
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: interdependency of green rust transformation and the partitioning and binding mode of arsenic†
C. M.
van Genuchten
 *ab,
T.
Behrends
b and
K.
Dideriksen‡
a
*ab,
T.
Behrends
b and
K.
Dideriksen‡
a
aGeological Survey of Denmark and Greenland (GEUS), Copenhagen, Denmark. E-mail: cvg@geus.dk
bDepartment of Earth Sciences – Geochemistry, Faculty of Geosciences, Utrecht University, Utrecht, The Netherlands
First published on 22nd July 2019
Abstract
We investigated the impact of aging-induced structural modifications of carbonate green rust (GR), a mixed valent Fe(II,III) (hydr)oxide with a high oxyanion sorption affinity, on the partitioning and binding mode of arsenic (As). Suspensions of carbonate GR were produced in the presence of As(V) or As(III) (i.e. co-precipitated with As(III) or As(V)) and aged in anoxic and oxic conditions for up to a year. We tracked aqueous As over time and characterized the solid phase by X-ray absorption spectroscopy (XAS). In experiments with initial As(V) (4500 μg L−1, As/Fe = 2 mol%), the fresh GR suspension sorbed >99% of the initial As, resulting in approximately 14 ± 8 μg L−1 residual dissolved As. Anoxic aging of the As(V)-laden GR for a month increased aqueous As to >60 μg L−1, which was coupled to an increase in GR structural order revealed by Fe K-edge XAS. Further anoxic aging up to a year transformed As(V)-laden GR into magnetite and decreased significantly the aqueous As to <2 μg L−1. The As binding mode was also modified during GR transformation to magnetite from sorption to GR particle edges to As substitution for tetrahedral Fe in the magnetite structure. These GR structural modifications altered the ratio of As partitioning to the solid (μg As/mg Fe) and liquid (μg As per L) phase from 2.0 to 0.4 to 14 L mg−1 for the fresh, month, and year aged suspensions, respectively. Similar trends in GR transformation and As partitioning during anoxic aging were observed for As(III)-laden suspensions, but occurred on more rapid timescales: As(III)-laden GR transformed to magnetite after a day of anoxic aging. In oxic aging experiments, rapid GR oxidation by dissolved oxygen to Fe(III) precipitates required only an hour for both As(V) and As(III) experiments, with lepidocrocite favored in As(V) experiments and hydrous ferric oxide favored in As(III) experiments. Aqueous As during GR oxidation decreased to <10 μg L−1 for both As(V) and As(III) series. Knowledge of this interdependence between GR aging products and oxyanion fate improves biogeochemical models of contaminant and nutrient dynamics during Fe cycling and can be used to design more effective arsenic remediation strategies that rely on arsenic sorption to GR.
Environmental significanceGreen rust (GR) is a mixed valent Fe(II,III) hydroxide that can modify contaminant (e.g. arsenic) and nutrient (e.g. phosphate) mobility by adsorption to particle edges. However, GR transforms readily into different Fe (oxyhydr)oxide phases with unknown impacts on chemical partitioning. We tracked the mineral phase, aqueous arsenic concentration, and solid phase arsenic speciation during aging of As(V)- and As(III)-laden GR from an hour to a year. When aged anoxically for a month, GR crystallized, which increased aqueous arsenic to >60 μg L−1. Further anoxic aging up to a year yielded magnetite and decreased aqueous arsenic to <5 μg L−1. Oxic aging rapidly (<1 hour) transformed GR to Fe(III) (oxyhydr)oxides, producing arsenic levels <1 μg L−1, well below the WHO recommended drinking water limit. |
1. Introduction
Green rust (GR) is a mixed valent Fe(II,III) hydroxide that consists of sheets of edge-sharing Fe(OH)6 octahedra with interlayer water and anions (e.g. Cl−, SO42−, CO32−).1,2 GR has been reported in suboxic soils and sediments that are exposed to high Fe(II) fluxes3,4 and it can be associated with microbial activity.5,6 The formation of GR is also frequently observed in engineered systems, particularly as a result of Fe(0) corrosion.7–9 In recent years, GR has gained attention because it is a powerful reductant capable of abiotically transforming many toxic organic10,11 and inorganic contaminants.12,13 In addition, the typically nanocrystalline structure of GR14 gives rise to a high density of surface sites that effectively bind oxyanions, such as arsenic (As).15,16 The abundance of reactive sorption sites on GR particle edges makes GR an ideal candidate for remediation of water and soils contaminated by As. For example, the controlled production of GR can be used to treat drinking water containing arsenic17,18 and in situ GR formation can be induced to improve the quality of soils or aquifers with high arsenic content.9,19,20 However, GR is only stable in a narrow window of redox potential, pH, and solution composition, and in most conditions in natural and engineered environments, GR transforms readily into other Fe (oxyhydr)oxide minerals.21,22 While GR has been shown to be an effective sorbent for As and other oxyanions, less is known about the partitioning of As during GR transformation and how the presence of As impacts the GR transformation products.Green rust can transform into a variety of products depending on aging time and solution conditions (i.e. the dissolved oxygen (DO) concentration and ionic composition). One of the most common transformation products in anoxic conditions is magnetite,5,23 a highly stable, mixed valent Fe(II,III) oxide that consists of Fe in both tetrahedral and octahedral coordination.24 Transformation under anoxic conditions is relevant in redoximorphic soils and sediments when redox conditions that facilitate GR formation remain stable. The anoxic transformation of GR to magnetite has been reported to occur on timescales of hours to weeks in abiotic systems,8,25 with microbial activity and alkaline pH increasing the transformation rates.5,26 However, strongly sorbing oxyanions, such as phosphate (P) and silicate (Si), have been shown to slow down the kinetics of magnetite formation from GR to over several months.23,25,27 Oxic transformation of GR to form different Fe(III) (oxyhydr)oxides occurs quite rapidly compared to anoxic transformation,1 which is attributed to the high instability of Fe(II) in the presence of DO. Transformation under oxic conditions is relevant to natural systems exposed to temporal redox variations, such as paddy soils. Complete GR oxidation by DO to goethite and lepidocrocite has been reported in less than an hour, with increased DO concentrations favoring lepidocrocite over goethite.26 Similar to anoxic GR transformation, strongly sorbing oxyanions can modify the kinetics and pathways of GR oxidation. Phosphate and Si decreased the rate of GR oxidation by DO, stabilizing the GR structure for several hours in air.27 Furthermore, P and Si modified the end products of GR oxidation by DO, with mixtures of poorly-crystalline hydrous ferric oxide and fully oxidized, ferric GR favored instead of lepidocrocite and goethite.23,27
These diverse transformations can alter key properties of the solid phase that govern the sorption reactivity, including specific surface area, coordination of surface sites, surface charge, and the average Fe oxidation state. Furthermore, if GR transforms into magnetite, aging would produce tetrahedral Fe sites in the mineral structure, which can be occupied by tetrahedral oxyanions, leading to strong, multinuclear sorption complexes.28–30 Despite the potential for changes in sorption reactivity induced by GR transformation, systematic investigations of the fate of oxyanions during GR aging and, in turn, the impact of oxyanions on GR transformation pathways, are absent from the literature. Understanding how As partitions to the solid or liquid phase and how the As sorption configuration changes during GR aging is critical since GR transformation could result in aqueous As levels below or above target drinking water limits (i.e. the World Health Organization 10 μg L−1 provisional limit) if GR is implemented in an Fe-based As remediation strategy.
In this work, we generated carbonate green rust (GR-CO3) in the presence of As(V) or As(III) and aged the As-laden suspensions in anoxic and oxic conditions from an hour to a year. Arsenic was selected as the oxyanion probe compound for several reasons. First, since As is redox active, changes in As oxidation state can provide information on possible redox reactions involving Fe that occur during GR transformation. Second, As yields high quality X-ray absorption spectra to facilitate identification of sorption configuration. Third, the sorption reactivity of As(V) and As(III) are comparable to other environmentally relevant oxyanions (i.e. P and Si), which aids in extending the results of this work to systems containing other oxyanions.31–33 We integrated aqueous As measurements during GR aging experiments with molecular-scale characterization of the Fe mineral phase and As bonding environment by Fe and As K-edge X-ray absorption spectroscopy. Our results advance the understanding of the fate of sorbed oxyanions during the transformation of GR. This information is critical to refine models of Fe and oxyanion cycling in natural systems exposed to spatial and temporal redox variation (i.e. paddy soils and marine sediments) and can be used to improve As remediation strategies that rely on GR as a reactive sorbent.
2. Methods
2.1. Synthesis of green rust suspensions
Suspensions of GR were generated by Fe(0) electrolysis, also called Fe(0) electrocoagulation (EC), following previously reported synthesis procedures.34 Briefly, GR suspensions were produced in 200 mL electrolyte solutions open to the atmosphere by generating 3 mM total Fe(II) by Fe(0) electrolysis. The initial solutions all contained 2 mM NaHCO3 and 10 mM NaCl, which were added from 0.3 M NaHCO3 and 0.5 M NaCl stock solutions, and the pH was set to 8 before electrolysis. The solutions also contained either 4500 μg L−1 As(V) (60 μM, As/Fe = 2 mol%) or 1125 μg L−1 As(III) (15 μM, As/Fe = 0.5 mol%). The initial DO of the electrolyte solution was set to 3.0 mg L−1 by bubbling N2(g) and was measured with a Hach Luminescent Dissolved Oxygen (LDO) probe. The production of GR was carried out by applying an electric current of 200 mA to the Fe(0) EC cell, which corresponds to an Fe(II) production rate of 300 μM Fe(II) per min (10 min total electrolysis time). The consumption of DO by electrochemically generated Fe(II) resulted in a final DO <0.1 mg L−1, which is required for mixed valent Fe(II,III) (hydr)oxide formation.34 During the production of GR by Fe(0) electrolysis, the pH was adjusted manually using pipettes with small additions of 0.01 or 0.1 M HCl or NaOH to maintain the initial value of 8.0 ± 0.3. Immediately after electrolysis, samples were collected (t = 0 in the aging series) to measure the aqueous As concentration and to characterize the initial solid phase. X-ray diffraction measurements (Fig. ESI 1†) confirmed that the initial As-laden suspensions consisted of GR-CO3 based on the positions of the basal Bragg peaks. No evidence of chloride GR (GR-Cl) nor arsenic GR (GR-As) was observed, consistent with previous work.342.2 Aging experiments
Several 200 mL batches of As(V) or As(III)-laden GR were prepared for the aging experiments because sampling for aqueous As measurements and solid phase characterization during aging (described below) required approximately 25–30 mL per time point. For anoxic aging experiments, GR suspensions were produced and transferred immediately to an anoxic chamber (95% N2, 5% H2). The 200 mL GR suspensions (3 mM total Fe) were divided into separate smaller 50 mL containers without any additional electrolyte solutions and were sealed and aged within the anoxic chamber. For oxic aging experiments, freshly prepared GR suspensions were transferred into 50 mL containers and the DO was set to ≈9.0 mg L−1 (air saturated conditions, verified using the LDO sensor). After sealing each 50 mL container, no additional methods were used to keep the suspensions sterile. The separate 50 mL containers of GR suspensions were well mixed with a rotary shaker at 150 ± 50 rpm. Samples for aqueous As measurements were collected at various time points from an hour to a year by withdrawing aliquots of the suspensions in the anoxic chamber (anoxic aging) or open to the atmosphere (oxic aging). Samples were passed through 0.22 or 0.45 μm filters and the filtered solution was reserved for analysis of aqueous As. Details on the aqueous arsenic measurements are provided in the ESI.† The dissolved Fe in the fresh GR suspensions was also measured. The solution pH was measured during anoxic and oxic aging and was allowed to drift from the initial value of 8.0 through the experiments. Each aging experiment was performed at least twice, with early time points (<1 month) replicated in independent experiments 3 or 4 times.2.3. X-ray absorption spectroscopy
As K-edge XAS data were collected at beam line 4-1 of the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, USA). Spectra were recorded at liquid nitrogen temperature (≈80 K) in fluorescence mode out to k of 13.5 or 14 Å−1 using a Lytle detector or solid-state PIPS detector. The vertical dimension of the X-ray beam during data collection was 1 mm and the horizontal dimension was 4 to 8 mm. To prevent second-order harmonics, the X-ray beam was detuned 50%. Beam calibration was performed by setting the maximum of the first derivative of an Au(0) foil to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 919 eV. The XANES was measured with 0.35 eV steps and step sizes of 0.05 Å−1 were used for the EXAFS region. Four to 10 scans were collected for each sample, depending on data quality.
919 eV. The XANES was measured with 0.35 eV steps and step sizes of 0.05 Å−1 were used for the EXAFS region. Four to 10 scans were collected for each sample, depending on data quality.
During data collection for both Fe and As K-edge spectra, changes in line shape and peak position indicative of beam-induced redox reactions were examined and no artifacts were observed. Spectra were aligned, averaged, and background-subtracted using SixPack software37 following standard methods described previously.38 The EXAFS spectra were extracted and given k3-weighting and were Fourier-transformed over the k-range 3 to 12 or 13 Å−1 using a Kaiser–Bessel window with dk of 3 Å−1.
860 to 11880 eV, with only non-negative percentages allowed, but without constraining the sum of the components to 1. Since the selection of particular references used in the LCFs can impact the fit-derived fractions,42 we performed three sets of XANES LCFs for each sample using three sets of references: As(III) and As(V) adsorbed to (i) green rust, (ii) 2-line ferrihydrite and (iii) magnetite. The fraction of As(III) and As(V) in each experimental sample is reported as the average and standard deviation of these three sets of LCFs. The spectra of the As(III) and As(V) adsorption references used in the LCFs were collected at the same beam line in identical conditions as the current data set. In addition to adsorption references, we report the spectrum of As(V) co-precipitated with magnetite (AsV Co-PPT), which was synthesized by mixing Fe(II) and Fe(III) salts in the presence of As(V) following previous procedures.43
45 and included As–O, As–O–O and As–Fe paths derived from the structure of scorodite.46 In preliminary fits, the CN and σ2 were found to be highly correlated, which produced high fit-derived standard errors in these fitting parameters. Therefore, following previous work,38,47 we constrained σ2 in the second shell fits (As–Fe path) to reduce the high correlations. We constrained σ2 to 0.009 for samples that contained a major fraction of green rust or magnetite in the Fe K-edge EXAFS data, whereas σ2 was set to 0.010 for samples that contained mainly Fe(III) precipitates. For all fits, the passive electron reduction parameter, S02, was set to 1.0, consistent with previous work.47,48 The goodness-of-fit was assessed based on the R-factor, which is reported along with fit-derived variables. The R-factor is defined as the mean square difference between the fit and the data on a point-by-point basis: 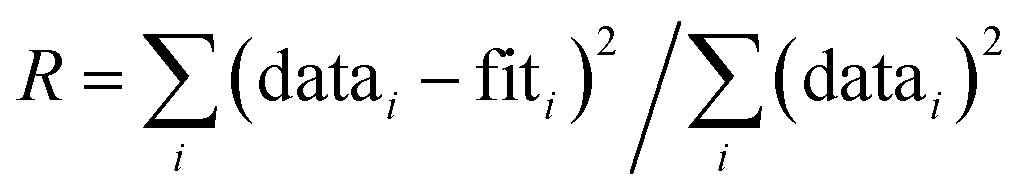 . An R-factor <0.05 is considered to reflect a reasonable fit.49
. An R-factor <0.05 is considered to reflect a reasonable fit.49
3. Results
3.1. Changes in aqueous arsenic during aging
Fig. 1 shows the changes in dissolved As concentration of As(V)- and As(III)-laden GR suspensions aged in anoxic and oxic conditions. For the As(V) series, the residual aqueous As of the fresh (i.e. t = 0) sample was 14 ± 8 μg L−1, which was >99% removal of the initial 4500 μg L−1 As(V) (60 μM). When the As(V)-laden GR was aged for a day anoxically, the aqueous As concentration increased to >60 μg L−1 and it remained at this level for up to a month of anoxic aging. However, after a year of anoxic aging, the aqueous As concentration decreased substantially to <2 μg L−1, the lowest As level in this aging series. These changes in aqueous As during GR aging correspond to ratios of As partitioning to the solid (μg As/mg Fe) and liquid (μg As per L) phase of 2.0, 0.4, and 14 L mg−1 for the fresh, month, and year aged samples, respectively. When As(V)-laden GR was aged in oxic conditions, the behavior of dissolved As differed from anoxic aging conditions. Rather than increasing on day and month timescales, dissolved As decreased rapidly from 14 ± 8 μg L−1 to <1 μg L−1 after an hour of aging (i.e. partitioning ratio increased from 2.0 to >25 L mg−1). The solution pH also decreased rapidly to <6 during oxic aging (Fig. ESI 2†) due to H+ production from Fe(II) oxidation, which contrasts the stable pH (7 to 8) in the anoxic aging series. The As concentration for the oxic aging series remained <1 μg L−1 throughout the duration of the experiment.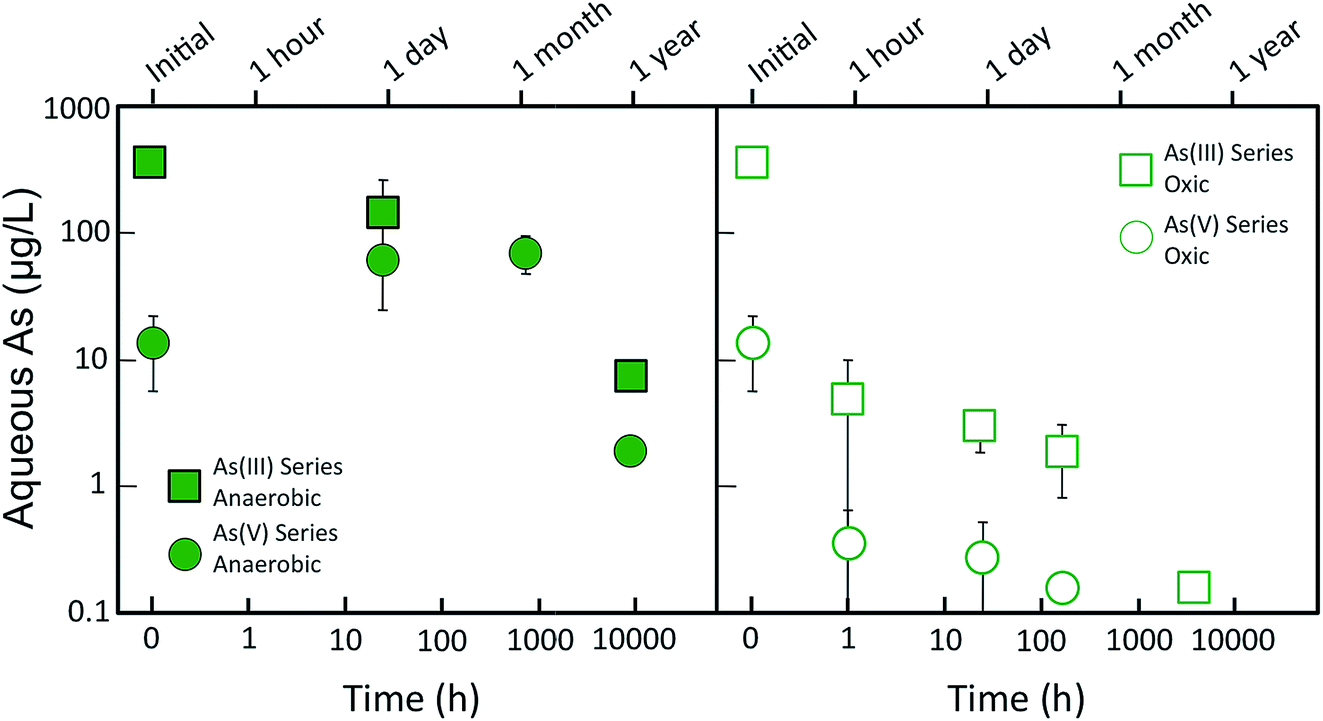 | ||
| Fig. 1 Aqueous As concentration during aging of green rust suspensions in anoxic (solid symbols) and oxic (open symbols) environments. The circles represent experiments with initial As(V) and the squares represent experiments with initial As(III). Data points and error bars represent the average and standard deviation of replicate aqueous As measurements, with some error bars smaller than the symbol. | ||
For the As(III) series, the residual As concentration for the fresh (i.e. t = 0) GR sample was 370 ± 16 μg L−1, which corresponded to 67% removal of the initial 1125 μg L−1 As(III) (15 μM). When As(III)-laden GR was aged anoxically for a day, the aqueous As concentration decreased to <150 μg L−1, which contrasts the behavior of aqueous As in the As(V) anoxic aging series. The As concentration decreased further when As(III)-laden GR was aged for a year, leading to a final As level below the WHO 10 μg L−1 limit. When the As(III) samples were aged oxically, aqueous As decreased to <10 μg L−1 after an hour. Although the pH of the anoxic As(III) series did not change significantly, the pH of the oxic aging series decreased to <6 after an hour (Fig. ESI 2†). Similar to the analogous As(V) samples, continued aging in oxic conditions further decreased aqueous As, with the lowest As concentration (<1 μg L−1) measured for the sample aged the longest (≈5 months).
3.2. Solid phase Fe speciation
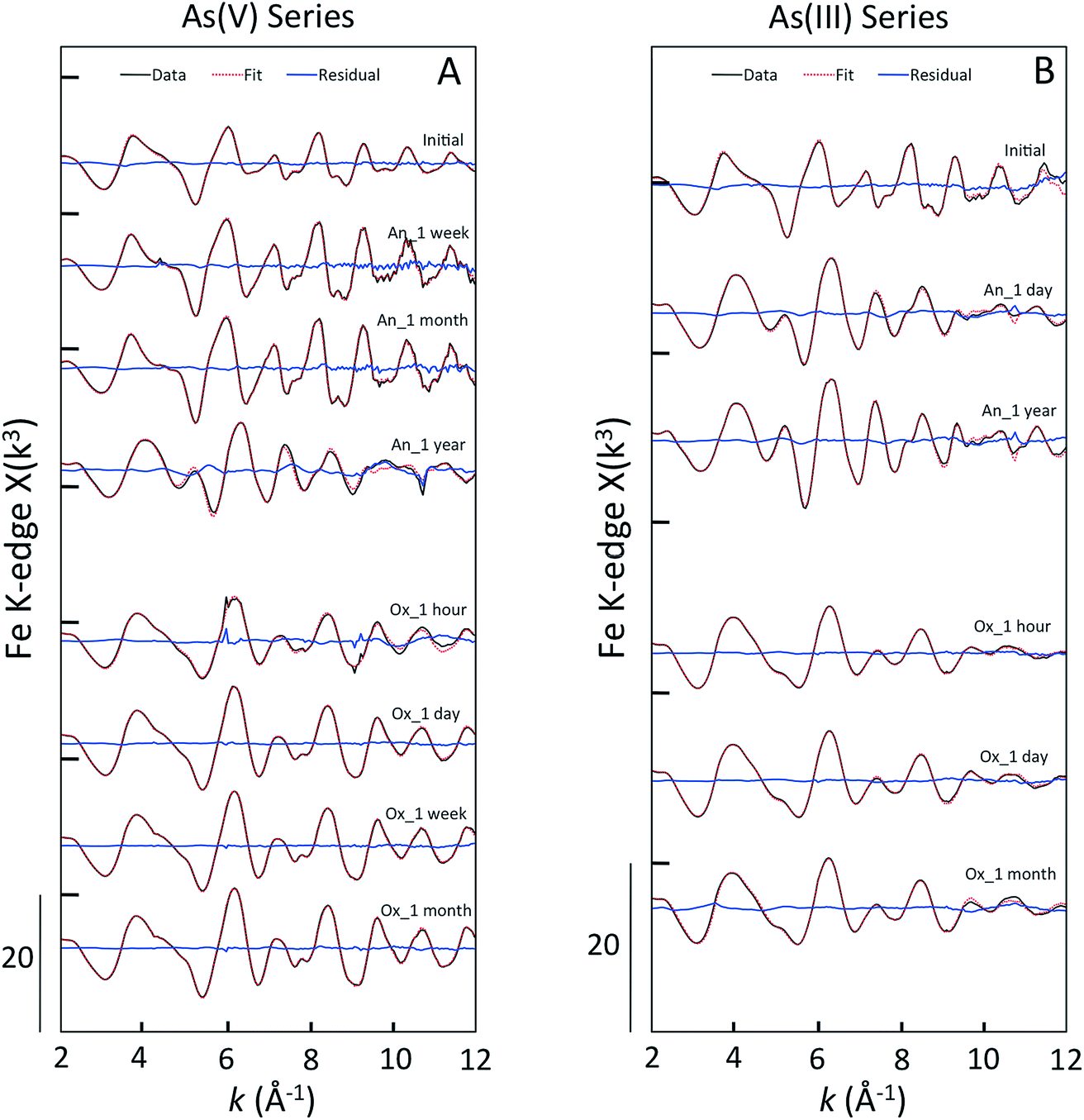 | ||
| Fig. 2 Fe K-edge EXAFS spectra of green rust aging samples in As(V) experiments (left panel) and As(III) experiments (right panel). The ITFA fitting output is shown in dotted red lines and the data is given in solid black lines. The fitting residual is given in solid blue lines. The anoxic aging series (e.g. An_1 week) are given above the oxic aging samples (e.g. Ox_1 hour). | ||
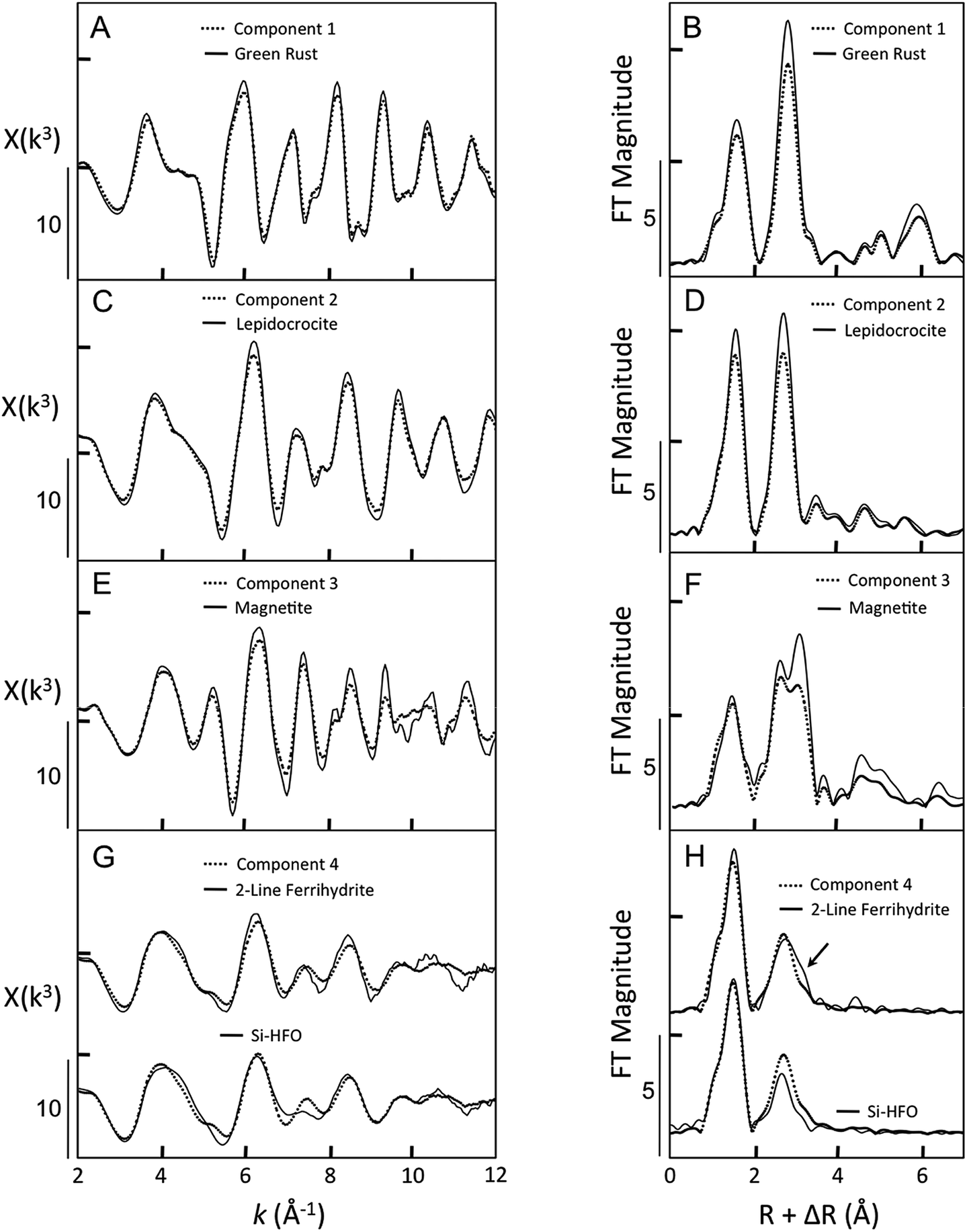 | ||
| Fig. 3 Comparison of ITFA-derived components and Fe K-edge EXAFS spectra of Fe-bearing reference materials. The EXAFS spectra are given in the left panel and the corresponding Fourier transforms are shown in the right panel. The dotted black lines represent the components and the solid black lines represent measured reference spectra. The arrow in H highlights features in the Fourier-transformed EXAFS spectrum of 2-line ferrihydrite that arise from corner-sharing Fe–Fe bonds. | ||
Component 1 was a clear match to the reference spectrum of GR based on several characteristic features (Fig. 3A), including the first asymmetric oscillation from 3.5 to 5.5 Å−1 and the subtle beat features near 7.8 Å−1 and 8.5 Å−1. The Fourier transforms of these spectra showed small differences in the amplitude of the second shell peak arising from Fe–Fe pairs from edge-sharing FeO6 octahedra, which suggests that Component 1 is a GR phase that is slightly less crystalline in the direction of the sheets than the reference GR. Component 2 strongly resembled the Fe K-edge EXAFS spectrum of magnetite, particularly the characteristic shape of the doublet in the first oscillation from 3.5 to 6.0 Å−1. In addition, Component 2 matched the subtle features of the magnetite reference spectrum at k > 8.0 Å−1, including the shoulder at 8.2 Å−1 and the peaks centered near 9.5, 10.7, and 11.3 Å−1. However, several oscillations in Component 2 had lower amplitudes relative to the magnetite reference spectrum. These differences are manifest in the Fourier transform as a lower amplitude of the shoulder at larger R in the second shell peak, which is indicative of Fe–Fe pairs linked through corner sharing polyhedra. Therefore, these results suggest that Component 2 is slightly less crystalline than the magnetite reference.
Component 3 reproduced all major and minor features of the EXAFS spectrum of lepidocrocite, such as the asymmetric first oscillation from 3.5 to 5.5 Å−1, the small beat near 7.8 Å−1, and the intense oscillation at k > 8.5 Å−1. The major peaks of the reference lepidocrocite spectrum have slightly larger amplitude than those of Component 3. This difference in amplitude corresponded to a lower amplitude of the first and second shell peaks of the Fourier transform, which is consistent with slightly higher crystallinity in the reference lepidocrocite spectrum relative to Component 3. Component 4 displayed several features characteristic of poorly-ordered Fe(III) precipitates, including a relatively symmetric first oscillation from 3.2 to 5.2 Å−1, a small beat feature near 5.5 Å−1, and broad oscillations with low amplitude at k > 8.5 Å−1. Although there is good agreement between Component 4 and the EXAFS spectrum of 2-line ferrihydrite (2LFh), some mismatches are present in the oscillations at 8.2 Å−1 and at k > 10 Å−1. In these regions, Component 4 is a better match to the Fe K-edge EXAFS spectrum of silicate-rich hydrous ferric oxide (Si-HFO), which was prepared by Fe(III) hydrolysis in the presence of strongly-sorbing Si. As shown in the Fourier transform, the shape and amplitude of the second shell peak of Component 4 lie intermediate between that of 2LFh and Si-HFO. Since the structures of 2LFh and Si-HFO differ mainly in the presence of more corner-sharing Fe–Fe bonds for 2LFh (indicated by the arrow in Fig. 3H), Component 4 represents a disordered Fe(III) precipitate with fewer corner-sharing Fe–Fe bonds than 2LFh, but higher Fe–Fe polymerization than Si-HFO.
Considering the similarity of the ITFA-derived independent components to the EXAFS spectra of Fe-bearing reference minerals, we herein refer to Component 1 as GR, Component 2 as magnetite, Component 3 as lepidocrocite, and Component 4 as hydrous ferric oxide (HFO).
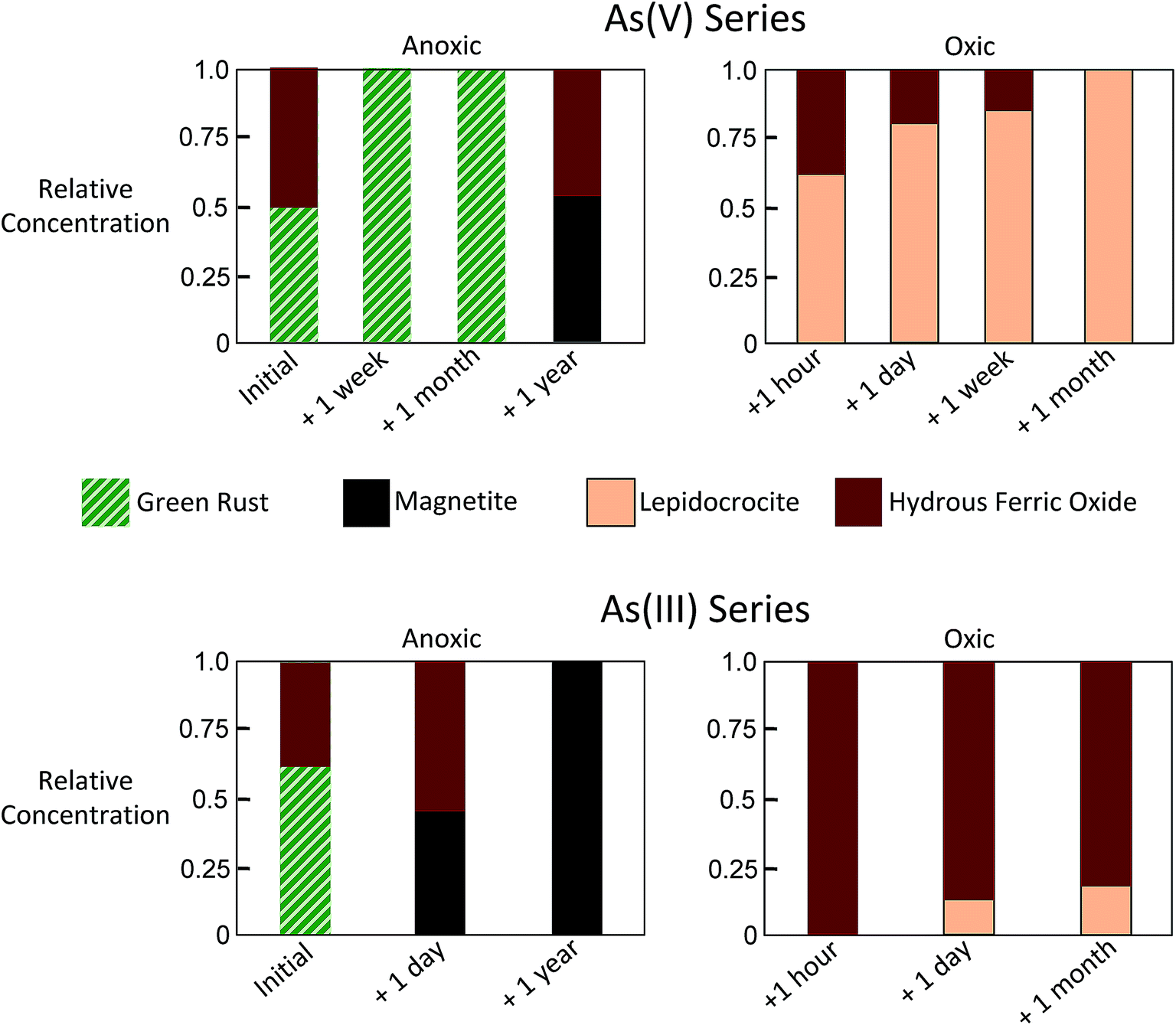 | ||
| Fig. 4 Relative concentrations of Component 1 (green rust), Component 2 (magnetite), Component 3 (lepidocrocite) and Component 4 (hydrous ferric oxide) derived by ITFA. | ||
For the As(III) series, the initial sample was found by ITFA to consist of mixtures of GR (0.61) and HFO (0.39), which is similar to the analogous As(V) sample. When As(III)-laden GR was aged anoxically for a day, ITFA indicated a transformation of the solids to magnetite (0.45) and HFO (0.55). When aged anoxically for a year, the relative concentration of magnetite detected by ITFA increased to 1.0, with no other Fe phases detected. Similar to the As(V) series, the As(III)-laden GR samples aged in oxic conditions transformed rapidly into Fe(III) precipitates, but the transformation products differed. In the As(III) series, the major phase making up the solids for all aging samples was HFO instead of lepidocrocite. After an hour, a day, and a month of aging in oxic conditions, the relative concentration of HFO in the As(III) series was 1.0, 0.87, and 0.82, respectively. Although still a minor fraction of solids in the As(III) series, lepidocrocite was detected at relative concentrations that increased systematically (0, 0.13, 0.18) with increased oxic aging (an hour, a day, a month), which was also observed in the analogous As(V) samples.
3.3. Arsenic oxidation state
For all samples in the As(V) anoxic aging series, the position of the XANES absorption maxima (11875 eV) matched that of the As(V) references (Fig. 5, ESI 4†). Shoulders in the XANES indicative of As(V) reduction to As(III) were not observed for the initial sample, nor for the samples aged in anoxic conditions for a week or a month. However, the As(V)-laden GR sample aged anoxically for a year displayed a clear shoulder in the absorption edge at 11871 eV, which is consistent with the presence of a significant fraction of As(III). In addition, this sample displayed unique post-edge oscillations that matched the As(V) co-precipitated with magnetite (AsV Co-PPT Mag) reference. The XANES LCFs for this aging series confirmed the predominance of As(V) in all samples (>97% As(V), Table 1) except for the sample aged for a year, which contained 44 ± 10% As(III). We also note that for this sample, which contained only As(V) in the initial electrolyte solution, the residual aqueous As is expected to be primarily As(III) since the sorption affinity of As(V) is orders of higher than As(III)33 and aqueous As(V) could displace sorbed As(III) if it were present in solution. The XANES spectra for all As(V)-laden GR samples aged in oxic conditions had a maximum near 11875 eV, consistent with As(V). The presence of As(III) was not supported by the XANES spectra of any As(V) sample aged oxically, with the LCFs yielding >98% As(V) for each sample.
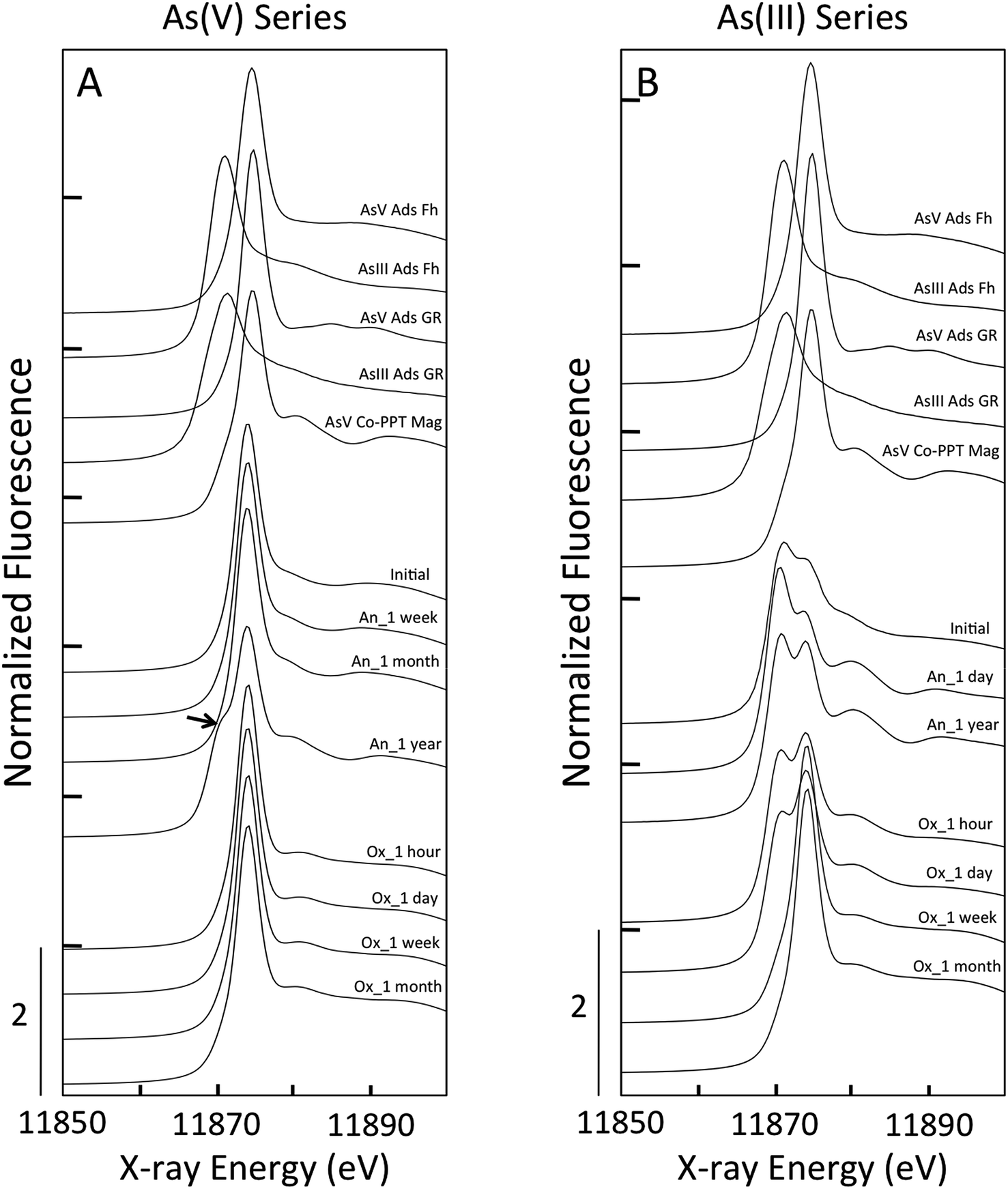 | ||
| Fig. 5 As K-edge XANES spectra for green rust aging samples in As(V) experiments (A) and As(III) experiments (B). The experimental data are given below As(III) and As(V) bearing reference materials. The anoxic aging series (e.g. An_1 week) are given above the oxic aging samples (e.g. Ox_1 hour). The arrow in A highlights a shoulder in the anoxic aging sample resulting from the presence of As(III). | ||
| Sample | Aging conditions | % As3+ | % As5+ |
|---|---|---|---|
| As(V) series | Initial | 7 ± 8 | 97 ± 10 |
| Anoxic +1 week | 6 ± 7 | 99 ± 9 | |
| Anoxic +1 month | 7 ± 7 | 98 ± 9 | |
| Anoxic +1 year | 44 ± 10 | 62 ± 11 | |
| Oxic + 1 hour | 5 ± 6 | 101 ± 10 | |
| Oxic + 1 day | 6 ± 7 | 100 ± 10 | |
| Oxic +1 week | 6 ± 7 | 100 ± 9 | |
| Oxic + 1 month | 8 ± 8 | 98 ± 12 | |
| As(III) series | Initial | 78 ± 7 | 25 ± 7 |
| Anoxic +1 day | 94 ± 6 | 9 ± 4 | |
| Anoxic +1 year | 82 ± 6 | 23 ± 5 | |
| Oxic + 1 hour | 73 ± 7 | 31 ± 6 | |
| Oxic + 1 day | 65 ± 7 | 40 ± 8 | |
| Oxic +1 week | 13 ± 12 | 93 ± 15 | |
| Oxic + 1 month | 9 ± 9 | 97 ± 12 |
In contrast to the As(V)-laden GR samples, the As(III) aging series in both anoxic and oxic systems consisted of samples having major fractions of As(III) and As(V), suggesting significant As(III) oxidation. For the initial sample in the As(III) series, which was produced by forming GR in the presence of As(III), the XANES spectrum displayed a double peak that was found by LCFs to contain 25 ± 7% As(V) (Table 1). This result is consistent with As(III) oxidation during GR formation by the reactive Fenton-type oxidants produced during Fe(II) oxidation by DO (i.e. Fe(IV)).50,51 The fractions of As(III) and As(V) determined by XANES LCFs varied slightly when As(III)-GR was aged in anoxic conditions (Table 1), but the fraction of As(V) in the sample aged a year (23 ± 5% As(V)) was consistent with the initial sample (25 ± 7% As(V)). Furthermore, both As(III) anoxic aging samples displayed the unique post-edge oscillations present in the As(V) Co-PPT magnetite reference and the As(V) sample aged for a year in anoxic conditions. This result suggests a change in the As coordination environment for both As(III) and As(V) samples when magnetite was detected in the Fe K-edge EXAFS spectra. The XANES spectra of the As(III)-laden GR samples aged under oxic conditions showed systematic changes consistent with As(III) oxidation. The amplitude of the XANES peak centered at 11871 eV, which is indicative of As(III), decreased continuously in the samples aged for an hour and a day in oxic conditions. By a week of oxic aging, the As(III) peak in the XANES spectrum was absent in favor of the As(V) peak at 11875 eV, which was similar to the sample aged a month in oxic conditions. The XANES LCFs confirmed the increase in As(V) for samples aged oxically for an hour (31 ± 5%), a day (40 ± 8%), a week (93 ± 15%) and a month (97 ± 12%).
3.4. Arsenic coordination environment
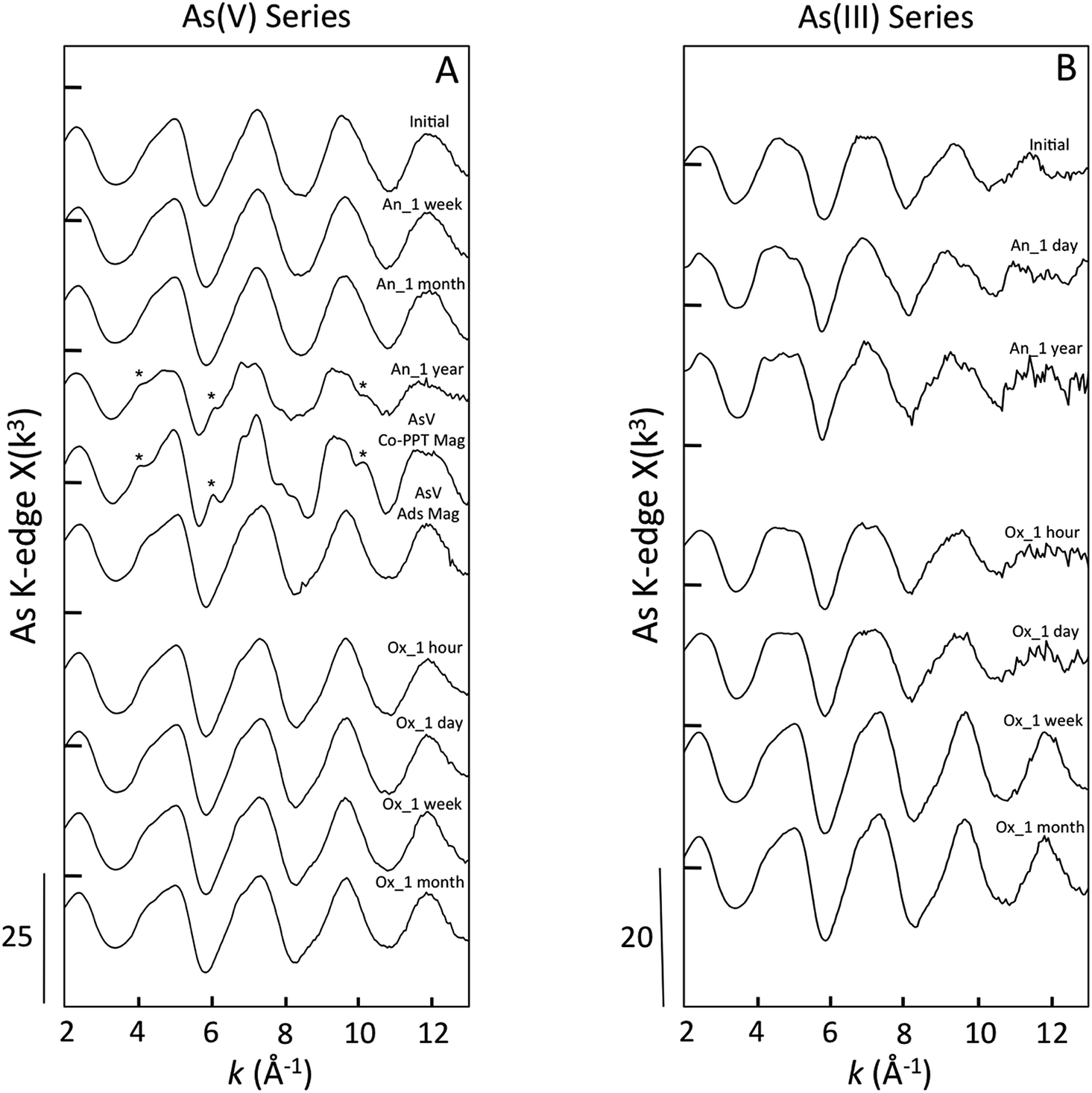 | ||
| Fig. 6 As K-edge EXAFS spectra of aging samples in As(V) experiments (A) and As(III) experiments (B). Reference spectra of As(V) co-precipitated with magnetite (AsV Co-PPT Mag) and adsorbed to magnetite (AsV Ads Mag) are given for comparison. The * symbols indicate EXAFS features consistent with As occupying the tetrahedral Fe site in magnetite. | ||
Several oscillations in the EXAFS spectra of As(III)-laden GR samples aged anoxically differed relative to the initial sample. For example, as aging increased to a year, the first oscillation flattened and the second oscillation grew more round with a shoulder near 6.2 Å−1. Since the XANES spectra did not show a significant change in the fractions of As(III) and As(V) between the initial sample and the anoxic aging samples, the differences in the EXAFS spectra are likely due to a difference in second shell (i.e. As–Fe) bonding environment. This result is consistent with the presence of unique post-edge oscillations in the XANES spectra for the aged samples that were absent from the initial sample. When aged in an oxic environment, the EXAFS spectra of samples aged an hour and a day were similar, consistent with the similar fractions of As(III) and As(V) in these samples (Table 1). The EXAFS spectra of samples aged oxically for a week and a month showed a narrowing of each oscillation, especially the last two peaks at 9.8 Å−1 and 11.8 Å−1. The samples aged oxically for a week and a month in both As(III) and As(V) series all exhibited similar EXAFS spectra, which is consistent with the similar predominance of As(V) in these samples, suggesting comparable coordination environments.
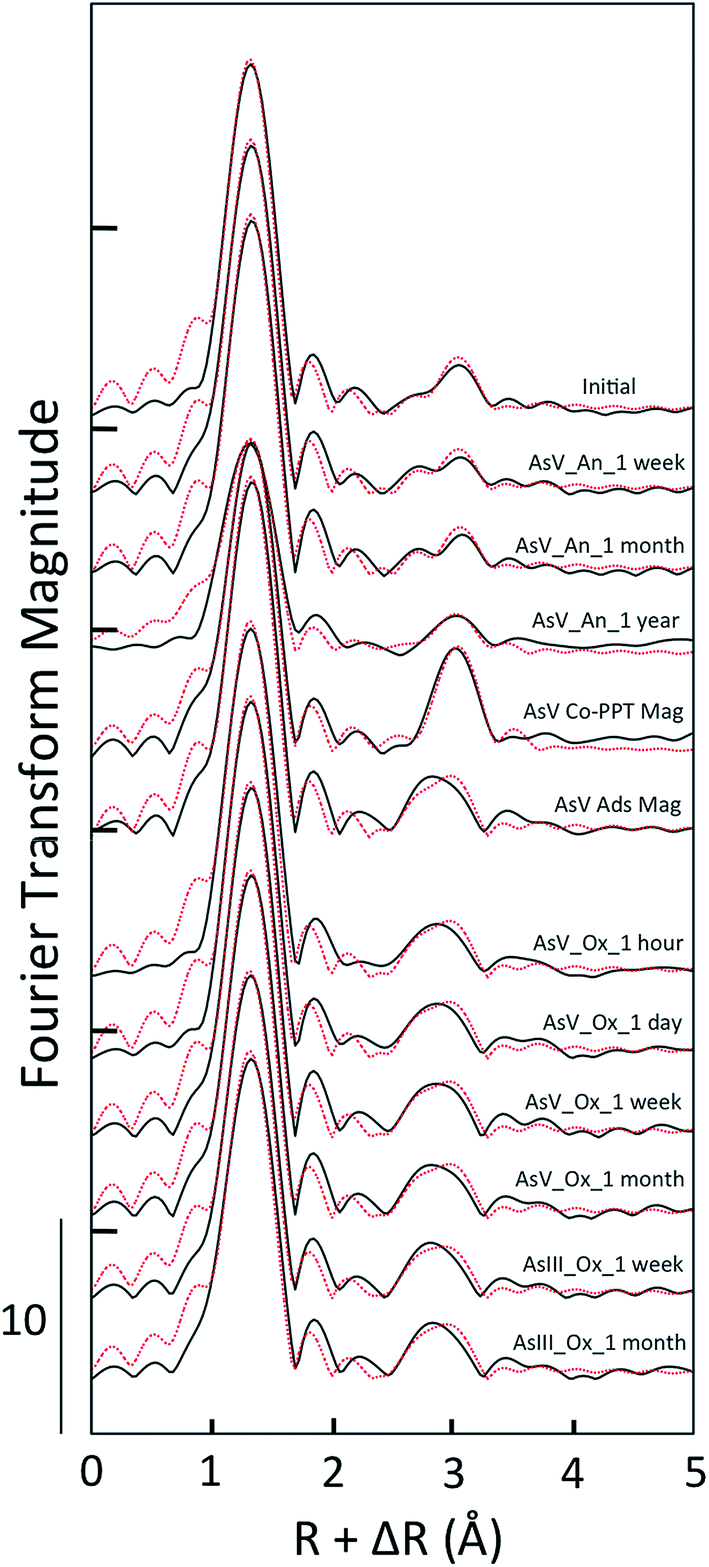 | ||
| Fig. 7 Fourier-transformed As K-edge EXAFS spectra of select aging samples in As(V) and As(III) experiments. The shell-by-shell fitting output (dotted red lines) is overlain to the data (solid black lines). In addition to aging samples, the fits of As(V) co-precipitated with magnetite (AsV Co-PPT Mag) and adsorbed to magnetite (AsV Ads Mag) are given. | ||
| Sample | Atomic pairs | CN | R (Å) | σ 2 (Å2) | ΔE0 (eV) | R-Factor |
|---|---|---|---|---|---|---|
| a CN represents the coordination number, R the interatomic distance, σ2 the mean squared atomic displacement and ΔE0 the change in threshold energy. Fitting parameters allowed to float are given with fit-determined standard errors in parenthesis, while constrained parameters appear without a parenthesis. | ||||||
| As(V)_Initial | As–O | 4.4 (0.5) | 1.69 (0.01) | 0.003(0.001) | 3.8 (2.0) | 0.019 |
| As–O–O | 12 | 1.82 (RAs-O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.1 (0.8) | 3.40 (0.03) | 0.009 | |||
| As(V)_Anoxic 1 week | As–O | 4.4 (0.5) | 1.69 (0.01) | 0.003 (0.001) | 5.4 (1.8) | 0.016 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 1.4 (0.7) | 3.39 (0.04) | 0.009 | |||
| As(V)_Anoxic 1 month | As–O | 4.3 (0.4) | 1.69 (0.01) | 0.002 (0.001) | 5.6 (1.8) | 0.016 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 1.5 (0.7) | 3.41 (0.03) | 0.009 | |||
| As(V)_Anoxic 1 year | As–O | 4.0 (0.6) | 1.71 (0.01) | 0.005 (0.001) | 1.9 (2.4) | 0.032 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.11 | σ 2 (As–O) | |||
| As–Fe1 | 2.7 (1.2) | 3.43 (0.03) | 0.009 | |||
| As–Fe2 | 1.3 (1.4) | 3.60 (0.07) | σ 2 (As–Fe1) | |||
| As(V) Co-PPT magnetite | As–O | 4.2 (0.5) | 1.70 (0.01) | 0.003 (0.001) | 4.5 (1.9) | 0.015 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.09 | σ 2 (As–O) | |||
| As–Fe1 | 7.7 (1.5) | 3.46 (0.02) | 0.009 | |||
| As–Fe2 | 3.9 (1.9) | 3.64 (0.03) | σ 2 (As–Fe1) | |||
| As(V) Ads magnetite | As–O | 4.2 (0.5) | 1.69 (0.01) | 0.002 (0.001) | 6.0 (2.0) | 0.019 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.2 (0.8) | 3.34 (0.02) | 0.009 | |||
| As(V)_Oxic 1 hour | As–O | 4.7 (0.6) | 1.69 (0.01) | 0.003 (0.001) | 4.4 (2.3) | 0.024 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.4 (1.0) | 3.34 (0.03) | 0.010 | |||
| As(V)_Oxic 1 day | As–O | 4.5 (0.5) | 1.69 (0.01) | 0.003 (0.001) | 4.5 (2.1) | 0.021 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.4 (0.9) | 3.34 (0.03) | 0.010 | |||
| As(V)_Oxic 1 week | As–O | 4.3 (0.05) | 1.69 (0.01) | 0.002 (0.002) | 4.3 (1.9) | 0.018 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.5 (0.8) | 3.34 (0.03) | 0.010 | |||
| As(V)_Oxic 1 month | As–O | 4.3 (0.5) | 1.69 (0.01) | 0.002 (0.001) | 4.0 (1.9) | 0.017 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.4 (0.8) | 3.34 (0.03) | 0.010 | |||
| As(III)_Oxic 1 week | As–O | 4.3 (0.5) | 1.69 (0.01) | 0.003 (0.001) | 2.9 (2.0) | 0.018 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.3 (0.8) | 3.32 (0.03) | 0.010 | |||
| As(III)_Oxic 1 month | As–O | 4.5 (0.5) | 1.69 (0.01) | 0.003 (0.001) | 3.7 (2.0) | 0.019 |
| As–O–O | 12 | 1.82 (RAs–O) = 3.08 | σ 2 (As–O) | |||
| As–Fe1 | 2.4 (0.8) | 3.33 (0.03) | 0.010 | |||
Consistent with the unique XANES and EXAFS features of As(V)-laden GR aged anoxically for a year, the shell fits for this sample differed relative to the other samples in this aging series. The CNAs–O returned by the fit (4.0 ± 0.6) was lower than the other aging samples, the disorder term (σAs–O2) was higher (0.005 ± 0.001 Å2) and the RAs–O was also higher (1.71 ± 0.01 Å), which is in agreement with the presence of a significant fraction of three-fold coordinated As(III) (Tables 1 and 2). Fits of the second shell of this sample required two As–Fe paths positioned at RAs–Fe1 = 3.43 ± 0.03 Å and RAs–Fe2 = 3.60 ± 0.07 Å. These RAs–Fe values are within the fit-derived standard error of those obtained in the fit of the As(V) Co-PPT magnetite reference (RAs–Fe1 = 3.46 ± 0.02 Å and RAs–Fe2 = 3.64 ± 0.03 Å) and are significantly longer than the fit-derived RAs–Fe for As(V) adsorbed to magnetite (3.34 ± 0.02 Å, Table 2). However, the sample aged anoxically for a year had lower CNs on these As–Fe paths than the As(V) Co-PPT magnetite reference, which could be a consequence of the high fraction of As(III) in the aging sample.
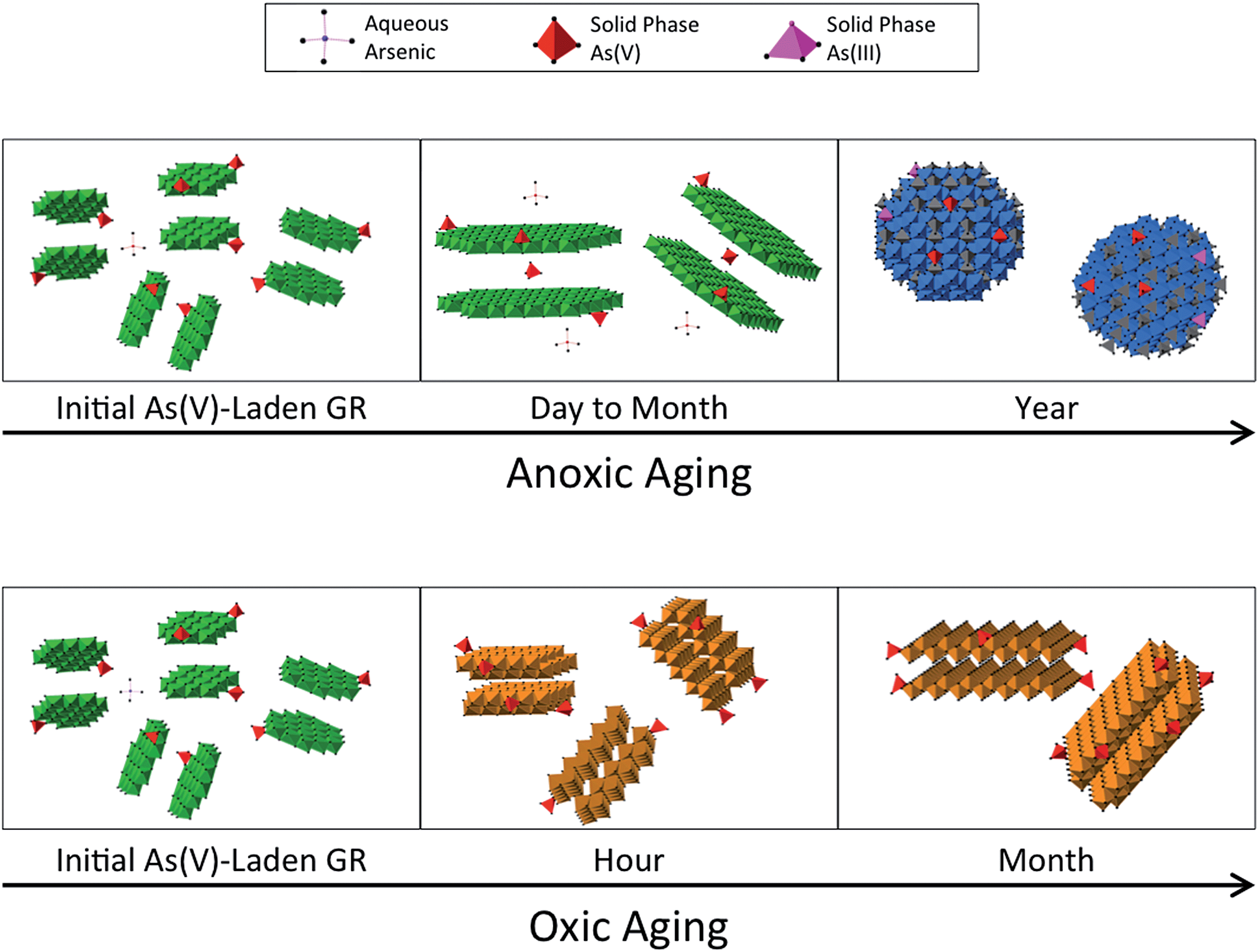 | ||
| Fig. 8 Models of the transformation of As(V)-laden green rust (GR) over time in anoxic (top panels) and oxic conditions (bottom panels). The initial GR transforms into magnetite (blue spherical crystals) in a year in the anoxic system, whereas lepidocrocite (brown layered crystals) forms in an hour in the oxic system. Experiments with As(III)-laden GR followed similar trends. | ||
Despite the differences in the As K-edge XAS data of the fresh As(V)- and As(III)-laden GR suspensions, these samples aged oxically produced identical shell-by-shell fits for almost every sample. The fits of the As(V)-laden GR samples aged in oxic conditions for an hour, a day, a week and a month all yielded RAs–O = 1.69 Å and CNAs–O = 4.3 to 4.7, which indicates As(V) in tetrahedral coordination. The second shell fits of these samples were indistinguishable within standard errors (RAs–Fe = 3.34 Å, CNAs–Fe = 2.4 to 2.5, Table 2), and are consistent with a similar 2C As(V) surface complex. For the As(III)-laden GR samples aged oxically for a week and a month, which were determined by XANES LCFs to consist of dominantly As(V), the shell-by-shell fits yielded similar structural parameters as the As(V) oxic aging samples. For the As(III) samples, the fits yielded CNAs–O = 4.3 to 4.5 and RAs–O = 1.69 Å for the first shell and CNAs–Fe = 2.3 to 2.4 and RAs–Fe = 3.32 to 3.33 Å for the second shell. These fitting parameters are consistent with As(V) binding in the 2C geometry to Fe(III) precipitate surfaces, which is similar to As(V) oxic aging samples.52,53
4. Discussion
4.1. Structural modifications of GR during aging
The characterization data indicated that GR transformation during aging depended on aging time and solution conditions (i.e. DO concentration and initial As oxidation state). Modifications of the initial GR suspensions included: (i) increased structural ordering, (ii) transformation of GR to magnetite and (iii) GR oxidation to form Fe(III) (oxyhydr)oxides. These three structural modifications during aging are discussed in the following subsections.It is noteworthy that when magnetite was first observed in both As(V) (a year) and As(III) (1 day) aging series, a fraction of HFO was also detected by ITFA. Consistent with our interpretation of the ITFA-derived fraction of HFO in the initial GR samples, the detection of magnetite and HFO by ITFA most likely reflects differences in the crystallinity between magnetite particles, rather than the formation of a mixture of two Fe phases. Finally, the Fe K-edge EXAFS analysis indicated that the magnetite formed by GR transformation was lower in crystallinity than the reference magnetite synthesized by mixing Fe(II) and Fe(III) salts, which produces nanoparticulate magnetite.54 The EXAFS spectrum of magnetite formed by GR transformation also matches well the EXAFS spectrum of a magnetite produced by Fe(0) electrolysis in the absence of dissolved carbonate, which had a crystallite size determined by transmission electron microscopy to be <10 nm.34 Therefore, we propose the magnetite formed by GR transformation in our study is nanoparticulate with a primarily crystallite size <10 nm.
Although GR was completely oxidized by DO at similar timescales in the As(V) and As(III) experiments, the reaction products in the As(V) and As(III) series were different. For the As(V) series, GR oxidation yielded primarily lepidocrocite, whereas the dominant end product of As(III)-laden GR oxidation was HFO. One explanation for the different end products in the As(V) and As(III) oxic aging experiments relates to the different sorption affinity of As(V) and As(III) for Fe(III) precipitates.33 The oxidation of Fe(II) by DO in the presence of strongly sorbing oxyanions, such as As(V) and P, has been shown to lead to the sequential formation of poorly-ordered solids that take up oxyanions at high As/Fe or P/Fe molar ratios, followed by moderately crystalline lepidocrocite once these oxyanions are removed from solution.60–62 By contrast, more weakly sorbing ions, such as As(III) and Si, are removed at lower oxyanion/Fe ratios, leaving the majority of Fe(II) to oxidize and polymerize in the presence of surface-poisoning oxyanions (i.e. As(III)) that prevent lepidocrocite formation.61,63 This interpretation is consistent with higher aqueous As concentration of the initial As(III)-laden GR sample (370 ± 16 μg L−1) relative to the initial As(V)-laden GR sample (14 ± 8 μg L−1), indicating Fe(III) polymerized in the presence of more surface-poisoning As in the As(III) oxic aging experiments.
4.2. Behavior of arsenic during green rust transformation
Modifications of the GR structure during aging depended on the initial As oxidation state and concentration, but in turn, these structural modification also resulted in changes in the partitioning and local bonding environment of As. These results suggest an interdependent relationship between the GR transformation pathway and the fate of As. When As(V)-laden GR was aged anoxically up to a month, the Fe K-edge EXAFS data suggested an increase in GR crystallinity, which corresponded with a significant increase in aqueous As (Fig. 1 and 8). These data are consistent with a decrease in reactive surface area per mass of GR during anoxic aging due to Ostwald ripening. The structural data suggest increased ordering along GR sheets during aging, which would lower the density of adsorption sites since As(V) binds primarily to GR particle edges.15,16 With respect to the As bonding environment, fits of the second shell of the Fourier-transformed As K-edge EXAFS spectra of As(V)-laden GR aged up to a month returned identical RAs–Fe values, but a slight decrease in the CNAs–Fe (2.1 ± 0.8 initially to 1.4 ± 0.7 after a month). These results could indicate that As(V) released from the particle edge during GR crystallization is associated with the solid phase by occupying interlayer sites. Intercalation of As(V) would not alter the average RAs–Fe because no Fe atoms would occur in the first few coordination shells of As, but the average CNAs–Fe would decrease, consistent with our data. However, we note that this interpretation is speculative because of the high fit-derived standard error on CNAs–Fe, which is a weakness of EXAFS spectroscopy.49,64The anoxic transformation of GR to magnetite was coupled to a dramatic decrease in the aqueous As concentration for both the As(V) and As(III) aging series. Based on systematic changes in the As K-edge XAS data, the formation of magnetite also coincided with a modification in the As coordination environment. For example, distinct post-edge oscillations appeared in the As K-edge XANES spectra and characteristic features in the As K-edge EXAFS data were present in all samples that contained magnetite. These unique XANES and EXAFS features arise from a change in the As bonding environment from adsorption in the 2C geometry at GR particle edges to occupation of the tetrahedral iron site in magnetite. Previous studies have shown that As(V) and As(III) can substitute for tetrahedral iron in magnetite, leading to strong multinuclear sorption complexes that more effectively remove As from solution than surface complexes.30,43,65 Therefore, the transformation of GR to magnetite enhances As uptake because magnetite has a higher density of sorption sites capable of binding As in presumably stronger uptake modes (i.e. multinuclear complexes).
In both As(V) and As(III) oxic aging series, the transformation of GR to Fe(III) precipitates decreased substantially the aqueous As concentration. Even though the Fe K-edge EXAFS analysis indicated an increased fraction of lepidocrocite with time for both aging series, which implies a decrease in the specific surface area of the solids, the As concentration still decreased with time. These results most likely reflect changes in the As oxidation state and solution pH during aging, and the larger specific surface area of the solid aging products compared to GR. For the As(V) oxic aging series, the As K-edge XAS data showed no evidence for changes in oxidation state with time. However, the shell-by-shell fits indicated a decrease in RAs–Fe from 3.40 ± 0.03 Å in the initial GR sample to 3.34 ± 0.03 Å, which is consistent with the difference in the average RFe–O between GR (2.08 Å) and lepidocrocite (2.00 Å). This result suggests As(V) binds to the initial GR and its oxic aging products in a similar 2C geometry.16,52 In contrast to the As(V) series, the As(III) samples showed changes in As oxidation during oxic aging, with complete As(III) oxidation in a week. The oxidation of As(III) in this aging series is attributed to a combination of Fenton-type oxidants produced by Fe(II) reactions with DO at early timescales (minutes to hours) and surface catalyzed As(III) oxidation by DO at longer timescales (hours to days).66 The orders of magnitude higher sorption affinity of As(V) than As(III) for Fe(III) precipitates33 is consistent with the decreased aqueous As in the As(III) aging series. Furthermore, the rapid drop in solution pH measured for both As(V) and As(III) oxic aging series, caused by H+ production during Fe(II) oxidation and Fe(III) polymerization,67 is also expected to improve As uptake especially for samples consisting of dominantly As(V). Sorption of negatively charged As(V) (pKa1 = 2.3, pKa2 = 6.8)67 to Fe (oxyhydr)oxides is most favorable at pH < 4,31,68,69 where the positive surface charge of these minerals (PZC = 6 to 8)70 enhances electrostatic attraction. Furthermore, lower pH would favor increased adsorption if As(V) and As(III) bind to the surface through a ligand exchange mechanism that generates OH−. Although the pH of the As(V) series was slightly higher than the As(III) series (Fig. ESI 2†), which could be due to increased buffering by H2AsO4−/HAsO42− or OH− production during adsorption, the pH of both oxic aging series decreased rapidly to <6. In particular, the pH of the As(III) oxic aging series was near the optimum pH for As(V) sorption after several weeks of aging, which explains the <1 μg L−1 residual As concentration in the final As(III) oxic aging sample consisting dominantly As(V) (Table 1).
4.3. Relevance to natural and engineered settings
Our results revealed the interdependent relationship between the GR transformation pathway and the fate of oxyanions, which must be considered in biogeochemical models of contaminant (e.g. As) and nutrient (e.g. P and Si) mobility during Fe cycling. Depending on aging time, redox conditions, and the oxidation state and concentration of As, GR transformed into a variety of end products, including poorly-ordered Fe(III) precipitates, lepidocrocite and magnetite. These structural modifications were tightly linked to aqueous As concentrations, with anoxic GR transformation to magnetite and oxic GR transformation to Fe(III) (oxyhydr)oxides both sequestering As more effectively than the initial GR phase. Since As(V) and As(III) are chemical analogues of P and Si with respect to charge and sorption reactivity with Fe (oxyhydr)oxides,33 our results suggest that GR transformation in anoxic conditions to magnetite would reduce the mobility and bioavailability of As, Si, and P, with the potential for enhanced oxyanion immobilization by occupying tetrahedral Fe sites.30,43 Furthermore, we expect that the presence of Si and P would lead to similar GR oxidation products (i.e. HFO), but these ions would decrease the As/Fe ratio of the solid phase because they compete effectively for sorption sites on Fe(III) precipitate surfaces. We also provide compelling evidence that GR transformation to magnetite can abiotically reduce As(V) to As(III). This constraint on the reduction capacity of GR is particularly important because GR minerals are thought to play an important role in the cycling of elements in Precambrian oceans.71,72 When GR was exposed to oxic conditions, our results suggest that rapid transformation to Fe(III) precipitates would also decrease oxyanion mobility by producing solids with higher sorption site density and by lowering solution pH. However, we note that the decrease in pH upon GR oxidation in our experiments, which likely enhanced As uptake, might not occur in natural systems with higher buffering capacity.With respect to As remediation, Fe-based engineered solutions can rely on mixed valent Fe(II,III) (oxyhydr)oxides to decrease aqueous As.8,9,18,20 In this study, we found that structural modifications of the initial GR resulted in differences between As concentrations spanning 10 μg L−1, the WHO provisional limit of As in drinking water. For example, in the As(V) series, the initial GR phase removed As to near the WHO limit, but after anoxic aging for a day and a month, aqueous As increased to concentrations ∼5 times higher than the WHO limit. Further anoxic aging for a year resulted in a decrease in aqueous As to levels ∼5 times lower than the WHO limit. This temporal variation in aqueous As during GR formation must be taken into account when designing GR-based remediation schemes, and especially the high potential for As release during GR crystallization before magnetite formation in anoxic systems. Another design consideration is that the release of As during GR crystallization might be desired in some situations. For example, it could be advantageous to employ a sorbent with properties that favor low cost and simple As sorption reversibility. Using a sorbent that releases As easily would decrease the amount of solid waste to be managed and would facilitate separating As from the solid phase, making waste reuse or reprocessing safer and simpler. The release of As from magnetite formed in the anoxic GR aging experiments is expected to be more challenging than GR, making reuse of the As-rich magnetite solids more difficult.
While we found that the initial GR phase removed high fractions of As from solution, the residual As concentration decreased when GR transformed to magnetite and Fe(III) precipitates. This conclusion appears to be inconsistent with previous studies designating GR as one of the most effective Fe-based sorbents for As. This discrepancy arises from comparing sorbent reactivity based on sorption capacity rather than the residual dissolved As concentration. For GR, the As sorption capacity has been reported to be larger than other Fe (oxyhydr)oxides, presumably because GR favors polymeric As surface species at high aqueous As concentrations.15 However, comparisons based on minimized residual As for a given sorbent amount (or for a given treatment cost) can be more meaningful than sorption capacity in the context of drinking water treatment since the sorption capacity is often obtained in laboratory experiments with unrealistically high As concentrations (i.e. several mg L−1). Therefore, if the type of Fe phase can be tuned, we propose that magnetite or Fe(III) (oxyhydr)oxides should generally be targeted to optimize As treatment, with a notable exception. Remediation schemes based on pre-synthesized Fe (oxyhydr)oxides coated on sands or used in filter columns could exploit the metastability of GR to improve As removal. Coating sand grains with stable Fe mineral phases would limit sorbent reactivity to the particle exterior. Since GR transforms to magnetite and Fe(III) precipitates likely by dissolution and reprecipitation,21 a high fraction of the Fe atoms making up the solid could be involved in binding As after GR transformation, and in the case of GR transformation to magnetite, the As uptake mode could shift from adsorption to GR particle edges to substitution for tetrahedral Fe and (partial) incorporation.30,43
5. Conclusions
In this study, we found an interdependent relationship between the pathways of GR transformation and the fate of As. The end products of GR transformation depended on the oxidation state and concentration of As (in addition to aging time and DO concentration), and the partitioning and uptake mode of As depended on the GR transformation products. In anoxic conditions, GR transformed to magnetite over timescales of a day (As(III) series) to a year (As(V) series), which decreased the aqueous As concentration to <10 μg L−1 and resulted in a transition from As adsorbed to GR particle edges to As taken up in multinuclear complexes by substituting for tetrahedral Fe. In oxic conditions, GR transformed to primarily lepidocrocite (As(V) series) or HFO (As(III) series) in an hour, which decreased the aqueous As concentration to <10 μg L−1, but did not modify the As uptake geometry (similar 2C As(V) surface complexes before and after oxic aging). These results advance our understanding of the impact of Fe cycling on oxyanion mobility in the environment and can be used to improve predictions of As removal efficiency in Fe-based As remediation strategies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding provided by the Dutch Organization for Scientific Research in a Veni Grant to CMvG (Project No. 14400). Synchrotron experiments were performed partly at the DUBBLE beam line at the ESRF, Grenoble, France, with assistance from Dipanjan Banerjee. We also thank Ryan Davis for technical support during synchrotron data collection at SSRL. Use of SSRL, SLAC National Accelerator Laboratory, was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Contract No. DE-AC02-76SF00515.References
- H. Hansen, Environmental Chemistry of Iron(II)-Iron(III) LDHs (Green Rusts), in Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova Science Publishers, 2001 Search PubMed.
- F. Trolard, G. Bourrie, M. Abdelmoula, P. Refait and F. Feder, Fougerite, a new mineral of the pyroaurite-iowaite group: Description and crystal structure, Clays Clay Miner., 2007, 55(3), 323–334 CrossRef CAS.
- G. Bourrie, F. Trolard, J. M. R. Genin, A. Jaffrezic, V. Maitre and M. Abdelmoula, Iron control by equilibria between hydroxy-Green Rusts and solutions in hydromorphic soils, Geochim. Cosmochim. Acta, 1999, 63(19–20), 3417–3427 CrossRef CAS.
- B. Christiansen, T. Balic-Zunic, K. Dideriksen and S. Stipp, Identification of Green Rust in Groundwater, Environ. Sci. Technol., 2009, 43(10), 3436–3441 CrossRef CAS PubMed.
- J. Miot, J. Li, K. Benzerara, M. T. Sougrati, G. Ona-Nguema, S. Bernard, J.-C. Jumas and F. Guyot, Formation of single domain magnetite by green rust oxidation promoted by microbial anaerobic nitrate-dependent iron oxidation, Geochim. Cosmochim. Acta, 2014, 139, 17 CrossRef.
- C. Hansel, S. Benner, J. Neiss, A. Dohnalkova, R. Kukkadapu and S. Fendorf, Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow, Geochim. Cosmochim. Acta, 2003, 2977–2992 CrossRef CAS.
- L. Legrand, M. Abdelmoula, A. Gehin, A. Chausse and J. Genin, Electrochemical formation of a new Fe(II)-Fe(III) hydroxy-carbonate green rust: characterisation and morphology, Electrochim. Acta, 2001, 46(12), 1815–1822 CrossRef CAS.
- K. L. Dubrawski, C. M. van Genuchten, C. Delaire, S. E. Amrose, A. J. Gadgil and M. Mohseni, Production and transformation of mixed-valent nanoparticles generated by Fe(0) electrocoagulation, Environ. Sci. Technol., 2015, 49(17), 2171–2179 CrossRef CAS PubMed.
- C. Su and R. W. Puls, Significance of iron(II,III) hydroxycarbonate green rust in arsenic remediation using zerovalent iron in laboratory column tests, Environ. Sci. Technol., 2004, 38(19), 5224–5231 CrossRef CAS PubMed.
- E. O'Loughlin and D. Burris, Reduction of halogenated ethanes by green rust, Environ. Toxicol. Chem., 2004, 23(1), 41–48 CrossRef.
- M. Elsner, R. Schwarzenbach and S. Haderlein, Reactivity of Fe(II)-bearing minerals toward reductive transformation of organic contaminants, Environ. Sci. Technol., 2004, 38(3), 799–807 CrossRef CAS PubMed.
- L. Skovbjerg, S. Stipp, S. Utsunomiya and R. Ewing, The mechanisms of reduction of hexavalent chromium by green rust sodium sulphate: Formation of Cr-goethite, Geochim. Cosmochim. Acta, 2006, 70(14), 3582–3592 CrossRef CAS.
- P. Refait, L. Simon and J. M. R. Genin, Reduction of SeO42- anions and anoxic formation of iron(II)-iron(III) hydroxy selenate green rust, Environ. Sci. Technol., 2000, 34(5), 819–825 CrossRef CAS.
- M. Usman, J. M. Byrne, A. Chaudhary, S. Orsetti, K. Hanna, C. Ruby, A. Kappler and S. B. Haderlein, Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals, Chem. Rev., 2018, 118(7), 3251–3304 CrossRef CAS PubMed.
- Y. H. Wang, G. Morin, G. Ona-Nguema, F. Juillot, F. Guyot, G. Calas and G. E. Brown, Evidence for Different Surface Speciation of Arsenite and Arsenate on Green Rust: An EXAFS and XANES Study, Environ. Sci. Technol., 2010, 44(1), 109–115 CrossRef CAS PubMed.
- J. Jonsson and D. M. Sherman, Sorption of As(III) and As(V) to siderite, green rust (fougerite) and magnetite: Implications for arsenic release in anoxic groundwaters, Chem. Geol., 2008, 255(1–2), 173–181 CrossRef CAS.
- P. A. Parga, V. Vazquez and H. Moreno, Thermodynamic studies of the arsenic adsorption on iron species generated by electrocoagulation, J. Metall., 2009, 2009, 286971 Search PubMed.
- M. Kobya, U. Gebologlu, F. Ulu, S. Oncel and E. Demirbas, Removal of arsenic from drinking water by the electrocoagulation using Fe and Al electrodes, Electrochim. Acta, 2011, 56(14), 5060–5070 CrossRef CAS.
- J. P. H. Perez, H. M. Freeman, J. A. Schuessler and L. G. Benning, The interfacial reactivity of arsenic species with green rust sulfate (GR), Sci. Total Environ., 2019, 648, 1161–1170 CrossRef CAS PubMed.
- Z. Lin and R. W. Puls, Potential indicators for the assessment of arsenic natural attenuation in the subsurface, Adv. Environ. Res., 2003, 7(4), 825–834 CrossRef CAS.
- A. Sumoondur, S. Shaw, I. Ahmed and L. Benning, Green rust as a precursor for magnetite: an in situ synchrotron based study, Mineral. Mag., 2008, 72(1), 201–204 CrossRef CAS.
- L. Legrand, L. Mazerolles and A. Chausse, The oxidation of carbonate green rust into ferric phases: solid-state reaction or transformation via solution, Geochim. Cosmochim. Acta, 2004, 68(17), 3497–3507 CrossRef CAS.
- O. Benali, M. Abdelmoula, P. Refait and J. Genin, Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate: The transformation of green rust to ferrihydrite, Geochim. Cosmochim. Acta, 2001, 65(11), 1715–1726 CrossRef CAS.
- W. Bragg, The structure of magnetite and the spinels, Nature, 1915, 95, 561 CrossRef.
- C. Ruby, A. Géhin, R. Aissa, J. Ghanbaja, M. Abdelmoula and J.-M. R. Génin, Chemical stability of hydroxysulphate green rust synthesized in the presence of foreign anions: carbonate, phosphate and silicate, Hyperfine Interact., 2006, 167, 5 CrossRef.
- X. Wang, F. Liu, W. Tan, X. Feng and L. K. Koopal, Transformation of hydroxycarbonate green rust into crystalline iron (hydr)oxides: Influences of reaction conditions and underlying mechanisms, Chem. Geol., 2013, 351, 9 CrossRef.
- X. Feng, X. Wang, M. Zhu, L. Koopal, H. Xu, Y. Wang and F. Liu, Effects of phosphate and silicate on the transformation of hydroxycarbonate green rust to ferric oxyhydroxides, Geochim. Cosmochim. Acta, 2015, 171, 1–14 CrossRef CAS.
- R. Prucek, J. Tuček, J. Kolařík, J. Filip, Z. Marušák, V. K. Sharma and R. Zbořil, Ferrate(VI)-induced arsenite and arsenate removal by in situ structural incorporation into magnetic iron(III) oxide nanoparticles, Environ. Sci. Technol., 2013, 47(7), 3283–3292 CrossRef CAS PubMed.
- R. P. Kralchevska, R. Prucek, J. Kolařík, J. Tuček, L. Machala, J. Filip, V. K. Sharma and R. Zbořil, Remarkable efficiency of phosphate removal: Ferrate(VI)-induced in situ sorption on core-shell nanoparticles, Water Res., 2016, 103, 83–91 CrossRef CAS PubMed.
- Y. Wang, G. Morin, G. Ona-Nguema and G. Brown, Arsenic(III) and Arsenic(V) Speciation during Transformation of Lepidocrocite to Magnetite, Environ. Sci. Technol., 2014, 48(24), 14282–14290 CrossRef CAS PubMed.
- M. Kanematsu, T. Young, K. Fukushi, P. Green and J. Darby, Arsenic(III, V) adsorption on a goethite-based adsorbent in the presence of major co-existing ions: Modeling competitive adsorption consistent with spectroscopic and molecular evidence, Geochim. Cosmochim. Acta, 2013, 106, 404–428 CrossRef CAS.
- X. G. Meng, S. Bang and G. P. Korfiatis, Effects of silicate, sulfate, and carbonate on arsenic removal by ferric chloride, Water Res., 2000, 34(4), 1255–1261 CrossRef CAS.
- L. C. Roberts, S. J. Hug, T. Ruettimann, M. Billah, A. W. Khan and M. T. Rahman, Arsenic removal with iron(II) and iron(III) in waters with high silicate and phosphate concentrations, Environ. Sci. Technol., 2004, 38(1), 307–315 CrossRef CAS PubMed.
- C. van Genuchten, T. Behrends, P. Kraal, S. Stipp and K. Dideriksen, Controls on the formation of Fe(II,III) (hydr)oxides by Fe(0) electrolysis, Electrochim. Acta, 2018, 286, 324–338 CrossRef CAS.
- M. Borsboom, W. Bras, I. Cerjak, D. Detollenaere, D. van Loon, P. Goedtkindt, M. Konijnenburg, P. Lassing, Y. Levine, B. Munneke, M. Oversluizen, R. van Tol and E. Vlieg, The Dutch-Belgian beamline at the ESRF, J. Synchrotron Radiat., 1998, 5, 518–520 CrossRef CAS PubMed.
- S. Nikitenko, A. Beale, A. van der Eerden, S. Jacques, O. Leynaud, M. O'Brien, D. Detollenaere, R. Kaptein, B. Weckhuysen and W. Bras, Implementation of a combined SAXS/WAXS/QEXAFS set-up for time-resolved in situ experiments, J. Synchrotron Radiat., 2008, 15, 632–640 CrossRef CAS PubMed.
- S. Webb, SIXPACK: a graphical user interface for XAS analysis using IFEFFIT, Phys. Scr., 2005, T115, 1011–1014 CrossRef CAS.
- C. van Genuchten, S. Addy, J. Pena and A. Gadgil, Removing arsenic from synthetic groundwater with iron electrocoagulation: An Fe and As K-edge EXAFS study, Environ. Sci. Technol., 2012, 46(2), 986–994 CrossRef CAS PubMed.
- A. Scheinost, A. Rossberg, D. Vantelon, I. Xifra, R. Kretzschmar, A. Leuz, H. Funke and C. Johnson, Quantitative antimony speciation in shooting-range soils by EXAFS spectroscopy, Geochim. Cosmochim. Acta, 2006, 70(13), 3299–3312 CrossRef CAS.
- A. Rossberg, K. Ulrich, S. Weiss, S. Tsushima, T. Hiemstra and A. Scheinost, Identification of Uranyl Surface Complexes an Ferrihydrite: Advanced EXAFS Data Analysis and CD-MUSIC Modeling, Environ. Sci. Technol., 2009, 43(5), 1400–1406 CrossRef CAS PubMed.
- A. Rossberg, T. Reich and G. Bernhard, Complexation of uranium(VI) with protocatechuic acid - application of iterative transformation factor analysis to EXAFS spectroscopy, Anal. Bioanal. Chem., 2003, 376(5), 631–638 CrossRef CAS PubMed.
- A. Manceau, M. Marcus and S. Grangeon, Determination of Mn valence states in mixed-valent manganates by XANES spectroscopy, Am. Mineral., 2012, 97(5–6), 816–827 CrossRef CAS.
- Y. Wang, G. Morin, G. Ona-Nguema, F. Juillot, G. Calas and G. Brown, Distinctive Arsenic(V) Trapping Modes by Magnetite Nanoparticles Induced by Different Sorption Processes, Environ. Sci. Technol., 2011, 45(17), 7258–7266 CrossRef CAS PubMed.
- M. Newville, IFEFFIT: interactive XAFS analysis and FEFF fitting, J. Synchrotron Radiat., 2001, 322–324 CrossRef CAS PubMed.
- J. Rehr, R. Albers and S. Zabinsky, High-order multiple-scattering calculations of X-ray absorption fine structure, Phys. Rev. Lett., 1992, 3397–3400 CrossRef CAS PubMed.
- K. Kitahama, R. Kiriyama and Y. Baba, Refinement of crystal-structure of scorodite, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 322–324 CrossRef CAS.
- C. Mikutta, J. Frommer, A. Voegelin, R. Kaegi and R. Kretzschmar, Effect of citrate on the local Fe coordination in ferrihydrite, arsenate binding, and ternary arsenate complex formation, Geochim. Cosmochim. Acta, 2010, 5574–5592 CrossRef CAS.
- D. Paktunc, J. Dutrizac and V. Gertsman, Synthesis and phase transformations involving scorodite, ferric arsenate and arsenical ferrihydrite: Implications for arsenic mobility, Geochim. Cosmochim. Acta, 2008, 2649–2672 CrossRef CAS.
- S. D. Kelly, D. Hesterberg and B. Ravel, Analysis of soils and minerals using X-ray absorption spectroscopy, in Methods of Soil Analysis. Part 5. Mineralogical Methods, SSA Book Series No. 5, 2008 Search PubMed.
- C. van Genuchten and J. Pena, Mn(II) Oxidation in Fenton and Fenton Type Systems: Identification of Reaction Efficiency and Reaction Products, Environ. Sci. Technol., 2017, 51(5), 2982–2991 CrossRef CAS PubMed.
- L. Li, C. M. van Genuchten, S. E. A. Addy, J. Yao, N. Gao and A. J. Gadgil, Modeling As(III) oxidation and removal with iron electrocoagulation in groundwater, Environ. Sci. Technol., 2012, 46(21), 12038–12045 CrossRef CAS PubMed.
- G. A. Waychunas, B. A. Rea, C. C. Fuller and J. A. Davis, Surface-chemistry of ferrihydrite .1. Exafs studies of the geometry of coprecipitated and adsorbed arsenate, Geochim. Cosmochim. Acta, 1993, 57(10), 2251–2269 CrossRef CAS.
- D. M. Sherman and S. R. Randall, Surface complexation of arsenie(V) to iron(III) (hydr)oxides: Structural mechanism from ab initio molecular geometries and EXAFS spectroscopy, Geochim. Cosmochim. Acta, 2003, 67(22), 4223–4230 CrossRef CAS.
- U. Schwertmann and R. M. Cornell, Iron oxides in the laboratory: preparation and characterization, VCH: Weinheim, New York, 1991, p. 137 Search PubMed.
- B. M. Toner, C. M. Santelli, M. A. Marcus, R. Wirth, C. S. Chan, T. McCollom, W. Bach and K. J. Edwards, Biogenic iron oxyhydroxide formation at mid-ocean ridge hydrothermal vents: Juan de Fuca Ridge, Geochim. Cosmochim. Acta, 2009, 73(2), 388–403 CrossRef CAS.
- R. W. G. Wyckoff, in Crystal Structures, Interscience Publishers: New York, New York, 1963, vol. 1, pp. 290–295 Search PubMed.
- G. Ona-Nguema, M. Abdelmoula, F. Jorand, O. Benali, A. Gehin, J. Block and J. Genin, Iron(II,III) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction, Environ. Sci. Technol., 2002, 36(1), 16–20 CrossRef CAS PubMed.
- J. M. R. Genin, P. Refait, G. Bourrie, M. Abdelmoula and F. Trolard, Structure and stability of the Fe(II)-Fe(III) green rust "fougerite" mineral and its potential for reducing pollutants in soil solutions, Appl. Geochem., 2001, 16(5), 559–570 CrossRef CAS.
- C. Ruby, M. Abdelmoula, S. Naille, A. Renard, V. Khare, G. Ona-Nguema, G. Morin and J. M. R. Genin, Oxidation modes and thermodynamics of FeII-III oxyhydroxycarbonate green rust: Dissolution-precipitation versus in situ deprotonation, Geochim. Cosmochim. Acta, 2010, 74(3), 953–966 CrossRef CAS.
- A. Voegelin, A. Senn, R. Kaegi, S. Hug and S. Mangold, Dynamic Fe-precipitate formation induced by Fe(II) oxidation in aerated phosphate-containing water, Geochim. Cosmochim. Acta, 2013, 117, 216–231 CrossRef CAS.
- A. Voegelin, R. Kaegi, J. Frommer, D. Vantelon and S. J. Hug, Effect of phosphate, silicate, and Ca on Fe(III)-precipitates formed in aerated Fe(II)- and As(III)-containing water studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74(1), 164–186 CrossRef CAS.
- C. M. van Genuchten, A. J. Gadgil and J. Pena, Fe(III) nucleation in the presence of bivalent cations and oxyanions leads to subnanoscale 7 A polymers, Environ. Sci. Technol., 2014, 48(20), 11828–11836 CrossRef CAS PubMed.
- A. Senn, R. Kaegi, S. Hug, J. Hering, S. Mangold and A. Voegelin, Composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in water at near-neutral pH: Interdependent effects of phosphate, silicate and Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- B. Ravel and S. Kelly, in The Difficult Chore of Measuring Coordination by EXAFS, AIP Conference Proceedings, 2007, 2007, pp. 150–152 Search PubMed.
- Y. H. Wang, G. Morin, G. Ona-Nguema, N. Menguy, F. Juillot, E. Aubry, F. Guyot, G. Calas and G. E. Brown, Arsenite sorption at the magnetite-water interface during aqueous precipitation of magnetite: EXAFS evidence for a new arsenite surface complex, Geochim. Cosmochim. Acta, 2008, 72(11), 2573–2586 CrossRef CAS.
- S. J. Hug and O. Leupin, Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the Fenton reaction, Environ. Sci. Technol., 2003, 37(12), 2734–2742 CrossRef CAS PubMed.
- M. M. Benjamin, Water chemistry, McGraw-Hill: Boston, 2002, p xix, 668 p Search PubMed.
- M. Stachowicz, T. Hiemstra and W. van Riemsdijk, Multi-competitive interaction of As(III) and As(V) oxyanions with Ca2+, Mg2+, PO43-, and CO32- ions on goethite, J. Colloid Interface Sci., 2008, 320(2), 400–414 CrossRef CAS PubMed.
- M. Stachowicz, T. Hiemstra and W. van Riemsdijk, Surface speciation of As(III) and As(V) in relation to charge distribution, J. Colloid Interface Sci., 2006, 302(1), 62–75 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The iron oxides : structure, properties, reactions, occurrence, and uses, VCH: Weinheim, New York, 1996, p. 573 Search PubMed.
- I. Halevy, M. Alesker, E. M. Schuster, R. Popovitz-Biro and Y. Feldman, A key role for green rust in the Precambrian oceans and the genesis of iron formations, Nat. Geosci., 2017, 10, 135–139 CrossRef CAS.
- E. Koeksoy, A. Sundman, J. M. Byrne, B. Lohmayer, B. Planer-Friedrich, I. Halevy, K. O. Konhauser and A. Kappler, Formation of green rust and elemental sulfur in an anlogue for oxygenated ferro-euxinic transition zones of Precambrian oceans, Geology, 2019, 47(3), 211–214 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00267g |
| ‡ Previous address: Department of Chemistry, University of Copenhagen, Copenhagen, Denmark. |
| This journal is © The Royal Society of Chemistry 2019 |