
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceS8-Catalyzed triple cleavage of bromodifluoro compounds for the assembly of N-containing heterocycles†
Shuilin
Deng
a,
Haohua
Chen
d,
Xingxing
Ma
a,
Yao
Zhou
 a,
Kai
Yang
b,
Yu
Lan
*de and
Qiuling
Song
*abc
a,
Kai
Yang
b,
Yu
Lan
*de and
Qiuling
Song
*abc
aInstitute of Next Generation Matter Transformation, College of Chemical Engineering, College of Material Sciences Engineering at Huaqiao University, 668 Jimei Boulevard, Xiamen, Fujian 361021, China. E-mail: qsong@hqu.edu.cn
bCollege of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, China
cState Key Laboratroy of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
dSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, P. R. China
eCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China
First published on 5th June 2019
Abstract
An unprecedented S8-catalyzed selective triple-cleavage of bromodifluoroacetamides is disclosed for the first time. Valuable 2-amido substituted benzimidazoles, benzoxazoles and benzothiazoles were obtained in good to excellent yields in a cascade protocol in this strategy. Mechanistic studies suggested that a C2 source was generated in situ by selective cleavage of three C–X bonds, including two inert C(sp3)–F bonds on bromodifluoroacetamides, while leaving C–C bonds intact. This strategy will undoubtedly further consummate the role of halo difluoro compounds and enrich both fluorine chemistry and pharmaceutical sciences.
The activation of C–F bonds concomitant with further transformations has emerged as a significant and ongoing theme in synthetic organic chemistry, which opens up new platforms for molecular construction.1 For the activation of C–F bonds, the strategies predominantly rely on metal catalysts and the formation of a B–F, Al–F, Si–F or Ge–F bond to surmount the high C–F bond dissociation energy.2 Although metal-mediated and -catalyzed C–F activation has attracted intensive attention, publications on metal-free activation of C–F bonds are rare.3 In the past few decades, halogenated difluoro compounds (XCF2R) have proven to be promising difluoroalkylation reagents by one C–X cleavage, enabling the rapid preparation of innumerable difluoroalkylated compounds4 (Scheme 1A). Aside from the single cleavage, halogenated difluoro compounds could also undergo the double cleavage of C–X and C–R bonds to be used as difluorocarbene precursors, which enable the introduction of difluorinated units into targeted molecules by difluoromethylation5 (Scheme 1A). Based on the bond strength, the above two cleavages are not a surprise, since the bond dissociation energy (BDE) of C–F is 485 kJ mol−1, which is much higher than that of the C–X bond (C–Cl: 328 kJ mol−1; C–Br: 276 kJ mol−1) and C–R bond (C–C: 332 kJ mol−1; C–H: 414 kJ mol−1). Very recently, our group developed an unprecedented strategy for the synthesis of N-containing compounds by using BrCF2COOEt (or ClCF2COONa) as a new C1 source, which represents the first example for quadruple cleavage of BrCF2COOEt (or ClCF2COONa)6 by breaking all four bonds attached to the fluorinated carbon (Scheme 1A). In light of our recent quadruple cleavage study of halo difluoro compounds, we envisage whether it is possible to achieve a transition-metal free triple cleavage of halo difluoro compounds by selectively breaking three C–X bonds, including two very stable C(sp3)–F bonds while keeping the C–C bond intact. If successful, we can willingly selectively cleave each of the four bonds on the fluorinated carbon in halo difluoro compounds based on the necessity of the transformation. It will undoubtedly further consummate the role of halo difluoro compounds and enrich fluorine chemistry.
 | ||
| Scheme 1 Various activation patterns for halo difluoroalkyl compounds: single cleavage, double cleavage, quadruple cleavage and the newly disclosed triple cleavage. | ||
However, in this scenario, several challenges need to be addressed in this transformation: (1) how to overcome the energy barrier under transition metal-free and strong base-free conditions because the C–F bond, based on fundamental knowledge, is too strong and too inert to be broken without transition-metal assistance or strong bases; (2) given the bond dissociation energy, how to selectively cleave three C–X bonds, including two very stable aliphatic C–F bonds (485.6 kJ mol−1) on the same carbon atom while keeping the weak C–C bond (332 kJ mol−1) untouched; and (3) how to harness the in situ generated C2 source and convert it into valuable products. In our previous work, the saponification of halo difluoro compounds was the initial step to achieve quadruple cleavage. We postulate that if the cleavage of the C–F bond preferentially takes place prior to the saponification under the set conditions, in other words, if the saponification of difluoroalkylating compounds is suppressed, the triple cleavage of halo difluoro compounds might be acquired at this point.
To test our assumption, the difluorobromoamides were selected as benchmark materials, since their saponification is more difficult than that of their ester counterparts. Recently we have been interested in sulfur chemistry and found elemental sulfur as a polyvalent synthon could bring forth unexpected reactivity and has been widely utilized to mediate/catalyze a wide array of reactions.7 Inspired by the peculiarity of elemental sulfur, when a catalytic amount of S8 was added to our system, the triple cleavage of halo difluoro compounds was indeed achieved, in which various 2-amido/ester substituted benzimidazoles, benzoxazoles and benzothiazoles were obtained. Although various tactics have been developed to assemble 2-aryl and 2-alkyl substituted benzimidazoles, benzoxazoles and benzothiazoles,8 effective methods for the construction of 2-amido/ester substituted benzimidazoles, benzoxazoles and benzothiazoles were sparsely documented and the only known synthetic method is tedious (four steps are required to access 2-amidobenzothiazoles),9 notwithstanding their prevailing structural motifs in bioactive molecules as shown in Scheme 1B.10 Herein, we would like to report our new discovery on efficient S8-catalyzed triple cleavage of halogenated difluoro compounds for the assembly of 2-amido/ester benzimidazoles, benzoxazoles and benzothiazoles, which represents the first example for triple cleavage of halogenated difluoro compounds under transition metal-free conditions (Scheme 1B); meanwhile it also provides pragmatic synthetic methods for high value N-containing heterocycles which are prevalent skeletons in drugs and pharmaceuticals.
Initially, we employed benzene-1,2-diamine (1a) and 2-bromo-2,2-difluoro-N-isopropylacetamide (2a) as benchmark substrates to explore the optimal reaction conditions (Table 1). Gratifyingly, the desired product N-isopropyl-1H-benzo[d]imidazole-2-carbox-amide (3a) was obtained in 89% yield when the reaction was performed by using S8 (20 mol%) and Na2CO3 (3 equiv.) in MeCN (1 mL) at 130 °C for 16 h, which was proven to be the optimum conditions (Table 1, entry 1). A sequence of alterations was also conducted to test the influence on the optimal results. No desired product was observed when the model reaction proceeded in the absence of S8 (entry 2), and replacing S8 with I2 just resulted in a sluggish reaction (entry 3), which indicated that S8 was a prerequisite for this transformation. When we reduced the reaction temperature from 130 °C to 120 °C or 100 °C, lower reactivities were achieved (entries 4 and 5); this may be necessary to reach a high enough temperature to overcome the energy barrier. The yield of 3a sharply decreased to 29% when the reaction was carried out under an air atmosphere (entry 6). The attempt to shorten the reaction time led to inferior results (entry 7). No improvements were acquired when other bases such as NaHCO3, CsF and K2HPO4 were utilized instead of K2CO3 (entries 8–10). Further solvent screening failed to deliver superior results, which showed that CH3CN was still the best reaction medium for this transformation (entries 11–13).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditons | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (1.2 equiv.), S8 (20% mmol), Na2CO3 (3 equiv.), and MeCN (1 mL) under N2 for 16 h under 130 °C. b Yield of isolated product. n.r. = no reaction. | ||
| 1 | None | 89 |
| 2 | Without S8 | n.r. |
| 3 | I2 instead of S8 | n.r. |
| 4 | At 120 °C | 17 |
| 5 | At 100 °C | Trace |
| 6 | Under air | 29 |
| 7 | 6 h instead of 16 h | 16 |
| 8 | NaHCO3 instead of Na2CO3 | 72 |
| 9 | CsF instead of Na2CO3 | 61 |
| 10 | K2HPO4 instead of Na2CO3 | Trace |
| 11 | Acetone instead of MeCN | 42 |
| 12 | DMF instead of MeCN | 82 |
| 13 | EtOH instead of MeCN | 84 |

Having the optimal reaction conditions in hand, we then evaluated the generality of this S8-promoted triple cleavage of bromodifluoro compounds for the synthesis of benzimidazoles, which is summarized in Scheme 2. First, we examined a range of difluorobromoacetamides to react with o-phenylenediamine. In addition to 2-bromo-2,2-difluoro-N-isopropylacetamide (2a), tert-butyl, benzyl, allyl, phenyl and cyclopropyl substituted bromodifluoroamides were also good donors in this S8-promoted triple cleavage, rendering the expected products (3a–3e) in 55–93% yields. Cyclopropyl is widely used in medicinal chemistry because of its unique properties, which may enhance the drug's efficacy, change lipophilicity and PK properties it is gratifying that 2-bromo-N-cyclopropyl-2,2-difluoroacetamide also gives the corresponding product (3f) in 32% yields.11 We then also investigated a number of N-disubstituted difluorobromoacetamide compounds. For instance, 2-bromo-2,2-difluoro-1-(pyrrolidin-1-yl)ethan-1-one (2g) was a good partner in this transformation, enabling the generation of 3g in 74% yield. N-Diethyl and N-dibenzyl bromodifluoroamides 2h and 2i could be engaged in this reaction as well, producing the benzimidazoles 3h and 3i in 45% and 91% yield, respectively. Subsequently, we focused on the substrate scope with regard to o-phenylenediamines. A series of o-phenylenediamines with different electronic properties were proven to be suitable substrates, delivering the targeted benzimidazoles (3j–3n) in 30–77% yields. The treatment of ethyl difluorobromoacetate with o-phenylenediamine was also successful and the desired ethyl 1H-benzo[d]imidazole-2-carboxylate (3o) could be obtained in modest yield, which highlights the generality of the current procedure. In order to verify the scalability of this protocol, we also carried out a gram-scale reaction of o-phenylenediamine (1a) and 2-bromo-2,2-difluoro-N-isopropylacetamide (2a) and 3a was readily obtained in 80% yield without loss of the efficiency.
 | ||
| Scheme 2 The scope of the synthesis of 2-amidobenzimidazoles.a | ||
The current S8-promoted triple cleavage of bromodifluoro compounds can also be extended to 2-aminophenols for the construction of 2-amido/ester benzoxazoles. As showcased in Scheme 3, a series of N-phenyl substituted difluorobromoamides having different substituents on the aromatic ring exhibited good reactivity in this transformation, affording the benzoxazoles (5a–5e) in 61–73% yields. Aside from N-phenyl substituted difluorobromoamides, various N-benzyl substituted difluorobromoamides were also good candidates, furnishing 5f–5l in decent yields. The structure of 5h was explicitly confirmed by X-ray crystallographic analysis. N-Alkylmonosubstituted difluorobromoamides could work smoothly in this transformation as well, producing the benzoxazoles 5m–5t in moderate yields. To our delight, more sterically hindered N-disubstituted difluorobromoamides were also compatible under identical conditions, furnishing the expected products 5u–5w in 38–63% yields. The optimal reaction conditions were also proven to be suitable with a number of 2-aminophenols which could be readily converted into the corresponding benzoxazoles (5x–5z). It was also pleasingly found that the reaction of ethyl bromodifluoroacetate and 2-aminophenol could produce 5za in moderate yield, which could either be hydrolyzed into carboxylic acid or converted into other derivatives.
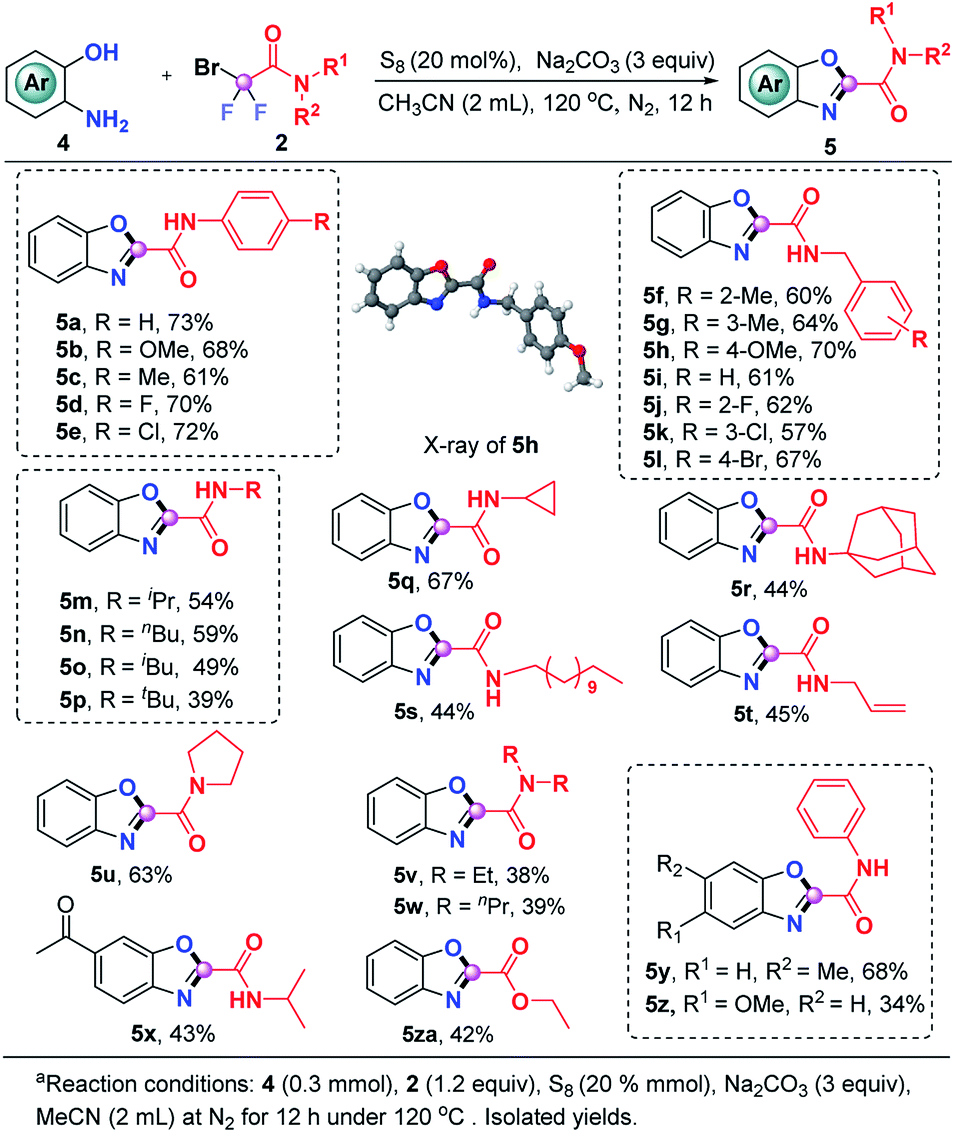 | ||
| Scheme 3 The scope of the synthesis of 2-amidobenzoxazoles.a | ||
To further broaden the substrate scope and limitations of this system, we subsequently investigated the reactions of 2-aminothiophenols with a variety of halogenated difluoro reagents.
We were glad to find that this S8-promoted triple cleavage of bromodifluoro compounds could also be employed to 2-aminothiophenols which reacted with numerous difluoro reagents to readily assemble various benzothiazoles in good yields (Scheme 4). A range of N-aryl and N-alkyl derived difluorobromoamides were amenable to this metal-free triple cleavage transformation, enabling the production of N-benzo[d]thiazole-2-carboxamides (7a–7m) in 54–90% yields. The structure of 7h was determined by single crystal X-ray diffraction analysis. In addition, N,N′-dialkyl substituted difluorobromoamides were also tolerated under the identified conditions, rendering the targeted products 7n–7s in moderate to good yields. Of note, the product 7q is a significant category of benzothiazoles which have anti-TB activity.10g Similar to o-phenylenediamine and 2-aminophenol, the reaction of ethyl difluorobromoacetate with 2-aminothiophenol was also successful, which gave rise to 7t and 7u in 75% and 46% yields, respectively.
 | ||
| Scheme 4 The scope of the synthesis of 2-amidobenzothiazoles.a | ||
To clarify the reaction mechanism, a string of control experiments was carried out. Not surprisingly, no desired heterocycles were detected when the reactions were conducted without S8 since S8 plays a prerequisite role in these transformations. Interestingly, 60% yield of 2-((2-aminophenyl)thio)-2,2-difluoro-N-isopropylacetamide (8c) was obtained when 2-aminothiophenol was used as the starting material (Scheme 5a). In order to figure out whether 8c is the intermediate, subsequently, the obtained 8c was subjected to the standard conditions; however, no reaction was observed (Scheme 5b), which indicated that 8c was not an intermediate in this transformation. In light of this result, we postulated that the free NH2 group should react with 2 preferentially. To test our hypothesis, aniline (9) was treated with 2a, using stoichiometric aniline, phenol and benzenethiol as additives (Scheme 5c). To our surprise and delight, the thioamide 10 was detected when phenol or benzenethiol was added to the reaction, which suggested that compound 10 might be the possible intermediate in this S8-catalyzed triple cleavage. Subsequently, thioamide 10 was subjected to aniline (9), phenol or benzenethiol under the standard conditions (Scheme 5d) and no further transformation was observed at this point, probably due to the difficulty of the intermolecular reaction pattern (in our cases, three transformations are intramolecular patterns) or it suggested that thioamide 10 was not the key intermediate in the transformations in this study. In order to verify whether the reaction was initiated from bromodifluoro compounds with S8, the reaction of 2-bromo-2,2-difluoro-N-isopropylacetamide (2a) and S8 was also conducted (Scheme 5e). However, no transformation was detected by the analysis of the reaction mixture and 18F-NMR. Therefore it excludes the possibility that S8 reacted with the difluoroalkylating reagent first, which further gave us a hint that S8 should be involved in the intermediate formation steps. To further gain insights into whether these are radical involved transformations, we also carried out radical trapping experiments and it was found that the reactions were not inhibited by the radical scavengers, indicating that this transformation was not a radical involved reaction regime (Scheme 5f).
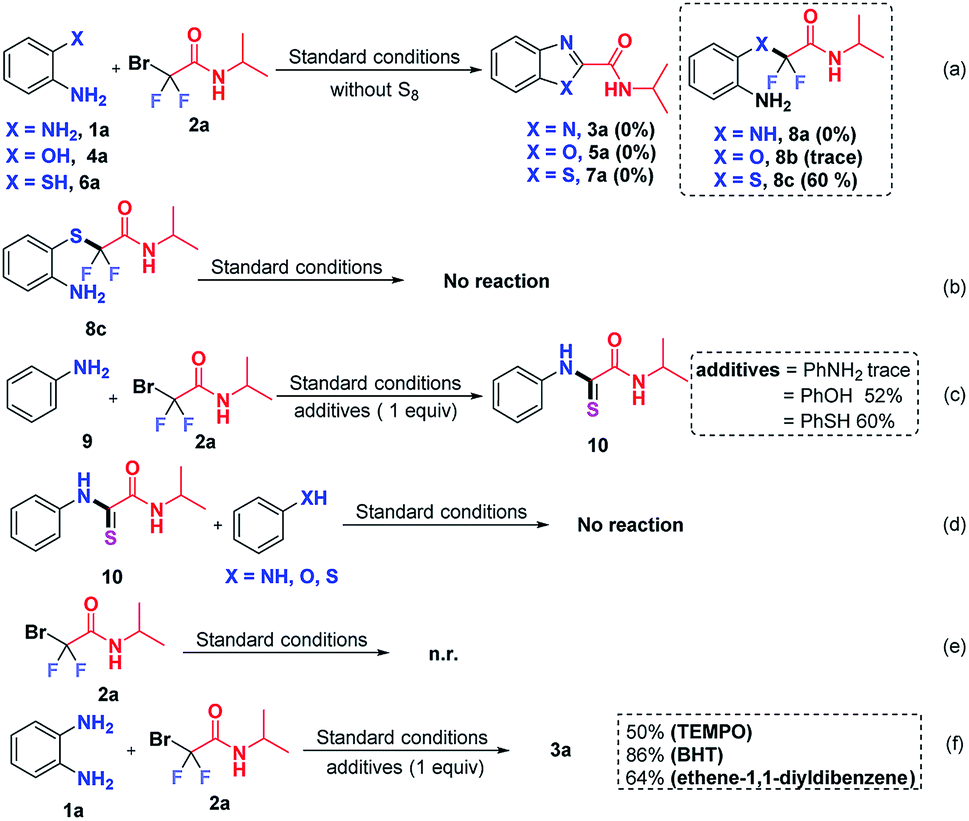 | ||
| Scheme 5 Control experiments for mechanistic studies. | ||
Combined with the above experimental observations and previous reports,12 the mechanism of S8-catalysed triple cleavage of halogenated difluoro compounds is proposed in Scheme 6. Deprotonation of sulfydryl of reactant 1 followed by nucleophilic attack on S8 affords the ring-opened sulfanyl anion I. Then intermolecular nucleophilic substitution of intermediate I with α-trihalogeno amide reactant 2 leads to the generation of a new C–N bond in amino amide II. The calculated free energy of this step is 28.2 kcal mol−1, which is considered to be the rate-determining step for the whole transformation. Subsequently, hydrogen fluoride elimination of intermediate II could afford monofluoride substituted imine III. The cleavage of the X–S bond can release S8 to finish the catalytic cycle by the release of anionic species IV. Then an outer sphere intramolecular nucleophilic addition of X to the imine moiety achieves the annulation in intermediate V, which can undergo further defluorination to provide the desired products. Further computational and experimental studies of the detailed mechanism are underway.
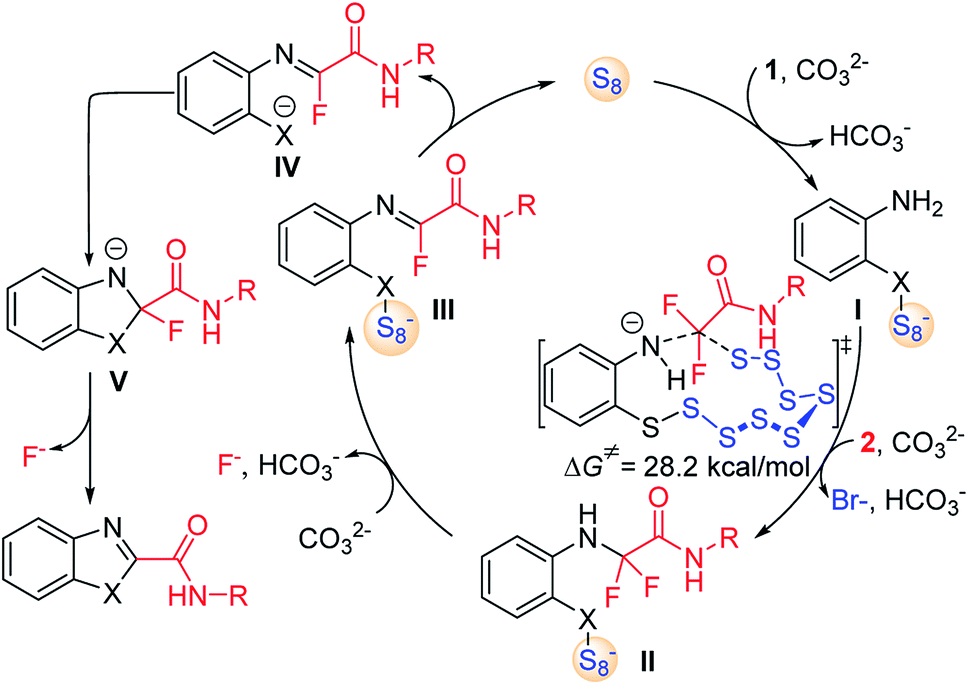 | ||
| Scheme 6 Proposed reaction mechanism. | ||
In summary, we have successfully developed an effective and direct S8-promoted synthesis of 2-amide/ester benzimidazoles, benzoxazoles and benzothiazoles from simple and readily available o-phenylenediamine, o-aminophenol and o-aminothiophenol via triple cleavage of halogenated difluoro compounds. The three halogen–carbon bonds of the halogenated difluoro compounds were simultaneously cleaved in this transformation in the absence of transition metal catalysts, ligands and external oxidants, which assembles the targeted N-containing heterocycles in good to excellent yields with a wide substrate scope. Further mechanistic studies of this S8 promoted triple cleavage of halogenated difluoro compounds and synthetic applications of this metal free protocol are in progress in our laboratory.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Natural Science Foundation (21772046) and the Natural Science Foundation of Fujian Province (2016J01064) is gratefully acknowledged. We also thank the Instrumental Analysis Center of Huaqiao University for analysis support. S. Deng thanks the Subsidized Project for Cultivating Postgraduates' Innovative Ability in Scientific Research of Huaqiao University.Notes and references
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) M. Hu, Z. He, B. Gao, L. Li, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 17302–17305 CrossRef CAS PubMed.
- G. Meißner, K. Kretschmar, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2017, 56, 16338–16341 CrossRef PubMed.
- (a) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2017, 115, 931–972 CrossRef PubMed; (b) M. Y. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061–9065 CrossRef CAS PubMed.
- (a) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS PubMed; (b) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS PubMed; (c) J. Wu, Q. Zhao, T. C. Wilson, S. Verhoog, L. Lu, V. Gouverneur and Q. Shen, Angew. Chem., Int. Ed., 2019, 58, 2413–2417 CrossRef CAS PubMed; (d) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270–12274 CrossRef CAS PubMed; (e) J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934–2938 CrossRef CAS PubMed; (f) M. Ke, Q. Feng, K. Yang and Q. Song, Org. Chem. Front., 2016, 3, 150–155 RSC; (g) M. Ke and Q. Song, J. Org. Chem., 2016, 81, 3654–3664 CrossRef CAS PubMed; (h) M. Ke and Q. Song, Chem. Commun., 2016, 53, 2222–2225 RSC; (i) M. Ke and Q. Song, Adv. Synth. Catal., 2017, 359, 384–389 CrossRef CAS; (j) W. Fu and Q. Song, Org. Lett., 2018, 20, 393–396 CrossRef CAS PubMed; (k) L. An, C. Xu and X. Zhang, Nat. Commun., 2017, 8, 1460–1468 CrossRef PubMed; (l) J. Xu, Z. Kuang and Q. Song, Chin. Chem. Lett., 2017, 29, 963–966 CrossRef; (m) C. Xu, W.-H. Guo, X. He, Y.-L. Guo, X.-Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170–1179 CrossRef.
- (a) M. Hu, C. Hu, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496–14501 CrossRef CAS PubMed; (b) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 125, 12616–12620 CrossRef; (c) Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918–923 CrossRef CAS PubMed; (d) C. Yu, J. Su, X. Ma, Y. Zhou and Q. Song, Asian J. Org. Chem., 2019, 8, 694–697 CrossRef CAS; (e) X. Ma, Q. Xuan and Q. Song, Acta Chim. Sin., 2018, 76, 972–976 CrossRef.
- (a) X. Ma, Y. Zhou and Q. Song, Org. Lett., 2018, 20, 4777–4781 CrossRef CAS PubMed; (b) X. Ma, S. Mai, Y. Zhou, G.-J. Cheng and Q. Song, Chem. Commun., 2018, 54, 8960–8963 RSC; (c) X. Ma, S. Deng and Q. Song, Org. Chem. Front., 2018, 5, 3505–3509 RSC.
- (a) J. Yu, J.-H. Lin and J.-C. Xiao, Angew. Chem., Int. Ed., 2017, 56, 16669–16673 CrossRef CAS PubMed; (b) Y. Xie, X. Chen, Z. Wang, H. Huang, B. Yi and G.-J. Deng, Green Chem., 2017, 19, 4294–4298 RSC; (c) J. Derendorf, C. Jenne and M. Keßler, Angew. Chem., Int. Ed., 2017, 56, 8281–8284 CrossRef CAS PubMed; (d) Z. Wang, X. Chen, H. Xie, D. Wang, H. Huang and G.-J. Deng, Org. Lett., 2018, 20, 5470–5473 CrossRef CAS; (e) T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, J. Am. Chem. Soc., 2018, 135, 118–121 CrossRef PubMed.
- (a) T. B. Nguyen, L. Ermolenko, P. Retailleau and A. Al-Mourabit, Angew. Chem., Int. Ed., 2014, 53, 13808–13812 CrossRef CAS; (b) X. Zhu, Y. Yang, G. Xiao, J. Song, Y. Liang and G. Deng, Chem. Commun., 2017, 53, 11917–11920 RSC; (c) G. Li, H. Xie, J. Chen, Y. Guo and G.-J. Deng, Green Chem., 2017, 19, 4043–4047 RSC; (d) Y. Huang, D. Yan, X. Wang, P. Zhou, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1742–1745 RSC; (e) Q. Song, Q. Feng and M. Zhou, Org. Lett., 2013, 15, 5990–5993 CrossRef CAS PubMed; (f) S.-Q. Zhu, Y.-L. Liu, H. Li, X.-H. Xu and F.-L. Qing, J. Am. Chem. Soc., 2018, 140, 11613–11617 CrossRef CAS PubMed.
- (a) Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086–9088 RSC; (b) Q. Feng and Q. Song, Adv. Synth. Catal., 2014, 356, 2445–2452 CrossRef CAS.
- (a) P. Shah, T. M. Dhameliya, R. Bansal, M. Nautiyal, D. N. Kommi, P. S. Jadhavar, J. P. Sridevi, P. Yogeeswari and D. Sriramb, Med. Chem. Commun., 2014, 5, 1489–1495 RSC; (b) X.-L. Chen, X. Li, L.-B. Qu, Y.-C. Tang, W.-P. Mai, D.-H. Wei, W.-Z. Bi, L.-K. Duan, K. Sun, J.-Y. Chen, D.-D. Ke and Y.-F. Zhao, J. Org. Chem., 2014, 79, 8407–8416 CrossRef CAS PubMed; (c) S. N. Mistry, J. Shonberg, C. J. Draper-Joyce, C. K. Herenbrink, M. Michino, L. Shi, A. Chris-topoulos, B. Capuano, P. J. Scammells and J. R. Lane, J. Med. Chem., 2015, 58, 6819–6843 CrossRef CAS PubMed; (d) Y. Nagasawa, Y. Tachikawa, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Adv. Synth. Catal., 2016, 358, 178–182 CrossRef CAS; (e) S. Pancholia, T. M. Dhameliya, P. Shah, P. S. Jadhavar, J. P. Sridevi, P. Yoge-shwari, D. Sriram and A. K. Chakraborti, Eur. J. Med. Chem., 2016, 116, 187–199 CrossRef CAS PubMed; (f) L.-T. Wu, Z. Jiang, J.-J. Shen, H. Yi, Y.-C. Zhan, M.-Q. Sha, Z. Wang, S.-T. Xue and Z.-R. Li, Eur. J. Med. Chem., 2016, 114, 328–336 CrossRef CAS PubMed; (g) T. M. Dhameliya, R. Tiwari, A. Banerjee, S. Pancholia, D. Sriram, D. Panda and A. Chakraborti, Eur. J. Med. Chem., 2018, 155, 364–380 CrossRef CAS PubMed.
- (a) Y. Kazuta, K. Hirano, K. Natsume, S. Yamada, R. Kimura, S. Matsumoto, K. Furuichi, A. Matsuda and S. Shuto, J. Med. Chem., 2013, 46, 1980–1988 CrossRef PubMed; (b) A. R. Renslo, P. Jaishankar, R. Venkatachalam, C. Hackbarth, S. Lopez, D. V. Patel and M. F. Gordeev, J. Med. Chem., 2005, 48, 5009–5024 CrossRef CAS PubMed; (c) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- (a) X. Chen, Z. Wang, H. Huang and G.-J. Deng, Adv. Synth. Catal., 2018, 360, 4017–4022 CrossRef CAS; (b) J. Zheng, R. Cheng, J.-H. Lin, D.-H. Yu, L. Ma, L. Jia, L. Zhang, L. Wang, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2017, 56, 3196–3200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1883275, 1883277 and 1875544. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01333d |
| This journal is © The Royal Society of Chemistry 2019 |