
Thin-walled hollow Au–Cu nanostructures with high efficiency in electrochemical reduction of CO2 to CO†
Jun-Hao
Zhou
,
Da-Wei
Lan
,
Sheng-Song
Yang
,
Yu
Guo
,
Kun
Yuan
,
Lin-Xiu
Dai
and
Ya-Wen
Zhang
 *
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: ywzhang@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 25th April 2018
Abstract
Bimetallic hollow structures can be efficiently used to improve the utilization of noble metals in electrocatalysts, but few studies have reported the application of bimetallic hollow nanocomposites for the electrochemical reduction of CO2. Herein, we demonstrated the synthesis of hollow Au–Cu nanoparticles of size ranging from 50 nm to 100 nm with a wall thickness of 4.95 ± 0.81 nm for efficient CO2 reduction reaction (CO2RR) via galvanic replacement and the Kirkendall effect between copper and gold. In the fabrication procedure, a gold precursor was pre-dissolved in TOP to lower the reduction potential of Au3+, which could then be reduced by Cu0. The obtained hollow Au–Cu NPs were then supported on carbon black for CO2RR measurement, and it exhibited a maximum CO FE of 53.3% at −0.7 V (vs. RHE), which was very close to that of Au NP/C, but the gold mass current density of hollow Au–Cu NP/C was much higher. The hollow Au–Cu NP/C also showed good stability in long-time measurements for CO2RR. To find out how the superior catalytic performance was achieved, hollow Au–Cu NP/C was then annealed in O2 and N2. It was found that after being annealed, the catalysts almost totally lost the activity towards CO2RR, which may be due to the partial segregation of Cu species on the nanoparticle surface as well as a decrease in the surface defect density with the improvement in crystallinity. Thus, it was concluded that high dispersion of more Au atoms on the nanoparticle surface together with the presence of more surface defect sites could account for the superior CO2RR performance of the as-synthesized hollow Au–Cu NP/C.
Introduction
Since the last decades, the increasing concentration of atmospheric CO2 and the heavy consumption of fossil fuels, which would result in global climate change and shortage of some energy sources, have become problems of major public concern.1 One promising method to lower the global carbon emission and alleviate energy crisis simultaneously is the electrochemical reduction of CO2 to value-added fuels and chemical feedstock using renewable energy sources.2 This solution seems to be perfect. However, the linear CO2 molecule is hard to be reduced due to its low electron affinity and large energy gap between its lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO).3 In fact, the first electron transfer needs potentials more negative than −1.97 V (vs. standard hydrogen electrode (SHE)) in dimethylformamide (DMF) or −1.90 V (vs. SHE) in water.4–6 Therefore, much effort has been devoted to developing a variety of metallic catalysts for boosting such a kinetically slow reaction.7–15It is commonly accepted that metal catalysts can be classified into three groups according to their different abilities to bind various intermediate and final products in the CO2 reduction reaction (CO2RR).16,17 As reported, the formation of CO2˙− intermediates is considered as a rate-determining step in most cases. For Sn, Hg, Pb, In, etc., which can hardly bind to the CO2˙− intermediate, the major product is formate.17 In addition, Au, Ag, Zn, Pd, etc., can bind *COOH tightly for further reaction, whereas the obtained *CO is weakly bound to the electrode surface and thus, CO directly desorbs from the surface and turns into the major product. Cu is the only metal that can bind both *COOH and *CO tightly. *CO adsorbed on the surface can be further converted into higher value-added fuels (such as hydrocarbons and alcohols).
Although numerous metallic electrocatalysts have been extensively investigated for CO2RR, this field still faces the challenges of large overpotential, low faradaic efficiency (FE) and poor product selectivity due to the competitive hydrogen evolution reaction (HER).18,19 One approach to enhance the catalytic performance for CO2RR is alloying the metallic electrocatalysts with heteroatoms.20–24 By alloying of various metals, we might be able to adjust the binding strength of the intermediates on the catalyst surface by means of synergistic geometric and electronic effects between heterometallic components. Once the binding strength of the intermediates is changed, the reaction kinetics as well as the final products may be quite different. For instance, Yang's group reported the synthesis of AuCu NPs with different proportions.11 It was found that by increasing the percentage of Cu in AuCu NPs, CO2 was converted into more kinds of value-added hydrocarbon products, but the total FE of carbon products was decreased.
Using a special fabrication procedure, bimetallic nanostructures with unique shape/morphology can be obtained.25–32 Among them, a hollow structure is quite attractive. In a typical catalytic process, the core of a solid catalyst has a neglectable role to play except for supporting the structure. In this respect, a hollow catalyst can minimize the usage of metals without sacrificing the catalytic activity. Many bimetallic hollow nanocomposites have been fabricated via sequential action of the galvanic replacement reaction and the Kirkendall effect.33–36
In 2015, Hahn et al. reported the synthesis of hollow Au–Cu nanostructures using a Cu2O template toward o-nitroaniline reduction to 1,2-benzenediamine.37 Besides, in 2016, Zhao and co-workers prepared hollow Au–Cu nanorods via galvanic replacement between AuCu3 nanorods and a gold precursor for p-nitrophenol reduction reaction with NaBH4.38 None of these hollow Au–Cu structures was applied for the electroreduction of CO2. We even found that few studies have reported the examination of the utilization of hollow bimetallic nanostructures in CO2RR field.
Herein, we reported a reliable method for the synthesis of thin-walled hollow Au–Cu nanoparticles (NPs) for CO2RR. Cu seed NPs were first synthesized. An Au precursor was dissolved in tri-n-octylphosphine (TOP) to decrease its reduction potential, and it was then injected into the Cu seed solution. Galvanic replacement occurred between Au3+ and Cu0, and the hollow structure was finally formed through the Kirkendall effect. The prepared hollow Au–Cu NPs were supported on carbon black and measured for the catalytic activity and selectivity towards CO2RR. It was found that this thin-walled hollow NP/C showed a higher gold mass current density and a very close FE of CO (53.3%) when compared with Au NP/C (54.6%), and it also exhibited good stability. Furthermore, to find out the origin of the superior catalytic performance towards CO2RR of the hollow Au–Cu NP/C, thermal treatment was carried out in O2 and N2. After being annealed, the catalytic activity was almost totally lost. This could be due to the segregation of Cu atoms on the surface and the improvement in crystallinity of NPs during the heating process. As a result, the presence of more Au atoms and more defects on the NP surfaces could account for the superior performance of the as-synthesized hollow NP/C.
Experimental section
Chemicals
The chemicals used in this experiment are HAuCl4·4H2O (A.R., Beijing Chemical Works, China), Cu(acac)2 (A.R., Sigma-Aldrich), oleylamine (>80%, J&K), oleic acid (>90%, Sigma-Aldrich), tri-n-octylphosphine (>90%, Alfa Aesar), acetic acid (A.R., Beijing Chemical Works, China), K2CO3 (≥99.995%, Shanghai Macklin Biochemical Co. Ltd, China), polyvinylidene fluoride (Mw ∼ 534![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Shanghai Macklin Biochemical Co. Ltd, China), N-methyl-2-pyrrolidone (A.R., Xilong Chemical Co. Ltd, China), dimethyl sulfoxide (≥99.8%, Aladdin), ethanol (A.R.), cyclohexane (A.R.), acetone (A.R.), and deionized water (Millipore, 18.2 MΩ cm).
000, Shanghai Macklin Biochemical Co. Ltd, China), N-methyl-2-pyrrolidone (A.R., Xilong Chemical Co. Ltd, China), dimethyl sulfoxide (≥99.8%, Aladdin), ethanol (A.R.), cyclohexane (A.R.), acetone (A.R.), and deionized water (Millipore, 18.2 MΩ cm).
Synthesis of Cu NPs and Au NPs
Herein, 0.25 mmol of Cu(acac)2 was pre-dissolved in 0.36 g of dry oleic acid (OA) and 1.6 g of dry oleylamine (OAm) at 85 °C in an oil bath. Dry OA (1.6 g) and 8.4 g of dry OAm were put into a 100 mL three-necked flask at room temperature. The solvent was heated to 140 °C, and it was evacuated at this temperature for 20 min to remove water and oxygen under magnetic stirring. The solvent was then heated to 280 °C. The pre-dissolved copper precursor was injected into the heated solvent inside the flask in 10 seconds using a 5 mL plastic syringe, and this was allowed to further react for 1 h at this temperature under an N2 atmosphere. When the reaction was complete, the solution was cooled down to room temperature, and an excess of absolute ethanol was added. The product was obtained by centrifugation (9500 rpm, 10 min) and washed repeatedly with ethanol and cyclohexane using redispersion–centrifugation cycles to remove excess OA and OAm. The final product was redispersed in cyclohexane. The synthesis procedure of Au NPs was similar to that of Cu NPs except that the reactant was 0.075 mmol of HAuCl4·4H2O pre-dissolved in 1.6 g of dry OAm, and the temperature for the reaction was 140 °C.Synthesis of hollow Au–Cu NPs
The synthesis process of Cu seed was the same as that of Cu NPs. When the reaction was complete at 280 °C, the mixture was cooled down to 140 °C. Then, a TOP solution of HAuCl4·4H2O (0.025 M, 1 mL) was injected into the solution using a 5 mL plastic syringe in 5 seconds. After 1 h, the mixture was cooled down to room temperature, and an excess of absolute ethanol was added. The product was obtained by centrifugation (9500 rpm, 10 min) and washed repeatedly with ethanol and cyclohexane. Finally, the product was redispersed in cyclohexane for further use.Synthesis of Au NP/C and Cu NP/C
Carbon black samples with weights 2 times that of Au and Cu NPs were added to the as-obtained Au and Cu cyclohexane dispersions. Excess cyclohexane was added until the volume of the solution reached 21 mL and then, the solution was mixed with 7 mL of acetone. The mixture was ultrasonically dispersed for 1 h and then, it was centrifuged at 9500 rpm for 10 min, followed by washing with cyclohexane; the mixture was then centrifuged for two times. Finally, the precipitated Au NP/C and Cu NP/C were redispersed in 3 mL of ethanol.Synthesis of hollow Au–Cu NP/C
A carbon black sample with a mass 2 times that of Au–Cu NPs was added to the as-obtained cyclohexane dispersion. Excess cyclohexane was added until the volume of the solution reached 21 mL and then, the solution was mixed with 7 mL of acetone. The mixture was ultrasonically dispersed for 1 h and then, it was centrifuged at 9500 rpm for 10 min, followed by washing with cyclohexane; the mixture was then centrifuged for two times. To dissolve Cu NPs mixed in hollow Au–Cu NPs and to remove OA and OAm adsorbed on the surface of Au–Cu NPs, the precipitate was dispersed in 17 mL of acetic acid, transferred to a 50 mL round flask, and heated at 70 °C in an oil bath for 12 h. The mixture was centrifuged at 9500 rpm for 10 min and then, it washed with ethanol for three times. Finally, the precipitated hollow Au–Cu NP/C was dried at 70 °C or redispersed in 3 mL of ethanol.Thermal treatment of hollow Au–Cu NP/C in O2/N2
For thermal treatment in an O2 or N2 atmosphere, an appropriate amount of the as-obtained dried hollow Au–Cu NP/C was annealed in air or N2 at 180 °C for 2 h and then, it was cooled down to room temperature.Electrochemical reduction of carbon dioxide
To prepare the working electrode, 1 mg of polyvinylidene fluoride (PVDF) and 2 drops of N-methyl-2-pyrrolidone were added to 1 mL of ethanol containing 1 mg of catalysts. The mixture was then dropped onto a glassy carbon substrate (1 × 1 cm2) and dried under an infrared lamp.Electrochemical reduction of CO2 was carried out in a gas-tight two-chamber reaction vessel separated by an anion exchange membrane (Nafion 117, Ion Power) on the CHI 760e electrochemical workstation (CH Instrument, USA). The experiment was performed in an aqueous solution of 0.5 M KHCO3, which was prepared from K2CO3 saturated with CO2. The working electrode compartment held 15 mL of electrolyte, whereas the counter electrode compartment held 20 mL of electrolyte. A platinum plate (1 × 1 cm2) was used as the counter electrode, and an Ag/AgCl electrode (saturated KCl) was used as the reference electrode. All potentials in this study were converted to reversible hydrogen electrode (RHE) scale using the formula E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 V + 0.0591 × pH.
To start the experiment, CO2 was first allowed to flow through the working compartment at a rate of 40 sccm for 30 min to saturate the 0.5 M KHCO3 solution (pH 7.3) and then, the flow rate of CO2 was changed to 8 sccm. Cyclic voltammetry (CV) was carried out at a scan rate of 100 mV s−1, whereas linear sweep voltammetry (LSV) was conducted at a scan rate of 5 mV s−1. During chrono-amperometry, the effluent gas from the cell was introduced directly into the gas sample loop of gas chromatograph (GC) for gas analysis. The gas chromatograph equipped with two of Porapak-N, MS-13X and HP-AL/S columns was used for quantifications. The gas-phase product was analyzed by GC every 15 min. CO in the gas products was analyzed using a thermal conductivity detector (TCD) with H2 as a carrier gas, whereas H2 in the gas products was analyzed using another TCD with N2 as a carrier gas, and the alkanes in the gas products were analyzed using a flame ionization detector (FID) with N2 as a carrier gas. The liquid products were characterized by 1H nuclear magnetic resonance (NMR) spectra, in which 0.5 mL electrolyte was mixed with 0.1 ml D2O, and 0.05 μL dimethyl sulfoxide (DMSO) was added as an internal standard. The one-dimensional 1H spectrum was recorded with water suppression using a solvent pre-saturation technique. The amount of charge passed to produce each product was divided by the total charge passed at a specific time or during the overall run to evaluate each FE (see ESI† for the details of calculation).
Instrumentation
Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were carried out using FEG-TEM (JEM-2100F, JEOL, Japan) operated at 200 kV. The samples for TEM observations were prepared by dropping the solution on nickel grids coated with amorphous carbon membranes and drying them naturally. Powder X-ray diffraction (PXRD) analysis was conducted using a D/MAX-2000 diffractometer (Rigaku, Japan) under Cu Kα irradiation. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed using a Profile Spec ICP-AES spectrometer (Leeman, USA). X-ray photoelectron spectroscopy (XPS) analysis was conducted using an Axi Ultra imaging photoelectron spectrometer (Kratos, UK). All GC spectrometry experiments were measured and recorded using GC-2014 (Shimadzu, Japan). 1H NMR spectroscopy was performed using Bruker-500 MHz NMR (Bruker, USA).Results and discussion
Synthesis and structural characterization of hollow Au–Cu NP/C
A typical synthesis procedure (Scheme 1) starts from the fabrication of Cu seed NPs at 280 °C with OA and OAm serving as solvent, reductant and capping agent. The Au precursor pre-dissolved in TOP was injected into the Cu seed solution at 140 °C. Galvanic replacement occurred between Cu0 and Au3+. Cu in the core was oxidized to Cu2+ and diffused into the solution. Accordingly, Au3+ in the solution was reduced to Au0 and diffused into NPs. During the redox process, the rate of Cu2+ diffusing out was higher than the rate of Au0 diffusing in, which is called the Kirkendall effect.39–43 Finally, the Cu core totally disappeared and hollow Au–Cu NPs were obtained, which were then supported on carbon black, followed by dispersion in acetic acid and heating in an oil bath at 70 °C for 12 h. The final product was referred to as hollow Au–Cu NP/C.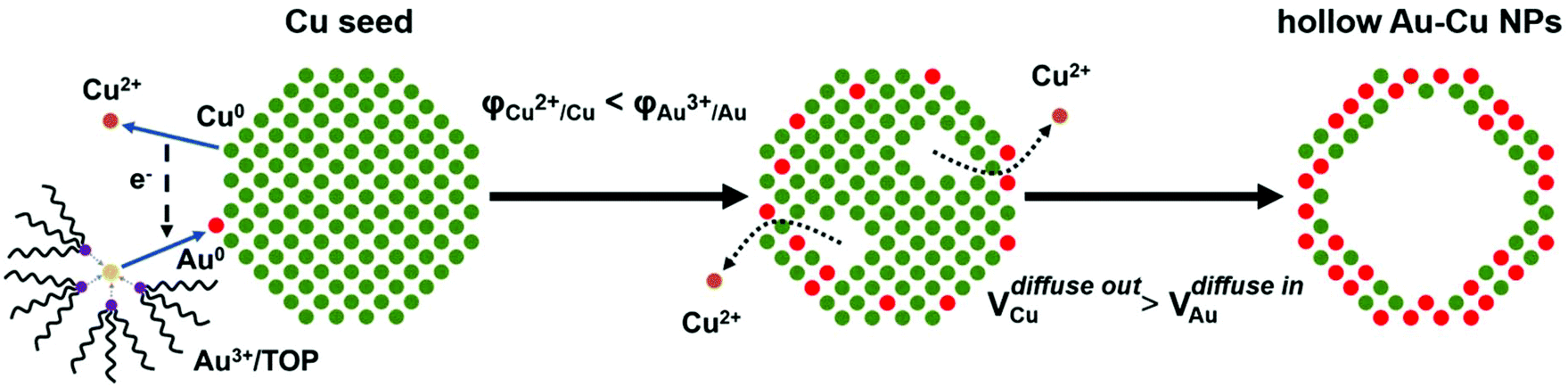 | ||
| Scheme 1 Growth mechanism of hollow Au–Cu NPs. | ||
The typical TEM image in Fig. 1a shows that the Au–Cu sample appeared in the shape of nanospheres of size ranging from 50 nm to 100 nm. It also revealed that Au–Cu NPs have a bright center and a dark edge, suggesting the presence of a hollow structure with a wall thickness of 4.95 ± 0.81 nm. The lattice spacing of a randomly selected single NP (Fig. 1b) was found to be 0.227 nm by HRTEM (Fig. 1b1 and b2), which was between the lattice spacing of the (111) plane of pure Au (0.235 nm) and that of Cu (0.208 nm), indicating that these NPs were composed of Au and Cu. HAADF-STEM-EDS was used to analyze the elemental distributions of Au and Cu in a single NP. HAADF-STEM image and EDS 2D elemental mapping, in agreement with the PXRD and HRTEM results, demonstrated that the Au element and the Cu element have uniform distribution (Fig. 1c), which confirmed the formation of Au–Cu alloy. The EDS linear scanning image of the hollow Au–Cu NP/C displayed the characteristics of a hollow structure (Fig. 1d), in which Au and Cu were mostly distributed on the shell of the nanosphere, whereas the core was almost completely hollow.
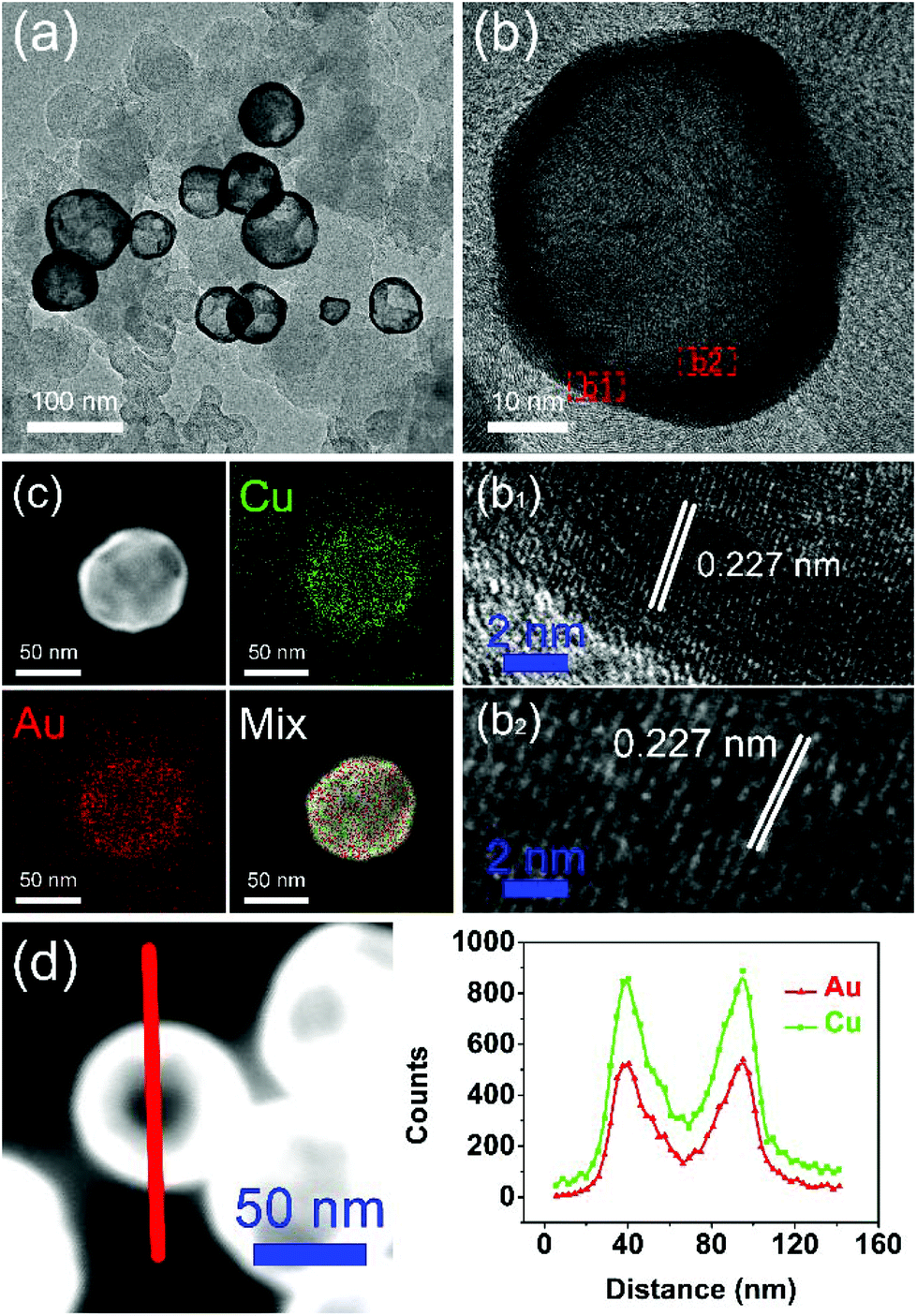 | ||
| Fig. 1 TEM image (a), HRTEM image (b), EDS 2D element mapping image (c) and EDS linear scanning image (d) of hollow Au–Cu NP/C. | ||
The solid solution structure of the hollow Au–Cu NP/C was characterized by PXRD analysis. From Fig. 2a, it can be observed that the peak positions of 111 and 200 diffractions were between those of monometallic fcc Au (JCPDS no. 04-0784) and monometallic fcc Cu (JCPDS no. 85-1326), which was consistent with the HRTEM result, suggesting the formation of Au–Cu nanoalloys.
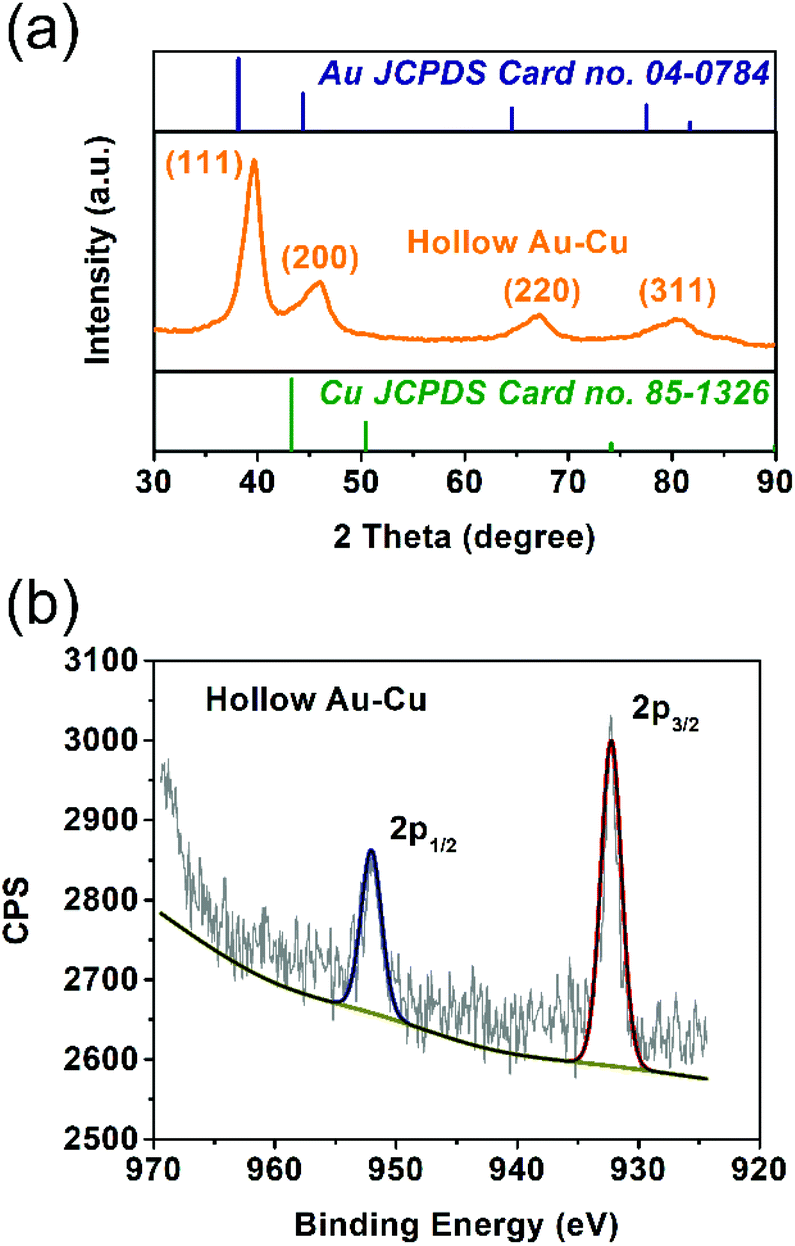 | ||
| Fig. 2 PXRD pattern (a) and Cu 2p XPS spectrum (b) of hollow Au–Cu NP/C. | ||
XPS spectroscopy was used to analyze the valence states and distributions of Au and Cu on the surface of hollow Au–Cu NP/C. Besides, Cu NP/C was also synthesized and characterized by XPS analysis as a comparison (see the Experimental section for details of synthesis). Fig. 2b and Fig. S1† present Cu 2p XPS spectra of hollow Au–Cu NP/C and Cu NP/C. We can observe that Cu NPs and hollow Au–Cu NPs exhibited nearly the same Cu 2p XPS spectra, in which the peaks appearing at ∼932.5 eV and ∼952.5 eV could be assigned to Cu 2p3/2 and Cu 2p1/2, respectively, and no satellite peaks were observed, indicating that CuO species was not detectable on the NP surface. The atomic ratio of Au and Cu in hollow Au–Cu NP/C calculated from the XPS spectrum (Au 4f XPS spectrum in Fig. S2 in ESI†) was 78:22, which was quite different from that obtained from ICP-AES (59:41). It seemed that Au was more likely to distribute on the surface, whereas Cu tended to be inside NPs. This may be due to the fact that these hollow NPs were prepared via galvanic replacement and the Kirkendall effect between the Cu seed and Au3+ dissolved in the solution. Although Cu diffused out and Au diffused in during the formation process of the hollow structure, more Cu species were still left in NPs and more Au still existed on the surface.
As mentioned above, the formation of the hollow structure occurs via galvanic replacement and the Kirkendall effect between Cu and Au. However, it is known that the reduction potential of Au3+ is much higher than that of Cu2+. If an Au3+ precursor is directly introduced into the Cu seed solution, it is quite possible that no galvanic replacement would occur. Yang et al. pointed out that if the Au precursor is pre-dissolved in OAm, it is reduced by OAm rapidly after it is injected into the Cu seed solution at the reaction temperature.44 The sharp increase in the concentration of Au atoms at the initial stage leads to a homogeneous nucleation reaction, and the Au atoms will nucleate and aggregate on their own. Finally, we will obtain a mixture of Au NPs and Cu NPs. Therefore, we must use a soft Lewis base such as TOP, which can bind to Au3+ strongly, according to hard and soft acids and bases (HSAB) theory, to replace OAm in dissolving the gold precursor.45 This is the only way by which the reduction potential of Au3+ can be lowered dramatically, and Au3+ cannot be easily reduced by OAm. Galvanic replacement will occur between Au3+ and Cu0, and hollow Au–Cu NPs will be obtained.
The TEM images of both NPs are shown in Fig. S3 in ESI.† Fig. S3a† shows that the samples prepared by pre-dissolving the gold precursor in TOP appeared in the shape of hollow nanospheres with 4.90 ± 0.98 nm thick wall. Except the hollow Au–Cu NPs, we could find some NPs with deeper contrast, which were identified as Cu NPs by HRTEM and PXRD (Fig. S4 in ESI†). Acetic acid was used to dissolve the intermingled Cu NPs and remove OA and OAm adsorbed on the surface. Both TEM and PXRD results indicated that after heating in acetic acid, no Cu NPs remained. Besides, the thickness of the wall did not change. Fig. S3b† shows that when the gold precursor was pre-dissolved in OAm, we obtained only a mixture of NPs with various sizes in which the relatively smaller ones were considered as monometallic Au, whereas the larger ones were considered as monometallic Cu.
Electrocatalytic performance towards CO2RR
The electrocatalytic performance towards CO2RR was tested for hollow Au–Cu NP/C. Besides, solid Au NP/C and Cu NP/C were also prepared and tested as a reference (see the Experimental section and Fig. S5–S6 in ESI† for details of synthesis and characterization). The surface redox potentials of hollow Au–Cu NP/C were studied by CV. Fig. S7a in ESI† gives the CV curve from −1.0 V (vs. RHE) to 0.5 V (vs. RHE) in the CO2-saturated 0.5 M KHCO3. No obvious cathodic peaks were found in the CV curves of hollow Au–Cu NP/C, which indicated that there was no oxide species on the surface, and this was in agreement with the XPS result.The electrochemical reductions of CO2 on hollow Au–Cu NP/C, Au NP/C and Cu NP/C were measured by LSV from −1.0 V (vs. RHE) to 0.3 V (vs. RHE) in the CO2-saturated 0.5 M KHCO3. First, the current density of these three kinds of catalysts was divided by the geometric surface area of the working electrode to calculate the geometric current density (Fig. S7b in ESI†). It was found that hollow Au–Cu NP/C exhibited a more negative onset potential and nearly the same current density when compared to Au NP/C, whereas Cu NP/C exhibited both the most negative onset potential and the least current density. Furthermore, if we normalize the current of Au NP/C and hollow Au–Cu NP/C with reference to the mass of gold present in each of these catalysts, we will obtain the Au mass current density (Fig. 3a). In this graph, we could observe that at the same potential applied, the current density of hollow Au–Cu NP/C was much higher than that of Au NP/C. This indicated that this thin-walled hollow alloy structure could reduce the amount of gold used without decreasing the current.
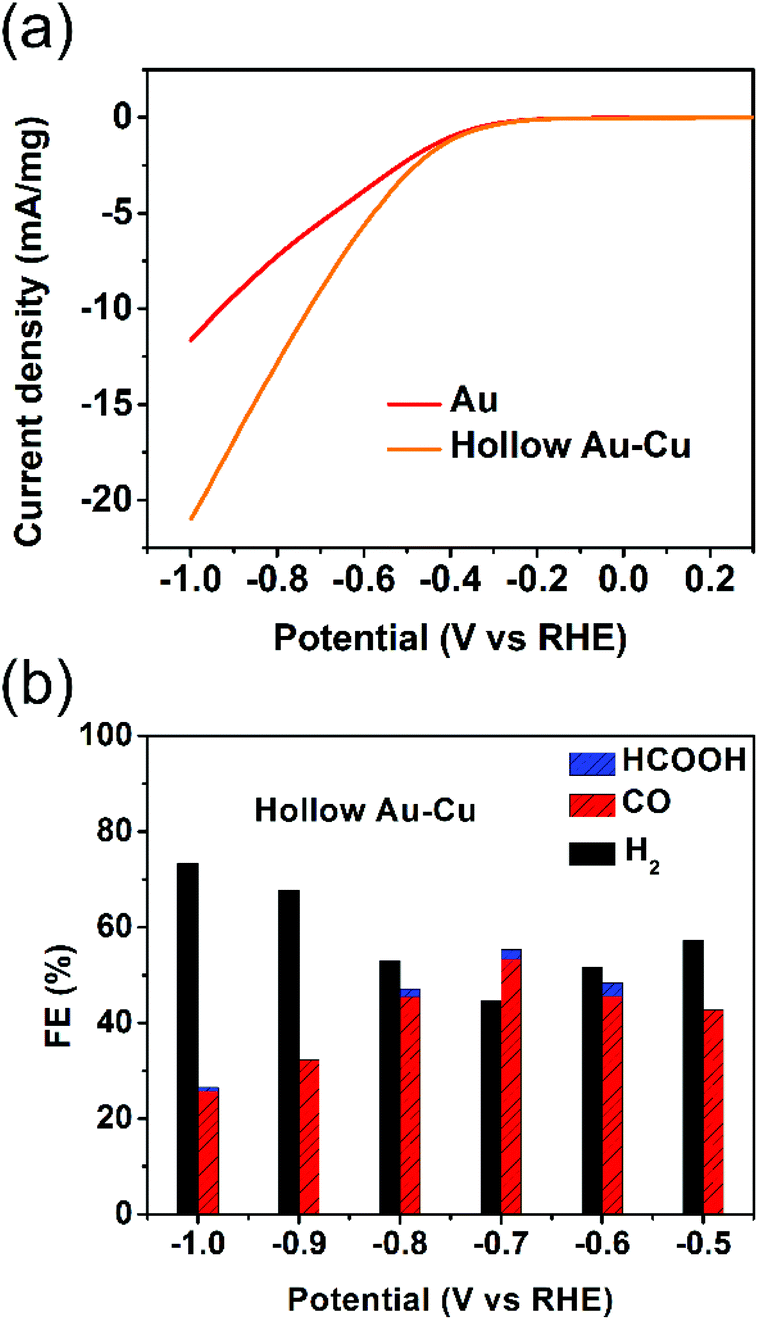 | ||
| Fig. 3 LSV curves of Au NP/C and hollow Au–Cu NP/C in the CO2-saturated 0.5 M KHCO3 at a scan rate of 5 mV s−1 (a); FEs of various products at different potentials measured on hollow Au–Cu NP/C (b). The current density of different catalysts was normalized with reference to the mass of gold they each contained. | ||
Controlled potential electrolysis of CO2 was further investigated on hollow Au–Cu NP/C, Au NP/C and Cu NP/C at potentials between −1.0 V (vs. RHE) and −0.5 V (vs. RHE) in the CO2-saturated 0.5 M KHCO3 at room temperature under atmospheric pressure. The results of hollow Au–Cu NP/C, Au NP/C and Cu NP/C are shown in Fig. 3b and Fig. S8 in ESI.† At different potentials applied, hollow Au–Cu NP/C generated a mixture of H2 and CO. With an increase in potential, CO FE increased at first and then decreased. At −0.7 V (vs. RHE), the CO selectivity reached the maximum of 53.3%. For Au NP/C, the maximum CO FE (54.6%), which appeared at −0.6 V (vs. RHE), was slightly higher than that of the as-synthesized hollow Au–Cu NPs. The maximum CO FE of the hollow Au–Cu NP/C was comparable with that of Au–Cu alloy (maximum CO FE of around 50%), as reported by Yang et al. in 2014, but it was lower than that of the ordered Au–Cu alloy (maximal CO FE of around 80%), as reported by Yang et al. in 2017.11,24 For hollow Au–Cu NP/C and Au NP/C, the selectivity of formate was very poor. However, Cu NP/C showed a quite different selectivity; it did not produce any CO, and CO2 was converted to HCOOH with a maximum FE of 18.3%.
The stability of hollow Au–Cu NP/C was also tested at −0.7 V (vs. RHE) for 12 h (Fig. 4). It was found that both the selectivity for CO and the current density decreased slightly after the measurement for 12 h. Besides, no obvious morphology change could be observed in the TEM images of the sample after the long-time measurements (Fig. S9 in ESI†). XPS analysis was also conducted on hollow Au–Cu NP/C after the stability test, and we could observe that Cu 2p3/2 and Cu 2p1/2 doublet peaks still appeared at ∼932.5 eV and ∼952.5 eV, and no satellite peaks appeared, which pointed out that the valency of Cu element in hollow Au–Cu was still zero after the stability test (Fig. S10 in ESI†). These results all indicated the good stability of hollow Au–Cu NP/C during the electrocatalytic test.
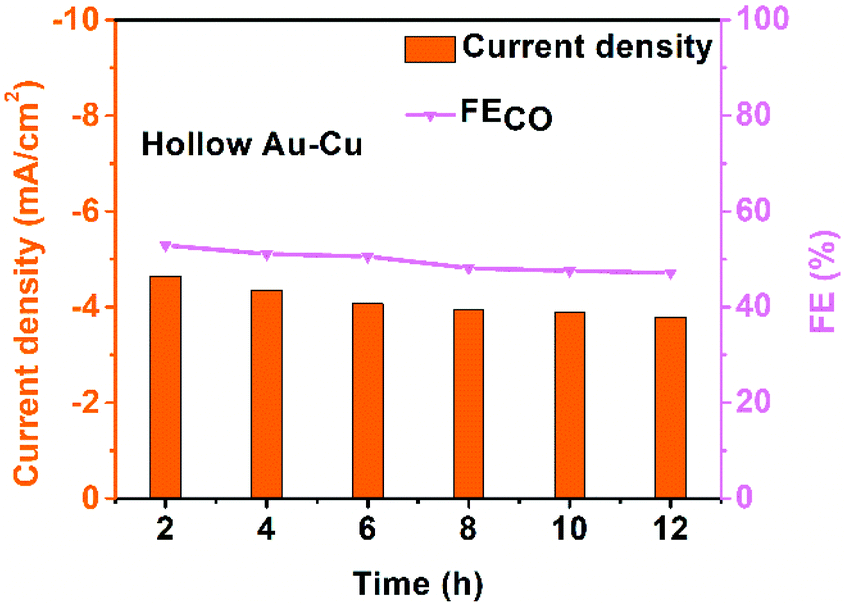 | ||
| Fig. 4 Chrono-amperometry results of hollow Au–Cu NP/C tested for CO2RR at −0.7 V (vs. RHE) for 12 h. | ||
To figure out why this hollow Au–Cu NP/C showed a relatively superior catalytic performance towards CO2RR, we investigated the defect state and the elementary distribution/valency on the surface of the catalyst, which were believed to be responsible for the catalytic activity and selectivity for CO2RR.7,46,47 Thus, thermal treatment was implemented for hollow Au–Cu NP/C in an O2 or N2 atmosphere at 180 °C for 2 h, and the corresponding electrocatalytic activity and selectivity towards CO2RR were tested.
LSV curves of the as-synthesized hollow Au–Cu NP/C and hollow Au–Cu NP/C annealed in O2 or N2 are shown in Fig. S11 in ESI.† It was found that thermal treatment improved the geometric current density, and the sample annealed in O2 exhibited the largest current density. However, after being annealed, the catalytic activity of hollow Au–Cu NP/C towards CO2RR decreased dramatically. Fig. 5a and b show that the maximum CO FE was 8.04% for hollow Au–Cu NP/C annealed in O2 and 4.69% for hollow Au–Cu NP/C annealed in N2. Although the selectivity towards formate was improved slightly after being annealed, it was still very poor.
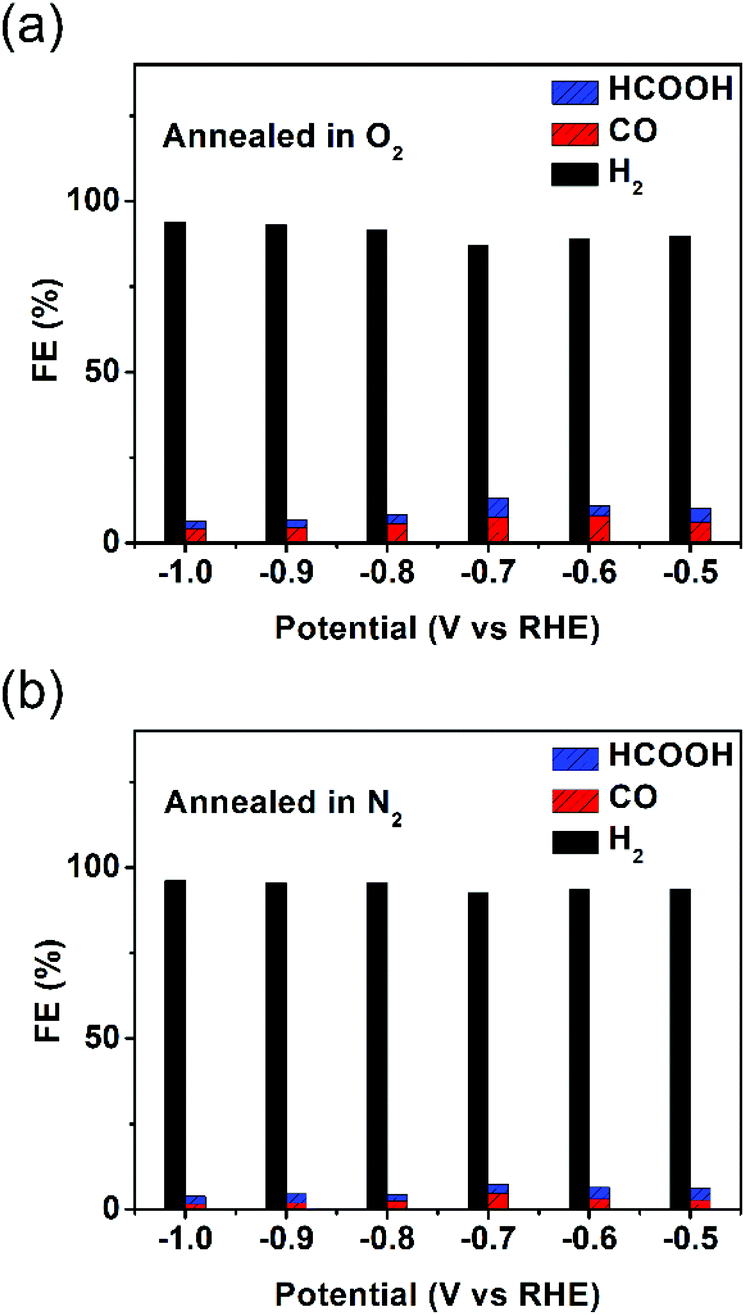 | ||
| Fig. 5 Reduction potential-dependent FEs for electrochemical reduction of CO2 at different potentials measured on hollow Au–Cu NP/C annealed in O2 (a) and N2 (b). | ||
Various characterization methods were implemented for the annealed hollow NPs to find out what happened during the heating process, due to which the catalytic activity was almost totally lost. Fig. S12 in ESI† shows the TEM and HRTEM images of hollow Au–Cu NP/C annealed in O2 or N2, and we could observe that hollow Au–Cu NP/C showed no clear morphological change. The HAADF-STEM and 2D element mapping images of hollow Au–Cu NP/C annealed in O2 or N2, shown in Fig. S13 and S14 in ESI,† indicate a uniform distribution of Au elements and Cu elements in NPs, proving that Au–Cu NPs have retained the alloy structures after the annealing treatment. However, the HRTEM images (Fig. S12b–c and S12e–f†) show that after thermal treatment, the lattice spacings of the hollow Au–Cu NPs increase slightly, and they are 0.229 nm for annealing in O2 and 0.228 nm for annealing in N2. Besides, in Fig. S12c and S12f,† we can see a very clear atomic diffraction lattice, which cannot be observed in the HRTEM image of the as-synthesized hollow Au–Cu NP/C (Fig. 1b1 and b2). The PXRD analysis shows that the 111 diffraction peaks shift from 39.64° to 39.35° and 39.20°, respectively, after the hollow Au–Cu NP/C is annealed in O2 or N2 (Fig. 6). In the Cu 2p XPS spectra (Fig. S15 in ESI†) of both hollow Au–Cu NP/C, the peaks appearing at ∼932.5 eV and ∼952.5 eV can be assigned to Cu(I)/Cu(0) species (indistinguishable). Besides, after being annealed in O2, Cu 2p3/2 and Cu 2p1/2 satellite peaks can be found at 938–945 eV and 960–965 eV, respectively, which can be associated with Cu(II). The compositions of these two kinds of hollow Au–Cu NP/C are determined by ICP-AES and XPS spectra (Au 4f XPS spectra can be seen in Fig. S16 in ESI†), and the results are listed in Table 1. The CV curve of hollow Au–Cu NP/C annealed in O2 (Fig. S17 in ESI†) shows a cathodic peak at −0.32 V (vs. RHE), which cannot be observed in the CV curve of hollow Au–Cu NP/C annealed in N2. Therefore, the characterization results indicate that the hollow structure is maintained after thermal treatment, but Cu atoms segregate on the surface of hollow Au–Cu NP/C; after annealing in O2, the extent of segregation is greater due to the formation of CuO. Besides, the crystallinity of the hollow NPs is improved after thermal treatment.
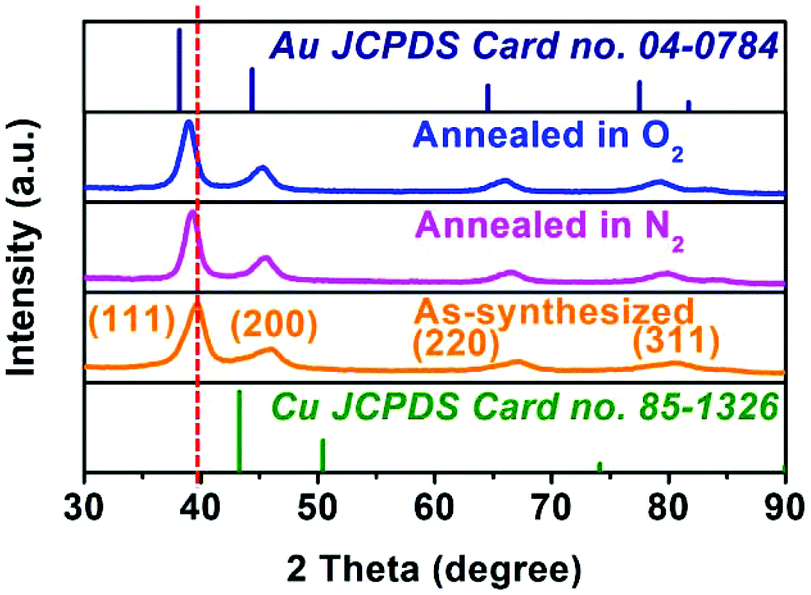 | ||
| Fig. 6 PXRD patterns of three kinds of hollow Au–Cu NP/C. | ||
:Cu obtained from ICP-AES and XPS analyses for different catalysts
| As-synthesized | Annealed in O2 | Annealed in N2 | |
|---|---|---|---|
| ICP-AES | Au0.59Cu0.41 | Au0.57Cu0.43 | Au0.61Cu0.39 |
| XPS | Au0.78Cu0.22 | Au0.48Cu0.52 | Au0.67Cu0.33 |
It was reported by Zhu's group that after thermal treatment in O2 at 250 °C for 1 h, the Cu component in fcc-AuCu alloy completely segregated on the surface, leaving a pure Au core with larger lattice spacing (0.235 nm).48 In our experiment, the temperature applied for thermal treatment in O2 was 180 °C, which was much less than 250 °C; thus, perhaps the amount of Cu segregated on the alloy surface was few, and that is why the lattice spacing and diffraction peaks changed slightly. As to being annealed in N2, the slightly more extent of segregation could be due to the fact that Cu could react with O2 to form CuO species and thus, Cu atom was more likely to diffuse to the surface when the hollow Au–Cu NP/C was heated in O2.49–51 The XPS and CV results of hollow Au–Cu NP/C confirmed the formation of CuO, and the only cathodic peak in the CV curve was due to the existence of minor amounts of CuO on the surface and thus, the two expected peaks (corresponding to the reduction of CuO to Cu2O and Cu2O to Cu) were not strong enough to be divided from each other.52 Finally, a clear atomic diffraction lattice appeared after thermal treatment, suggesting improvement in crystallinity of NPs.
To clarify the relationship between the catalytic performance towards CO2RR and the change in structure/composition of hollow Au–Cu NP/C on the basis of the above-mentioned various characterization results, three points are presented below: first, we point out that the elemental distribution on the surface of the catalyst plays an important role in CO2RR. According to the atomic ratios of Au and Cu obtained from ICP-AES and XPS spectra, the surface atomic ratio of Au and Cu is 78:22 for the as-synthesized hollow Au–Cu NP/C. There are quite a lot of Au atoms on the surface and thus, hollow NP/C acts like Au NP/C and can still maintain the FE of CO. However, because of the segregation of Cu, this ratio changes to 48:52 and 67:33 after being annealed in O2 and N2, respectively, which makes the hollow NP/C act more like Cu NP/C; thus, the selectivity towards CO decreases sharply.
Second, surface defects also influenced the catalytic performance towards CO2RR. It can be found in Fig. 1b2 that there were a certain amount of disordered defects on the surface of the as-synthesized hollow Au–Cu NP/C, which could be active sites for the catalytic reaction. After annealing in O2 or N2, there was improvement in crystallization, which in turn decreased the number of defects (this can be found in Fig. S12c and S12f†); thus, the CO selectivity also decreased. This finding was consistent with that reported by Zhang et al., in which they stated that the lattice defects associated with the sulphide-derived Bi were likely to have a positive influence on CO2RR.47 Besides, the electroconductivity of hollow NP/C also increased due to a decrease in the defect density, which could be responsible for the increase in the current density.
Finally, there was another finding while comparing the catalytic performances of the hollow Au–Cu NPs annealed in O2 and N2. In the former, there were more Cu atoms segregated on the surface, but the amount of CO2 reduced was slightly more than the amount of CO2 reduced by hollow Au–Cu NP/C annealed in N2. This could be due to the oxidation of copper segregated on the surface when annealed in O2. As reported by Kanan et al., the unique surface structures of Cu particles formed by reducing thick Cu2O layers promoted the catalytic reduction of CO2.46 Although the CuO formed after annealing in O2 was less, it was reduced to Cu0 and slightly improved the catalytic activity for CO2 reduction.
To sum up, the superior catalytic performance of the as-synthesized hollow Au–Cu NP/C toward CO2RR could be due to more Au atoms and more defect sites distributed on the NP surfaces.
Conclusions
In this study, hollow Au–Cu NP/C with 4.95 ± 0.81 nm thick wall was prepared via galvanic replacement and the Kirkendall effect between copper and gold for electrochemical reduction of CO2. In this process, TOP was used to dissolve the gold precursor to lower the reduction potential of Au3+. Then, hollow Au–Cu NPs were supported on carbon black and tested for the catalytic activity and selectivity towards CO2RR. The thin-walled hollow structure showed a maximum CO FE of 53.3% at −0.7 V (vs. RHE) and decreased the amount of Au used when compared with solid Au NP/C; besides, it also showed good stability during long-time measurements for CO2RR. The superior performance of this bimetallic hollow catalyst was due to more Au atoms and more defect sites distributed on the NP surfaces, which was proved by annealing experiments in O2 and N2. After thermal treatment, the catalytic activity was almost lost, which was partially due to the segregation of Cu on the surface. Besides, the decrease in the defect density caused by the crystallinity improvement during the heating process could also partially account for the loss of catalytic performance. This study sheds new light on the design and utilization of bimetallic thin-walled hollow NPs in the electrochemical reduction of CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Natural Science Foundation of China (No. 21573005, 21771009, and 21621061), the National Key Research and Development Program of MOST of China (No. 2016YFB0701100), and the Beijing Natural Science Foundation (No. 2162019).Notes and references
- C. N. Waters, J. Zalasiewicz, C. Summerhayes, A. D. Barnosky, C. Poirier, A. Galuszka, A. Cearreta, M. Edgeworth, E. C. Ellis, M. Ellis, C. Jeandel, R. Leinfelder, J. R. McNeill, D. Richter, W. Steffen, J. Syvitski, D. Vidas, M. Wagreich, M. Williams, A. Zhisheng, J. Grinevald, E. Odada, N. Oreskes and A. P. Wolfe, Science, 2016, 351, 2622 CrossRef PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467 CrossRef CAS PubMed.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- E. Lamy, L. Nadjo and J. M. Saveant, J. Electroanal. Chem. Interfacial Electrochem., 1977, 78, 403–407 CrossRef CAS.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68 CrossRef CAS PubMed.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen and B. Han, Angew. Chem., 2018, 130, 2451–2455 CrossRef.
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- J. P. Jones, P. G. K. Surya and O. G. A, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- H. Tao, X. Sun, S. Back, Z. Han, Q. Zhu, A. Robertson, T. Ma, Q. Fan, B. Han, Y. Jung and Z. Sun, Chem. Sci., 2018, 9, 483–487 RSC.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- P. J. Straney, L. E. Marbella, C. M. Andolina, N. T. Nuhfer and J. E. Millstone, J. Am. Chem. Soc., 2014, 136, 7873–7876 CrossRef CAS PubMed.
- N. Lu, J. Wang, S. Xie, Y. Xia and M. J. Kim, Chem. Commun., 2013, 49, 11806–11808 RSC.
- S. Ham, H. J. Jang, Y. Song, L. Shuford Kevin and S. Park, Angew. Chem., Int. Ed., 2015, 54, 9025–9028 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- Y. Imura, S. Koizumi, R. Akiyama, C. Morita-Imura and T. Kawai, Langmuir, 2017, 33, 4313–4318 CrossRef CAS PubMed.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- E. González, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377 CrossRef PubMed.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef CAS PubMed.
- H. Liang, Y. Guo, H. Zhang, J. Hu, L. Wan and C. Bai, Chem. Commun., 2004, 13, 1496–1497 RSC.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- D. Van Thuan, N. Tri Khoa, S. W. Kim, E. J. Kim and S. H. Hahn, J. Catal., 2015, 329, 144–150 CrossRef CAS.
- S. Thota, S. Chen and J. Zhao, Chem. Commun., 2016, 52, 5593–5596 RSC.
- H. Nakajima, JOM, 1997, 49, 15–19 CrossRef CAS.
- A.-A. El Mel, R. Nakamura and C. Bittencourt, Beilstein J. Nanotechnol., 2015, 6, 1348–1361 CrossRef CAS PubMed.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS PubMed.
- K.-Y. Niu, J. Park, H. Zheng and A. P. Alivisatos, Nano Lett., 2013, 13, 5715–5719 CrossRef CAS PubMed.
- H. Jin fan, M. Knez, R. Scholz, K. Nielsch, E. Pippel, D. Hesse, M. Zacharias and U. Gösele, Nat. Mater., 2006, 5, 627 CrossRef PubMed.
- Z. Niu, F. Cui, Y. Yu, N. Becknell, Y. Sun, G. Khanarian, D. Kim, L. Dou, A. Dehestani, K. Schierle-Arndt and P. Yang, J. Am. Chem. Soc., 2017, 139, 7348–7354 CrossRef CAS PubMed.
- T.-L. Ho, in Hard and Soft Acids and Bases Principle in Organic Chemistry, Academic Press, New York, 1977 Search PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- Y. Zhang, F. Li, X. Zhang, T. Williams, C. D. Easton, A. M. Bond and J. Zhang, J. Mater. Chem. A, 2018, 6, 4714–4720 CAS.
- W. Zhan, J. Wang, H. Wang, J. Zhang, X. Liu, P. Zhang, M. Chi, Y. Guo, Y. Guo, G. Lu, S. Sun, S. Dai and H. Zhu, J. Am. Chem. Soc., 2017, 139, 8846–8854 CrossRef CAS PubMed.
- X. Liu, A. Wang, X. Wang, C.-Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–3189 RSC.
- A. Sandoval, C. Louis and R. Zanella, Appl. Catal., B, 2013, 140–141, 363–377 CrossRef CAS.
- X. Liu, A. Wang, L. Li, T. Zhang, C.-Y. Mou and J.-F. Lee, J. Catal., 2011, 278, 288–296 CrossRef CAS.
- Y. Wan, Y. Zhang, X. Wang and Q. Wang, Electrochem. Commun., 2013, 36, 99–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: More data on TEM images, PXRD and XPS analyses, and electrocatalytic measurements of Au/Cu nanoparticles. See DOI: 10.1039/c8qi00297e |
| This journal is © the Partner Organisations 2018 |