
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox processes in sodium vanadium phosphate cathodes – insights from operando magnetometry†
Gregor
Klinser
 *a,
Roman
Zettl
b,
Martin
Wilkening
bd,
Heinz
Krenn
c,
Ilie
Hanzu
bd and
Roland
Würschum
a
*a,
Roman
Zettl
b,
Martin
Wilkening
bd,
Heinz
Krenn
c,
Ilie
Hanzu
bd and
Roland
Würschum
a
aInstitute of Material Physics, Graz University of Technology, Petersgasse 16, A-8010 Graz, Austria. E-mail: wuerschum@tugraz.at; g.klinser@tugraz.at; Fax: +43 316 873 108480; Tel: +43 316 873 8481
bInstitute for Chemistry and Technology of Materials, Christian Doppler Laboratory for Lithium Batteries, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria
cInstitute of Physics, University of Graz, Universitätsplatz 5, A-8010 Graz, Austria
dALISTORE – European Research Institute, CNRS FR3104, Hub de l'Energie, Rue Baudelocque, 80039 Amiens, France
First published on 4th September 2019
Abstract
Operando magnetic susceptibility measurements of sodium ion cathode materials during repetitive electrochemical cycling enable a continuous and bulk sensitive monitoring of the transition metal oxidation states. Such measurements on NaxV2(PO4)3 identified vanadium to be the only ion undergoing oxidation/reduction processes upon battery operation. For the initial battery charging–discharging cycle as well as for the first cycle after prolonged room temperature storage, however, peculiarities within the magnetic susceptibility measurements indicate parasitic side reactions, likely on the cathode surface.
1 Introduction
Transition metal compounds, such as oxides and phosphates, are universally used as positive electrode (cathode) materials in advanced batteries. For instance, lithium vanadium phosphate (LVP, Li3V2(PO4)3) was proven as a highly stable, very fast cycling active material with applications ranging from high-power batteries to battery-supercapacitor hybrid devices.1–3 Its sodium analogue, the sodium vanadium phosphate (NVP, Na3V2(PO4)3) shows good performance as cathode material in sodium ion batteries.4 As with most transition metal oxides used for lithium or sodium storage, the transition metal center undergoes oxidation or reduction upon charging or discharging of the battery electrode.Magnetometry has proven to be a powerful tool for studying such oxidation/reduction processes in layered cathode materials with Li as the mobile ionic species (e.g.ref. 5–15). Combining magnetic measurements with operando techniques enables a continuous and bulk sensitive monitoring of the oxidation and reduction processes during electrochemical cycling.16–19 As the magnetic susceptibility serves as highly sensitive fingerprint to the oxidation state of the transition metal, irrespectively of the ion species, the technique of magnetometry will also give valuable insights for the sodium analogue.
The present study aims at operando magnetic susceptibility measurements of sodium ion cathode materials. In particular, the oxidation and reduction processes in sodium vanadium phosphate upon electrochemical cycling are studied.
2 Results
The electrochemical behavior of the prepared NVP carbon composite (NVP/C) was investigated by cyclic voltammetry (CV) and galvanostatic cycling with potential limitation (GCPL). CV accurately shows the characteristic oxidation peak at 3.5 V vs. Na/Na+ as well as the reduction peak at 3.3 V vs. Na+/Na (Fig. 1a). These values fit nicely to previous studies on NVP.20,21 The electrochemical process is clearly reversible since almost symmetrical peaks occur at the oxidation and reduction side. The small visible shoulders in the oxidation and reduction peaks (indicated by arrows in Fig. 1a) indicate structural reorganization22 which is related to a change in sodium ion occupancy.23 | ||
| Fig. 1 (a) Cyclic voltammetry of NVP carbon composite (NVP/C) with a cycling rate of 0.05 mV s−1. A different behavior in the first cycle is observed in CV, see text for further explanation. (b) Specific capacity and coulombic efficiency of NVP/C during galvanostatic cycling between 2.8 and 4.0 V for 175 cycles at different C-rates: C/10, C/5, C/2, C, 2C and 5C. Note: the voltage profiles for these measurements can be found in Fig. S1, ESI.† | ||
In the potential range of 2.8–4.0 V (vs. Na/Na+) a fully reversible two-phase electrochemical reaction occurs where two sodium ions can be extracted and inserted, respectively.‡ Nevertheless, the first cycle is slightly different compared to the subsequent cycles (see Fig. 1a). On the one hand, there is a small shift in potential towards more negative values. On the other hand, the CV slope significantly deviates for the first cycle (more pronounced in the oxidation peak) (see Fig. S2, ESI† for different sweep rates). This could be related to a slightly lower stoichiometry of sodium in NVP as revealed by inductively coupled plasma mass spectrometry (see Table S1, ESI†). However, deviations within the first charging cycle are also seen in magnetic susceptibility measurements which cannot be explained with a lower stoichiometry of sodium in NVP (see below).
Fig. 1b shows the cycling behavior of NVP/C half-cell. After each 25 cycles the C-rates where increased from C/10, C/5, C/2, C, 2C to finally 5C. The last 15 cycles serve as a control as they are conducted again with a slow C-rate of C/10. GCPL revealed that NVP/C shows almost no capacity loss within a certain cycling rate. The drop in capacity upon increasing the cycling rate is related to the increase of ohmic drops at higher currents. The drops, however, are relatively small, a sign of the good electronic conductivity introduced by the carbon phase of the NVP/C composite. Interestingly the capacity is still as high as 52 mA h g−1 even for cycling the cell at the highest rate of 5C.
The electrode electrochemical potential E and the change in the magnetic susceptibility Δχ (with respect to the magnetic signal of the pristine sample) as a function of charging time t for two independent measurements are shown in Fig. 2. In Fig. 2a and b the first three cycles of a freshly prepared cell are shown. With the onset of charging, χ immediately starts to decrease. This decrease prevails monotonously until the upper cut-off potential of 4.0 V is reached after 8.0 h of charging. Reversing the current flow results in an instantaneous linear re-increase of χ, which lasts until the end of the discharging cycle at t = 15.6 h. The second and third cycle qualitatively show the same linear variation of the magnetic susceptibility. While Fig. 2a and b display the first three cycles of a battery cell, Fig. 2c and d show cycles 25 to 28 of a different cell. It should be noted that the cell rested under open circuit conditions for 6 days between cycle 24 and 25. The observed susceptibility variation in Fig. 2d can be described as above (Fig. 2b). The susceptibility immediately starts to decrease with the onset of charging. This decrease prevails linearly until the end of charging and re-increases instantaneously as soon as discharging starts.
 | ||
| Fig. 2 (a) Electrochemical electrode potential E with the cut off potentials 4.0 and 2.5 V and (b) change in the magnetic susceptibility Δχ as a function of charging time t for three consecutive cycles of charging and discharging of the cathode material NaxV2(PO4)3 (C/8 rate). (c), (d) same as (a), (b) for a cell which has been cycled 24 times before the magnetic measurement. The straight dashed line in (b) and in (d) are calculated according to eqn (1) and (2) assuming pure V3+(S = 1) → V4+(S = 1/2) oxidation. | ||
The data represented in Fig. 2a and b are replotted in Fig. 3 as a function of the sodium content x, which is calculated for the nominal cathode mass using Faraday's law and the assumption that the entire current is consumed for Na extraction and insertion, respectively. While the electrode potential curve of different charging and discharging cycles overlaps (see Fig. 3a), the change in the magnetic susceptibility (displayed in Fig. 3b) differs between the first and second cycle. However, from the second cycle onwards Δχ perfectly overlaps for the subsequent charging and discharging cycles.
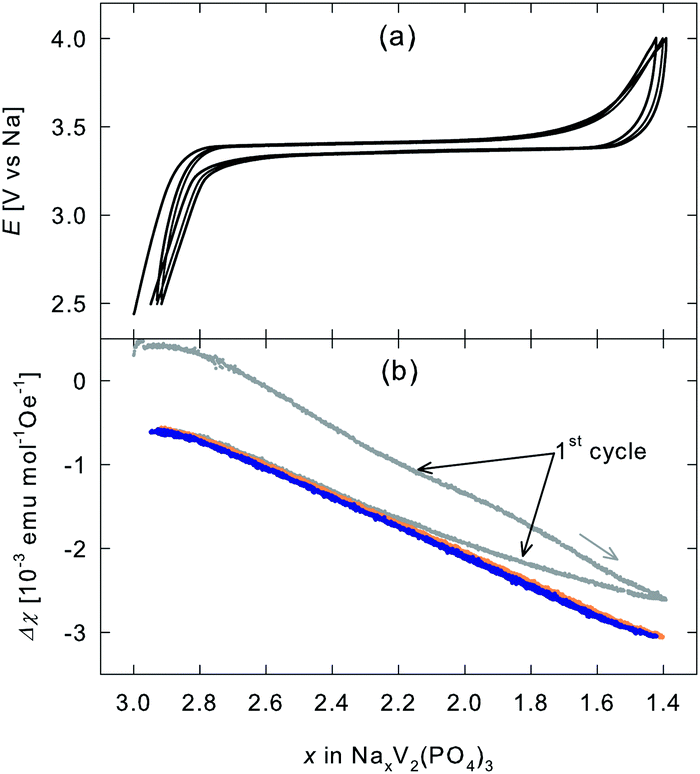 | ||
| Fig. 3 (a) Electrode potential E (cut off potentials 4.0 and 2.5 V) and (b) change in the magnetic susceptibility Δχ as a function of Na-content x of the cathode material NaxV2(PO4)3. | ||
Ex situ magnetic susceptibility measurement on the same cathode material, NaxV2(PO4)3 with x = 2 (Fig. S3, ESI†) show a linear variation of 1/χ as a function of temperature as characteristic for pure Langevin paramagnetism.
3 Discussion
As ex situ magnetic susceptibility measurement revealed pure Langevin paramagnetism at room temperature, charging induced variation of the susceptibility χ(t) can be calculated according to the Curie–Weiss law:17,19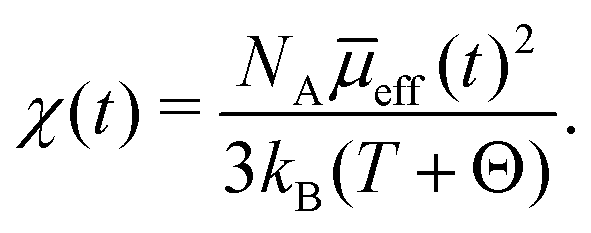 | (1) |
![[small mu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cd.gif) eff the mean effective magnetic moment. For Θ a value of 50 K was determined by the ex situ magnetic susceptibility measurement. eff can be calculated from the effective magnetic moment of the transition metal ions in its respective oxidation state μi and their corresponding concentration ci:17,19
eff the mean effective magnetic moment. For Θ a value of 50 K was determined by the ex situ magnetic susceptibility measurement. eff can be calculated from the effective magnetic moment of the transition metal ions in its respective oxidation state μi and their corresponding concentration ci:17,19 | (2) |
As vanadium is the only transition metal ion in this compound, the χ-variation is considered to arise from V3+(S = 1, μi = 2.83 μB)§ ↔ V4+(S = 1/2, μi = 1.73 μB) oxidation/reduction during charging/discharging. The variation of the magnetic susceptibility, as predicted according to eqn (1) and (2) for pure vanadium oxidation/reduction, is plotted as a function of charging time in Fig. 2b and d as black dashed line. Apart from the first cycle, the experimentally observed χ-variation can indeed be well described by the χ-variation expected for the V3+/V4+ oxidation/reduction (Fig. 2b and d). Deviations within the first cycle are only seen in the magnetic susceptibility data but not within the electrode potential during galvanostatic cycling (Fig. 3). However, some small deviations of the electrochemical response occur in the first cycle of a cyclic voltammetry experiment (Fig. 1a).
It seems likely that the observed deviations in the magnetic susceptibility measurements are related to some chemical reactions (potential assisted) at the surface of the electrode/electrolyte, e.g. the formation of a passivation layer. Indeed an unstable passivation layer on the same cathode material in 1 M NaFSI in EC/PC (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was reported by Manohar et alref. 25. Upon such a reaction, Na ions may be extracted from the cathode which do not contribute to battery charging similar to the formation of surface layer compounds proposed for LiMn2O4.26 Due to charge neutrality conditions, Na ions which are lost in the passivation layer formation process will cause V3+ → V4+ oxidation. Thus, during the first charging cycle this parasitic effects contributes to vanadium oxidation which results in a more pronounced χ-decrease compared to the predicted model (see Fig. 2b dashed lines). The less pronounced increase at the beginning of the first discharging cycle could be due to the same reason. Here, the reduction of vanadium ions due to battery discharging is superimposed by oxidation process caused by the passivation layer formation. After around 12 hours of battery operation (see Fig. 2b) the measured and predicted χ-variation according to eqn (1) and (2) agree, indicating that the reaction not contributing to the battery cycling has vanished (is in equilibrium). The deviations at the beginning of the 25th cycle in Fig. 2d may arise for the very same reason. The cell was under open circuit condition for 6 days before measured within the SQUID magnetometer. It is reasonable to think that after 6 days the material relaxes toward a different equilibrium state that, to some extent, involves a return to the original situation (before any cycling) as the passivation layer is found to be unstable in organic electrolytes.25 It should be noted that magnetic ordering was reported on the similar Li NASICON phase Li3Fe2(PO4)3.27 As this ordering phenomena occur at low temperatures it is not believed to have any influence on the observed room temperature χ-variation of this work.
:1) was reported by Manohar et alref. 25. Upon such a reaction, Na ions may be extracted from the cathode which do not contribute to battery charging similar to the formation of surface layer compounds proposed for LiMn2O4.26 Due to charge neutrality conditions, Na ions which are lost in the passivation layer formation process will cause V3+ → V4+ oxidation. Thus, during the first charging cycle this parasitic effects contributes to vanadium oxidation which results in a more pronounced χ-decrease compared to the predicted model (see Fig. 2b dashed lines). The less pronounced increase at the beginning of the first discharging cycle could be due to the same reason. Here, the reduction of vanadium ions due to battery discharging is superimposed by oxidation process caused by the passivation layer formation. After around 12 hours of battery operation (see Fig. 2b) the measured and predicted χ-variation according to eqn (1) and (2) agree, indicating that the reaction not contributing to the battery cycling has vanished (is in equilibrium). The deviations at the beginning of the 25th cycle in Fig. 2d may arise for the very same reason. The cell was under open circuit condition for 6 days before measured within the SQUID magnetometer. It is reasonable to think that after 6 days the material relaxes toward a different equilibrium state that, to some extent, involves a return to the original situation (before any cycling) as the passivation layer is found to be unstable in organic electrolytes.25 It should be noted that magnetic ordering was reported on the similar Li NASICON phase Li3Fe2(PO4)3.27 As this ordering phenomena occur at low temperatures it is not believed to have any influence on the observed room temperature χ-variation of this work.
4 Conclusion and summary
Operando magnetic susceptibility measurements can serve as highly sensitive fingerprint for the charge compensation process in battery cathodes as the magnetic moment of the transition metal ions is directly related to its oxidation state. For the cathode material NaxV2(PO4)3 the charge compensation process was confirmed to be entirely due to V3+(S = 1) ↔ V4+(S = 1/2) oxidation/reduction during charging/discharging. Cyclic voltammetry and magnetic measurements clearly revealed deviations for the first charging cycle which could be related to the formation of an electrode–electrolyte interphase layer on the NVP cathode.5 Experimental
Synthesis of NVP/C
Sodium vanadium phosphate–carbon composite (NVP/C) was synthesized via a sol–gel synthesis analogous to the synthesis of LVP/C (lithium vanadium phosphate, Li3V2(PO4)3–carbon composite).28 Here, the precursor LiH2PO4 was replaced with NaH2PO4. Citric acid as gel-forming agent and NH4VO3 were separately dissolved in 50 ml of deionized water and then mixed in a beaker. The molar ratio of citric acid to vanadium was 1:1. The reaction mixture was slowly heated to 60 °C. NaH2PO4 in stoichiometric amounts was dissolved in 50 ml deionized water and was added slowly (drop-wise in approximately 1 h) from a separation funnel to the solution of NH4VO3 and citric acid at 60 °C. The solution was then heated to 80 °C to evaporate the water on a hot plate until a gel formed. The obtained gel was subsequently dried overnight in an oven at 80 °C under air. Afterward, it was first ground in an agate mortar and then thermally treated under argon in a quartz tube placed in a tube oven at 700° for 8 h. The heating ramp was set at 4 °C min−1, the cooling ramp being the natural cooling rate of the oven, which is approximately 8–12 °C min−1 above 350 °C. In these synthesis conditions, the citric acid converts to conductive carbon and forms a composite with the NVP. At last two grounding processes of the NVP carbon composite (using an agate mortar) were performed with an sintering step in between. The XRD pattern of the obtained crystalline black powered can be seen in Fig. S4 (ESI†).
Electrochemical characterization
The electrodes were prepared by the slurry cast method. The slurry contained the active material (NVP/C, 800 mg), conductive carbon (Super C65, 100 mg) and a binder (Kynar 761, 100 mg) resulting in a mass ratio of 8:1:1 respectively. Additionally, 3 g of solvent (1-methyl-2-pyrrolidinone (NMP)) were added. The components were mixed together by ball milling at 400 rpm 4 times for 15 minutes. The well dispersed slurry was cast on an Al current collector which was, before casting, etched with a 5 wt% KOH solution at 40 °C. The doctor blade casting was carried out at a velocity of 5 mm s−1 and using a casting gap of 150 μm. The cast films were then dried at 60 °C in air, electrode disks were cut (10 mm in diam.), vacuum dried in a Büchi oven and then transferred to an Ar-filled glovebox with water and oxygen contents smaller than 0.5 ppm. The electrodes, with a mass loading of 1.68 mg cm−2, were assembled in Swagelok-type cells with metallic Na as reference and counter electrode. The electrolyte used in all experiments was 1 M NaFSI in EC:DEC (volume ratio 4:6). Electrochemical measurements were carried out on VMP3 or MPG2 potentiostat/galvanostat instruments from Bio-Logic controlled by the EC-Lab software. Cyclic voltammetry (CV) was performed at different increasing scan rates, i.e. 0.05 mV s−1, 0.1 mV s−1, 0.2 mV s−1 and 0.5 mV s−1. The samples were cycled between 2.8 V to 4.0 V vs. Na/Na+. Galvanostatic cycling with potential limitation was carried out between 2.8 V to 4.0 V vs. Na/Na+. The cycling rates were C/10, C/5, C/2, C, 2C, 5C (each 25 cycles) and again C/10 (15 cycles). The currents corresponding to the chosen C-rates were calculated considering the theoretical capacity of NVP, which is 118 mA h g−1.
Operando magnetometry
The cathodes for the operando magnetic measurements were prepared from the same electrode slurry used for electrochemical characterization (see above). A flattened end of an aluminum wire was first dip-coated, then pre-dried in air and subsequently dried at 370 K under vacuum conditions (10−5 mbar) for 24 hours. The dip-coated cathodes were mounted in a self-developed purpose-built electrochemical cell suitable for the use in a SQUID magnetometer.16,29 This cell enables an electrochemical measurement in a 3-electrode geometry and ensures that the measured magnetic signal variation arise originate solely from the cathode material. Please note that with the developed cell design the magnetic signal of both the anode and cathode is actually measured. However, sodium plating and dissolving stripping on the anode side contributes less than 1% to the total magnetic signal variation. Thus, the variation of the magnetic susceptibility can entirely be attributed to the variation of the susceptibility of the cathode material. In the present case, the cell compartment was a polyethylene tube. Metallic sodium pressed on copper wires served as anode and reference electrode. The electrolyte was the same as for the electrochemical characterization (vide supra). Charging and discharging was performed with a constant current of 18.4 μA (Fig. 2a and b) and 25.7 μA (Fig. 2c and d) corresponding to a C-rate of about C/8 based on the specific theoretical cathode capacity of 118 mA h g−1 and the coating masses of 1.56 mg and 2.73 mg using a single channel potentiostat/galvanostat instrument potentiostat (Autolab PGSTAT 128N).The cathode electrode film for the ex situ magnetometry measurement was prepared in the same way as for the electrochemical measurements. A pouch cells was used in which the reference electrode and the counter electrode was made of metallic sodium foil. The square 4 cm2 electrode containing 8.5 mg of NVP/C active mass was cycled between 2.8 V and 3.8 V vs. Na/Na+ and subsequently stopped in the 4th charging cycle at an approximate sodium content of x = 2 (NaxV2(PO4)3). Afterward the cell was dismantled, the NVP-containing cathode rinsed with dimethyl carbonate (DMC) and dried under in vacuum conditions at room temperature before placing it in an airtight SQUID sample holder with minimized magnetic contribution. In addition, the measured signal was also corrected for the magnetic signal of the additives (carbon and binder) and the aluminum substrate both of which in sum contribute by about 9% to the overall signal. The whole process of cathode preparation, handling and transfer to the SQUID magnetometer was performed under protective Ar-atmosphere.
Magnetic susceptibility measurements were performed at an applied magnetic field of 5000 Oe using a SQUID magnetometer (Quantum Design MPMS-XL-7) in the same manner as described elsewhere.18 In the case of operando studies, the temperature was controlled and kept at 300 K, whereas the ex situ samples were measured as a function of temperature (field-cooling, 300 K to 10 K).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in the framework of the inter-university cooperation of TU Graz and Uni Graz on natural science (NAWI Graz). This work was financially supported by the Austrian Research Promotion Agency (FFG) through the project SolaBat (grant no. 853627).References
- N. Böckenfeld and A. Balducci, J. Power Sources, 2013, 235, 265–273 CrossRef.
- X. Rui, N. Ding, J. Liu, C. Li and C. Chen, Electrochim. Acta, 2010, 55, 2384–2390 CrossRef CAS.
- M. Secchiaroli, G. Giuli, B. Fuchs, R. Marassi, M. Wohlfahrt-Mehrens and S. Dsoke, J. Mater. Chem. A, 2015, 3, 11807–11816 RSC.
- Q. Wang, J. Xu, W. Zhang, M. Mao, Z. Wei, L. Wang, C. Cui, Y. Zhu and J. Ma, J. Mater. Chem. A, 2018, 6, 8815–8838 RSC.
- A. Mauger, F. Gendron and C. Julien, J. Alloys Compd., 2012, 520, 42–51 CrossRef CAS.
- Z. Li, N. A. Chernova, M. Roppolo, S. Upreti, C. Petersburg, F. M. Alamgir and M. S. Whittingham, J. Electrochem. Soc., 2011, 158, A516 CrossRef CAS.
- M. Labrini, I. Saadoune, F. Scheiba, A. Almaggoussi, J. Elhaskouri, P. Amoros, H. Ehrenberg and J. Brötz, Electrochim. Acta, 2013, 111, 567 CrossRef CAS.
- D. Mohanty and H. Gabrisch, J. Power Sources, 2012, 220, 405 CrossRef CAS.
- G. Klinser, S. Topolovec, H. Kren, S. Koller, H. Krenn and R. Würschum, Solid State Ionics, 2016, 293, 64 CrossRef CAS.
- D. Kellerman, V. Galakhov, A. Semenova, Y. Blinovskov and O. Leonidova, Phys. Solid State, 2006, 48, 548–556 CrossRef CAS.
- J. T. Hertz, Q. Huang, T. McQueen, T. Klimczuk, J. W. G. Bos, L. Viciu and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075119 CrossRef.
- K. Miyoshi, C. Iwai, H. Kondo, M. Miura, S. Nishigori and J. Takeuchi, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 075113 CrossRef.
- J. Sugiyama, H. Nozaki, J. H. Brewer, E. J. Ansaldo, G. D. Morris and C. Delmas, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 144424 CrossRef.
- K. Mukai, Y. Ikedo, H. Nozaki, J. Sugiyama, K. Nishiyama, D. Andreica, A. Amato, P. L. Russo, E. J. Ansaldo, J. H. Brewer, K. H. Chow, K. Ariyoshi and T. Ohzuku, Phys. Rev. Lett., 2007, 99, 087601 CrossRef CAS PubMed.
- T. Motohashi, T. Ono, Y. Sugimoto, Y. Masubuchi, S. Kikkawa, R. Kanno, M. Karppinen and H. Yamauchi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 165114 CrossRef.
- S. Topolovec, H. Kren, G. Klinser, S. Koller, H. Krenn and R. Würschum, J. Solid State Electrochem., 2016, 20, 1491–1496 CrossRef CAS.
- G. Klinser, S. Topolovec, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, Appl. Phys. Lett., 2016, 109, 213901 CrossRef.
- G. Klinser, M. Stückler, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, J. Power Sources, 2018, 396, 791–795 CrossRef CAS.
- G. Klinser, S. Topolovec, H. Krenn and R. Würschum, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 849–855 Search PubMed.
- L. Si, Z. Yuan, L. Hu, Y. Zhu and Y. Qian, J. Power Sources, 2014, 272, 880–885 CrossRef CAS.
- N. Böckenfeld and A. Balducci, J. Solid State Electrochem., 2014, 18, 959–964 CrossRef.
- E. Wang, M. Chen, X. Liu, Y. Liu, H. Guo, Z. Wu, W. Xiang, B. Zhong, X. Guo, S. Chou and S.-X. Dou, Small Methods, 2019, 3, 1800169 CrossRef CAS.
- W. Song, X. Ji, Z. Wu, Y. Zhu, Y. Yang, J. Chen, M. Jing, F. Li and C. E. Banks, J. Mater. Chem. A, 2014, 2, 5358–5362 RSC.
- Y. Fang, J. Zhang, L. Xiao, X. Ai, Y. Cao and H. Yang, Adv. Sci., 2017, 4, 1600392 CrossRef PubMed.
- C. Manohar, A. Raj K, M. Kar, M. Forsyth, D. R. MacFarlane and S. Mitra, Sustainable Energy Fuels, 2018, 2, 566–576 RSC.
- T. Eriksson, A. M. Andersson, C. Gejke, T. Gustafsson and J. O. Thomas, Langmuir, 2002, 18, 3609–3619 CrossRef CAS.
- G. Rousse, J. Rodríguez-Carvajal, C. Wurm and C. Masquelier, Chem. Mater., 2001, 13, 4527–4536 CrossRef CAS.
- C. Huang, D. Chen, Y. Huang and Y. Guo, Electrochim. Acta, 2013, 100, 1–9 CrossRef CAS.
- S. Topolovec, H. Krenn and R. Würschum, Rev. Sci. Instrum., 2015, 86, 063903 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04045e |
| ‡ The remaining sodium ion, residing on a different crystallographic position, is much stronger bound and its electrochemical extraction is not possible in this potential range.24 |
§ In the spin only case  with the spin state Si and g = 2. with the spin state Si and g = 2. |
| This journal is © the Owner Societies 2019 |