
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodynamic therapy in 3D cancer models and the utilisation of nanodelivery systems
Layla
Mohammad-Hadi
 *,
Alexander J.
MacRobert
,
Marilena
Loizidou
and
Elnaz
Yaghini
*
*,
Alexander J.
MacRobert
,
Marilena
Loizidou
and
Elnaz
Yaghini
*
Division of Surgery and Interventional Science, Department of Nanotechnology, University College London, Royal Free Campus, Rowland Hill St, London NW3 2PE, UK. E-mail: layla.hadi.13@ucl.ac.uk; elnaz.yaghini@ucl.ac.uk
First published on 8th January 2018
Abstract
Photodynamic therapy (PDT) is the subject of considerable research in experimental cancer models mainly for the treatment of solid cancerous tumours. Recent studies on the use of nanoparticles as photosensitiser carriers have demonstrated improved PDT efficacy in experimental cancer therapy. Experiments typically employ conventional monolayer cell culture but there is increasing interest in testing PDT using three dimensional (3D) cancer models. 3D cancer models can better mimic in vivo models than 2D cultures by for example enabling cancer cell interactions with a surrounding extracellular matrix which should enable the treatment to be optimised prior to in vivo studies. The aim of this review is to discuss recent research using PDT in different types of 3D cancer models, from spheroids to nano-fibrous scaffolds, using a range of photosensitisers on their own or incorporated in nanoparticles and nanodelivery systems.
 Layla Mohammad-Hadi | Layla Mohammad-Hadi is a doctoral student researching in nanomedicine and cancer therapy at University College London in the Dept. of Nanotechnology, Division of Surgery and Interventional Science. She graduated in Pharmacology and then completed a Masters degree in Reproductive Medicine and Women's Health. Her doctoral research is mainly focused on Photodynamic Therapy and the use of Photochemical internalisation for the delivery anti-cancer drugs to their target sites of action in various 3D models of breast and ovarian cancer. |
 Alexander J. MacRobert | Alexander (Sandy) MacRobert is a biophysical chemist and is Professor of Photochemistry and Photobiology at University College London. He was educated at the University of Cambridge and then carried out his PhD and postdoctoral research on free radical spectroscopy and kinetics in London at QMUL and Imperial College, Oxford University, and Bielefeld University, Germany. His research is focused on biomedical optical spectroscopy and imaging, and therapeutic applications of reactive oxygen species ranging from cancer to antimicrobial treatments. |
 Marilena Loizidou | Marilena Loizidou is Professor of Cancer Nanotechnology and Deputy Director of the Division of Surgery and Interventional Science (SIS) at University College London. She graduated in Biochemistry from McMasters University, Canada followed by a PhD in Solid Tumour Biology from the University of Southampton. She became lead clinical scientist at the University of Southampton/NHS and moved to UCL in 1993 becoming Head of Department at the Royal Free Campus in 2013. Her research focuses on targeting cancer using tissue engineered 3D in vitro models of cancer from patient derived cells to determine responses to specific treatments. |
 Elnaz Yaghini | Elnaz Yaghini is a senior research associate in the Division of Surgery and Interventional Science at University College London (UCL), and a member of the UCL Institute for Healthcare Engineering. She graduated in Clinical Medicine from Iran, followed by a PhD in Nanomedicine from UCL in 2011. In her research at UCL, she has investigated the potential of various nanoparticles and photosensitising dye molecules for preclinical diagnosis and treatment of cancer, and photochemical internalisation including the application of 3D models. Her research is primarily focused on the development of novel targeted fluorescent nanoparticles for cancer imaging and intraoperative image-guided surgery. |
Introduction
Photodynamic therapy (PDT) is a minimally invasive treatment for various cancers and for certain non-malignant lesions.1 PDT requires administration of a photosensitiser, which is then activated by blue, red or near infrared (NIR) light resulting in the generation of cytotoxic reactive oxygen species (ROS). This method is clinically approved for treating several types of solid cancerous tumours for example prostate,2 head and neck skin and oesophagus.3 PDT has also been utilised to tackle non-cancerous conditions such as age-related macular degeneration, atherosclerosis and bacterial infections.4 PDT treatment is advantageous for both the patient and the clinician since it minimises the need for major surgery, reduces recovery periods, promotes good healing and conserves integrity and function of organs with relatively little risk of local and systemic treatment-related morbidity and side-effects.5–8 Furthermore PDT can be repeated and even used after various treatments such as surgery, chemotherapy and radiotherapy without inducing any immunosuppressive or myelosuppressive effects.9Two dimensional (2D) cell culture where cells are grown on flat surfaces as monolayers and in vivo models have been the main means of conducting cancer and drug discovery studies until recently.10 The 2D monolayer cultures present several advantages such as easy preparation, maintenance and monitoring in addition to being amenable for microscopic and molecular investigations.11,12 However the growth of cells on a flat surface does not properly integrate important interactions between the cells and the surrounding extracellular matrix (ECM) that is present in vivo which consists largely of structural proteins, predominantly type 1 collagen fibrils.13 Moreover cell-to-cell interaction is restricted in 2D models since the main interaction is with the host surface, normally plastic. The absence of such interactions in 2D models may cause adhesion properties and organization of cancer cells to deviate from their in vivo counterparts thereby affecting proliferation and signal transduction as well as cellular responses to drug and radiation therapies.14,15 Moreover, when nanoparticles and drugs are applied to monolayer cell cultures they are able to reach cells without encountering any physical limitations whereas nanoparticles delivered in vivo experience hindrance by the ECM surrounding the cancer cells.16 Thus exposure of the cancer cells in monolayer culture to a uniform environment with a steady supply of oxygen and nutrients prevents them from mimicking in vivo cancer tissues which are normally affected by nutrient and oxygen gradients17 and can lead to altered gene expression patterns. The ECM is the major component of in vivo connective tissue,18,19 and in solid tumours (e.g. colon, prostate, breast), the cancer cells grow and proliferate in contact with the surrounding connective tissue, which is also called the ‘stroma’. The 3D models incorporating ECM materials such as collagen are therefore capable of mimicking cancer growth within its local environment, and provide a scaffold for cellular organisation in 3D and adoption of shape and structural features seen in vivo.20–22 The stromal interactions exhibited in the 3D models can also influence therapeutic response,23 drug and nanoparticle penetration, anti-apoptotic signalling as well as multicellular resistance and hypoxia.16,24,25
In this review we will be specifically discussing the use of PDT in different 3D cancer models with a focus on the possible application of nanoparticles in the PDT treatment of such models as seen in some of the studies mentioned later.
How PDT works
The mechanism of action of PDT and the nature of cell death depend on several factors such as genotype of cells, PDT dosimetry (e.g. light intensity) and the localisation of the photosensitiser.26,27 Since most photosensitisers do not have the tendency to accumulate within the nuclei,28 PDT rarely induces DNA related damage, mutations or carcinogenesis.27 Photosensitisers, which localise within the mitochondria mainly, stimulate apoptosis,29whereas photosensitisers that localise within the plasma membrane mostly induce necrosis upon exposure to light.30 Overall the manner in which cell death is triggered shifts from apoptotic to necrotic as the intensity of the damage caused to the cell becomes excessive resulting in swift cell lysis instead of an orderly programmed type of cell death.31 Photodamage may also result in a cytoprotective response termed autophagy.32In terms of the photophysical processes involved in PDT, photosensitisers possess a stable singlet state as their lowest energy level33 and upon light activation the photosensitiser is raised to a singlet excited state which is short-lived.34,35 The photosensitiser may then return to ground state through internal conversion or radiatively via fluorescence which can be used for photodetection purposes in a clinical setting,36 or be converted via intersystem crossing to the longer lived triplet state.5 In this case, providing that a sufficient supply of oxygen is available, the excited triplet state photosensitiser can undergo either a type I or type II reaction process to produce ROS.37 In a type I reaction the photosensitiser interacts with a substrate in a direct manner through proton or electron transfer to produce radicals.38 In a type II reaction, resonant energy transfer occurs to generate the singlet oxygen species.38 Due to the high reactivity and thus short diffusion distance of singlet oxygen, damage is locally confined,39 and if the light field is well controlled, some selectivity of damage can be achieved to the target lesions with minimal damage occurring to the adjacent normal tissue.38 However, singlet oxygen is still able to transverse and escape from inert matrices of nanocarriers such as mesoporous silica,40 therefore photosensitisers incorporated within the nanoparticles can retain their photosensitisation properties, as discussed later. An extensive range of laser and non-laser sources of light can be used in photodynamic therapy.41 Solid state semiconductor diode lasers provide cost-effective and high power delivery with the advantages of being portable and simple to operate.42 Light delivery using optical fiber technology can either be directed onto the surface of the lesion using an endoscope or interstitially using multiple fibre-optic catheters implanted percutaneously within solid tumours.43,44
Photosensitisers used in PDT
An ideal photosensitiser should display chemical, photophysical as well as biological characteristics, which allow it to be taken up by tumours, undergo fast clearance and have a large absorption peak at red to near infrared wavelengths where tissue is relatively transparent.39 Photosensitisers for PDT are normally categorised into two groups: tetrapyrrole derivatives (e.g. porphyrins) and non-tetrapyrroles (e.g. methylene blue).27,45First and second-generation photosensitisers
First generation photosensitisers include haematoporphyrin derivatives (HpD) and a purified form of HpD known as porfimer sodium (Photofrin™). HpD has shown to localise within the tumours and produce a good tumouricidal response when activated by red light. Porfimer sodium has obtained FDA approval for treating lung and oesophageal using PDT.46,47 Although porfimer sodium absorbs weakly around 630 nm,47 it is still commonly used in clinical applications, however its more widespread use has been undermined by its retention in the skin resulting in long-term cutaneous photosensitivity.5,48To address the limitations of these original photosensitisers, newer second-generation photosensitiers have been developed.27 In comparison with the first generation agents, these photosensitisers are generally more chemically pure, capable of absorbing light at longer wavelengths, possess higher singlet oxygen quantum yields and induce lower skin photosensitivity.39 The chlorin photosensitiser m-tetrahydroxyphenylchlorin (mTHPC, temoporfin) is one of the most widely studied second-generation photosensitisers.49 Another widely studied agent for PDT particularly in dermatology is 5-aminolaevulinic acid (ALA) which is a pro-drug for protoporphyrin IX (PPIX). However mTHPC, has a higher potency than either of the porphyrin-based approaches using ALA,39 or porfimer sodium50 and requires lower light and drug doses for treating tumours.51 Moreover mTHPC absorbs light at 652 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52,53 in comparison to the weak absorption peak at 630 nm of both porfimer sodium and ALA induced PPIX which enables deeper excitation in the tissue.53 For very thin lesions such as actinic keratoses in the skin, blue light excitation is sufficient and is used clinically with ALA. The search for drugs that are more tumour specific and can undergo activation with light of a longer wavelength with a shorter period of photosensitivity is still ongoing.54 Other photosensitisers that have been tested in clinical trials include tin ethyl etiopurpurin (SnET2),55 mono-L-aspartyl chlorin e6 (MACE),56 benzoporphyrin derivative mono acid, ring A (BPD, verteporfin),57 and lutetium texaphyrin (Lu-Tex).58 These compounds have absorption peaks at higher wavelengths of 660 nm,59 664 nm,56 690 nm60 and 732 nm respectively compared to first as well as second generation photosensitisers and lead to mild and brief skin photosensitivity. For clinical applications, a liposomal formulation (Visudyne™) of BPD is generally used.61,62 The clinical formulation of mTHPC is known as Foscan™. Liposomal formulations of mTHPC have also been tested preclinically49 known as Foslip™ and a pegylated version known as Fospeg™.
52,53 in comparison to the weak absorption peak at 630 nm of both porfimer sodium and ALA induced PPIX which enables deeper excitation in the tissue.53 For very thin lesions such as actinic keratoses in the skin, blue light excitation is sufficient and is used clinically with ALA. The search for drugs that are more tumour specific and can undergo activation with light of a longer wavelength with a shorter period of photosensitivity is still ongoing.54 Other photosensitisers that have been tested in clinical trials include tin ethyl etiopurpurin (SnET2),55 mono-L-aspartyl chlorin e6 (MACE),56 benzoporphyrin derivative mono acid, ring A (BPD, verteporfin),57 and lutetium texaphyrin (Lu-Tex).58 These compounds have absorption peaks at higher wavelengths of 660 nm,59 664 nm,56 690 nm60 and 732 nm respectively compared to first as well as second generation photosensitisers and lead to mild and brief skin photosensitivity. For clinical applications, a liposomal formulation (Visudyne™) of BPD is generally used.61,62 The clinical formulation of mTHPC is known as Foscan™. Liposomal formulations of mTHPC have also been tested preclinically49 known as Foslip™ and a pegylated version known as Fospeg™.
Description of different types of 3D cell culture models
3D culture models can be created using various formats such as multicellular aggregates, culturing cells on inserts or embedding cells in an artificial nanofibrous matrix or scaffold developed from natural or synthetic material.63 The characteristics of different types of 3D models are summarised as follows.Spheroids
Spheroids are well-rounded 3D cancer models typically several hundred μm in diameter that can also be referred to as spheres, nodules and micronodules.64–66 Such 3D models can be created either by growing cells in low adhesion conditions (e.g. plates, hanging drop methods etc.) where they assume naturally a spherical aggregate conformation67–69 or by embedding cells in a 3D matrix. The closely packed arrangement where cell to cell contact is dominant and the reduced rate of diffusion of drugs and oxygen through the spheroids makes them comparable to in vivo tissues.70 Spheroid 3D models are used for the assessment of various specific 3D properties such as the development of invasive characteristics, changes in the dependence of cells on growth factors, increased luminal survival because of the stimulation of anti-apoptotic and pro-proliferative signals and the capability to avoid growth arrest because of these pro-proliferative signals.20,71 Spheroid 3D models have been widely used for a variety of photosensitiser-PDT studies including those involving two photon excitation (Table 1).| Photosensitiser | Description of 3D cancer model | Cancer cell line | Ref. |
|---|---|---|---|
| Methylene blue | Spheres on (poly 2-hydroxyethyl methacrylate) (polyHEMA) coated microwells in a microfluidic device | Human breast carcinoma (T47D) | 91 |
| Benzoporphyrin Derivative (BPD) | Micronodules on Matrigel matrix | Human ovarian carcinoma (OVCAR5) | 92 |
| 5-Ethylamino-9-diethylaminobenzo[a] phenothiazinium chloride (EtNBS) | Nodules on growth factor reduced (GFR) Matrigel matrix | Human ovarian carcinoma (OVCAR5) | 93 |
| BPD in DMSO | Micronodules on Matrigel matrix | Human ovarian carcinoma (OVCAR5) | 94 |
| BPD | Micronodules on Matrigel matrix | Human ovarian carcinoma (OVCAR5) | 95 |
| 5-Aminolevulinic acid (5-ALA) | Nodules on Matrigel matrix | Human epidermal carcinoma (A431) | 35 |
| BPD, mono-N-aspartyl derivative of chlorin e6 (MACE) | Mammary architecture and micro-environment engineering (MAME) of breast cancer | Human breast carcinoma (SUM149, MDA-MB-231, Hs578T) | 96 |
| Spheroids formed on glass cover slips coated with reconstituted basement membrane | |||
| Tetraphenyl disulfonated porphyrin (TPPS2a), disulfonated tetraphenyl chlorin (TPCS2a) | Single cells seeded in hydrogel (collagen) scaffold | Human prostate adenocarcinoma (PC3) | 97 |
| Tetraphenyl disulfonated porphyrin (TPPS2a), disulfonated tetraphenyl chlorin (TPCS2a) | Single cells seeded in hydrogel (collagen) scaffold | Human head and neck squamous cell carcinoma (PCI30) | 98 |
| mTHPC | Single cells seeded in hydrogel (collagen) scaffold | Human breast carcinoma (MCF-7) | 99 |
| BPD | Nodules on Matrigel | Human ovarian carcinoma (OVCAR5) | 100 |
| Hypericin | Spheroid on agarose coating | Human bladder carcinoma (RT-112) | 101 |
| Ruthenium(II) polypyridyl complexes (Ru1–Ru3) | Spheroid on agarose coating | Human cervical carcinoma (HeLa) | 102 |
| Ruthenium(II) polypyridyl complexes (RuL1–RuL4) | Spheroid on agarose coating | Human cervical carcinoma (HeLa) | 103 |
| Fluorinated ruthenium(II) complexes (Ru1–Ru5) | Spheroid using liquid overlay method | Human cervical carcinoma (HeLa) | 104 |
Cell derived matrices (CDMs)
CDMs are commonly produced by culturing cells that excrete ECM proteins on pre-coated scaffold surfaces or as a monolayer (2D), or multicellular aggregates (3D) to allow the deposition of adequate ECM. Upon depositing sufficient ECM, decellularisation processing can be used to disrupt as well as remove the cellular component from the ECM. Such processing is essential for minimising the risk of encountering adverse immunological responses.72CDMs are known for providing a shared ECM based feedback to cancer cell signalling plus an enhanced cell–matrix adhesion, motility as well as proliferation compared to 2D cultures.71 Such differences between CDMs and 2D cultures may arise due to expanded dimension or the activities of growth factors present in the CDMs, which are not detected in the 2D environments.65,71 Moreover cells that are cultured on CDMs have similar morphologies to those observed in vivo since they can form specified 3D matrix adhesions, which are also found within in vivo models.73
Microfluidic devices
The microfluidic technology also known as Lab-on-a-chip (LOC)74 provides the opportunity for the development of 3D cell cultures and cell based assays in complex microenvironments that can be controlled, reproduced and optimised.75 This type of technology possesses several key features: (1) it exhibits micro-scale dimensions which have great compatibility to the microstructures found in the microenvironments of in vivo cancer models; (2) it is very cost-effective as a small quantity of the samples are required, keeping reagent consumption low thus reducing the costs of bioanalysis as well as drug discovery and development; (3) some of the substrates used in microfluidic devices have high O2 permeability which has an important impact on cell proliferation and (4) this technology allows the integration of numerous features namely cell culture and sampling, control of fluids, capturing of cells, cell lysis as well as mixing and detection all on one single device.76,77 The various microfluidic platforms that have been previously used in 3D cell cultures include glass/silicon-based, polymer-based together with paper-based platforms, which have been named depending on the substrates used in the fabrication of the micro-device.75 Such technology has also been used for photo-oxidation reaction studies related to PDT.78Scaffolds
3D scaffolds consisting of a nano-fibrous matrix create an environment, which supports the proliferation, growth as well as migration of cancer cells allowing such aspects to be investigated.79 Scaffolds present several advantages over spheroids in particular the capability of mimicking tumour heterogeneity as well as the control of the 3D dimensions. Furthermore the extent of migration, proliferation and aggregation of the cells can be controlled by the surface properties as well as the composition, configuration and the porosity of the scaffolds.79,80 These properties also make such models good candidates for nanocarrier delivery studies as shown by the work of Lopez et al. (2016)81 which focused on the diffusion properties of liposomes and micelles in a 3D collagen scaffold model.81 Scaffolds can be subdivided into natural or synthetic scaffolds based on the materials incorporated therein.82Natural scaffolds are largely hydrogels containing mostly water83 and natural components such as collagen type 1 (Fig. 2), matrigel, agarose, elastin, laminin and hyaluronic acid.71,84 Although mechanically weak due to the high volume of excess fluid with them, hydrogel models permit the movement and proliferation of cells within a biological environment.79 However their low density does not reflect the dense environment surrounding tumour cells in vivo.79 This problem can be resolved in part by the remodelling of the hydrogel to assist cell–matrix interaction studies and a rise in matrix density due to contraction.85 The development of the plastic compression technology has resulted in the production of improved biomimetic scaffolds with increased cell and collagen density through the removal of interstitial fluid from the hydrogel model.86 The collagen stiffness in these scaffolds not only influences rate of cell growth as well as morphologies,87 but owing to the higher collagen density and reduced oxygen diffusion also enables the hypoxic core that is normally observed in vivo to be imitated in vitro.79Fig. 2 exemplifies the stages involved in the formation of the compressed 3D model. Recent studies with compressed collagen hydrogels have utilised ‘tumouroid’ models consisting of a cancer mass surrounded by a multi-cellular stroma to study the uptake of nanoparticles in addition to their use in enhancing drug delivery.81,88
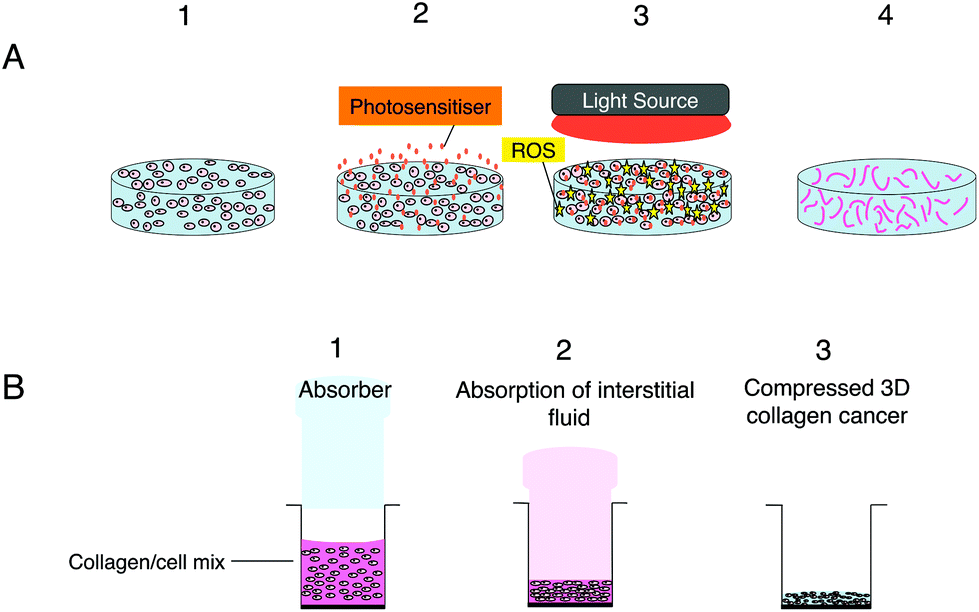 | ||
| Fig. 1 (A) PDT using nanoparticles in 3D hydrogel model consisting of cancer cells surrounded by a matrix (e.g. type 1 rat tail collagen) illustrated with sequential procedures; step 1: seeding of cancer cells in the hydrogel scaffold; step 2, addition of a photosensitiser for cell uptake followed by washing; step 3: the irradiation of the 3D model with light and generation of ROS. Step 4: the dead cancer cells in the hydrogel scaffold. (B) Construction of compressed 3D collagen cancer models with higher collagen density for therapeutic studies illustrated sequentially. Step 1, the formation of collagen/cell mix prior to undergoing the plastic compression stage using an insertable absorber to extract fluid from the hydrogel; step 2 shows the gradual absorption of the interstitial fluid from the collagen/cell mix by the absorber and the slow compression of the model; step 3 demonstrates the thinner (typically 200 μm depth) compressed 3D collagen cancer model following the absorption of the fluid from the model resulting in a ten-fold higher collagen density. | ||
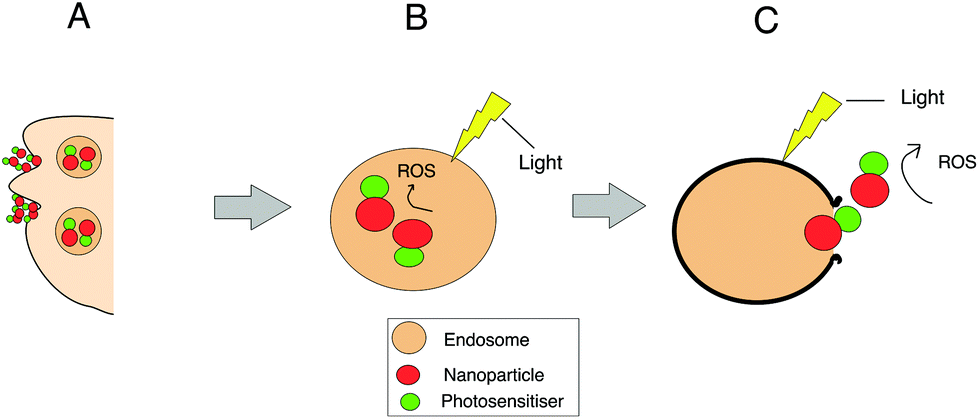 | ||
| Fig. 2 Endocytic uptake and reactive oxygen species (ROS) generation for nanoparticles conjugated with the photosensitiser. (A) Uptake of photosensitiser conjugated nanoparticles by the cell via endocytosis and their localisation within endosomes and subsequently lysosomes. (B) Light-induced generation of ROS within the endosome containing the conjugates where they interact with the membrane and other components inside the organelle. Some ROS may also migrate through the membrane of the endososome where they can oxidise other substrates in the cell. (C) Photo-induced rupture of the endo/lysosomal membrane after prolonged irradiation via interaction of ROS with the membrane, leading to the escape of the nanoparticle into the cytosol and production of ROS at other intracellular sites. | ||
Synthetic scaffolds can also be developed from polymers e.g. polyactide (PLA), polyglycolide (PGA) and co-polymers (PLGA).82 These polymers are biodegradable and can be manipulated to create a variety of structures such as mesh, fibers and sponge.89 Synthetic scaffolds have a stronger mechanical structure than natural scaffolds and can specifically replicate biomolecular structures seen in vivo.90 Cell adhesions are unfortunately weaker on these polymers and therefore surface modifications are required to overcome this problem.79
PDT applications in 3D cancer models
There is growing interest in the use of 3D models to better replicate the cellular response to PDT and overcome some of the limitations of conventional monolayer culture. A further motivation is that the use of 3D models and their rapid screening capability could reduce the need expensive in vivo experimentation. 3D cancer models, particularly using spheroid cultures have contributed to photodynamic research by allowing the uptake and the therapeutic efficacy of various photosensitisers to be examined.66 More recently, microfluidic technology has been applied to PDT.91 Since PDT efficacy is dependent on an adequate oxygen supply, investigation of the PDT response at lower oxygen levels found in solid tumours is particularly relevant. Moreover the PDT process consumes molecular oxygen as molecular substrates become oxidised, thus diffusion and consumption of oxygen within a tissue construct can mimic the in vivo dynamics.13 Regarding visible light penetration within the 3D models, since the constructs are relatively thin (typically <1 mm) partly in order to permit oxygen replenishment, attenuation of the light for PDT activation is negligible.Table 1 presents a summary of experimental data obtained using 3D in vitro cancer models and a range of photosensitisers administered in solution form. The description of the 3D cancer model (e.g. spheroid, micronodule etc.) uses the terminology given in the quoted reference articles.
In several studies differences were noted between cellular responses to PDT in 2D and 3D culture models. For example, Chen et al. (2015)91 used the microfluidic platform to create a breast cancer sphere culture environment on a chip using T47D cells. The formation of such spheres was a consequence of aggregation of cells in each individual microwell. The 3D cultures were incubated with photosensitiser methylene blue at a concentration of 10 μM for 1 hour before undergoing illumination for different periods. The results showed that after 10 minutes under an exposure dose of 7.3 J cm−2 approximately 50% cell kill was attained in the 2D cell monolayer while in the 3D spheres a majority of the cells were viable. Even after the longest illumination period of 1 hour and highest light dose (44 J cm−2) a portion of the T47D cells within the centre of the spheres were still viable. The larger spheres demonstrated more resistance towards PDT under the same therapeutic conditions than small spheres indicating the effect of sphere size on PDT efficacy. The penetration of the photosensitiser and the lower oxygen levels prevalent at the interior of the spheroid are both factors limiting the PDT efficacy in larger spheroids.
A different study carried out by Rizvi et al. (2013)95 focused on using BPD-PDT in 3D tumour models of ovarian cancer, which consisted of micronodules of OVCAR5 human cancer cultures developed on growth factor reduced (GFR) Matrigel. The models were incubated with 0.25 μM, 1 μM and 10 μM BPD for 90 minutes prior to undergoing irradiation using a 690 nm fibre-coupled diode laser. Significant reduction in cell viability occurred in nodules that were treated with 0.25 μM BPD-PDT mainly after 72 hours and 96 hours post treatment compared to nodules treated with 1 μM and 10 μM BPD-PDT. However nodules that underwent therapy with a higher concentration of 10 μM BPD-PDT exhibited the poorest response. This result may have been due to aggregation of the photosensitiser, which can impair its photosensitising efficacy, as well as other microenvironmental factors.
In another recent study conducted by Evans et al. (2011),93 the ovarian cancer cell line OVCAR5 was again used to develop spheroids on GFR containing Matrigel to investigate the effect of PDT using photosensitiser 5-ethylamino-9-diethylaminobenzo[a] phenothiazinium chloride (EtNBS) on hypoxic cell populations within 3D tumour models of ovarian cancer. The spheroids were incubated with 500 nM EtNBS for 4.5 hours to allow the photosensitiser to concentrate into the core cell populations within the spheroid. At a light dose of 5 J cm−2, EtNBS managed to selectively destroy the spheroid core cells. This shows EtNBS is able to both concentrate into and destroy the hypoxic cell populations that are normally difficult to treat. At higher doses of light EtNBS-PDT was capable of causing cell killing across the entire model indicating that such therapy is effective against both hypoxic and normoxic regions of a tumour. Interestingly the uptake and cytotoxicity of EtNBS was found to increase with expansion of spheroid size mainly owing to the rise in the hypoxic populations found in the larger spheroids. These results demonstrate the utility of 3D models for testing PDT efficacy under lower oxygen partial pressures that apply in vivo in contrast to conventional 2D model testing, and together with Chen et al.,91 exemplify the advantages of using the 3D architecture of these biomimetic models to obtain a clearer understanding of processes relevant to in vivo experimentation.
The use of nanoparticles in PDT and their applications in 3D cancer models
Localised treatment with minimal side effects as well as the lack of mutagenicity and mild allergenicity are in principle key advantages of using photosensitisers. However it is recognized that there are several limitations such as the lack of sufficient tumour selectivity and poor bioavailability which has led to the development of nanoparticle delivery systems in PDT.61,105 For example, the bioavailability of otherwise water insoluble photosensitisers may be improved using a water soluble ‘nanocarrier’ thereby enabling intravenous administration. Nanocarriers can be “actively” targeted to the diseased site using surface-conjugated ligands that bind to overexpressed receptors or antigens on the target tissue.105–110 Improved tumour targeting is also possible via the enhanced permeability and retention (EPR) properties of nanoparticles, a process that is termed “passive” targeting selectivity. In this case, the targeting of photosensitisers can be commonly achieved through encapsulation or conjugation to nanocarriers such as liposomes or polymeric particles. Alternatively, the photosensitisers can be covalently bound to the surface of the nanoparticle (e.g. silica) or within the matrix via either a stable bond or a biodegradable linkage so that the photosensitiser can migrate elsewhere within the cell following uptake of the nanoparticle. If the matrix of the nanoparticle is biodegradable, the photosensitiser release occurs either within the cell or during circulation. Owing to their size, nanoparticles are generally taken up via endocytosis as shown in Fig. 2. If the photosensitiser remains entrapped with the nanoparticle during cell uptake, ROS are then photogenerated within the endo/lysosomes, as depicted in Fig. 2. With a sufficient light dose the membrane will be ruptured enabling the nanoparticles to migrate from the endo/lysosomes. Nanoparticles can also act as photocatalytic agents or photosensitisers themselves, in effect as ‘nanophotosenstisers’ to generate reactive oxygen species.111–113 Examples include metallic or semiconducting nanoparticles and organic fullerenes.111,112 In combination with a dye, Förster resonance energy transfer (FRET) can also occur with light-activated nanoparticle.112,114,115 A wide range of nanocarriers have been tested, from inorganic to organic compositions. Example of inorganic nanocarriers include single-wall carbon nanotubes (SWCNTs),116 upconversion nanoparticles (NaYF4:Yb3+,Er3+)117 and self-lighting nanoparticles (NaYF4:Yb3+,BaFBr:Eu2+,Mn2+,LaF3:Tb3+).116 Nanoparticles can also be sub-classified into either biodegradable (alginate, chitosan, cyclodextrin, albumin PLA, PLGA)118,119 or non-biodegradable (polyacrylamide, silica, gold (Au), iron oxide) nanoparticles.In 3D cultures, both oxygen and nanoparticle delivery are controlled by diffusion to the cells through the matrix following administration to the surface of the construct. In principle therefore 3D models are well suited to studying the efficacy of nanoparticles for PDT. Incorporation of a vascular network is still in its infancy and remains a longer term goal.120 Numerous studies have investigated the properties of nanoparticles to improve the therapeutic effects obtained from PDT treatment of various cell types in 3D models. Table 2 provides examples of nanoparticles used in PDT studies that have been carried out in 3D cancer models. A key benefit of employing 3D models for PDT studies involving nanoparticles is their ability to incorporate the cell penetration problems experienced by nanoparticles in contrast with 2D monolayer cultures.25
| Photosensitiser | Nanoparticle | Description of 3D cancer model | Cancer cell line | Ref. |
|---|---|---|---|---|
| 5-Aminolaevulinic acid (5-ALA) induced PPIX | Gold nanoparticle (AuNPs) | Nodules on microfluidic device | Human breast carcinoma (MCF-7) | 121 |
| Pheophorbide A (Pheo) | Micelles poly(ethyleneoxide-b-3-caprolactone) | Spheroid on ultra-low attachment well plates | Human colorectal carcinoma (HCT-116), human squamous cell carcinoma (FaDu) | 68 |
| m-Tetrahydroxyphenylchlorin (mTHPC) | Lipidots | Spheroid using hanging drop method | Human tongue squamous cell carcinoma (CAL-33) | 69 |
| EtNBS | Poly(lactic-co-glycolic acid) (PLGA) | Spheroid on Matrigel | Human ovarian carcinoma (OvCAR) | 122 |
| cis–Bis(2, 2′-bipyridine)dichlororuthenium(II) hydrate | SWCNTs | Spheroid using liquid overlay method | Human cervical carcinoma (HeLa) | 123 |
| Chlorin e6 (Ce6) | Reduced graphene oxide (rGO) | Spheroid | Human brain carcinoma (U87) | 124 |
| m-THPC | Liposome (Foslip™ and Fospeg™) | Spheroid using liquid overlay method | Human cervical carcinoma (HeLa) | 125 |
| Zinc phthalocyanine (ZnPc) | Liposome | Spheroid on agarose coating | Human cervix adenocarcinoma (HeLa cells) and mouse Mus musculus colon carcinoma (CT26) | 126 |
| Photofrin | Liposome | Spheroid using spinner flasks on a stir-plate | Human bladder carcinoma (MGHU3) | 127 |
| Indocyanine green | PLGA/lipid | Spheroid on agarose coating | Mouse breast carcinoma (4T1) | 128 |
The various types of photosensitisers in nanoparticle form that have been studied in 3D cancer models are listed in Table 2. The nanoparticles used range from inorganic (gold), micelles, polymeric carriers, liposomes and two types of carbon-based particles. Studies shown in some cases included comparisons between 2D and 3D models. For example, Yang et al. (2015)121 employed a 3D breast cancer tissue model comprised of human breast cancer cells (MCF-7) as well as primary adipose-derived stromal cells (ASCs) using a microfluidic device (Fig. 3) to study the synergistic enhancement of PDT using a combination of 5-ALA and AuNPs. The distribution profiles of the agents and influence of light penetration on PDT according to depth of the cancer tissue were also investigated. The breast cancer tissues underwent incubation with 1 mM 5-ALA dissolved in a serum free medium either with or without AuNPs for 4 hours. For comparison the same PDT treatment conditions were also applied on MCF-7 cells in 2D. When 5-ALA was administered alone, PDT gave approximately 50% cell kill in 2D monolayer culture and 17% cell kill in 3D culture compared to higher cell kill of 70% and 50% achieved respectively in the presence of AuNPs. By extending the illumination period most of the cells in the 2D monolayer were destroyed with or without the AuNPs, whereas in the 3D culture with 5-ALA alone was less effective with 50% kill compared to 90% kill with the AuNPS present. Generally much higher cell destruction was observed in the 2D culture compared to the 3D model throughout all illumination periods. Such evidence for synergistic cell kill with 5-ALA/AuNP combined PDT has also been confirmed by Benito et al. (2013).129 Yang et al. (2015)121 also observed a more homogenous response where the dead cells were found to be distributed across the full thickness of the cancer tissue in the 3D model post-treatment with 5-ALA and AuNPs combined, whereas with 5-ALA-only PDT, a majority of the dead cells were identified within the superficial parts of the cancer tissue.
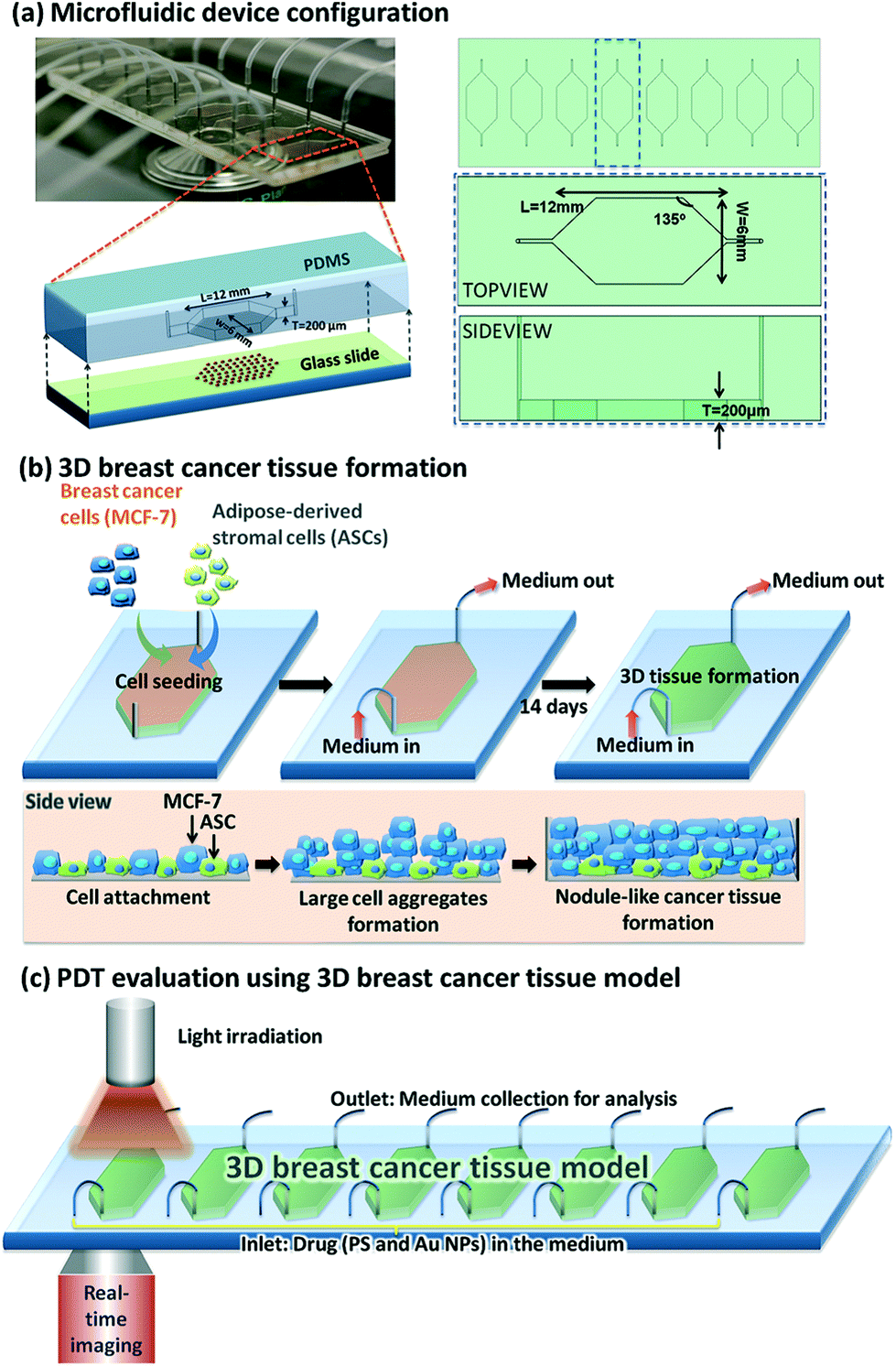 | ||
| Fig. 3 Microfluidic device design and PDT application using Au NPs. (a) Left top: Optically transparent microfluidic device on the stage of a time-lapse microscope within a climate control incubator. Left bottom: Configuration of our microfluidic device for 3D breast cancer tissue formation in which polydimethylsiloxane serves as the housing material and glass slide serves as the substrate. Right: Schematic layout of eight chambers on a 3′′ × 1′′ glass slide with defined parameters for the chamber design. (b) Conceptual illustration of the development of MCF-7 and ASCs cells into 3D breast cancer tissue within the microfluidic chambers. (c) Experimental setup for PDT evaluation using the microfluidic breast cancer tissue model, in which the photosensitiser (PS) and Au NPs are introduced into the system together with a cell culture medium via the inlet, and light irradiation is realised by exposing the transparent device to the light source from the top. Reprinted with permission from ref. 121 (Fig. 1, Y. Yang, X. Yang, J. Zou, C. Jia, Y. Hu, H. Du, H. Wang, Lab Chip, 2015, 15, 735–44). | ||
Till et al. (2016)68 on the other hand used 3D spheroid models developed from human colorectal carcinoma cell line (HCT-116) and human squamous cell carcinoma cell line (FaDu) on ultra-low attachment plates for investigating PDT efficacy. This study compared PDT using a pheophorbide photosensitiser (Pheo) in free solution form to the use of encapsulated photosensitiser in crosslinked polymeric micellar self-assemblies versus uncrosslinked micellar counterparts. In this experiment the spheroids were incubated with photosensitiser in free or encapsulated form for 30 minutes before undergoing irradiation for 8 minutes. The spheroids were exposed to two further cycles of 8-minute irradiation every 24 hours for two days after the first cycle of irradiation. Using the photosensitiser in free form led to low PDT efficiency as a result of its aggregation in water, with a lower response in FaDu spheroids and in HCT-116 spheroids only a 35% decrease in spheroid size was achieved after the third cycle of irradiation. PDT treatment of the spheroids using encapsulated pheophorbide was more effective than free photosensitiser, and the crosslinked systems were more effective compared to the uncrosslinked nanovectors. Different results were obtained in 2D at low concentration where the uncrosslinked micelles were found to elicit better cell kill. The authors noted that in vivo studies of crosslinked vs. uncrosslinked micelles bearing chemotherapeutics have shown that the crosslinked micelles are more effective, which supports their contention that the 3D model better simulated the in vivo response.
In an attempt to minimise the dark toxicity of EtNBS and examine its potential as a PDT photosensitser, Hung et al. (2016)122 encapsulated EtNBS in PLGA NPs and tested this formulation in OVCAR5 monolayer and spheroid models. In monolayer culture, EtNBS loaded nanoparticles showed reduced dark toxicity compared to cells treated with free EtNBS. Uptake studies in the spheroids demonstrated the diffusion of PLGA-EtNBS throughout the spheroids in the same way as free EtNBS and the release of the photosensitiser from PLGA upon illumination with laser light of 635 nm wavelength. PDT using PLGA-EtNBS delivery was still effective even in hypoxic cellular microenvironments present in the spheroid models, with comparable efficacy to free EtNBS. Zhang et al. (2015)123 studied Ru(II) complex loaded SWCNTs (Ru@SWCNTs) as an approach for inducing photothermal and two-photon photodynamic therapy (PTT/TPPDT) effects in HeLa cervical cancer spheroid models. The closely packed cells in spheroids make this type of model particularly suitable for demonstrating the optical sectioning capabilities of multiphoton PDT. After incubation with Ru@SWCNTs (50 μg ml−1) excitation with 808 nm laser irradiation led to a reduction in the mean diameter of the spheroids and significant loss in cell viability. Liu et al. (2017)124 investigated loading chlorin e6 onto rGO to study PTT and PDT efficacy in 3D U87 cell spheroid models. Although both therapies proved effective on 2D cultures of U87 cells, only PTT demonstrated a considerable treatment efficacy in the spheroid models.
Owing to their ability to encapsulate either lipophilic and hydrophilic compounds and other advantages, liposomes have been used in several PDT studies on 3D cancer spheroid models.125–127 For example Gaio et al. (2016)125 investigated the photo-induced damage of two liposomal formulations of m-THPC, Foslip® and Fospeg® compared to Foscan® in 3D HeLa spheroid models. Confocal fluorescence microscopy showed that m-THPC penetration was limited and mainly confined to the external cell layers of spheroids with a slightly higher accumulation of Foslip® and Fospeg® with respect to Foscan®. A significant reduction in cell viability of the spheroids was observed in models incubated in the dark with Foscan (8 μM) while the spheroids treated with the liposomal formulations did not elicit dark toxicity. After PDT the cell viabilities of the spheroids were greatly reduced with all three different formulations with Foslip® showing the most reduction in viability at each time point.
As mentioned in the Introduction,16,25 a key advantage of 3D models is their utility for studying drug and nanoparticle penetration. López-Dávila et al.81 used confocal fluorescence microscopy of a Nile Red dye and DNA photosensitiser in free form (DMSO solution) or incorporated in micelles and liposomes to study the depth of dye penetration over a 24 hours period into compressed collagen hydrogels containing a colorectal cancer mass. The rate of diffusion of the free dye determined using fluorescence imaging was significantly higher than observed with either of the nanoparticles, which had similar distributions. An advantage of using a compressed hydrogel (Fig. 1b) for penetration and diffusion studies is that the collagen density (weight/volume) is much higher and closer to physiological levels, whereas for a standard uncompressed collagen hydrogel the density is <0.5%. Most of the nanoparticle PDT studies mentioned in Table 2 have been carried out on spheroid models and useful new information has been obtained with these models. However replication of drug and medium perfusion through tissue in 3D models remains challenging although microfluidic models appear to provide a promising approach, although further studies are needed.
Conclusions
The development of new photosensitisers that can be activated at longer wavelengths and their incorporation in nanoparticles for PDT has led to a surge of interest in this field over recent years. 3D cancer models provide the opportunity to carry out more physiologically realistic studies of minimally invasive methods such as photodynamic therapy and help bridge the gap between in vitro and in vivo studies. As exemplified by the studies covered in this review, exploiting the biomimetic properties of 3D models can counter overestimation of photosensitising efficacy observed with 2D monolayer model studies by taking account of the limited drug/nanoparticle penetration through the extracellular matrix.PDT is generally used for treatment of solid tumours where hypoxia and treatment-induced oxygen consumption may limit treatment efficacy. The ability of 3D models to simulate normoxic vs. hypoxic conditions and light dosimetry planning are all factors that will encourage further use of 3D models of solid cancer for PDT studies. In this review we have focused on biomimetic constructs, which are much cheaper to prepare than carrying out rodent tumour model studies. However we note that another approach, which is also relatively inexpensive, based on zebrafish tumour models has been tested using nanoparticle-mediated PDT recently that may prove useful in future.130
Future prospects
With rapid advances being made in developing more sophisticated 3D tissue culture systems, their application will be increasingly exploited for PDT and related studies relying on photoactivation. The development of perfusable vascularised constructs will be particularly useful for PDT studies since vascular damage induced by PDT plays a key role for many photosensitisers. There is increasing interest in the combination of PDT as part of a combination therapy in particular its use with other agents such as chemotherapeutics,106 where 3D models may prove useful since the cellular response to the chemotherapeutics may also be better simulated in the 3D model. In this approach the photosensitiser and agent may be co-delivered using one nanocarrier and comparison of these methods versus separate administration should be studied using 3D models. In a related development, PDT may also be used to enhance drug delivery using a technique known as photochemical internalisation (PCI), which relies on the photosensitiser being localised in endolysosomal membranes. PCI can improve the intracellular delivery of a macromolecular drug or nanocarrier entrapped within endolysosomes to the cytosol potentially leading to a higher therapeutic effect than using PDT alone.131,132 From the mechanistic point of view, the interaction between tumour and stromal components such as tumour-associated fibroblasts and their contribution to the tumour cell response may also be elucidated better using 3D model technology. This approach will also be useful for non-PDT photoactivatable agents such as a ruthenium-caged photosensitiser for cathepsin K inhibition, where the spatiotemporal control over activation that can be exerted using a 3D model has been demonstrated recently.133 Two photon activation of photosensitisers using ultrafast laser excitation is another area that will benefit from the use of 3D models since it will be easier to demonstrate the optical sectioning selectivity conferred using two photon versus one photon excitation.134Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge support for EY from Innovate UK and the Medical Research Council under the Biomedical Catalyst scheme (grant number 101875) and funding from the NIHR i4i Scheme (grant number II-LA-0813-20002). We also thank Amir Afrashtehpour for his help in the preparation of the manuscript.References
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- A. R. Azzouzi, S. Vincendeau, E. Barret, A. Cicco, F. Kleinclauss, H. G. van der Poel, C. G. Stief, J. Rassweiler, G. Salomon, E. Solsona, A. Alcaraz, T. T. Tammela, D. J. Rosario, F. Gomez-Veiga, G. Ahlgren, F. Benzaghou, B. Gaillac, B. Amzal, F. M. Debruyne, G. Fromont, C. Gratzke and M. Emberton, Lancet Oncol., 2017, 18, 181–191 CrossRef CAS PubMed.
- S. Mallidi, S. Anbil, A. L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458–2487 CrossRef CAS PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- C. Hopper, Lancet Oncol., 2000, 1, 212–219 CrossRef CAS PubMed.
- H. Nyst, I. Tan, F. Stewart and A. Balm, Photodiagn. Photodyn. Ther., 2009, 6, 3–11 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS PubMed.
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discovery Today, 2010, 15, 842–850 CrossRef CAS PubMed.
- A. Nyga, M. Loizidou, M. Emberton and U. Cheema, Acta Biomater., 2013, 9, 7917–7926 CrossRef CAS PubMed.
- C. Hsieh, Y. Chen, S. Huang, H. Wang and M. Wu, BioMed Res. Int., 2015, 2015, 1–10 Search PubMed.
- H. L. Ma, Q. Jiang, S. Han, Y. Wu, J. Cui Tomshine, D. Wang, Y. Gan, G. Zou and X. J. Liang, Mol. Imaging, 2012, 11, 487–498 CAS.
- M. Alemany-Ribes, M. Garcia-Diaz, M. Busom, S. Nonell and C. E. Semino, Tissue Eng., Part A, 2013, 19, 1665–1674 CrossRef CAS PubMed.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS PubMed.
- M. Zhang, P. Boughton, B. Rose, C. Lee and A. Hong, Int. J. Biomater., 2013, 2013, 1–9 CrossRef PubMed.
- X. Xu, C. R. Sabanayagam, D. A. Harrington, M. C. Farach-Carson and X. A. Jia, Biomaterials, 2014, 35, 3319–3330 CrossRef CAS PubMed.
- B. Keith and M. C. Simon, Cell, 2007, 129, 465–472 CrossRef CAS PubMed.
- R. Edmondson, J. Broglie, A. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- K. M. Yamada and E. Cukierman, Cell, 2007, 130, 601–610 CrossRef CAS PubMed.
- D. Antoni, H. Burckel, E. Josset and G. Noel, Int. J. Mol. Sci., 2015, 16, 5517–5527 CrossRef CAS PubMed.
- V. van Duinen, S. J. Trietsch, J. Joore, P. Vulto and T. Hankemeier, Curr. Opin. Biotechnol., 2015, 35, 118–126 CrossRef CAS PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- C. S. Shin, B. Kwak, B. Han and K. Park, Mol. Pharmaceutics., 2013, 10, 2167–2175 CrossRef CAS PubMed.
- J. P. Celli, I. Rizvi, A. R. Blanden, I. Massodi, M. D. Glidden and B. W. Pogue, Sci. Rep., 2014, 4, 3751 CrossRef PubMed.
- J. Zhao, H. Lu, B. Sandy Wong, M. Lu, P. Xiaob and M. H. Stenze, Polym. Chem., 2017, 8, 3317–3326 RSC.
- P. Acedo, J. C. Stockert, M. Canete and A. Villanueva, Cell Death Dis., 2014, 5, 1122 CrossRef PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- T. A. Slastnikova, A. A. Rosenkranz, T. N. Lupanova, P. V. Gulak, N. V. Gnuchev and A. S. Sobolev, Dokl. Biochem. Biophys., 2012, 446, 235–237 CrossRef CAS PubMed.
- D. Kessel, Photochem. Photobiol. Sci., 2015, 14, 1397–1402 CAS.
- D. Kessel and N. L. Oleinick, Methods Mol. Biol., 2010, 635, 35–46 CAS.
- S. Marchal, A. Fadloun, E. Maugain, M. D'Hallewin, F. Guillemin and L. Bezdetnaya, Biochem. Pharmacol., 2005, 69, 1167–1176 CrossRef CAS PubMed.
- D. Kessel, Autophagy, 2015, 11, 1941–1943 CrossRef CAS PubMed.
- I. O. Bacellar, T. M. Tsubone, C. Pavani and M. S. Baptista, Int. J. Mol. Sci., 2015, 16, 20523–20559 CrossRef CAS PubMed.
- A. Juarranz, P. Jaen, F. Sanz-Rodriguez, J. Cuevas and S. Gonzalez, Clin. Transl. Oncol., 2008, 10, 148–154 CrossRef CAS PubMed.
- J. Hempstead, D. P. Jones, A. Ziouche, G. M. Cramer, L. Rizvi and S. Arnason, Sci. Rep., 2015, 5, 10093 CrossRef CAS PubMed.
- T. Kushibiki, T. Tajiri, Y. Tomioka and K. Awazu, Int. J. Clin. Exp. Med., 2010, 3, 110–114 CAS.
- H. Ding, H. Yu, Y. Dong, R. Tian, G. Huang and D. A. Boothman, J. Controlled Release, 2011, 156, 276–280 CrossRef CAS PubMed.
- R. Yin, T. Agrawal, U. Khan, G. K. Gupta, V. Rai, Y. Y. Huang and M. R. Hamblin, Nanomedicine, 2015, 10, 2379–2404 CrossRef CAS PubMed.
- M. Triesscheijn, P. Baas, J. H. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034–1044 CrossRef CAS PubMed.
- F. Selvestrel, F. Moret, D. Segat, J. H. Woodhams, G. Fracasso, I. M. Rio Echevarria, L. Bau, F. Rastrelli, C. Compagnin, E. Reddi, C. Fedeli, E. Papini, R. Tavano, A. Mackenzie, M. Bovis, E. Yaghini, A. J. MacRobert, S. Zanini, A. Boscaini, M. Colombatti and F. Mancin, Nanoscale, 2013, 5, 6106–6116 RSC.
- R. M. Valentine, K. Wood, C. T. Brown, S. H. Ibbotson and H. Moseley, Phys. Med. Biol., 2012, 57, 6327–6345 CrossRef CAS PubMed.
- S. Mallidi, Z. Mai, I. Rizvi, J. Hempstead, S. Arnason, J. Celli and T. Hasan, J. Biomed. Opt., 2015, 20, 048003 CrossRef PubMed.
- S. Parker, Br. Dent. J., 2013, 215, 167–171 CrossRef CAS PubMed.
- R. Toftegaard, J. Arnbjerg, K. Daasbjerg, P. R. Ogilby, A. Dmitriev, D. S. Sutherland and L. Poulsen, Angew. Chem., Int. Ed., 2008, 47, 6025–6027 CrossRef CAS PubMed.
- Y. Y. Huang, S. K. Sharma, R. Yin, T. Agrawal, L. Y. Chiang and M. R. Hamblin, J. Biomed. Nanotechnol., 2014, 10, 1918–1936 CrossRef CAS PubMed.
- A. Oseroff, L. Blumenson, B. Wilson, T. Mang, D. Bellnier, J. Parsons, N. Frawley, M. Cooper, N. Zeitouni and T. A. Dougherty, Lasers Surg. Med., 2006, 38, 417–426 CrossRef PubMed.
- J. Usuda, H. Kato, T. Okunaka, K. Furukawa, H. Tsutsui and K. Yamada, J. Thorac. Oncol., 2006, 1, 489–493 CrossRef PubMed.
- M. Tanaka, H. Kataoka, M. Mabuchi, S. Sakuma, S. Takahashi and R. Tujii, Anticancer Res., 2011, 31, 763–769 CAS.
- M. J. Bovis, J. H. Woodhams, M. Loizidou, D. Schegglmann, S. G. Bown and A. J. MacRobert, J. Controlled Release, 2012, 157, 196–205 CrossRef CAS PubMed.
- B. J. Qumseya, W. David and H. C. Wolfsen, Clin. Endosc., 2013, 46, 30–37 CrossRef PubMed.
- C. Hopper, A. Kubler, H. Lewis, I. B. Tan and G. Putnam, Int. J. Cancer., 2004, 111, 138–146 CrossRef CAS PubMed.
- D. Meier, C. Campanile, S. M. Botter, W. Born and B. Fuchs, J. Visualized Exp., 2014, 18 DOI:10.3791/51213.
- W. Jerjes, Z. Hamdoon and C. Hopper, Head Neck Oncol., 2012, 4, 16 CrossRef PubMed.
- L. B. Josefsen and R. W. Boyle, Br. J. Pharmacol., 2008, 154, 1–3 CrossRef CAS PubMed.
- Z. Huang, Technol. Cancer Res. Treat., 2005, 4, 283–293 CrossRef CAS PubMed.
- I. Yoon, J. Z. Li and Y. K. Shim, Clin. Endosc., 2013, 46, 7–23 CrossRef PubMed.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS PubMed.
- A. K. Bhatta, U. Keyal and X. L. Wang, Photodiagn. Photodyn. Ther., 2016, 15, 228–235 CrossRef CAS PubMed.
- C. Poriel, D. Kessel and M. G. Vicente, Photochem. Photobiol., 2005, 81, 149–153 CrossRef CAS PubMed.
- R. R. Allison, G. H. Downie, R. Cuenca, X. H. Hu, C. J. Childs and C. H. Sibata, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS PubMed.
- G. Obaid, M. Broekgaarden, A. L. Bulin, H. C. Huang, J. Kuriakose, J. Liu and T. Hasan, Nanoscale, 2016, 8, 12471–12503 RSC.
- A. Puri, Pharmaceutics, 2013, 6, 1–25 CrossRef PubMed.
- L. C. Kimlin, G. Casagrande and V. M. Virador, Mol. Carcinog., 2013, 52, 167–182 CrossRef PubMed.
- L. B. Weiswald, D. Bellet and V. Dangles-Marie, Neoplasia, 2015, 17, 1–15 CrossRef PubMed.
- M. L. Kutys, A. D. Doyle and K. M. Yamada, Exp. Cell Res., 2013, 319, 2434–2439 CrossRef CAS PubMed.
- C. Evans, Front. Phys., 2015, 3 DOI:10.3389/fphy.2015.00015.
- K. Charoen, B. Fallica, Y. Colson, M. Zaman and M. Grinstaff, Biomaterials, 2014, 35, 2264–2271 CrossRef CAS PubMed.
- U. Till, L. Gibot, P. Vicendo, M. Rols, M. Gaucher, F. Violleau and A. Mingotaud, RSC Adv., 2016, 6, 69984–69998 RSC.
- D. Hinger, F. Navarro, A. Käch, J. Thomann, F. Mittler, A. Couffin and C. Maake, J. Nanobiotechnol., 2016, 14 DOI:10.1186/s12951-016-0221-x.
- G. Mehta, A. Hsiao, M. Ingram, G. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- D. Herrmann, J. R. Conway, C. Vennin, A. Magenau, W. E. Hughes and J. P. Morton, Carcinogenesis, 2014, 35, 1671–1679 CrossRef CAS PubMed.
- L. E. Fitzpatrick and T. C. McDevitt, Biomater. Sci., 2015, 3, 12–24 RSC.
- S. Geraldo, A. Simon, N. Elkhatib, D. Louvard, L. Fetler and D. Vignjevic, Eur. J. Cell Biol., 2012, 91, 63–81 CrossRef PubMed.
- L. Ying and Q. Wang, BMC Biotechnol., 2013, 13, 76 CrossRef PubMed.
- X. J. Li, A. V. Valadez, P. Zuo and Z. Nie, Bioanalysis, 2012, 4, 1509–1525 CrossRef CAS PubMed.
- S. Halldorsson, E. Lucumi, R. Gomez-Sjoberg and R. M. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- N. Gupta, J. Liu, B. Patel, D. Solomon, B. Vaidya and V. Gupta, Bioeng. Transl. Med., 2016, 1, 63–81 Search PubMed.
- E. K. Lumley, C. E. Dyer, N. Pamme and R. W. Boyle, Org. Lett., 2012, 14, 5724–5727 CrossRef CAS PubMed.
- A. Nyga, U. Cheema and M. Loizidou, J. Cell Commun. Signal., 2011, 5, 239–248 CrossRef PubMed.
- R. Ng, R. Zang, K. Yang, N. Liu and S. Yang, RSC Adv., 2012, 2, 10110 RSC.
- V. López-Dávila, H. Welch, M. V. Dwek, I. Uchegbu and M. Loizidou, Nanomedicine, 2016, 11, 331–344 CrossRef PubMed.
- A. Asti and L. Gioglio, Int. J. Artif. Organs, 2014, 37, 187–205 Search PubMed.
- H. Geckil, F. Xu, X. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- U. Cheema, S. Y. Yang, V. Mudera, G. G. Goldspink and R. A. Brown, Cell Motil. Cytoskeleton, 2003, 54, 226–236 CrossRef CAS PubMed.
- R. Brown, M. Wiseman, C. Chuo, U. Cheema and S. Nazhat, Adv. Funct. Mater., 2005, 15, 1762–1770 CrossRef CAS.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg and A. Gefen, Cancer Cell, 2005, 8, 241–254 CrossRef CAS PubMed.
- K. Ricketts, U. Cheema, A. Nyga, A. Castoldi, C. Guazzoni, T. Magdeldin, M. Emberton, A. Gibson, G. Royle and M. Loizidou, Small, 2014, 10, 3954–3961 CrossRef CAS PubMed.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. Kumar, Int. J. Polym. Sci., 2011, 2011, 1–19 CrossRef.
- R. Perez-Castillejos, Mater. Today, 2010, 13, 32–41 CrossRef CAS.
- Y. C. Chen, X. Lou, Z. Zhang, P. Ingram and E. Yoon, Sci. Rep., 2015, 5, 12175 CrossRef PubMed.
- I. Rizvi, J. P. Celli, C. L. Evans, A. O. Abu-Yousif, A. Muzikansky, B. W. Pogue, D. Finkelstein and T. Hasan, Cancer Res., 2010, 70, 9319–9328 CrossRef CAS PubMed.
- C. L. Evans, A. O. Abu-Yousif, Y. J. Park, O. J. Klein, J. P. Celli, I. Rizvi, X. Zheng and T. Hasan, PLoS One, 2011, 6, 23434 Search PubMed.
- C. J. Rowlands, J. Wu, S. G. Uzel, O. Klein, C. L. Evans and P. T. So, Laser Phys. Lett., 2014, 11 DOI:10.1088/612-2011/11/11/115605.
- I. Rizvi, S. Anbil, N. Alagic, J. Celli, L. Z. Zheng, A. Palanisami, M. D. Glidden, B. W. Pogue and T. Hasan, Photochem. Photobiol., 2013, 89, 942–952 CrossRef CAS PubMed.
- N. Aggarwal, A. M. Santiago, D. Kessel and B. F. Sloane, Breast Cancer Res. Treat., 2015, 154, 251–262 CrossRef CAS PubMed.
- A. Martinez de Pinillos Bayona, J. Woodhams, H. Pyea, R. A. Hamoudi, C. M. Moore and A. J. MacRobert, Cancer Lett., 2017, 393, 68–75 CrossRef CAS PubMed.
- C. O'Rourke, C. Hopper, A. J. MacRobert, J. B. Phillips and J. H. Woodhams, Int. J. Pharm., 2017, 528, 133–143 CrossRef PubMed.
- K. E. Wright, E. Liniker, M. Loizidou, C. Moore, A. J. Macrobert and J. B. Phillips, Br. J. Cancer, 2009, 101, 658–665 CrossRef CAS PubMed.
- S. Anbil, I. Rizvi, J. P. Celli, N. Alagic, B. W. Pogue and T. Hasan, J. Biomed. Opt., 2013, 18, 098004 CrossRef PubMed.
- A. Huygens, D. Huyghe, G. Bormans, A. Verbruggen, A. R. Kamuhabwa, T. Roskams and P. A. de Witte, Photochem. Photobiol., 2003, 78, 607–614 CrossRef CAS PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS PubMed.
- K. Qiu, J. Wang, C. Song, L. Wang, H. Zhu, H. Huang, J. Huang, H. Wang, L. Ji and H. Chao, ACS Appl Mater Interfaces, 2017, 9, 18482–18492 CAS.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- B. Q. Spring, R. B. Sears, L. Z. Zheng, Z. Mai, R. Watanabe, M. E. Sherwood, D. A. Schoenfeld, B. W. Pogue, S. P. Pereira, E. Villa and T. Hasan, Nat. Nanotechnol., 2016, 11, 378–387 CrossRef CAS PubMed.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS PubMed.
- T. T. Wu and S. H. Zhou, Int. J. Med. Sci., 2015, 12, 187–200 CrossRef PubMed.
- Y. X. Zhu, H. R. Jia, Z. Chen and F. G. Wu, Nanoscale, 2017, 9, 12874–12884 RSC.
- S. Bamrungsap, Z. Zhao, T. Chen, L. Wang, C. Li, T. Fu and W. Tan, Nanomedicine, 2012, 7, 1253–1271 CrossRef CAS PubMed.
- W. Zhang, Y. Li, J. Niu and Y. Chen, Langmuir, 2013, 29, 4647–4651 CrossRef CAS PubMed.
- E. Yaghini, A. M. Seifalian and A. J. MacRobert, Nanomedicine, 2009, 4, 353–363 CrossRef CAS PubMed.
- A. S. Stasheuski, V. A. Galievsky, A. P. Stupak, B. M. Dzhagarov, M. J. Choi, B. H. Chung and J. Y. Jeong, Photochem. Photobiol., 2014, 90, 997–1003 CAS.
- A. Kamkaew, F. Chen, Y. Zhan, R. Majewski and W. Cai, ACS Nano, 2016, 10, 3918–3935 CrossRef CAS PubMed.
- E. Yaghini, F. Giuntini, I. M. Eggleston, K. Suhling, A. M. Seifalian and A. J. MacRobert, Small, 2014, 10, 782–792 CrossRef CAS PubMed.
- R. Bazak, M. Houri, S. E. Achy, W. Hussein and T. Refaat, Mol. Clin. Oncol., 2014, 2, 904–908 CrossRef PubMed.
- M. K. Gnanasammandhan, N. M. Idris, A. Bansal, K. Huang and Y. Zhang, Nat. Protoc., 2016, 11, 688–713 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- A. Mahapatro and D. K. Singh, J. Nanobiotechnol., 2011, 9, 55 CrossRef CAS PubMed.
- T. Magdeldin, V. Lopez-Davila, J. Pape, G. W. Cameron, M. Emberton, M. Loizidou and U. Cheema, Sci. Rep., 2017, 7, 44045 CrossRef PubMed.
- Y. Yang, X. Yang, J. Zou, C. Jia, Y. Hu, H. Du and H. Wang, Lab Chip, 2015, 15, 735–744 RSC.
- H. Hung, O. Klein, S. Peterson, S. Rokosh, S. Osseiran, N. Nowell and C. Evans, Sci. Rep., 2016, 6, 33234 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces., 2015, 7, 23278–23290 CAS.
- J. Liu, K. Liu, L. Feng, Z. Liu and L. Xu, Biomater. Sci., 2017, 5, 331–340 RSC.
- E. Gaio, D. Scheglmann, E. Reddi and F. Moret, J. Photochem. Photobiol., B, 2016, 161, 244–252 CrossRef CAS PubMed.
- J. Lee, J. Kim, M. Jeong, H. Lee, U. Goh, H. Kim, B. Kim and J. H. Park, Nano Lett., 2015, 15, 2938–2944 CrossRef CAS PubMed.
- Z. Xiao, C. B. Hansen, T. M. Allen, G. G. Miller and R. B. Moore, J. Pharm. Pharm. Sci., 2005, 8, 536–543 CAS.
- Y. Wang, Y. Xie, J. Li, Z. H. Peng, Y. Sheinin, J. Zhou and D. Oupicky, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS PubMed.
- M. Benito, V. Martin, M. D. Blanco, J. M. Teijon and C. Gomez, J. Pharm. Sci., 2013, 102, 2760–2769 CrossRef CAS PubMed.
- J. Zhang, Y. C. Liang, X. Lin, X. Zhu, L. Yan, S. Li, X. Yang, G. Zhu, A. L. Rogach, P. K. N. Yu, P. Shi, L. C. Tu, C. C. Chang, X. Zhang, X. Chen, W. Zhang and C. S. Lee, ACS Nano, 2015, 9, 9741–9756 CrossRef CAS PubMed.
- A. A. Sultan, W. Jerjes, K. Berg, A. Hogset, C. A. Mosse, R. Hamoudi, Z. Hamdoon, Z. Simeon, D. Camell, M. Forster and C. Hopper, Lancet Oncol., 2016, 17, 1217–1229 CrossRef CAS PubMed.
- A. Martinez de Pinillos Bayona, C. M. Moore, M. Loizidou, A. J. MacRobert and J. H. Woodhams, Int. J. Cancer, 2016, 138, 1049–1057 CrossRef CAS PubMed.
- M. K. Herroon, R. Sharma, E. Rajagurubandara, C. Turro, J. J. Kodanko and I. Podgorski, Biol. Chem., 2016, 397, 571–582 CrossRef CAS PubMed.
- Y. Shen, A. J. Shuhendler, D. Ye, J. J. Xu and H. Y. Chen, Chem. Soc. Rev., 2016, 45, 6725–6741 RSC.
| This journal is © The Royal Society of Chemistry 2018 |