
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Living β-selective cyclopolymerization using Ru dithiolate catalysts†
Kijung
Jung
a,
Tonia S.
Ahmed
b,
Jaeho
Lee
 a,
Jong-Chan
Sung
a,
Hyeyun
Keum
a,
Robert H.
Grubbs
b and
Tae-Lim
Choi
*a
a,
Jong-Chan
Sung
a,
Hyeyun
Keum
a,
Robert H.
Grubbs
b and
Tae-Lim
Choi
*a
aDepartment of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: tlc@snu.ac.kr
bThe Arnold and Mabel Beckman Laboratory of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA
First published on 22nd July 2019
Abstract
Cyclopolymerization (CP) of 1,6-heptadiyne derivatives is a powerful method for synthesizing conjugated polyenes containing five- or six-membered rings via α- or β-addition, respectively. Fifteen years of studies on CP have revealed that user-friendly Ru-based catalysts promoted only α-addition; however, we recently achieved β-selective regiocontrol to produce polyenes containing six-membered-rings, using a dithiolate-chelated Ru-based catalyst. Unfortunately, slow initiation and relatively low catalyst stability inevitably led to uncontrolled polymerization. Nevertheless, this investigation gave us some clues to how successful living polymerization could be achieved. Herein, we report living β-selective CP by rational engineering of the steric factor on monomer or catalyst structures. As a result, the molecular weight of the conjugated polymers from various monomers could be controlled with narrow dispersities, according to the catalyst loading. A mechanistic investigation by in situ kinetic studies using 1H NMR spectroscopy revealed that with appropriate pyridine additives, imposing a steric demand on either the monomer or the catalyst significantly improved the stability of the propagating carbene as well as the relative rates of initiation over propagation, thereby achieving living polymerization. Furthermore, we successfully prepared diblock and even triblock copolymers with a broad monomer scope.
Introduction
Cyclopolymerization (CP) of terminal diynes via olefin metathesis is a powerful method for preparing conjugated polymers containing cycloalkene repeat units.1 After extensive early research on CP using ill-defined catalysts, such as Ziegler–Natta,2,3 MoCl5, and WCl6 catalysts,4–8 the first breakthrough on CP came with the development of well-defined catalysts by the Schrock group who reported the first living CP and the related comprehensive mechanistic details.9,10 The whole field expanded rapidly when Ru-based Grubbs11–14 and modified Grubbs catalysts (e.g., Hoveyda–Grubbs15,16 and Buchmeiser catalysts17–22) showed excellent activity, selectivity, and stability for CP.To develop a more powerful CP, one should be able to control the reactivity to obtain high-molecular-weight (MW) polymers with a narrow distribution, as well as the regioselectivity to synthesize polymers with regular and predictable structures. As shown in Scheme 1a, there are two possible pathways in CP, α-addition and β-addition, resulting in a five- and six-membered ring repeat unit, respectively. The reaction pathway is determined by the orientation of the approaching metal alkylidenes to the terminal alkynes (Scheme 1a).10 Although the early-stage catalysts produced ill-defined regiorandom polyenes,1 the Buchmeiser group successfully demonstrated the first selective CP to generate five-membered rings via exclusive α-addition using Mo-based Schrock catalysts,23–27 and later, Ru-based Buchmeiser catalysts.17–22,28,29 Afterward, our group reported α-selective living CP to prepare various soluble polyacetylenes with complex architectures,30–33 and the Xie group demonstrated CP of functionalized monomers to generate polyacetylenes exerting high ionic conductivity,34–36 by using a user-friendly, fast-initiating third-generation Grubbs catalyst.11–14
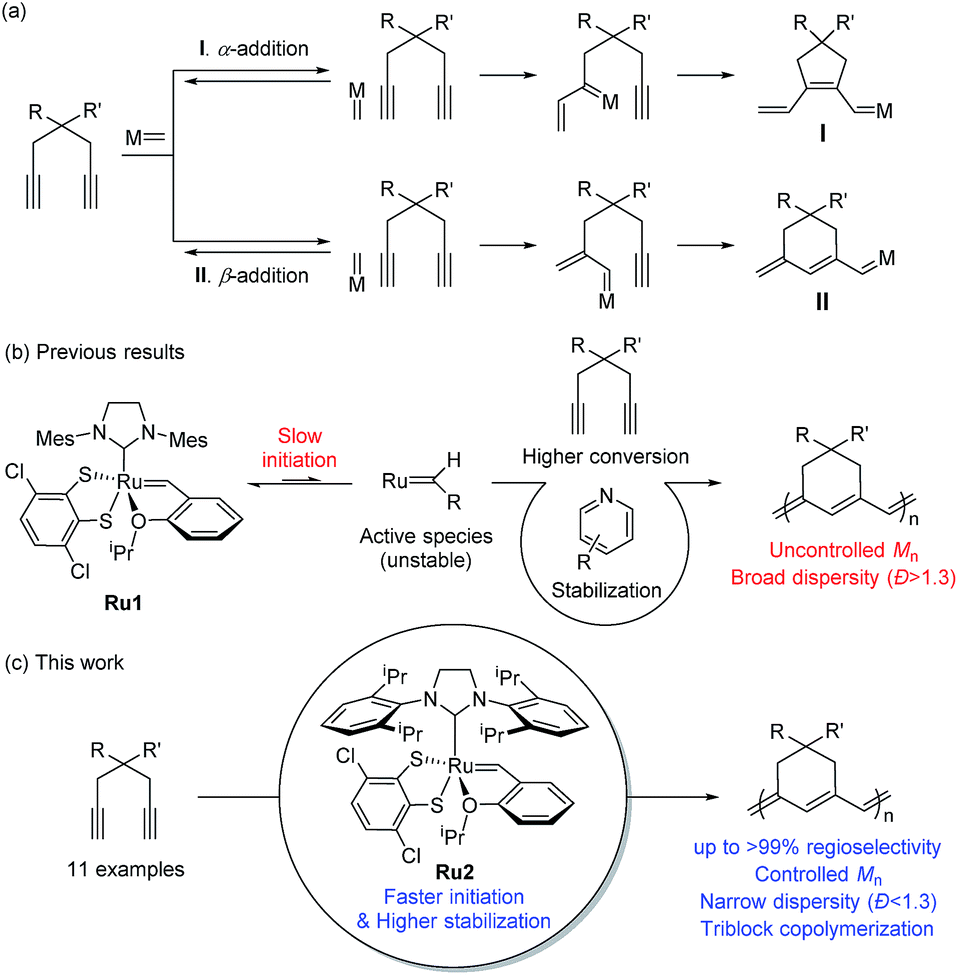 | ||
| Scheme 1 (a) Two possible pathways for CP of 1,6-heptadiynes, (b) proposed scheme showing the effects of the pyridine additive and the limitations of this method, and (c) living β-selective CP using fast-initiating Ru2. | ||
The first β-selective CP producing six-membered rings was reported by the Schrock group in 1996, using modified Mo catalysts,37,38 but no follow-up studies have been reported. Recently, our group demonstrated the first β-selective CP using a user-friendly, Ru-based Grubbs Z-selective catalyst39 to generate conjugated polymers containing six-membered rings, with 67–95% β-selectivity.40 Notably, a new catalyst Ru1 containing a dithiolate ligand, developed by the Hoveyda group,41 exerted far higher β-selectivity in CP of 1,6-heptadiyne monomers (85–99% β-selectivity), generating mainly six-membered conjugated polyenes bearing comparable thermal properties to the five-membered counterparts.42 However, these new polymers showed lower optical band-gaps than the five-membered analogues, thereby making them potentially more attractive materials in the electronic application.42 We also examined the origin of the exceptional regioselectivity using DFT calculations, concluding that Ru1 which had adopted trigonal bipyramidal geometry would prefer β-addition due to electronic effects.43 However, both catalysts showed very slow initiation rate (ki) and relatively fast propagation rate (kp) leading to low ki/kp values and poor MW control. Furthermore, the relatively low stability of the propagating carbene seemed to result in fast termination and broad dispersity (Đ) (Scheme 1b).42
β-Selective living CP is a much more challenging area, as there is only one example, using Schrock's Mo catalyst and just one monomer.37,38 Herein, we introduce two strategies to achieve β-selective living CP with user-friendly Ru catalysts: lowering kp by introducing pyridine additives and sterically bulky substituents on monomers, and dramatically increasing ki by employing a catalyst with a bulkier ligand. These strategies, combined with a synergetic effect of stabilized propagating species by steric demand, led to successful controlled polymerization with a broad monomer scope. Furthermore, we successfully demonstrated fully β-selective diblock and triblock copolymerizations (Scheme 1c). Finally, a mechanistic investigation using in situ kinetic experiments clarified the role of pyridine additives and allowed quantification of their effects by direct comparison of ki/kp values.
Results and discussion
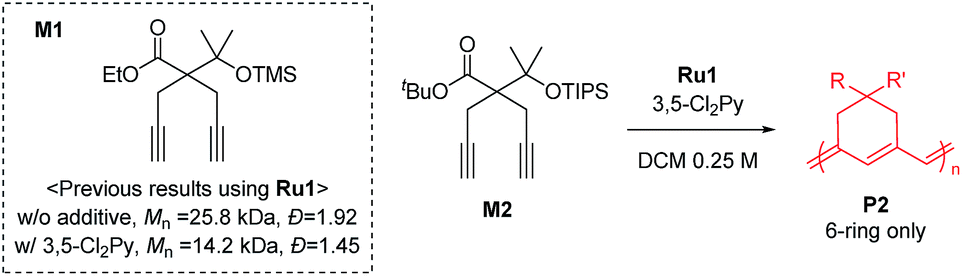
To achieve living polymerization, high stability of the propagating species and a high ki/kp value are crucial. In previous studies of ours and others, pyridine derivatives were found to coordinate to Grubbs catalysts,11,44 and in CP using Ru1, we discovered that pyridine additives coordinate to Ru1 competitively with the monomer, thereby slowing down the polymerization and stabilizing the propagating species.42 However, living polymerization was not achieved, presumably due to low ki and decomposition of Ru1. Since CP of monomer M1 containing a gem-dimethyl group showed narrower dispersity (Đ) with the addition of 3,5-Cl2Py (reduced from 1.92 to 1.45),42 we expected that living polymerization might be achieved by introducing an even bulkier side chain, which would increase the stabilization on the propagating species and the ki/kp value. Therefore, we synthesized M2, which replaced the ethyl ester and TMS side chains of M1 with the sterically bulky tert-butyl ester and TIPS substituents (Table 1), and obtained conjugated polyene P2 containing six-membered rings via exclusive β-addition. Without additives, CP of M2 using Ru1 at RT in DCM showed poor reactivity with less than 10% conversion (Table 1, entry 1). Although carrying out the reaction in THF at 70 °C increased the conversion to 94%, a broad Đ of 1.69 still implied uncontrolled CP (entry 2). Gratifyingly, with the addition of 3,5-Cl2Py, P2 was synthesized at RT with higher conversion, and excellent molecular weight control was achieved, with a linear increase in Mn from 7.3 to 38.2 kDa with M/I between 15 and 75 (entries 3–7, Fig. 1a). Furthermore, the dispersities were less than 1.23, implying a successfully controlled polymerization, except for the highest-MW polymer (M/I of 75) which showed some tailing in size-exclusion chromatography (SEC) traces (entry 7, Fig. 1b).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | M/I/add | Temp. (°C) | Time (h) | Conv.a (%) | Yieldb (%) | M n (kDa) | Đ |
| a Determined by 1H NMR. b Precipitated in MeOH at −78 °C. c Determined by THF SEC calibrated using polystyrene standards. d Conducted in THF. | |||||||
| 1 | 30/1/— | 25 | 1 | <10 | — | — | — |
| 2d | 30/1/— | 70 | 3 | 94 | 69 | 18.2 | 1.69 |
| 3 | 15/1/10 | 25 | 1 | >99 | 50 | 7.3 | 1.18 |
| 4 | 30/1/10 | 25 | 3 | >99 | 81 | 13.0 | 1.19 |
| 5 | 45/1/15 | 25 | 3 | >99 | 89 | 22.0 | 1.23 |
| 6 | 60/1/20 | 25 | 3 | >99 | 78 | 30.4 | 1.20 |
| 7 | 75/1/25 | 20 | 8 | 98 | 79 | 38.2 | 1.39 |
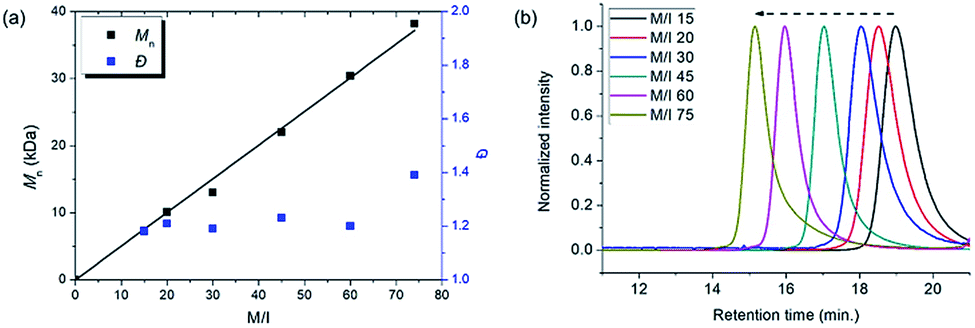 | ||
| Fig. 1 (a) Plots of the obtained Mnvs. M/I (solid line shows a fit of the data) and the corresponding Đ values for P2, and (b) SEC traces of P2 from entries 3–7 in Table 1. | ||
Having achieved β-selective living polymerization, we attempted diblock copolymerization at RT using M2 as the first monomer (Scheme 2), because any of β-selective block copolymerization has never been reported. After complete consumption of 15 equiv. of M2, we added another 15 equiv. of M3, containing the di-tert-butyl malonate moiety, as the second monomer to prepare the fully conjugated polymer P2-b-P3 by β-addition (Scheme 2a). Block copolymerization was confirmed by SEC analyses showing the complete shift of the traces from the P2 homopolymer (7.3 kDa) to the block copolymer (12.6 kDa), with a narrow dispersity (1.30, Scheme 2b).
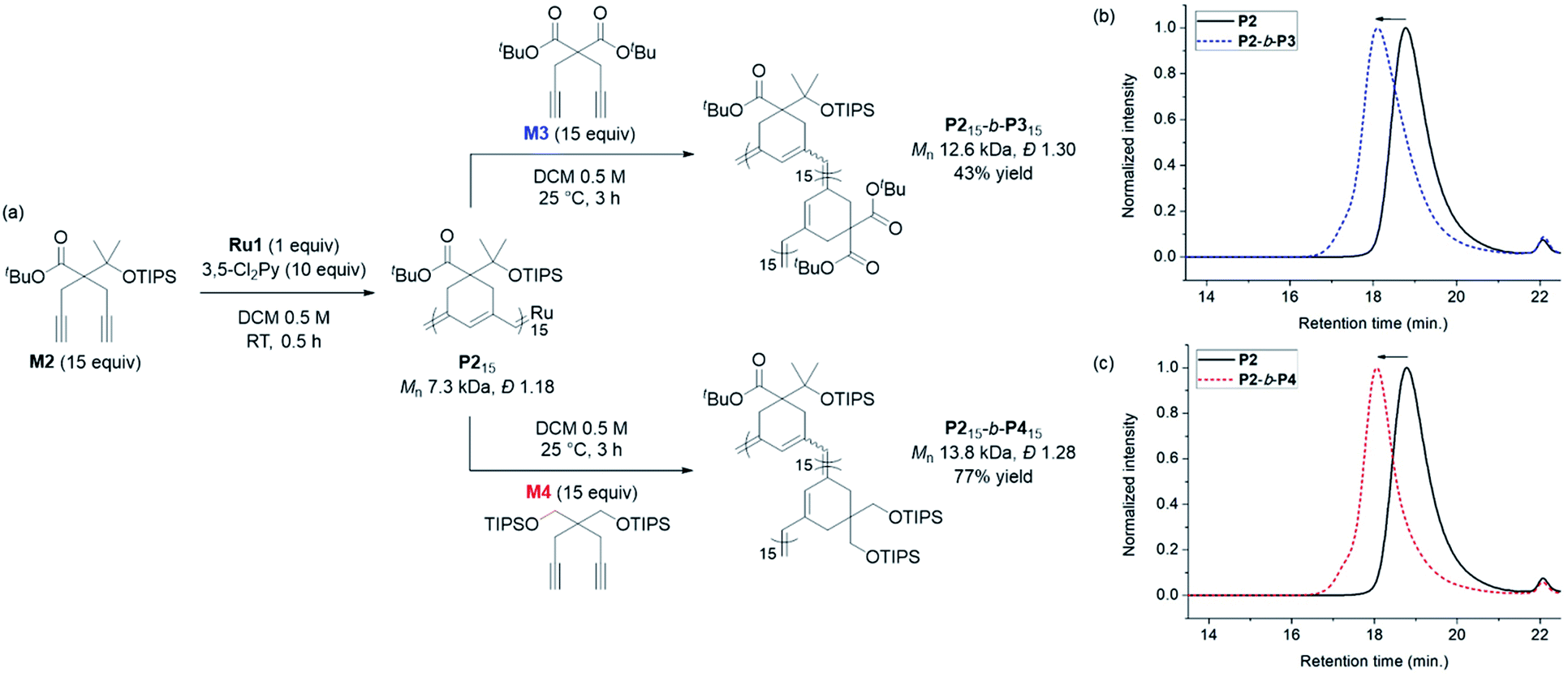 | ||
| Scheme 2 (a) Diblock copolymerization of with M2 as the first monomer, and M3 (above) and M4 (below) as the second monomers. SEC traces of homopolymer P2 and diblock copolymers: (b) P2-b-P3, and (c) P2-b-P4. | ||
An analogous diblock copolymerization was successful as well when 15 equiv. of M4, containing bis-silylether, was introduced as the second monomer, to afford P2-b-P4, with a Mn of 13.8 kDa and Đ of 1.28, which was verified by SEC analyses (Scheme 2c). Remarkably, these two diblock copolymers showed perfect β-selectivity, confirmed by 13C NMR measurements. Although living homopolymerizations of M3 and M4 using Ru1 were not possible in our previous work,42 we were able to prepare well-defined diblock copolymers from the P2 macroinitiator as the initiation and stability of the living chain end were established in the first block.
To understand the origin of the successful living polymerization, we conducted a mechanistic investigation using in situ NMR analysis by monitoring initiation and propagation of Ru1 during CP of M2 (M/I = 20 in 0.1 M DCM-d2). With the pyridine additives, the signal intensity of the new propagating carbene proton gradually increased by up to 74% during the first 40 minutes, whereas that from a smaller monomer, diethyl malonate-derived M5, increased by only 56% in 5 minutes and then decreased continuously to 32% at 25 minutes (see ESI† for details).‡ This result supports our hypothesis that a bulkier monomer enhances the stability of the propagating species. Furthermore, from this in situ NMR monitoring, ki and kp for CP of M2 were obtained with and without the 3-ClPy additive (Fig. S3a and b†). With 7 equiv. of 3-ClPy, kp was approximately three times lower than without the pyridine additive (0.15 vs. 0.05 min−1), due to competitive coordination to form a dormant 18e− species, while ki did not significantly change. Therefore, the overall ki/kp value increased by 2.3 times with the use of 3-ClPy (3.07 vs. 1.31). SEC analyses of the resulting P2s demonstrated that an Mn of 10.1 kDa (close to the theoretical value, 8.1 kDa) with a narrow Đ (1.21) was obtained using 3-ClPy, whereas an unusually high Mn of 38.0 kDa and a broad Đ (2.05) were found without addition of 3-ClPy (Fig. S3c†). In short, sterically bulky monomers and pyridine additives increase the stability of the propagating species (from Ru1) as well as the ki/kp value, thereby promoting living polymerization.
However, living polymerization of monomers with smaller substituents was not possible using this approach.42 We then envisioned that a faster initiating β-selective catalyst would be necessary to increase the scope of suitable monomers for living polymerization. The Wagener group improved the initiation efficiencies of Grubbs and Hoveyda-Grubbs catalysts by replacing mesityl groups in the N-heterocyclic carbene (NHC) ligand with much bulkier N-2,6-diisopropylphenyl (DIPP) groups, facilitating dissociation of the Ru–O bond.45 This inspired us to use new dithiolate catalyst Ru246 containing the bulky DIPP NHC ligand (Fig. 2a) to promote living CP with an even higher β-selectivity with a broader monomer scope, as a result of steric repulsion between the DIPP group and monomer substituents in the metallacyclobutene intermediates (Fig. 2b).
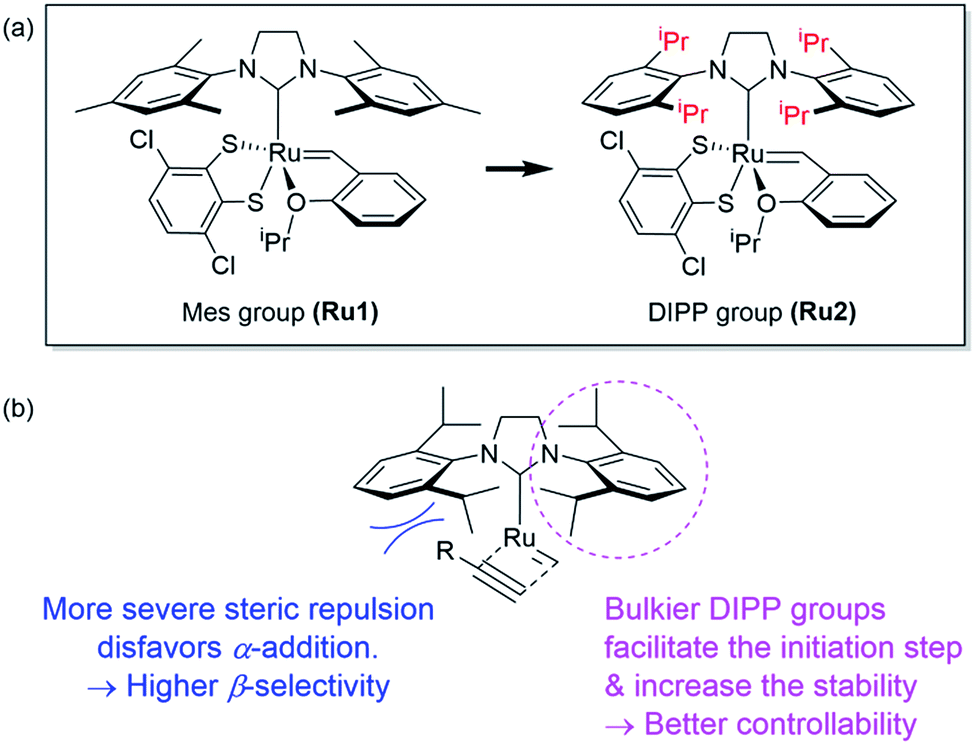 | ||
| Fig. 2 (a) Modifying ligands for living polymerization, and (b) model for improved β-selectivity and controllability of CP using Ru2. | ||
To explore the effect of the DIPP group on the initiation, we measured the initiation rate of Ru1 (ki,Ru1) to compare it with the recently reported value of Ru2 (ki,Ru2).46 Following the reported protocol, the consumption of Ru1 was monitored by 1H NMR spectroscopy upon addition of butyl vinyl ether at −20 °C, and ki,Ru1 was determined to be 1.53 × 10−5 s−1, which is about 400 times slower than ki,Ru2 measured under identical conditions (Fig. 3). Therefore, much faster-initiating Ru2 should be an effective catalyst for living β-selective CP.
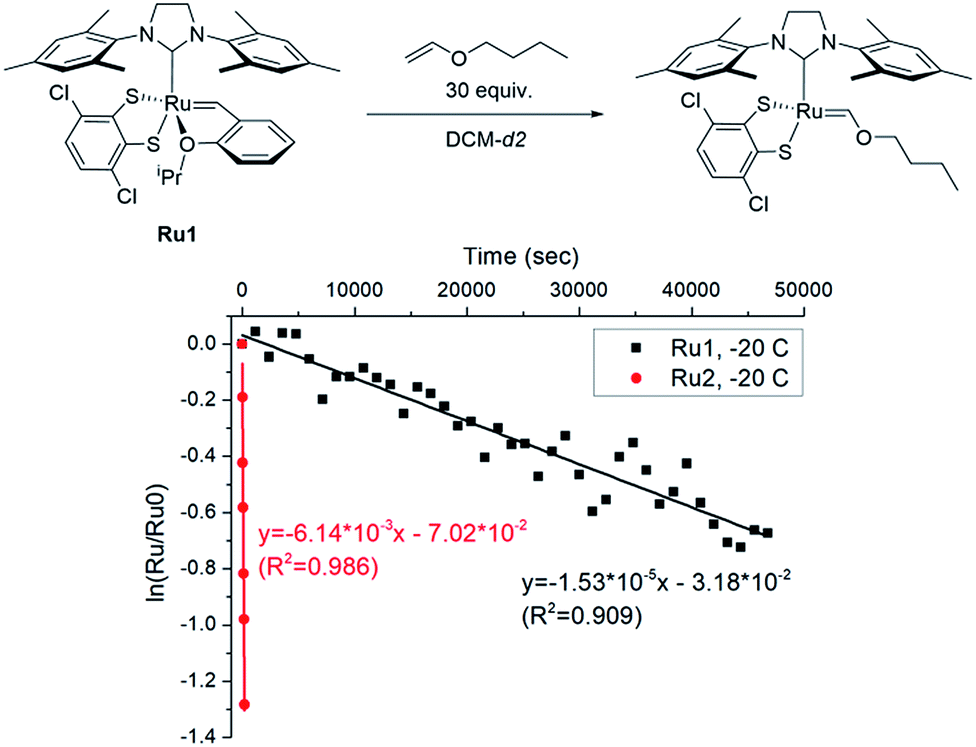 | ||
| Fig. 3 The plot of ln([Ru]/[Ru]0) vs. time to measure the initiation rates of Ru1-246 at −20 °C by monitoring the disappearance of the benzylidene signal using 1H NMR. | ||
To test β-selective CP using Ru2, we chose diethyl dipropargyl malonate (M5) as a model monomer because its β-selectivity can be easily measured using 1H NMR (Table 2). The reaction with an M/I of 30, without an additive, was completed in just one minute with a high β-selectivity of 95% (Table 2, entry 1). This indicated that Ru2 was more active and highly β-selective compared with Ru1, which, when used under the same reaction conditions, resulted in a 76% conversion and 85% β-selectivity after 1 hour.42 However, the propagation of CP using Ru2, compared to the initiation, was too fast for controlled polymerization; thus, Mn and Đ values were higher than expected. To lower the kp, 6 equiv. (with respect to the catalyst), of 3,5-Cl2Py, the optimal additive for Ru1, was used, but Đ was still broad (1.41), implying that 3,5-Cl2Py was not effective for Ru2 (entry 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Conv.a (%) | Yieldb (%) | M n (kDa) | Đ | β-selectivitya (%) |
| a Determined by 1H NMR. b Precipitated in hexane at −78 °C. c Determined by THF SEC, calibrated using polystyrene standards. | ||||||
| 1 | — | >99 | 99 | 13.6 | 1.86 | 95 |
| 2 | 3,5-Cl2Py | >99 | 99 | 14.2 | 1.41 | 95 |
| 3 | 3-ClPy | >99 | 92 | 10.8 | 1.19 | 96 |
| 4 | 3-IPy | >99 | 72 | 9.6 | 1.19 | 94 |
| 5 | Pyridine | >99 | 77 | 9.6 | 1.11 | 97 |
| 6 | 4-MeOPy | >99 | 78 | 9.1 | 1.10 | 95 |
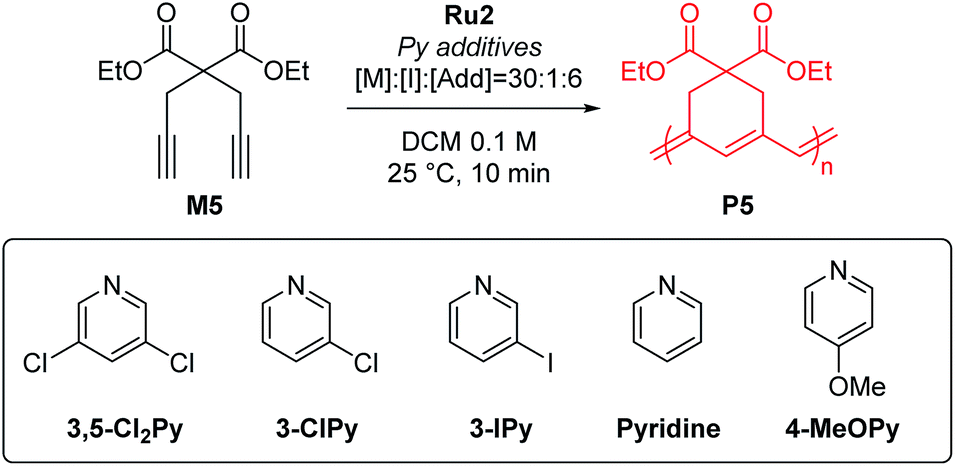
To our delight, the use of sterically less bulky and more strongly-binding mono-halogenated pyridine derivatives such as 3-ClPy and 3-IPy led to much narrower dispersities (Đ of 1.19, entries 3 and 4). These results led us to speculate that the binding affinity of the additives affected the controllability of CP, so we tried more basic ligands such as pyridine and 4-MeOPy. As a result, we observed controlled polymerizations with even narrower dispersities (Đ of 1.11 and 1.10, respectively, entries 5 and 6), and the highest β-selectivity, 97% in the pyridine case.
To investigate how various pyridine additives with different electronic properties affected the efficiency and selectivity of CP, we conducted in situ kinetic studies using 1H NMR, and monitored changes in the propagating carbene protons with four pyridine additives (M/I = 20 in 0.1 M DCM-d2, Fig. 4). First, upon addition of 3,5-Cl2Py to the Ru2, the carbene proton signal for Ru2 at 14.47 ppm decreased to 71% without generating a new carbene proton signal. After the addition of the monomer M5, Ru2 fully initiated with complete conversion of M5 in 80 seconds. This indicates superior reactivity of Ru2 to CP since for Ru1, only half of the catalyst initiated after 80 seconds, and it took 10 minutes for the complete conversion of M5 (see ESI† for details). However, virtually no propagating carbene proton or Fischer carbene proton signal was detected during polymerization or after quenching with ether vinyl ether (EVE), suggesting complete decomposition of the catalyst. Therefore, we concluded that coordination of the bulkier and less basic 3,5-Cl2Py to Ru2 was inefficient, resulting in a broad dispersity due to the failure of stabilizing the active species (Đ of 1.86, Fig. 5b).
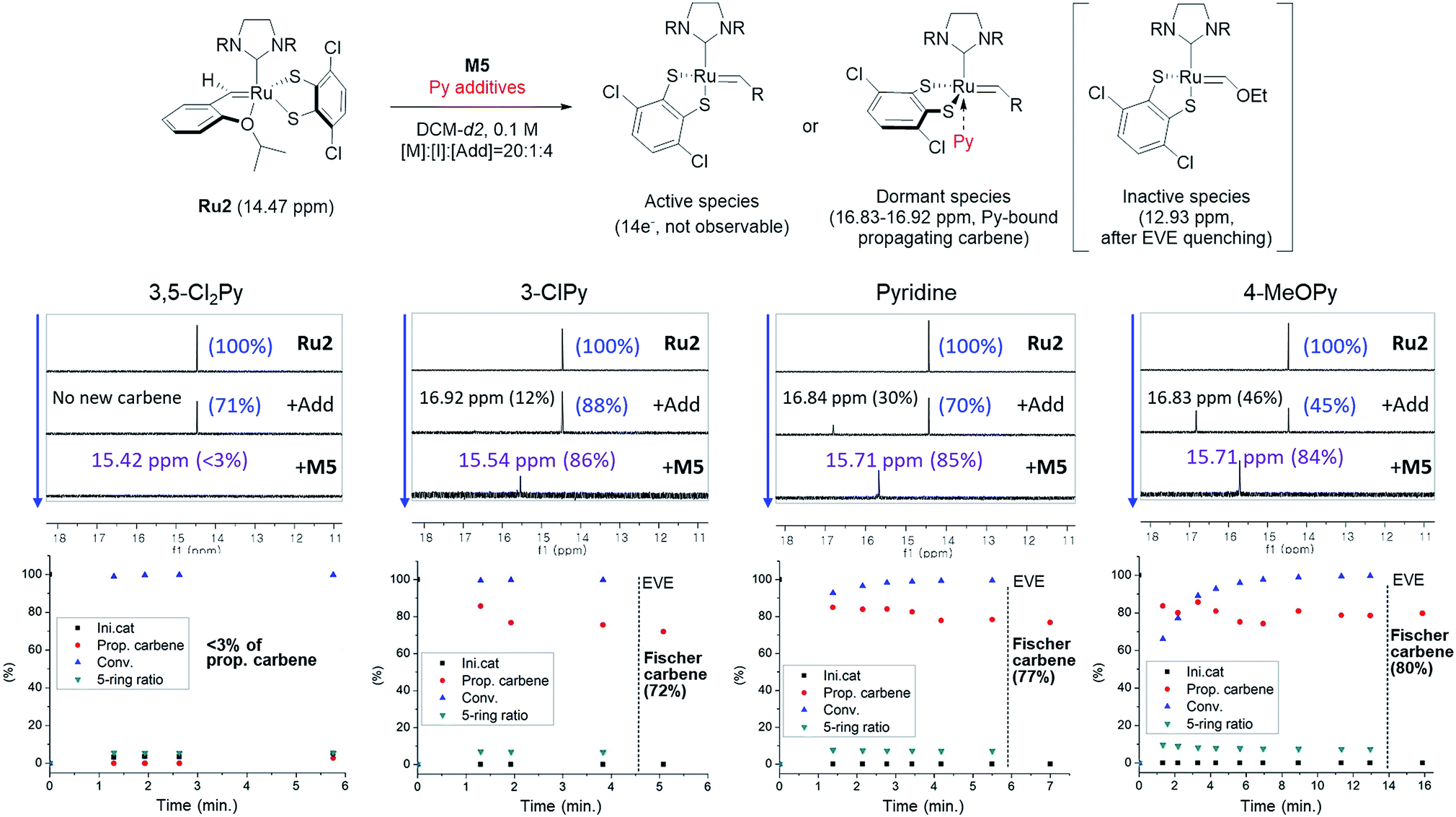 | ||
| Fig. 4 Scheme for the 1H NMR kinetic experiments (top), monitoring the changes in carbene proton signals during CP of M5 using Ru2 (middle) and their corresponding plots in real time (bottom). | ||
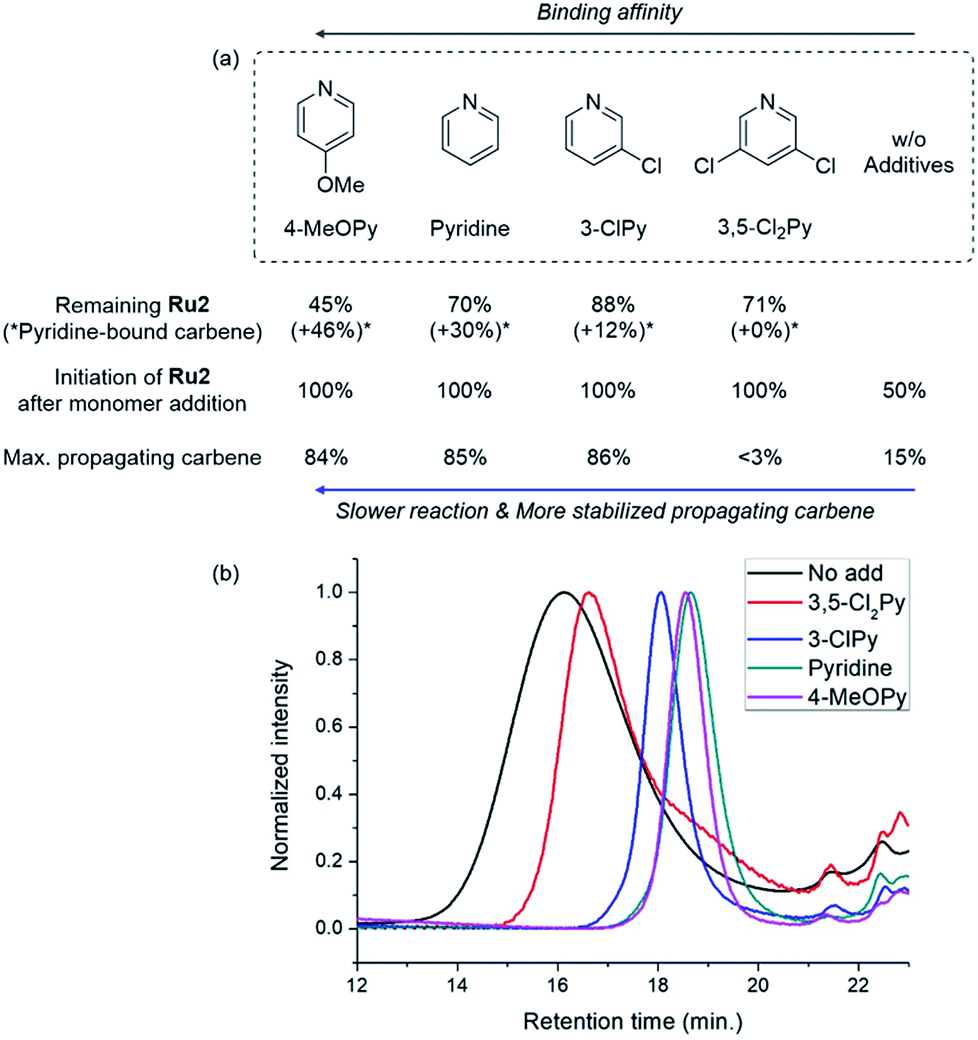 | ||
| Fig. 5 (a) Summary of the results from the kinetic experiments showing the relationship with the binding affinity of the additives and (b) THF-SEC traces of the corresponding polymers. | ||
When the stronger ligand 3-ClPy was added, the amount of Ru2 dropped to 88% while at the same time, a new carbene proton signal equivalent to 12% of the initial Ru2 signal appeared at 16.92 ppm. This new carbene is thought to be pyridine-bound Ru2, given the downfield shift, and the sum of two carbene proton signals was 100%, suggesting no decomposition of the catalyst. Upon the addition of M5, the signals of M5 and both carbene protons disappeared in 90 seconds, and a new propagating carbene proton signal appeared at 15.54 ppm, with 86% intensity relative to the initial Ru2 signal. When we added the more strongly binding pyridine and 4-methoxypyridine to Ru2, new carbene proton signals at 16.8 ppm were observed with higher relative intensities of 30% and 46%, respectively. These two effective ligands slowed down the propagation and stabilized the resulting propagating carbenes (15.71 ppm), with 85% and 84% intensities, respectively, which persisted throughout the reactions. Given that the propagating carbene proton signal in the reaction with Ru1 and M5 reached only 56% of the initial Ru1 signal intensity, the higher values (up to 86%) observed with Ru2 strongly support that the bulky DIPP ligand efficiently stabilizes the propagating species, thereby improving the controllability, with Đ as low as 1.11. A summary of the kinetic experiments is shown in Fig. 6a, demonstrating that the stronger electron-donating pyridine additives tend to stabilize the propagating carbenes more efficiently by forming 16 or 18e− dormant states. This is well-reflected in the corresponding SEC traces showing a narrow Gaussian distribution for pyridine and 4-MeOPy, but broad dispersities with long tailings due to chain termination for the cases with no additive or weakly coordinating 3,5-Cl2Py (Fig. 5b). We selected pyridine as the optimal additive because it gave the highest β-selectivity as well as a low Đ (Table 2).
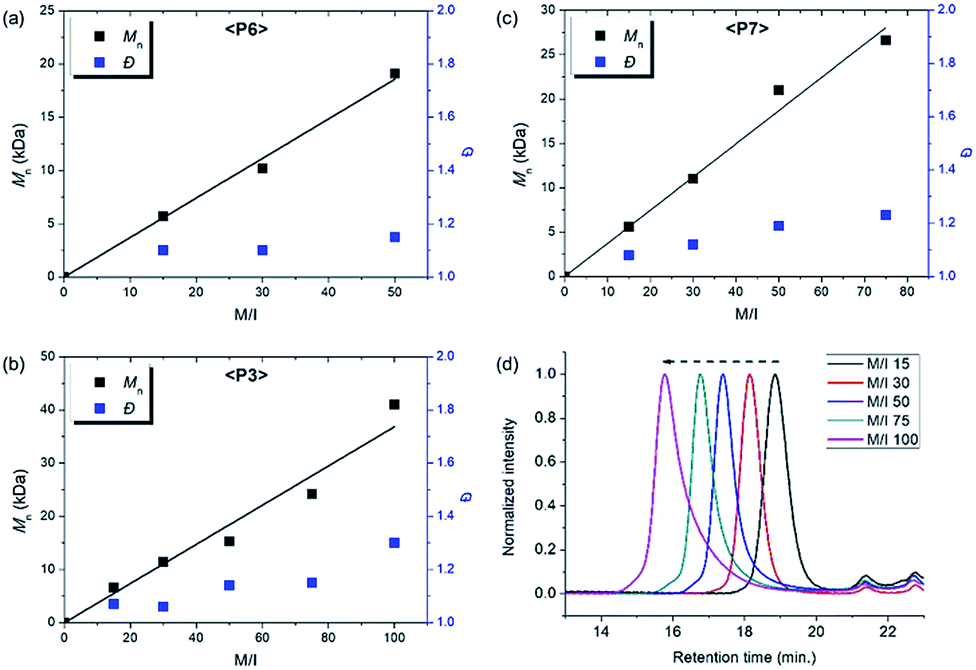 | ||
| Fig. 6 Plots of Mnvs. M/I and corresponding Đ values of (a) P6, (b) P3, (c) P7, and (d) SEC traces of P3 from entries 7–11. | ||
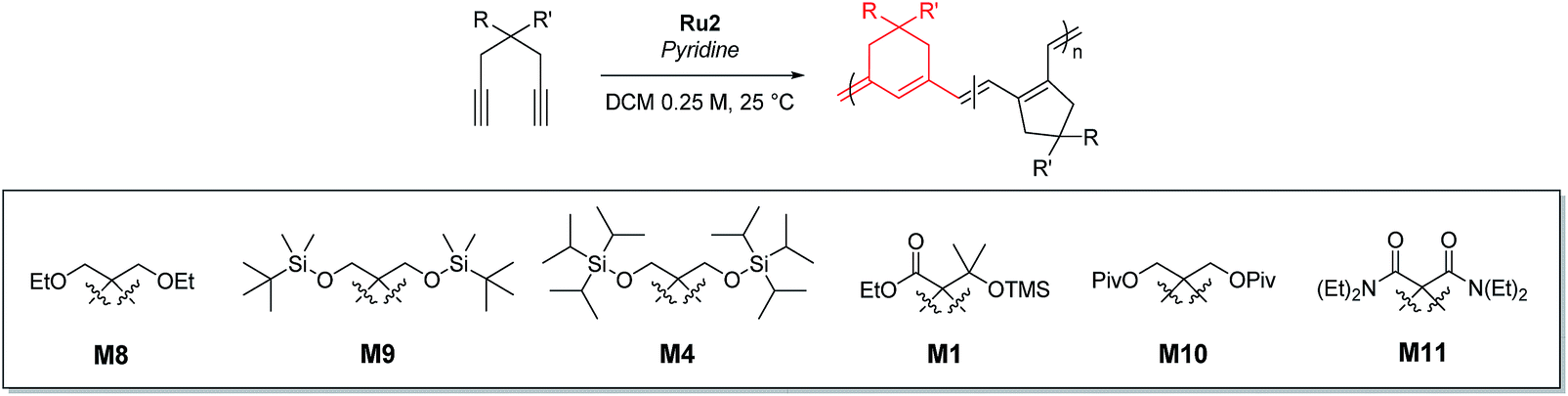
Using pyridine as an additive, we investigated controlled β-selective CP of various malonate-type monomers (Table 3). We observed the complete conversion of M5, with controlled Mn and narrow dispersity (1.15) for M/Is of up to 50, and 13C NMR analysis confirmed the high β-selectivity (94%), showing the six-membered ring on the polymer backbone (entry 1). For the higher M/I of 75, we obtained P5 with an expected Mn of 21 kDa, but a broadening appeared in the SEC trace with Đ of 1.50, even after several optimizations (entry 2). Additionally, a higher-molecular-weight shoulder corresponding to doubling of the molecular weight appeared in the SEC trace of the resulting polymer, presumably due to the bimolecular decomposition of Ru2 after prolonged reaction time.47 We were able to solve this problem by reducing the reaction time or concentration (see ESI† for details). Using monomer M6, containing an isopropyl group, CP proceeded with high reactivity for M/Is of 15 to 75 and a corresponding linear increase in Mn from 5.7 to 24.4 kDa, while retaining high β-selectivity (94%) and narrow Đ values (<1.15), except for the highest DP polymer (M/I of 75) where a severe broadening in the SEC trace was observed (entries 3–6, Fig. 6a). Taking a lesson from our previous work that introducing a sterically bulkier substituent improved both the stability of the propagating species and β-selectivity, we chose monomer M3 with a much bulkier tert-butyl group, which was polymerized to yield P3 with a higher β-selectivity of 97%. More importantly, we observed improved polymerization efficiency and controllability, generating P3 with a linear increase in Mn up to 41 kDa and narrow dispersities (Đ of 1.06–1.30 for M/Is 15–100, entries 7–11, Fig. 6b and d). Maximizing the steric bulkiness by introducing an adamantyl group in M7, we successfully conducted CP with complete conversion, producing P7 with a linear increase in Mn (6–27 kDa) and narrow Đ (1.12–1.23) for M/Is of 15 to 75 (entries 12–15, Fig. 6c). Notably, P7 contained only six-membered ring repeat units via exclusive β-addition, as determined by 13C NMR analysis. To broaden the monomer scope, we tested CP using Ru2 and monomers containing various non-malonate functional groups for M/Is of 30 and 50 (Table 4). CP of M8, containing the small ethyl ether group, showed high reactivity with complete conversion, and a high β-selectivity of 93%, but uncontrolled Mn with relatively broad dispersities (entries 1 and 2). Bulky M9 and M4, containing silylether groups, showed not only excellent reactivity and β-selectivity (99%) but also great controllability with narrow Đs, except for a slight broadening in the CP of M4, with a M/I of 50 (entries 3–6). M1 was another successful example with an excellent conversion for both M/Is of 30 and 50, producing P1 with a controlled Mn and narrow dispersity (<1.20), along with an excellent β-selectivity of 99% (entries 7 and 8). Furthermore, M10, with a pivaloyl group, reacted efficiently with excellent conversion, controlled Mn, and narrow dispersity (<1.20) for both M/Is of 30 and 50, despite showing a moderate β-selectivity of 74% (entries 9 and 10). Lastly, amide group-containing M11 showed complete conversion for an M/I of 30 with a narrow Đ and a good β-selectivity of 84%, but the reactivity decreased at higher M/I, resulting in only 74% conversion and uncontrolled Mn and Đ (entries 11 and 12).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Monomer | M/I/add | Temp. (°C) | Time | Conv.a (%) | Yieldb (%) | M n,theo (kDa) | M n (kDa) | Đ | β-selectivityd (%) |
| a Determined by 1H NMR. b Precipitated in hexane at −78 °C. c Determined by THF SEC calibrated using polystyrene standards. d Determined by 13C NMR. | ||||||||||
| 1 | M5 | 50/1/10 | 25 | 15 min | >99 | 86 | 11.8 | 15.5 | 1.15 | 94 |
| 2 | 75/1/10 | 20 | 30 min | 92 | 87 | 16.3 | 20.7 | 1.50 | ||
| 3 | M6 | 15/1/5 | 25 | 3 min | >99 | 86 | 4.0 | 5.7 | 1.10 | |
| 4 | 30/1/10 | 25 | 10 min | >99 | 64 | 7.9 | 10.2 | 1.10 | ||
| 5 | 50/1/10 | 20 | 30 min | >99 | 85 | 13.2 | 19.1 | 1.15 | 94 | |
| 6 | 75/1/10 | 15 | 4 h | 85 | 75 | 16.9 | 24.4 | 1.94 | ||
| 7 | M3 | 15/1/5 | 25 | 10 min | >99 | 56 | 4.4 | 6.6 | 1.07 | |
| 8 | 30/1/10 | 25 | 30 min | >99 | 70 | 8.8 | 11.4 | 1.06 | ||
| 9 | 50/1/15 | 25 | 2 h | >99 | 81 | 14.6 | 15.3 | 1.14 | 97 | |
| 10 | 75/1/20 | 25 | 3 h | >99 | 82 | 21.9 | 24.2 | 1.15 | ||
| 11 | 100/1/25 | 25 | 4 h | >99 | 97 | 29.2 | 41.0 | 1.30 | ||
| 12 | M7 | 15/1/5 | 25 | 10 min | >99 | 62 | 6.7 | 5.6 | 1.08 | |
| 13 | 30/1/10 | 25 | 20 min | >99 | 72 | 13.5 | 11.0 | 1.12 | ||
| 14 | 50/1/15 | 25 | 2 h | >99 | 86 | 22.4 | 21.0 | 1.19 | >99 | |
| 15 | 75/1/10 | 15 | 4 h | >99 | 76 | 33.7 | 26.6 | 1.23 | ||

|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Monomer | M/I/add | Time (h) | Conv.a (%) | Yieldb (%) | M n,theo (kDa) | M n (kDa) | Đ | β-selectivityd (%) |
| a Determined by 1H NMR. b Precipitated in MeOH at −78 °C. c Determined by THF SEC calibrated using polystyrene standards. d Determined by 13C NMR. e Precipitated in hexane at −78 °C. f Conducted at 15 °C. | |||||||||
| 1 | M8 | 30/1/10 | 0.5 | >99 | 83 | 6.3 | 8.1 | 1.36 | 93 |
| 2 | 50/1/15 | 1 | >99 | 85 | 10.4 | 9.4 | 1.65 | ||
| 3 | M9 | 30/1/10 | 1 | >99 | 82 | 11.4 | 15.4 | 1.11 | >99 |
| 4 | 50/1/15 | 2 | >99 | 73 | 19.0 | 26.5 | 1.17 | ||
| 5 | M4 | 30/1/10 | 2 | >99 | 66 | 14.0 | 16.3 | 1.11 | >99 |
| 6 | 50/1/15 | 3 | 97 | 60 | 22.6 | 26.2 | 1.35 | ||
| 7 | M1 | 30/1/10 | 1 | >99 | 61 | 8.8 | 7.8 | 1.20 | >99 |
| 8 | 50/1/15 | 3 | >99 | 77 | 14.7 | 14.2 | 1.18 | ||
| 9 | M10 | 30/1/10 | 2 | >99 | 75 | 9.6 | 11.3 | 1.11 | 74 |
| 10 | 50/1/15 | 4 | >99 | 78 | 16.0 | 21.7 | 1.18 | ||
| 11e | M11 | 30/1/10 | 1.5 | >99 | 99 | 8.7 | 8.4 | 1.18 | 84 |
| 12e,f | 50/1/15 | 2 | 74 | 65 | 10.8 | 7.7 | 1.37 | ||
Based on the successfully controlled polymerization, we tried another block copolymerization to determine if living polymerization was feasible with Ru2. Initially, M9 was used as the first monomer to form P9 with an Mn of 5.4 kDa and Đ of 1.09, after which 15 equiv. of M3 was added to successfully produce P9-b-P3 (Mn 15.7 kDa, Đ 1.17), detected by SEC analysis with a complete shift of the traces (Scheme 3a and b). Using the same procedure, we synthesized another diblock copolymer using M3 as the first monomer, followed by the addition of 15 equiv. of M7 as the second monomer, to form P3-b-P7 with an Mn of 11.6 kDa and a narrow dispersity of 1.08 (Scheme 3c). To this living polymer end, we further added 15 equiv. of a third monomer, M11, which resulted in complete conversion to produce P3-b-P7-b-P11, showing a complete shift of SEC trace, with Mn of 15.2 kDa and Đ of 1.08 in a good yield of 88% (Scheme 3d). This remarkable success with diblock and triblock copolymerizations with a broad monomer scope and narrow Đ suggests the superior versatility of Ru2 in β-selective living/controlled polymerization, compared with Ru1, which had a narrower scope.
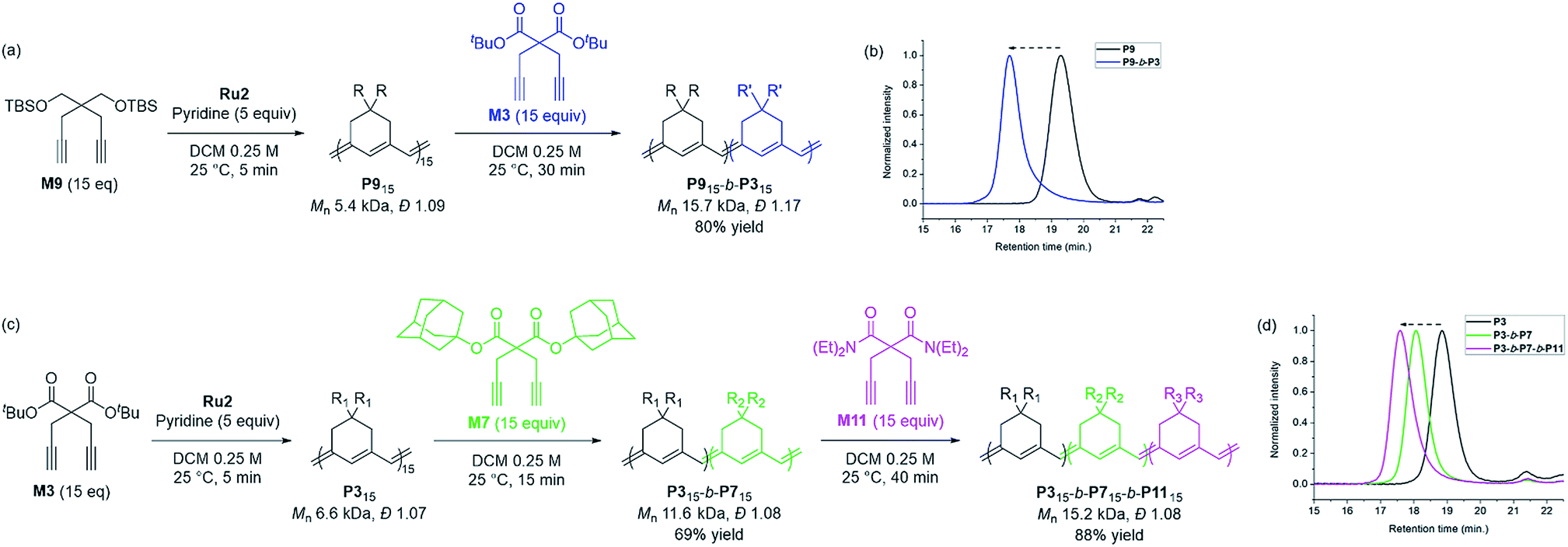 | ||
| Scheme 3 Diblock and triblock copolymerization for exclusively β-selective conjugated polyenes (a and c), and THF-SEC traces of the corresponding polymers (b and d). | ||
Conclusions
In conclusion, we successfully performed β-selective living/controlled CP using two Ru dithiolate catalysts to prepare various conjugated polyenes, bearing mostly six-membered ring repeat units. The high controllability was achieved by maximizing the steric demands on either the monomer or the catalyst, which improved the stability of the propagating species with the aid of pyridine additives. Ru1 containing less bulky NHC ligands required the extremely bulky monomer M2 for controlled polymerization. On the other hand, Ru2, already containing a bulky ligand, demonstrated much faster initiation and intrinsically greater stabilization of the propagating species, whereby a versatile living polymerization with a broader monomer scope was possible. Furthermore, we systematically studied the effect of pyridine additives and changing the catalyst by in situ1H NMR kinetic experiments. Particularly, we found that ligands which coordinate more strongly to Ru2 better stabilized the propagating species and promoted better living/controlled CP. More significantly, we achieved several β-selective diblock and triblock copolymerizations, for the first time. In short, we achieved a rare β-selective living CP by analyzing the mechanistic details and kinetic parameters, and we expect this study to increase the insight into and versatility of Ru-catalyzed polymerizations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from Creative Research Initiative Program and the Nano-Material Technology Development Program through NRF is acknowledged. We thank NCIRF at SNU for in situ kinetic experiments using 1H NMR.Notes and references
- G. I. Peterson, S. Yang and T.-L. Choi, Acc. Chem. Res., 2019, 52, 4994 CrossRef PubMed.
- J. K. Stille and D. A. Frey, J. Am. Chem. Soc., 1961, 83, 1697 CrossRef CAS.
- H. W. Gibson, F. C. Bailey, A. J. Epstein, H. Rommelmann, S. Kaplan, J. Harbour, X. Q. Yang, D. B. Tanner and J. M. Pochan, J. Am. Chem. Soc., 1983, 105, 4417 CrossRef CAS.
- Y. H. Kim, Y. S. Gal, U. Y. Kim and S. K. Choi, Macromolecules, 1988, 21, 1991 CrossRef CAS.
- M. S. Ryoo, W. C. Lee and S. K. Choi, Macromolecules, 1990, 23, 3029 CrossRef CAS.
- M. S. Jang, S. K. Kwon and S. K. Choi, Macromolecules, 1990, 23, 4135 CrossRef CAS.
- K. L. Kang, H. N. Cho, K. Y. Choi, S. K. Choi and S. H. Kim, Macromolecules, 1993, 26, 4539 CrossRef CAS.
- Y. S. Gal, S. H. Jin, J. W. Park, W. C. Lee, H. S. Lee and S. Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4101 CrossRef CAS.
- H. H. Fox and R. R. Schrock, Organometallics, 1992, 11, 2763 CrossRef CAS.
- H. H. Fox, M. O. Wolf, R. O'Dell, B. L. Lin, R. R. Schrock and M. S. Wrighton, J. Am. Chem. Soc., 1994, 116, 2827 CrossRef CAS.
- E.-H. Kang, I. S. Lee and T.-L. Choi, J. Am. Chem. Soc., 2011, 133, 11904 CrossRef CAS PubMed.
- I. S. Lee, E.-H. Kang, H. Park and T.-L. Choi, Chem. Sci., 2012, 3, 761 RSC.
- E.-H. Kang, S. Y. Yu, I. S. Lee, S. E. Park and T.-L. Choi, J. Am. Chem. Soc., 2014, 136, 10508 CrossRef CAS PubMed.
- C. Kang, E.-H. Kang and T.-L. Choi, Macromolecules, 2017, 50, 3153 CrossRef CAS.
- J.-A. Song, S. Park, T.-S. Kim and T.-L. Choi, ACS Macro Lett., 2014, 3, 795 CrossRef CAS.
- J. A. Song and T.-L. Choi, Macromolecules, 2017, 50, 2724 CrossRef CAS.
- J. O. Krause, M. T. Zarka, U. Anders, R. Weberskirch, O. Nuyken and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965 CrossRef CAS PubMed.
- J. O. Krause, O. Nuyken and M. R. Buchmeiser, Chem.–Eur. J., 2004, 10, 2029 CrossRef CAS PubMed.
- T. S. Halbach, J. O. Krause, O. Nuyken and M. R. Buchmeiser, Macromol. Rapid Commun., 2005, 26, 784 CrossRef CAS.
- M. G. Mayershofer, O. Nuyken and M. R. Buchmeiser, Macromolecules, 2006, 39, 3484 CrossRef CAS.
- P. S. Kumar, K. Wurst and M. R. Buchmeiser, J. Am. Chem. Soc., 2009, 131, 387 CrossRef CAS PubMed.
- M. Sudheendran, M. Horecha, A. Kiriy, S. A. Gevorgyan, F. C. Krebs and M. R. Buchmeiser, Polym. Chem., 2013, 4, 1590 RSC.
- U. Anders, O. Nuyken, M. R. Buchmeiser and K. Wurst, Angew. Chem., Int. Ed., 2002, 41, 4044 CrossRef CAS PubMed.
- U. Anders, M. Wagner, O. Nuyken and M. R. Buchmeiser, Macromolecules, 2003, 36, 2668 CrossRef CAS.
- M. R. Buchmeiser, S. Sen, J. Unold and W. Frey, Angew. Chem., Int. Ed., 2014, 53, 9384 CrossRef CAS PubMed.
- K. Herz, J. Unold, J. Hänle, R. Schowner, S. Sen, W. Frey and M. R. Buchmeiser, Macromolecules, 2015, 48, 4768 CrossRef CAS.
- M. R. Buchmeiser, Chem.–Eur. J., 2018, 24, 14295 CrossRef CAS PubMed.
- Y. S. Vygodskii, A. S. Shaplov, E. I. Lozinskaya, P. S. Vlasov, I. A. Malyshkina, N. D. Gavrilova, P. Santhosh Kumar and M. R. Buchmeiser, Macromolecules, 2008, 41, 1919 CrossRef CAS.
- B. Autenrieth, E. B. Anderson, D. Wang and M. R. Buchmeiser, Macromol. Chem. Phys., 2013, 214, 33 CrossRef CAS.
- J. Kim, E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2012, 1, 1090 CrossRef CAS.
- E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2013, 2, 780 CrossRef CAS.
- S. Yang, S. Shin, I. Choi, J. Lee and T.-L. Choi, J. Am. Chem. Soc., 2017, 139, 3082 CrossRef CAS PubMed.
- S. Shin, M.-L. Gu, C.-Y. Yu, J. Jeon, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 475 CrossRef CAS PubMed.
- W. Song, H. Han, X. Liao, R. Sun, J. Wu and M. Xie, Macromolecules, 2014, 47, 6181 CrossRef CAS.
- W. Liu, X. Liao, Y. Li, Q. Zhao, M. Xie and R. Sun, Chem. Commun., 2015, 51, 15320 RSC.
- J. Wu, H. Li, D. Zhou, X. Liao, M. Xie and R. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 485 CrossRef CAS.
- F. J. Schattenmann, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1996, 118, 3295 CrossRef CAS.
- F. J. Schattenmann and R. R. Schrock, Macromolecules, 1996, 29, 8990 CrossRef CAS.
- B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693 CrossRef CAS PubMed.
- K. Jung, E.-H. Kang, J.-H. Sohn and T.-L. Choi, J. Am. Chem. Soc., 2016, 138, 11227 CrossRef CAS PubMed.
- M. J. Koh, R. K. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, Nature, 2015, 517, 181 CrossRef CAS PubMed.
- K. Jung, K. Kim, J.-C. Sung, T. S. Ahmed, S. H. Hong, R. H. Grubbs and T.-L. Choi, Macromolecules, 2018, 51, 4564 CrossRef CAS.
- H. Jung, K. Jung, M. Hong, S. Kwon, K. Kim, S. H. Hong, T.-L. Choi and M. H. Baik, J. Am. Chem. Soc., 2018, 140, 834 CrossRef CAS PubMed.
- S. J. P'Poo and H.-J. Schanz, J. Am. Chem. Soc., 2007, 129, 14200 CrossRef PubMed.
- F. C. Courchay, J. C. Sworen and K. B. Wagener, Macromolecules, 2003, 36, 8231 CrossRef CAS.
- T. S. Ahmed and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 11213 CrossRef CAS PubMed.
- G. A. Bailey, M. Foscato, C. S. Higman, C. S. Day, V. R. Jensen and D. E. Fogg, J. Am. Chem. Soc., 2018, 140, 6931 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterizations, 1H and 13C NMR spectra for new compounds and polymers, SEC traces, and other supporting experi-ments. See DOI: 10.1039/c9sc01326a |
| ‡ Without pyridine additives, the propagating carbenes from Ru1 were not observable, so it was impossible to compare the stability of the propagating carbene with and without additives. |
| This journal is © The Royal Society of Chemistry 2019 |