
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction of an arsinoamide with chloro tetrylenes: substitution and As–N bond insertion†
Xiao
Chen
,
Thomas
Simler
 ,
Ravi
Yadav
,
Michael T.
Gamer
,
Ralf
Köppe
and
Peter W.
Roesky
*
,
Ravi
Yadav
,
Michael T.
Gamer
,
Ralf
Köppe
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 16th July 2019
Abstract
Reaction of the arsinoamide [(Mes2AsNPh){Li(OEt2)2}] with the low-valent group 14 compounds, [{PhC(tBuN)2}ECl] (E = Si, Ge) and GeCl2·dioxane, resulted in two different reaction pathways: simple substitution or substitution accompanied by an insertion step. As a result, either insertion products with an As–Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N unit and an As–Ge bond, or substitution products, in which the intact arsinoamide binds to the group 14 elements via the N atom, were obtained.
N unit and an As–Ge bond, or substitution products, in which the intact arsinoamide binds to the group 14 elements via the N atom, were obtained.
Although P–N ligands such as bis(phosphino)amines R2P–N(H)–PR2 and aminophosphines R2P–N(H)R′, and their anionic derivatives (R2P–N–PR2)− and (R2P–NR′)−, are well-established ligands for a wide range of metals,1 the coordination chemistry of the heavier arsenic congener R2As–N(H)R′ is virtually unknown. Lately, we reported the synthesis of the aminoarsane Mes2AsN(H)Ph and its alkali metal derivatives [(Mes2AsNPh){Li(OEt2)2}], [(Mes2AsNPh){Na(OEt2)}]2 and [(Mes2AsNPh){K(THF)}]2.2 Herein, we showcase the first reactivity studies of these arsinoamides by salt metathesis reactions of the Li salt with low-valent group 14 compounds. Different reaction pathways were observed depending on the nature of the group 14 element and its substituent.
Low-valent group 14 compounds were chosen as suitable precursors because they possess a dual Lewis acid–base character, due to the presence of a lone pair and a vacant p orbital on the same atom.3 These features allow for a wide range of reactivity, e.g. low-valent group 14 compounds have attracted increasing attention in the context of small molecule activation (e.g. H2, NH3, N2O, and CO2).4 The usual mechanism involves the donation from the filled σ orbital of the small molecule into the vacant p orbital of low-valent group 14 elements and the back-donation from the lone pair of low-valent group 14 elements to the empty σ* orbital of the small molecule.5 Because of their ability to split both single bonds, e.g. E–H (E = H, C, N, P etc.),4d,6 and double bonds, e.g. CX (X = C, O, S etc.),7 low-valent group 14 compounds possess a great potential application for catalytic reactions, in a similar way as transition metal compounds.4c,8
Reaction of the silylene [LSiCl] (L = PhC(tBuN)2) with the arsinoamide [(Mes2AsNPh){Li(OEt2)2}] at room temperature led to the formation of [LSi(NPh)(AsMes2)] (1) (Scheme 1), which corresponds to the insertion product of the silylene into the As–N bond of the arsinoamide. Compound 1 was isolated in 61% crystalline yield. The insertion of the Si atom into the As–N bond resulted in the formal oxidation of the Si centre, from +2 to +4, which is reflected by the 29Si{1H} NMR spectrum of 1. In comparison to the precursor [LSiCl] (δ(29Si{1H}) = 14.6 ppm),9 the 29Si{1H} NMR signal of 1 is upfield shifted to −68.9 ppm, which fits well with the published values for four-fold coordinated silicon compounds, e.g. [LSi(NAd)X] (X = NPh2 or NMe2, Ad = adamantyl).10
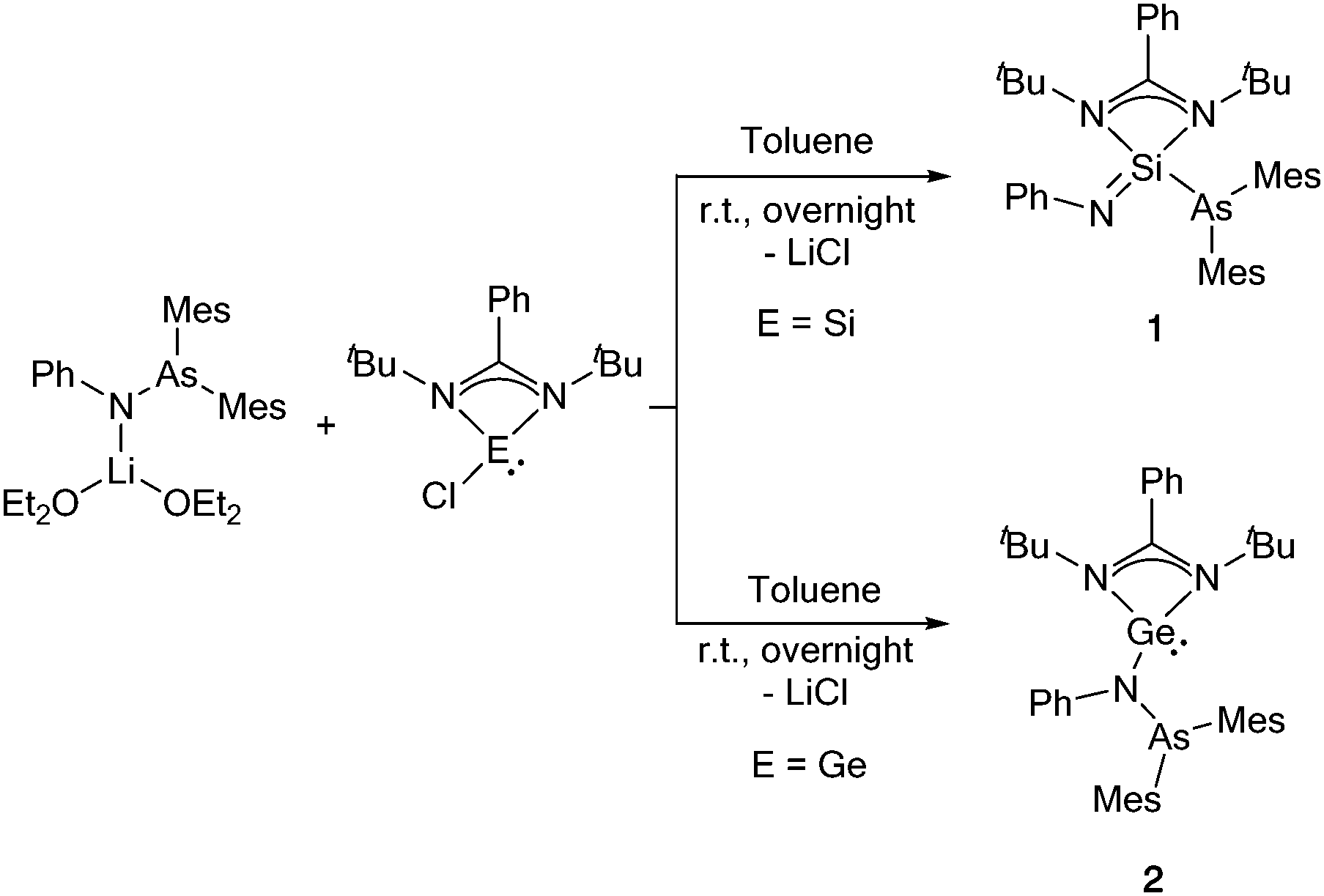 | ||
| Scheme 1 Reaction of the arsinoamide [(Mes2AsNPh){Li(OEt2)2}] with amidinate silylene and germylene [{PhC(tBuN)2}ECl] (E = Si, Ge). | ||
The molecular structure of 1 further reveals an unexpected As–N bond cleavage, which occurred through an oxidative addition step, forming a NSi–As fragment (Fig. 1). As a result, one Si(IV)–As(III) bond with a distance of 2.3948(6) Å is formed. The observed bond distance is consistent with that of other silicon–arsenic single bonds, e.g. 2.352(1) Å in [Mo(CO)4(Me2AsSiMe2(CH2)2AsMe2)].11 In addition, the SiN3 bond length (1.570(2) Å) agrees well with the reported value for a silicon–nitrogen double bond (e.g. 1.545(2) Å in the silaimine [LSi(Cl)N-(2,6-iPr2C6H3)]).12 The N3Si–As angle formed after the insertion of Si into the As–N bond is 133.76(8)°, resulting in a distorted tetrahedral geometry for the four-coordinated silicon atom. Interestingly, 1H NMR monitoring of the reaction between [(Mes2AsNPh){Li(OEt2)2}] and LSiCl revealed the formation of an intermediate, possibly corresponding to the substitution product before the insertion step, that cleanly converts into 1 within 1 h at room temperature (see Fig. S10 and S11, ESI†). As the product was purified by crystallisation, the moderate isolated yield of 1 can be traced back to losses during the crystallisation process. Very recently, a related insertion of [LSiCl] into the P–N bond of the phosphinoamide [(Ph2PNDipp)Li] (Dipp = 2,6-diisopropylphenyl) has been reported. However, much harsher conditions (80 °C, 18 h) were required for the completion of this reaction.13
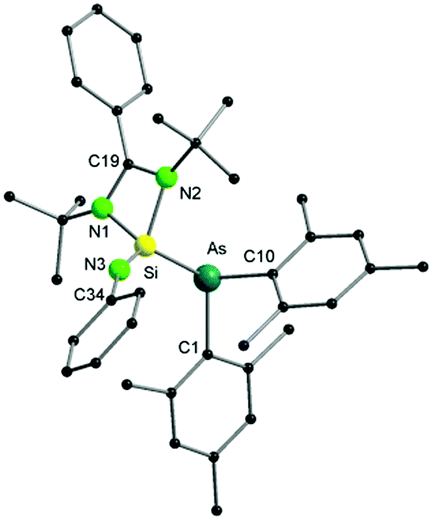 | ||
| Fig. 1 Molecular structure of compound 1 in the solid state. Hydrogen atoms have been omitted for clarity. Selected bond distances and angles are given in the ESI.† | ||
Motivated by the unexpected results obtained for the reaction of the silylene, we moved on to the heavier tetrylene analogue for comparison. After the reaction of [LGeCl] with [(Mes2AsNPh){Li(OEt2)2}] under identical reaction conditions as for the synthesis of 1, [LGe(Mes2AsNPh)] (2) was isolated in 63% crystalline yield (Scheme 1). Compared to [(Mes2AsNPh){Li(OEt2)2}], the signals of the phenyl group of the arsinoamide are downfield shifted in the 1H NMR spectrum of 2 (7.79–7.16 ppm vs. 7.33–6.63 ppm in [(Mes2AsNPh){Li(OEt2)2}]). In the 13C{1H} NMR spectrum, the characteristic signal of the NCN carbon atom in the amidinate unit of 2 is upfield shifted (167.4 ppm) compared to that of [LGeCl] (173.3 ppm).14
Single crystal X-ray diffraction of 2 (Fig. 2) reveals that, in contrast to [LSiCl], the germylene analogue [LGeCl] did not insert into the As–N bond under the same conditions as used for the synthesis of 1. In compound 2, the Ge atom persists in the divalent state. The nitrogen atom of the arsinoamide is bound to both the Ge centre and the As atom, the latter further coordinated by the two mesityl groups. The Ge–N3 bond of 2 is slightly shorter (ca. 0.1 Å) than the Ge–N1 and Ge–N2 bonds with the amidinate ligand. However, all three Ge–N bond lengths are in the typical range for single bonds (e.g. from 1.847(3) to 1.981(3) Å in amidinate–germylenes).15 As seen in [(Mes2AsNPh){Li(OEt2)2}], the arsinoamide in 2 exhibits a trans conformation, as defined by the relative orientation of the substituents on the N and As atoms.16 The three-fold coordinated germanium atom shows a pyramidal coordination geometry (sum of bond angles around Ge of 267.87°), which indicates the presence of a lone pair on the germanium atom. An additional lone pair is located on the As atom, rendering compound 2 a potentially useful ligand for coordination chemistry. The reactivity difference of [LGeCl] vs. [LSiCl] towards the As–N bond may be the result of the lower reduction potential of GeII compared to SiII and the energetic stabilization of the lone pair in heavier group 14 elements (the inert pair effect). Formation of 1 may result from the rearrangement of a silicon intermediate, structurally similar to 2, through the nucleophilic attack of the Si-lone pair on the As–N bond. However, the exact mechanism remains elusive. In case of the phosphinoamide ligand [(Ph2PNDipp)]− (see above), a divalent silicon compound, which may be structurally similar to the intermediate leading to 1, was isolated and slowly isomerised via insertion of Si into the P–N bond.13 An attempted thermolysis reaction monitored by 1H NMR spectroscopy revealed that 2 does not undergo a similar isomerisation even at elevated temperatures (up to 100 °C).
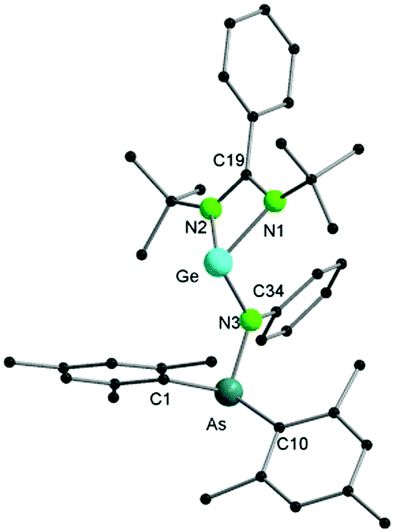 | ||
| Fig. 2 Molecular structure of compound 2 in the solid state. Hydrogen atoms have been omitted for clarity. Selected bond distances and angles are given in the ESI.† | ||
To gain insight into the different reactions of the silylene and germylene, quantum chemical calculations were performed (see ESI† for details). We were able to confirm that 1 is indeed thermodynamically by 49 kJ mol−1 more stable with respect to the hypothetical arsinoamide adduct [LSi(Mes2AsNPh)]. In contrast, in the case of the germanium compound, 2 is 52 kJ mol−1 more stable than the insertion product [LGe(NPh)(AsMes2)] possessing an As–GeN unit. The reason for this different behaviour is explained by the bonding strengths of the EN and E–As bonds in the case of the insertion products 1 and [LSi(Mes2AsNPh)] on the one hand and those of the E–N and As–N bonds of the adducts [LGe(NPh)(AsMes2)] and 2 on the other hand. The results of a Ahlrichs–Heinzmann population analysis based on occupation numbers clearly show that, based on the shared electron numbers (SEN), the SiN double bond in 1 is distinctly strengthened over the Si–N single bond in [LSi(Mes2AsNPh)] (SEN (Si–N) = 1.19 vs. SEN (SiN) = 1.62 in 1), while this effect is less pronounced in the germanium compounds, [LGe (NPh)(AsMes2)] (SEN (GeN) = 1.41) and 2 (SEN (Ge–N) = 1.02). Instead, in the germanium compounds, the As–N bond in 2 is stronger than the Ge–As bond in [LGe(NPh)(AsMes2)] (SEN (Ge–As) = 0.83 vs. SEN (As–N) = 0.91 in 2). Such an effect is not found in the silicon compounds (1: SEN (Si–As) = 0.92; [LSi(Mes2AsNPh)]: SEN (As–N) = 0.89). From these results we conclude that the weaker GeN bond with respect to the SiN bond is responsible for the preference of structure 2 over [LGe (NPh)(AsMes2)] and 1 over [LSi(Mes2AsNPh)]. The lower bond angle ![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) (GeN–C) = 132.6° and the higher partial charge at N (Q(N) = −0.35) in [LGe(NPh)(AsMes2)] compared to 1 ((SiN–C) = 159.3°, Q(N) = −0.25) also confirms the different double bond and lone pair characters in both insertion products (1 and [LGe(NPh)(AsMes2)]). This hypothesis is furthermore confirmed by means of a comparative plot of localized molecular orbitals (LMO) of interest (Fig. S20, ESI†).
(GeN–C) = 132.6° and the higher partial charge at N (Q(N) = −0.35) in [LGe(NPh)(AsMes2)] compared to 1 ((SiN–C) = 159.3°, Q(N) = −0.25) also confirms the different double bond and lone pair characters in both insertion products (1 and [LGe(NPh)(AsMes2)]). This hypothesis is furthermore confirmed by means of a comparative plot of localized molecular orbitals (LMO) of interest (Fig. S20, ESI†).
To investigate the effect of the amidinate supporting ligand on the observed As–N bond insertion, GeCl2·dioxane was employed as a precursor in the reaction (Scheme 2). The treatment of GeCl2·dioxane with 1 equiv. of [(Mes2AsNPh){Li(OEt2)2}] at room temperature resulted in the formation of the insertion product [{Mes2As}(Cl)Ge(μ-NPh)]2 (3), which was isolated in 36% crystalline yield. The insertion leads to a formally tetravalent Ge species (Scheme 2). As a result, the NMR spectra of 3 show significant shifts of all signals. In the 1H NMR spectrum of 3, the resonances of the o- and p-methyl groups (2.23 and 2.22 ppm) of the mesityl ring are highly shifted compared to the starting arsinoamide [(Mes2AsNPh){Li(OEt2)2}] (2.66 and 2.12 ppm).2
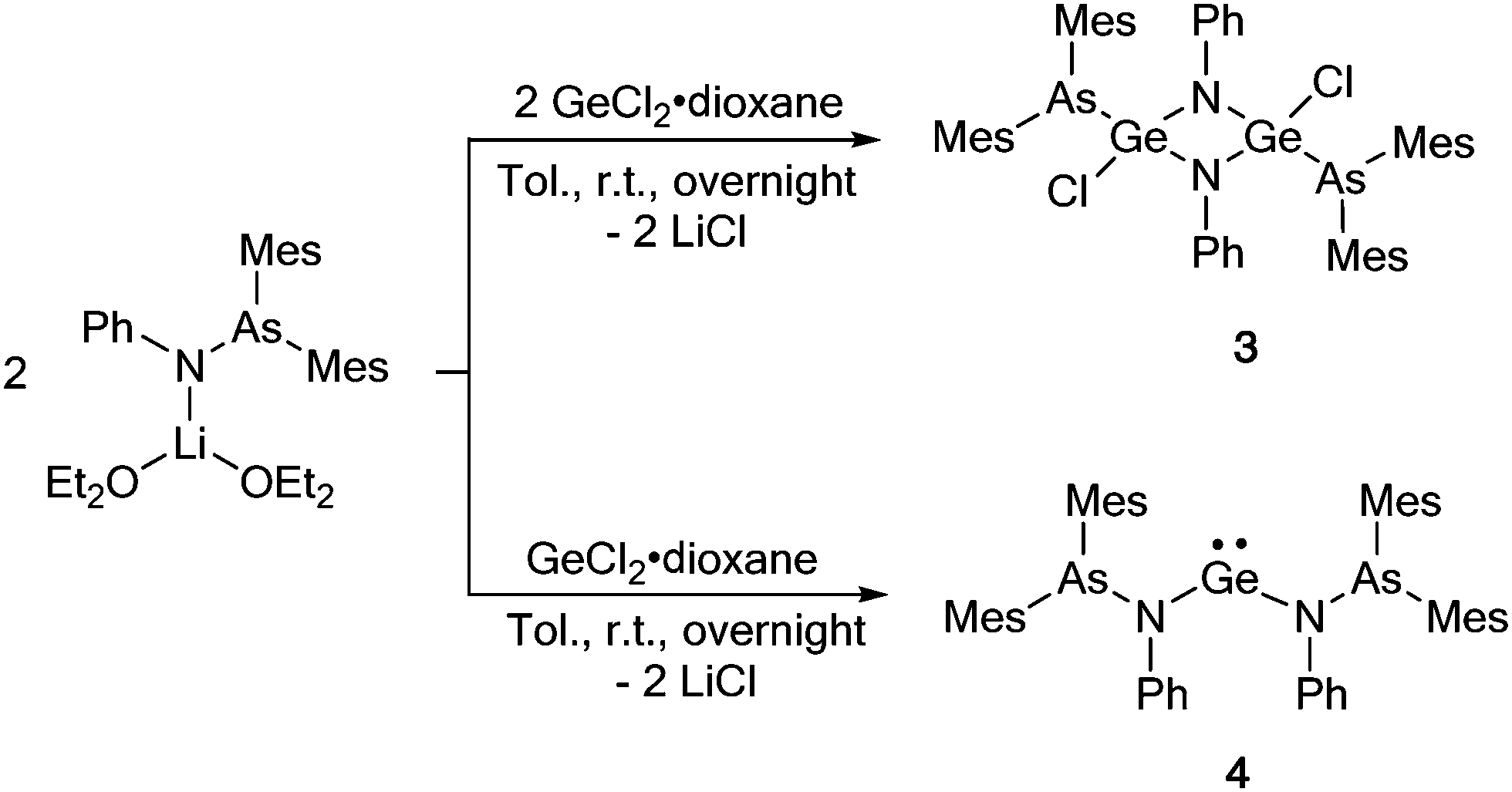 | ||
Scheme 2 Reaction of the arsinoamide [(Mes2AsNPh){Li(OEt2)2}] with GeCl2·dioxane in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 ratio. :1 and 2:1 ratio. | ||
According to single crystal X-ray diffraction studies, compound 3 reveals a dimeric arrangement (Fig. 3), in which the germanium atom is inserted into the As–N bond, forming a rare Ge–As bond. Only a limited number of compounds containing a Ge–As bond have been reported so far.1a,17 In the solid state, 3 forms a perfect planar Ge–N–Ge–N four-membered ring with four-fold coordinated germanium atoms in a distorted tetrahedral geometry. The (Mes2As)− groups and the chloride anions are arranged in a trans fashion across this ring. The distance of the Ge(IV)–As(III) bond is 2.4316(7) Å, which is in good agreement with that in 1,3-diarsa-2-sila-4-germacyclobutanes (2.457(1) Å)17a and in lithioarsinoorganogermanes (2.442(2) Å).17b In comparison to [LGeCl], GeCl2·dioxane features a higher reactivity towards activation of the As–N bond, which may be a result of the absence of the amidinate ligand, which is well known to stabilize low-valent group 14 compounds.14,18 It is noteworthy that, to the best of our knowledge, no P–N bond insertion has ever been observed in germylene phosphinoamide compounds.19
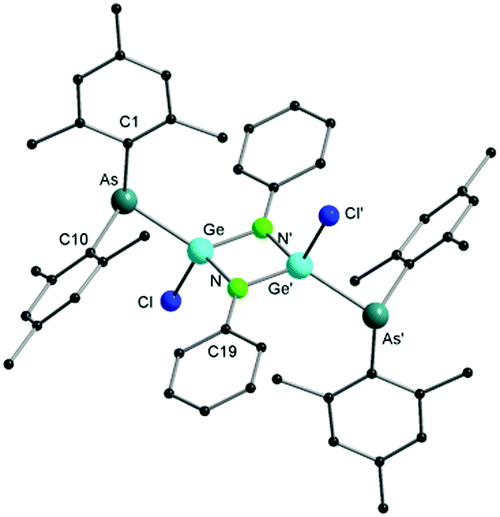 | ||
| Fig. 3 Molecular structure of compound 3 in the solid state. Hydrogen atoms have been omitted for clarity. Selected bond distances and angles are given in the ESI.† | ||
Interestingly, the reaction of 2 equiv. of [(Mes2AsNPh){Li(OEt2)2}] with GeCl2·dioxane does not result in an As–N bond cleavage, indicating that the stoichiometry of the reactants plays a major role in the As–N bond activation. Instead, the di(arsinoamide)germylene [(Mes2AsNPh)2Ge] (4) was isolated as a bright yellow solid in high yields (87%) (Scheme 2). The 1H and 13C{1H} spectra NMR indicate a highly symmetric species in solution.
Single crystal X-ray diffraction studies confirmed the formation of a di(arsinoamide) species (Fig. 4). We assume that the presence of two bulky arsinoamides stabilizes the divalent oxidation state of the Ge atom due to both steric and electronic effects. The Ge–N bond distances (1.879(2) Å) fit well with the reported values, e.g. 1.854(5) Å and 1.869(5) Å in a benzannulated germylene.20 The N–Ge–N bond angle (101.27(13)°) is smaller than that observed in the analogue bis(phosphinoamide)germylene complex (107.1(2)°),19a which may relate to the different steric demands of the ligands. In view of the sum of the bonding angles (359.5°), the nitrogen atoms in 4 adopt an almost ideally trigonal planar coordination geometry. The arsinoamides exhibit trans conformations, similar to that observed in the starting lithium arsinoamide.
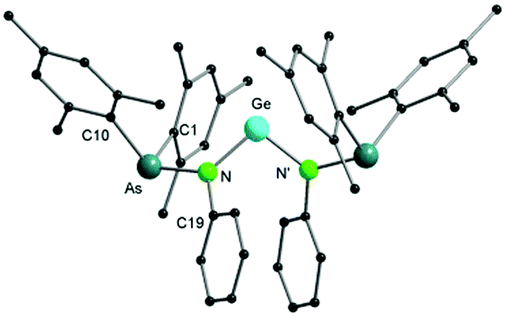 | ||
| Fig. 4 Molecular structure of compound 4 in the solid state. Hydrogen atoms have been omitted for clarity. Selected bond distances and angles are given in the ESI.† | ||
In summary, the reaction of the arsinoamide [(Mes2AsNPh){Li(OEt2)2}] with divalent Si and Ge species was investigated. In case of the silylene [LSiCl], an insertion into the As–N bond was exclusively observed. However, a more diverse reactivity arises in the case of germylenes. Depending on the stoichiometry of the reactants and the presence of amidinate supporting ligands, either arsinoamide-substituted germylenes (2 and 4), in which the As–N bond stays intact, or a tetravalent germanium compound (3) were isolated. In the latter case, the insertion into the As–N bond occurred under mild conditions. Obviously, the substituents on the germylene centre significantly influence the facility of the As–N bond insertion. Compounds 2 and 4 represent the first examples of successful salt metathesis reactions of an arsinoamide without decomposition. Due to the presence of lone pairs on both Ge and As, these new germylene compounds are potential ligands for the further synthesis of binuclear heteroatomic complexes.
X. C. thanks the China Scholarship Council (No. 201506250062) for generous support. T. S. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. The authors acknowledge computational support by the state of Baden-Württemberg through bwHPC and the Deutsche Forschungsgemeinschaft (DFG) through grant no. INST 40/467-1 FUGG.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. M. Godfrey, I. Mushtaq and R. G. Pritchard, J. Chem. Soc., Dalton Trans., 1999, 1319 RSC; (b) T. G. Wetzel, S. Dehnen and P. W. Roesky, Angew. Chem., Int. Ed., 1999, 38, 1086 CrossRef CAS; (c) P. W. Roesky, Heteroat. Chem., 2002, 13, 514 CrossRef CAS; (d) P. W. Roesky, M. T. Gamer and N. Marinos, Chem. – Eur. J., 2004, 10, 3537 CrossRef CAS; (e) L. M. Broomfield, Y. Wu, E. Martin and A. Shafir, Adv. Synth. Catal., 2015, 357, 3538 CrossRef CAS; (f) C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14 CrossRef CAS; (g) J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115 RSC; (h) C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237 CrossRef CAS; (i) N. I. Saper, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Polyhedron, 2016, 114, 88 CrossRef CAS; (j) H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498 CrossRef CAS; (k) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 9516 CrossRef CAS.
- X. Chen, M. T. Gamer and P. W. Roesky, Dalton Trans., 2018, 47, 12521 RSC.
- (a) L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1 CrossRef; (b) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479 CrossRef CAS; (c) F. Breher, Coord. Chem. Rev., 2007, 251, 1007 CrossRef CAS.
- (a) G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS; (b) P. P. Power, Acc. Chem. Res., 2011, 44, 627 CrossRef CAS PubMed; (c) S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2011, 45, 298 CrossRef; (d) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS.
- (a) Y. Wang and J. Ma, J. Organomet. Chem., 2009, 694, 2567 CrossRef CAS; (b) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268 CrossRef CAS; (c) R. H. Crabtree, Acc. Chem. Res., 1990, 23, 95 CrossRef CAS; (d) D. M. Heinekey, A. Lledós and J. M. Lluch, Chem. Soc. Rev., 2004, 33, 175 RSC; (e) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS.
- (a) F. Diab, F. S. Aicher, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, Chem. – Eur. J., 2019, 25, 4426 CrossRef CAS; (b) R. C. Turnell-Ritson, J. S. Sapsford, R. T. Cooper, S. S. Lee, T. Földes, P. A. Hunt, I. Pápai and A. E. Ashley, Chem. Sci., 2018, 9, 8716 RSC; (c) C. Präsang, M. Stoelzel, S. Inoue, A. Meltzer and M. Driess, Angew. Chem., Int. Ed., 2010, 49, 10002 CrossRef; (d) M.-D. Su and S.-Y. Chu, Inorg. Chem., 1999, 38, 4819 CrossRef CAS; (e) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272 CrossRef CAS PubMed.
- (a) P. Jutzi and A. Möhrke, Angew. Chem., Int. Ed. Engl., 1989, 28, 762 CrossRef; (b) P. Jutzi, D. Eikenberg, A. Möhrke, B. Neumann and H.-G. Stammler, Organometallics, 1996, 15, 753 CrossRef CAS; (c) F. M. Mueck, J. A. Baus, M. Nutz, C. Burschka, J. Poater, F. M. Bickelhaupt and R. Tacke, Chem. – Eur. J., 2015, 21, 16665 CrossRef CAS; (d) A. Schmidpeter, S. Lochschmidt and A. Willhalm, Angew. Chem., Int. Ed. Engl., 1983, 22, 545 CrossRef.
- (a) M. F. Lappert and R. S. Rowe, Coord. Chem. Rev., 1990, 100, 267 CrossRef CAS; (b) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS; (c) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS; (d) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176 RSC; (e) M. K. Sharma, D. Singh, P. Mahawar, R. Yadav and S. Nagendran, Dalton Trans., 2018, 47, 5943 RSC.
- C. W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948 CrossRef CAS.
- R. Azhakar, H. W. Roesky, J. J. Holstein, K. Pröpper and B. Dittrich, Organometallics, 2013, 32, 358 CrossRef CAS.
- J. Grobe, S. Göbelbecker, B. Krebs and M. Läge, Z. Anorg. Allg. Chem., 1992, 611, 11 CrossRef CAS.
- S. Khan, S. S. Sen, D. Kratzert, G. Tavčar, H. W. Roesky and D. Stalke, Chem. – Eur. J., 2011, 17, 4283 CrossRef CAS.
- C.-W. So and M. L. B. Ismail, Chem. Commun., 2019, 55, 2074 RSC.
- S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459 CrossRef CAS.
- J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and D. Polo, Chem. – Eur. J., 2014, 20, 8654 CrossRef CAS.
- (a) G. Trinquier and M. T. Ashby, Inorg. Chem., 1994, 33, 1306 CrossRef CAS; (b) N. Poetschke, M. Nieger, M. A. Khan, E. Niecke and M. T. Ashby, Inorg. Chem., 1997, 36, 4087 CrossRef CAS.
- (a) M. Drieß and H. Pritzkow, Chem. Ber., 1994, 127, 477 CrossRef; (b) L. Zsolnai, G. Huttner and M. Driess, Angew. Chem., Int. Ed. Engl., 1993, 32, 1439 CrossRef; (c) M. F. Davis, W. Levason, G. Reid and M. Webster, Dalton Trans., 2008, 2261 RSC; (d) F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2009, 49, 752 CrossRef; (e) S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Chem. Commun., 2015, 51, 3866 RSC; (f) S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2016, 7, 10480 CrossRef CAS; (g) S. Yao, Y. Grossheim, A. Kostenko, E. Ballestero-Martínez, S. Schutte, M. Bispinghoff, H. Grützmacher and M. Driess, Angew. Chem., 2017, 129, 7573 CrossRef; (h) K. Izod, P. Evans and P. G. Waddell, Chem. Commun., 2018, 54, 2526 RSC.
- (a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2009, 132, 1123 CrossRef; (b) S. S. Sen, M. P. Kritzler-Kosch, S. Nagendran, H. W. Roesky, T. Beck, A. Pal and R. Herbst-Irmer, Eur. J. Inorg. Chem., 2010, 5304 CrossRef CAS.
- (a) S. Pal, R. Dasgupta and S. Khan, Organometallics, 2016, 35, 3635 CrossRef CAS; (b) N. Parvin, S. Pal, V. C. Rojisha, S. De, P. Parameswaran and S. Khan, ChemistrySelect, 2016, 1, 1991 CrossRef CAS; (c) T. Böttcher and C. Jones, Main Group Met. Chem., 2015, 38, 165 Search PubMed.
- R. Aysin, L. Leites, S. Bukalov, A. Zabula and R. West, Inorg. Chem., 2016, 55, 4698 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of 1–4 including crystallographic data. CCDC 1922573–1922576. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc04530a |
| This journal is © The Royal Society of Chemistry 2019 |