of the cooled solution was transferred to a separate vial as a reference for later TEM images. The first reaction step “on surface” was the nucleophilic substitution of the bromides in the polymer platelets of P2 by potassium thioacetate to P3. Afterwards, a small amount of sample was taken and the solvent was removed at reduced pressure. The dried polymer platelets were then dissolved in deuterated chloroform and 1H NMR spectroscopy proved the characteristic resonances at 3.17 ppm (methylene group next to the thioacetate group) and 2.35 ppm (methyl group of the thioacetate group) for P3 (Fig. S11†) for a successful modification. When the same reaction was conducted in solution with P2, the NMR spectra of the product proved the same resonances for P3 (Fig. S9†). The 31P NMR spectrum shows a single signal at −1.13 ppm (Fig. S10†) confirming only one phosphorus species present. The TEM images in Fig. S12 and S13† prove the unchanged lateral dimensions of the polymer platelets of P2 and P3 before and after the “on surface” reaction. In DSC measurements, P3 shows a Tg of −49 °C and a Tm of 58 °C (Fig. 3). The second reaction step was the hydrolysis of the 2-acetylthioethyl protective group, which was conducted “on surface” as well. The 2-acetylthioethyl ester group is a protective group for phosphoric acid (P–OH-groups) and can be cleaved under acidic or basic conditions to release P–OH functionality, which should alter the surface properties of the polymer platelets.38
500 g mol−1, Mw/Mn = 2.07), the purified polymer was then hydrogenated using a Pd/C catalyst to give P2 (Mn = 15900, Mw/Mn = 1.67). To prepare the dispersion of the polymer platelets of P2, the polymer was dissolved in ethyl acetate at 60 °C at a concentration of 1 mg mL−1. Then the solution was cooled in a temperature bath, which was set to 20 °C. A part of the cooled solution was transferred to a separate vial as a reference for later TEM images. The first reaction step “on surface” was the nucleophilic substitution of the bromides in the polymer platelets of P2 by potassium thioacetate to P3. Afterwards, a small amount of sample was taken and the solvent was removed at reduced pressure. The dried polymer platelets were then dissolved in deuterated chloroform and 1H NMR spectroscopy proved the characteristic resonances at 3.17 ppm (methylene group next to the thioacetate group) and 2.35 ppm (methyl group of the thioacetate group) for P3 (Fig. S11†) for a successful modification. When the same reaction was conducted in solution with P2, the NMR spectra of the product proved the same resonances for P3 (Fig. S9†). The 31P NMR spectrum shows a single signal at −1.13 ppm (Fig. S10†) confirming only one phosphorus species present. The TEM images in Fig. S12 and S13† prove the unchanged lateral dimensions of the polymer platelets of P2 and P3 before and after the “on surface” reaction. In DSC measurements, P3 shows a Tg of −49 °C and a Tm of 58 °C (Fig. 3). The second reaction step was the hydrolysis of the 2-acetylthioethyl protective group, which was conducted “on surface” as well. The 2-acetylthioethyl ester group is a protective group for phosphoric acid (P–OH-groups) and can be cleaved under acidic or basic conditions to release P–OH functionality, which should alter the surface properties of the polymer platelets.38
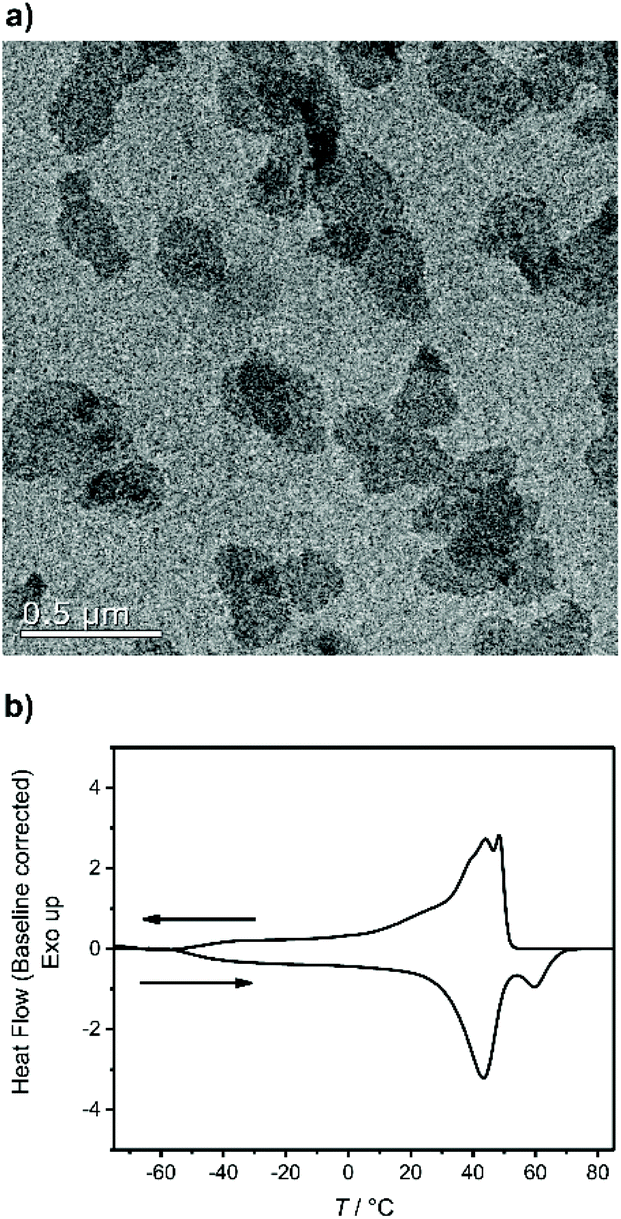 | ||
| Fig. 3 (a) Transmission electron microscope micrograph of the single P3 crystals (0.1 mg mL−1). (b) Differential scanning calorimetry (baseline corrected) of P3 showing the second heating and cooling curve with a Tg at −49 °C and Tm at 58 °C determined in the second heating curve. | ||
The dispersion was cooled to 0 °C for 15 min before starting stirring and adding the hydrazine in ethanol. After ca. 45 min, aggregation of the crystals was visible. The solvent was allowed to evaporate while keeping the temperature at 0 °C. The product (P4) was not soluble in any solvent, as expected for a polyphosphodiester.31
Fig. 4 shows the TEM images of the crystal dispersion before (P3) (Fig. 4a and b) and after (poly6) (Fig. 4c and d) the addition of hydrazine. Before the treatment, the crystals exhibited a size of around 128 ± 40 nm and are crystalline as it is apparent from the diffraction pattern. After the addition of the hydrazine, the crystal size remained at around 132 ± 46 nm and the diffraction pattern still proves their crystallinity. The P–OH groups on the crystal surface induced hydrogen bonds, which lead to the stacking of the crystals, with P–OH as an H-bond donor and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as an H-bond acceptor.31Fig. 4c shows a TEM image of the crystals stacking on top of each other.
O as an H-bond acceptor.31Fig. 4c shows a TEM image of the crystals stacking on top of each other.
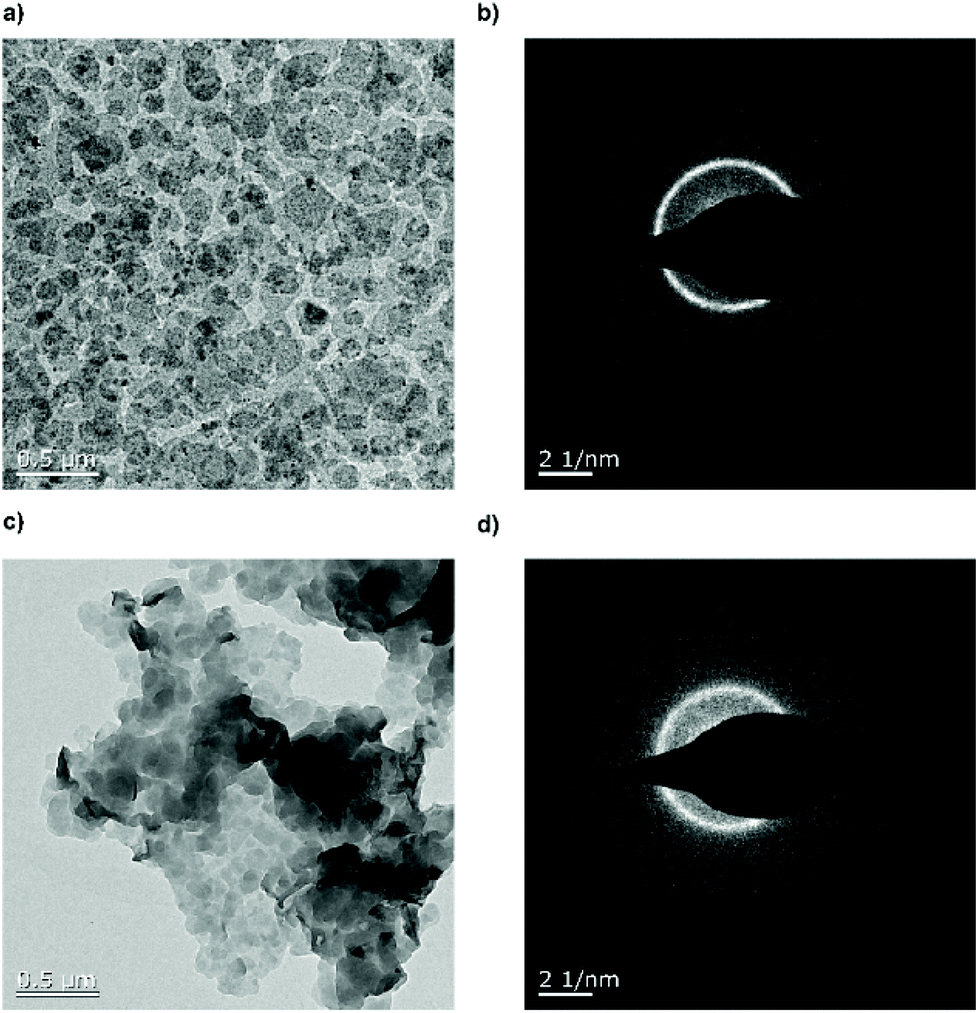 | ||
| Fig. 4 (a) EM micrograph of 1 mg mL−1P3. (b) EM diffraction pattern of 1 mg mL−1P3. (c) Transmission electron microscopy image of 1 mg mL−1P3. (b) Transmission electron microscopy diffraction pattern of 1 mg mL−1P4. | ||
IR spectroscopy proved the successful cleavage of the thioacetyl-protective group and the formation of the P–OH groups as shown in Fig. 5. The CO stretching frequency at 1694 cm−1 and the C(O)–S stretching frequency at 625 cm−1, which are characteristic for thioacetates,39 vanished and a new resonance at 1200 cm−1 appeared, which is characteristic for PO stretching in acidic phosphates.40
 | ||
| Fig. 5 FTIR spectrum of P3 and P4 (after two step reaction) highlighting the important frequencies. | ||
Experimental section
Materials and methods
All chemicals were purchased from commercial suppliers as reagent grade and used without further purification.The crystallization behavior of these polymers was studied by drop-cast TEM measurement. A TEM specimen of solution-grown polymer crystals was prepared from a 1 mg mL−1 or 0.1 mg mL−1 dispersion in ethyl acetate. The solution was heated and cooled according to the different temperature profiles. One drop of each dispersion was applied to a carbon-coated TEM grid, excess liquid was blotted off with a filter paper and the specimen was allowed to dry under ambient conditions.
for the analytical description of the 31P DQ build-up, where τDQ is the experimental DQ excitation time and the dipolar 31P second Moment M2 the only free parameter.36
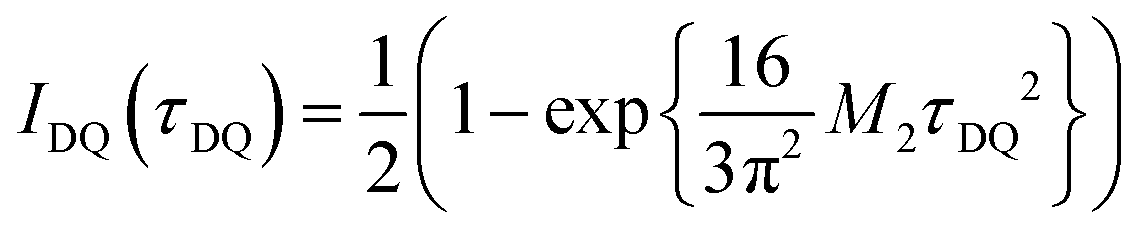
Syntheses
1H NMR (300 MHz, Chloroform-d, δ) 5.82 (ddt, J = 17.0, 10.3, 6.7 Hz, 2H), 4.97 (dd, J = 18.3, 13.6 Hz, 4H), 4.31 (q, J = 6.8 Hz, 2H), 4.06 (p, J = 6.4, 6.0 Hz, 4H), 3.55 (t, J = 6.3 Hz, 2H), 2.05 (q, J = 7.0 Hz, 4H), 1.70 (p, J = 6.7 Hz, 4H), 1.30 (s, 24H). 31P {H} NMR (121 MHz, Chloroform-d, δ) −1.25 (s, 1P).
P2a: M2 (500 mg, 0.98 mmol) was placed in a 25 mL Schlenk tube equipped with a stir bar and the system was degassed by three consecutive Argon/vacuum cycles. 8 mg of Grubbs catalyst 1st generation (0.1 mol%) were added and polymerization was carried out at 40 °C at reduced pressure (0.1 mbar). Increasing viscosity of the solution indicated ongoing polymerization. After 18 h, the second portion of Grubbs catalyst 1st generation was added and the polymerization was continued for an additional 24 h. The solution was allowed to cool down to room temperature, then 2 mL CH2Cl2 were added to dissolve the polymer and 200 μL ethyl vinyl ether were added to quench the catalyst. The mixture was kept stirring for 2 h while its color changed from purple to orange. Further CH2Cl2 was added to dilute the solution and the polymer was precipitated from methanol. After centrifugation, the product was isolated and dried under vacuum to yield a dark yellow, honey-like polymer (67% yield).
SEC (PS standard, THF as the eluent): Mn = 20500 g mol−1, Mw = 42500 g mol−1, Mw/Mn = 2.07.
1H NMR (300 MHz, CDCl3): δ = 5.36 (b, 2H, –CH2–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 4.29 (q, J = 6.9 Hz, 2H, –O–C2–CH2–Br), 4.05 (q, J = 6.8 Hz, 4H, –OPO3–C2–), 3.53 (t, J = 6.3 Hz, 2H, –O–CH2–C2–Br), 1.98 (m, 4H, –C2–CH), 1.68 (m, 4H, –OPO3–CH2–C2 1.41–1.24 (m, 24H, alkyl). 13C NMR (75 MHz, CDCl3) δ = 130.32, 68.15 (d, J = 6.2 Hz), 66.47 (d, J = 5.3 Hz), 32.62, 30.26 (d, J = 6.8 Hz), 29.78–29.16 (m), 27.23, 25.42. 31P NMR (121 MHz, CDCl3) δ = −1.24.
), 4.29 (q, J = 6.9 Hz, 2H, –O–C2–CH2–Br), 4.05 (q, J = 6.8 Hz, 4H, –OPO3–C2–), 3.53 (t, J = 6.3 Hz, 2H, –O–CH2–C2–Br), 1.98 (m, 4H, –C2–CH), 1.68 (m, 4H, –OPO3–CH2–C2 1.41–1.24 (m, 24H, alkyl). 13C NMR (75 MHz, CDCl3) δ = 130.32, 68.15 (d, J = 6.2 Hz), 66.47 (d, J = 5.3 Hz), 32.62, 30.26 (d, J = 6.8 Hz), 29.78–29.16 (m), 27.23, 25.42. 31P NMR (121 MHz, CDCl3) δ = −1.24.
P2: P2a (260 mg, 0.54 mmol) was dissolved in 10 mL dry toluene in a glass vessel equipped with a stirring bar. Argon was bubbled through the solution for 10 min to remove oxygen. 52 mg of 10 wt% Pd/C was added, and the glass vessel was charged in a 250 mL ROTH autoclave. Hydrogenation was performed at 50 °C and 50 bar of H2 for 19 h. Pd/C was removed by filtration and the polymer was obtained after solvent evaporation as an off-white, hard material (quantitative yield).
SEC (PS standard, THF as the eluent): Mn = 15900 g mol−1, Mw = 26500 g mol−1, Mw/Mn = 1.67.
1H NMR (300 MHz, CDCl3): δ 4.29 (q, J = 8.4, 6.3 Hz, 2H, –O–C2–CH2–Br), 4.04 (q, J = 6.4 Hz, 4H, –OPO3–C2–), 3.53 (t, J = 6.3 Hz, 2H, –O–CH2–C2–Br), 1.68 (p, J = 6.7 Hz, 4H, –OPO3–CH2–C2), 1.25 (s, 32H, alkyl), 0.87. 13C NMR (75 MHz, CDCl3) δ = 68.16 (d, J = 6.2 Hz), 66.47 (d, J = 5.3 Hz), 30.26 (d, J = 6.8 Hz), 29.76–29.44 (m), 29.15, 25.43. 31P NMR (121 MHz, CDCl3) δ = −1.25.
P3: A Schlenk tube was charged with P2 (48.1 mg, 93.7 μmol, 1 eq.) and potassium thioacetate (12.8 mg, 112.4 μmol, 1.2 eq.), dissolved in dry THF (3 mL) under an argon atmosphere. The reaction was stirred overnight at 40 °C. The crude mixture was filtered, concentrated at reduced pressure, and dissolved in dichloromethane. The organic phase was washed with distilled water. The organic layer was dried over magnesium sulfate, filtered, and concentrated at reduced pressure to give a yellowish powder.
1H NMR (250 MHz, Chloroform-d, δ) 4.07 (m, 6H), 3.17 (t, J = 6.5 Hz, 2H), 2.35 (s, 3H), 1.67 (t, J = 7.0 Hz, 4H), 1.25 (m, 32H). 31P {H} NMR (202 MHz, Chloroform-d, δ) −1.13 (s, 1P).
P4: A vial was charged with P3 (30 mg, 59.0 μmol, 1 eq.) and ethyl acetate (30 mL). The dispersion was heated to 60 °C and allowed to cool to R.T. for 4–5 h. Then the crystal dispersion was cooled to 0 °C and hold at this temperature for 15 min. The stirring was turned on and 1 M hydrazine solution in ethanol (2 mL, 2 mmol, 40 eq.) was added while keeping the temperature at 0 °C. After 24 h the solvent was evaporated with a flow of nitrogen while still keeping the temperature at 0 °C.
Conclusion
We utilized phosphorus chemistry and simple polycondensation to introduce precisely spaced defects in polyethylene like polymers. We could demonstrate, that the relatively large size of the phosphate groups allow the control of the chain-folding during crystallization. The molecular arrangement in the crystal was studied by TEM and solid-state NMR, which proved a pseudohexagonal crystal structure with the phosphate groups emanating from the two opposing surfaces of the crystal. With chemical functionality in the pendant phosphoester groups, we pave the way for conducting chemistry “on surface” of anisotropic nanoplatelets, which was proven by nucleophilic substitution and hydrolysis reaction. The long-chain PE-like polyphosphates can be used as a general platform to design chemically functional anisotropic nanomaterials for various applications from the biofield to materials science, with the possibility of degradation of the phosphoester bonds combined with the crystallinity of polyethylene.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG WU 750/8-1) for funding. The authors thank Christine Rosenauer for the DLS measurements. The authors thank Prof. Dr Katharina Landfester (MPI-P, Germany) for her continuous support. Open Access funding provided by the Max Planck Society.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- T. E. Nowlin, Business and Technology of the Global Polyethylene Industry: An In-depth Look at the History, Technology, Catalysts, and Modern Commercial Manufacture of Polyethylene and Its Products, Wiley, 2014 Search PubMed.
- T. M. Karlsson, L. Arneborg, G. Broström, B. C. Almroth, L. Gipperth and M. Hassellöv, Mar. Pollut. Bull., 2018, 129, 52–60 CrossRef CAS.
- J. M. Kelly, J. Macromol. Sci., Part B: Phys., 2002, 42, 355–371 CrossRef.
- S. Hambir and J. P. Jog, Bull. Mater. Sci., 2000, 23, 221–226 CrossRef CAS.
- B. M. Sole and A. Ball, Tribol. Int., 1996, 29, 457–465 CrossRef CAS.
- G. Liu, Y. Chen and H. Li, Wear, 2004, 256, 1088–1094 CrossRef CAS.
- L. A. Goettler, K. Y. Lee and H. Thakkar, Polym. Rev., 2007, 47, 291–317 CrossRef CAS.
- A. Osichow, C. Rabe, K. Vogtt, T. Narayanan, L. Harnau, M. Drechsler, M. Ballauff and S. Mecking, J. Am. Chem. Soc., 2013, 135, 11645–11650 CrossRef CAS.
- B. Scheinhardt, J. Trzaskowski, M. C. Baier, B. Stempfle, A. Oppermann, D. Wöll and S. Mecking, Macromolecules, 2013, 46, 7902–7910 CrossRef CAS.
- K. Busse, C. Fuchs, N. Hasan, M. Pulst and J. Kressler, Langmuir, 2018, 34, 12759–12763 CrossRef CAS.
- Y.-R. Zheng, H. T. Tee, Y. Wei, X.-L. Wu, M. Mezger, S. Yan, K. Landfester, K. Wagener, F. R. Wurm and I. Lieberwirth, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS.
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef CAS.
- C. Pelosi, M. R. Tinè and F. R. Wurm, Eur. Polym. J., 2020, 141, 110079 CrossRef CAS.
- T. Haider, O. Shyshov, O. Suraeva, I. Lieberwirth, M. von Delius and F. R. Wurm, Macromolecules, 2019, 52, 2411–2420 CrossRef CAS.
- N. Hasan, C. Schwieger, H. T. Tee, F. R. Wurm, K. Busse and J. Kressler, Eur. Polym. J., 2018, 101, 350–357 CrossRef CAS.
- J. Mandal, S. Krishna Prasad, D. S. S. Rao and S. Ramakrishnan, J. Am. Chem. Soc., 2014, 136, 2538–2545 CrossRef CAS.
- P. H. Till Jr., J. Polym. Sci., 1957, 24, 301–306 CrossRef.
- D. J. Blundell, A. Keller and A. J. Kovacs, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 481–486 CrossRef CAS.
- S. A. E. Boyer and J. M. Haudin, Polym. Test., 2010, 29, 445–452 CrossRef CAS.
- T. Miyata and T. Masuko, Polymer, 1998, 39, 5515–5521 CrossRef CAS.
- N. Bosq, N. Guigo, E. Zhuravlev and N. Sbirrazzuoli, J. Phys. Chem. B, 2013, 117, 3407–3415 CrossRef CAS.
- L. Mandelkern, J. Appl. Phys., 1955, 26, 443–451 CrossRef CAS.
- G. Guerin, G. Molev, D. Pichugin, P. A. Rupar, F. Qi, M. Cruz, I. Manners and M. A. Winnik, Macromolecules, 2019, 52, 208–216 CrossRef CAS.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- T. Steinbach, E. M. Alexandrino, C. Wahlen, K. Landfester and F. R. Wurm, Macromolecules, 2014, 47, 4884–4893 CrossRef CAS.
- T. Steinbach, E. M. Alexandrino and F. R. Wurm, Polym. Chem., 2013, 4, 3800–3806 RSC.
- M. Steinmann, J. Markwart and F. R. Wurm, Macromolecules, 2014, 47, 8506–8513 CrossRef CAS.
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- H. T. Tee, K. Koynov, T. Reichel and F. R. Wurm, ACS Omega, 2019, 4, 9324–9332 CrossRef CAS.
- K. N. Bauer, H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2016, 49, 3761–3768 CrossRef CAS.
- T. Haider, O. Suraeva, M. L. O'Duill, J. Mars, M. Mezger, I. Lieberwirth and F. R. Wurm, Polym. Chem., 2020, 11, 3404–3415 RSC.
- O. Ruiz de Ballesteros, F. Auriemma, G. Guerra and P. Corradini, Macromolecules, 1996, 29, 7141–7148 CrossRef.
- K. Saalwächter, in Modern Magnetic Resonance, ed. G. A. Webb, Springer International Publishing, Cham, 2018, pp. 755–781, DOI:10.1007/978-3-319-28388-3_59.
- K. Saalwächter, F. Lange, K. Matyjaszewski, C.-F. Huang and R. Graf, J. Magn. Reson., 2011, 212, 204–215 CrossRef.
- P. A. Abragam and A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, 1961 Search PubMed.
- J. C. Markwart and F. R. Wurm, Tetrahedron, 2018, 74, 7426–7430 CrossRef CAS.
- A. M. M. El-Assar, C. P. Nash and L. L. Ingraham, Biochemistry, 1982, 21, 1972–1976 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 2004 Search PubMed.
- K. Saalwächter, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 1–35 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01352h |
| This journal is © The Royal Society of Chemistry 2020 |