
Synthesis and self-assembly of unconventional C3-symmetrical trisubstituted triphenylenes†
Yang
Li
a,
Yi-Xuan
Wang
 a,
Xiang-Kui
Ren
b and
Long
Chen
*a
a,
Xiang-Kui
Ren
b and
Long
Chen
*a
aTianjin Key Laboratory of Molecular Optoelectronic Science, School of Science, Tianjin University, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China. E-mail: long.chen@tju.edu.cn
bSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, P. R. China
First published on 19th September 2017
Abstract
Triphenylene-based self-assembly modules, bearing three amide groups at the 1-, 5-, and 9-positions, were designed and synthesized. Supramolecular gels could be constructed by combining hydrogen bonding, π–π stacking and van der Waals interactions. In addition, the assembly exhibited liquid crystalline mesophases by tuning of the length of the alkyl chains. This work provides an essential representative of functional assembly based on C3-symmetric triphenylenes.
 Long Chen | Long Chen completed his bachelor's degree at Shanghai University (2003) and Master's degree at Shanghai Jiao Tong University with Prof. Wanbin Zhang (2006). Afterwards, he received his PhD in 2009 under the supervision of Prof. Donglin Jiang at the Institute for Molecular Science (IMS, Japan). He then joined Prof. Klaus Müllen's group at Max Planck Institute for Polymer Research (MPIP, Germany) as an Alexander von Humboldt research fellow (2010–2012) and was later appointed as a project leader in the same group (2012–2014). Currently he is a faculty member at Tianjin University. His research focuses on the synthesis and self-organization of functional π-conjugated molecules, conjugated porous polymers for catalysis and energy conversion. |
Introduction
Supramolecular self-assembly based on a rigid π-conjugated core is an efficient approach to fascinating soft materials with well-ordered superstructures.1,2 The molecular building blocks can self-assemble into nano- or micro-scale architectures3,4via multiple non-covalent interactions (such as hydrogen bonding,5,6 metal coordination,7 van der Waals force,8 and π–π stacking9), and subsequently evolve into macroscopic materials.10 The presence of noncovalent interactions endows supramolecular materials with inherent responsiveness to external stimuli, and makes them suitable for practical applications.11 Recently, disk-shaped conjugated molecules have attracted increasing attention due to their excellent assembly abilities, and potential utilities for optical, magnetic, and electrochemical materials.12–15Triphenylene, one of the classical disk-shaped chromophores, has been versatilely functionalized and employed to construct supramolecular materials, in which the π–π stacking of triphenylene moieties restricts molecular movements and yields assemblies with good stability. Other noncovalent interactions, especially hydrogen bonds, have also been incorporated to promote such regulation due to their good directionality. Therefore, the stacking of a triphenylene core becomes more compact. For instance, E. W. Meijer and co-workers developed a series of symmetrical molecules that associate into supramolecular stacks via π–π interactions and hydrogen-bonds.12 S. Shinkai and co-workers developed a symmetrical organogelator which has six long alkyl chains as tail groups and six amide groups as hydrogen-bonding sites.16 The resulting excimer emission clearly proved the eclipsed stacking between triphenylene moieties within the supramolecular structures. S. V. Bhosale and co-workers successfully prepared long flexible nanoscopic fibrous assemblies based on naphthalene diimides, through the combination of π–π interactions with hydrogen-bonds.17 It is well known that the molecular configuration greatly affects the structure and functions of the self-assembled materials.18,19 To date, most of the symmetrical triphenylenes with hydrogen-bonding sites were introduced via linkage with amide groups at the 2-, 3-, 6-, 7-, 10- and 11-positions (Chart 1). This is because substitution in triphenylene is directed by a steric effect, leading to higher reaction activity at the 2- and 3-positions than that at the 1-position.20 To balance the crystallization and dissolution, flexible spacers (e.g., ether chains) have to be introduced between the conjugated cores and amide groups.
 | ||
| Chart 1 Chemical structures of triphenylene derivatives discussed in this work. | ||
Herein, symmetrical trisubstituted triphenylene derivatives were synthesized and demonstrated as efficient organogelator and liquid crystalline mesogens. The number of amide groups was reduced to prevent excessive aggregation, and therefore, the extra spacer was not necessary. Our design not only simplified the synthetic procedure, but also optimized the molecular configuration of triphenylene-based modules for supramolecular self-assembly. To the best of our knowledge, the assembly properties of such 1,5,9-trisubstituted triphenylenes have been rarely explored (Scheme 1).20
 | ||
| Scheme 1 Synthetic routes for triphenylene derivatives 1, 2, and 3. | ||
Experimental
Materials
All starting chemical materials were directly used from commercial suppliers without further purification. Solvents were purified according to the standard procedures. The intermediates were synthesized according to the reported literature (see the ESI†), including triphenylene-1,5,9-triamine (S-3), 4-(dodecyloxy)benzoic acid (S-5), 4-(dodecyloxy)benzoyl chloride (S-6), N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)decanamide (S-7) and 1,5,9-tribromotriphenylene (S-8).Characterization
1H NMR and 13C NMR spectra were recorded in deuterated solvents on a Bruker ADVANCE 400 operating at 400 MHz (1H NMR) and 100 MHz (13C NMR). Chemical shifts are indicated in ppm in relation to the particular internal standard. MALDI-TOF mass spectra were recorded on a Bruker New Autoflex Speed LIN Spectrometer using a 337 nm nitrogen laser with dithranol as a matrix. High resolution mass spectra (HRMS) were recorded from a Perkin-Elmer TURBOMASS instrument. The UV-Vis absorption spectra were obtained on a PerkinElmer Lambda 750 spectrophotometer. FT-IR spectra were collected in transmission mode on a Bruker Alpha spectrometer with a scan range of 400–4000 cm−1. The morphologies of the gels were investigated using field emission scanning electron microscopy (FE-SEM) which was performed on a Hitachi Limited model S-4800 microscope operating at an accelerating voltage of 3.0 kV. The samples were prepared on a silicon wafer. The silicon wafer was then coated with a thin layer by soaking in the gels. As for compounds 2 and 3, the dry samples were suspended in acetonitrile, and heated until all solids were completely dissolved to form a clear solution, then dropped onto a silicon wafer and cooled gradually to room temperature. All samples were then coated with a 2 nm layer of gold using a KYKY SBC-12 automated sputter coater. Transmission electron microscopy (TEM) imaging was carried out using a Tecnai G2F20S-TWIN at 200 kV. The gels for TEM examination were coated with a thin layer by soaking in the gels on a copper specimen grid supported by a holey carbon film. Atomic force microscope (AFM) images of the gels were recorded in tapping mode by a Bruker MultiMode 8 SPM. The substrate of the samples was silicon wafer. The samples were then coated with a thin layer by soaking in the gels. Powder X-ray diffraction (XRD) measurement was carried out on X-ray diffractometer model FLlex 600 diffractometer (Japan) at 40 kV, 40 mA with a Cu-target tube and a graphite monochromator. A high-flux small-angle X-ray scattering instrument (SAXSess, Anton Paar) equipped with a Kratky block-collimation system and a Philips PW3830 sealed-tube X-ray generator (Cu Kα) was employed to simultaneously measure the small-angle and wide-angle scattering of the dried sample. The variable-temperature X-ray diffraction patterns were recorded on an imaging-plate with a pixel size of 42.3 × 42.3 μm2 which extends to the high-angle range (the q range covered by the imaging plate is from 0.06 to 29 nm−1, q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, where λ is the wavelength of 0.1542 nm and 2θ is the scattering angle). The diffraction peak positions were calibrated with silver behenate. The original experimental data such as data acquisition, background subtraction, and data reduction, were handled by Anton Paar SAXSquant 1.01 software and the PCG software package. A temperature control unit (Anton Paar TCS300) in conjunction with the SAXSess was utilized to study the structure evolution as a function of temperature. The 2D WAXD experiment of the oriented sample of 1b (prepared by mechanical shearing) was recorded on a Bruker D8 Discover diffractometer with a Vantec500 as the 2D detector. The 2D diffraction pattern was recorded in transmission mode with the point-focused X-ray beam aligned perpendicular to the shear direction. The background scattering was recorded and subtracted from the sample pattern. The thermal stabilities were evaluated using thermogravimetric analysis (TGA) using a differential thermal analysis instrument (NETZSCH STA 409PC analyzer) over the temperature range from 25 °C to 800 °C under a nitrogen atmosphere with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were carried out with a TA DSC-Q20 under a nitrogen atmosphere at a heating rate of 10 °C min−1. Polarizing optical microscopy (POM) was performed on a Nikon microscope (ECLIPSE LV100ND) with a temperature controlled stage (Linkam LTS420) and a microscope camera (Nikon DS-Ri2). Theoretical calculations were implemented in the Gaussian 09 program. The B3LYP functions and the 6-31G(d) basis set were used for all the atoms in the DFT calculations.
sinθ/λ, where λ is the wavelength of 0.1542 nm and 2θ is the scattering angle). The diffraction peak positions were calibrated with silver behenate. The original experimental data such as data acquisition, background subtraction, and data reduction, were handled by Anton Paar SAXSquant 1.01 software and the PCG software package. A temperature control unit (Anton Paar TCS300) in conjunction with the SAXSess was utilized to study the structure evolution as a function of temperature. The 2D WAXD experiment of the oriented sample of 1b (prepared by mechanical shearing) was recorded on a Bruker D8 Discover diffractometer with a Vantec500 as the 2D detector. The 2D diffraction pattern was recorded in transmission mode with the point-focused X-ray beam aligned perpendicular to the shear direction. The background scattering was recorded and subtracted from the sample pattern. The thermal stabilities were evaluated using thermogravimetric analysis (TGA) using a differential thermal analysis instrument (NETZSCH STA 409PC analyzer) over the temperature range from 25 °C to 800 °C under a nitrogen atmosphere with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were carried out with a TA DSC-Q20 under a nitrogen atmosphere at a heating rate of 10 °C min−1. Polarizing optical microscopy (POM) was performed on a Nikon microscope (ECLIPSE LV100ND) with a temperature controlled stage (Linkam LTS420) and a microscope camera (Nikon DS-Ri2). Theoretical calculations were implemented in the Gaussian 09 program. The B3LYP functions and the 6-31G(d) basis set were used for all the atoms in the DFT calculations.
Synthesis
The triphenylene derivatives 1a, 1b, and 1c, as shown in Chart 1, were composed of a rigid triphenylene core, three secondary amide groups, and three flexible alkyl chains, which could provide strong π–π stacking interactions, hydrogen bonds, and van der Waals interactions, respectively. Meanwhile, the flexible alkyl chains could assist in achieving a balance between crystallization and dissolution, which is indispensable for the formation of ordered assembly structures. The synthetic routes to the targeted triphenylenes 1a–c, and 2 are quite straightforward by employing an amidation reaction as the key step (Scheme 1).21To shed more light on the effect of the aryl amide group on assembly behaviour, compound 3, with an extended aromatic core and the amide linkage located at the periphery,22,23 was synthesized from 1,5,9-tribromotriphenylene via Suzuki cross-coupling with the corresponding borate ester (compound S-5, Scheme 1). All the target molecules were unambiguously characterized by 1H and 13C NMR spectroscopy, and mass spectroscopy.
Results and discussion
To prepare a physical gel, a mixture of the powdered compound and an appropriate organic solvent was firstly heated to afford a homogeneous solution, which was then allowed to cool at room temperature to form gels. The resulting gels could stand for more than one week (Fig. S1, see the ESI†), and the sol–gel transition could be repeated many times. As shown in Fig. 1, compounds 1b and 1c showed excellent gelation abilities in many apolar solvents, including toluene, xylene, chlorobenzene, dichlorobenzene and tetrachloromethane. The critical gelator concentrations (CGCs) of these two gelators in chlorobenzene were tested by using the classical method (see the ESI†).24 The values were determined as 8.5 mM for 1b, and 7.5 mM for 1c, indicating that each triphenylene molecule could entrap more than 1100 solvent molecules. However, compound 1a bearing shorter alkyl chains only forms a partial gel (Fig. S2, see the ESI†), probably due to its poor solubility. In order to explore the relationship between the π-conjugated molecular structure and gelation property, compounds 2 and 3 (attached with additional phenyl rings in different ways) were synthesized for comparison, and their self-assembly behavior was also investigated in detail. As shown in Fig. S3 (see the ESI†), neither of these compounds form supramolecular gels under identical conditions, implying that the peripheral aromatic rings may influence the stacking behavior during the self-assembly.20,24 | ||
| Fig. 1 Photographs of gels and partial gels taken under natural light, (a) 1b and (b) 1c (15 mM), in various solvents after cooling at room temperature (from left to right: toluene, xylene, chlorobenzene, dichlorobenzene, tetrachloromethane, dichloromethane, n-hexane and cyclohexane). | ||
Systematic characterization of the triphenylene derivatives by UV-Vis, photoluminescence (PL), FT-IR and 1H NMR spectroscopies was conducted to determine the driving forces for the constitution of the organogels.25,26 Compared with the film, the dilute THF solution of 1b or 1c exhibited red-shifted UV-Vis absorption peaks (ca. 269 nm for the film and 279 nm for solution, Fig. S4, see the ESI†), indicating that the π–π stacking of the conjugated cores play a significant role in the process of self-assembly.27 Such a red-shift (ca. 10 nm) of the π–π* transition band from the film-state to the solution-state was also observed for 1a (Fig. S4a, ESI†). In contrast, for compounds 2 and 3, the shifts were ambiguous, which confirms the weaker π–π interactions in compounds 2 and 3. This might result from the steric repulsion of the rotatable phenyl ring. The temperature-dependent UV-Vis spectra of the 1c gelator were further recorded in situ to monitor the driving forces for the assembly. The absorption of 1c clearly showed a blue-shift from 282 nm to 271 nm on decreasing the temperature and a great broadening was observed (Fig. 2). However, there were no obvious changes for compounds 2 and 3 in comparable conditions, again revealing the weaker π–π stacking interactions (Fig. S5, see the ESI†). As shown in the temperature-dependent photoluminescence (PL) spectra, the maximum emission of 1c was blue-shifted from 404 nm to 390 nm as the temperature decreased (Fig. S6, see the ESI†). These blue shifts in both the absorption and emission bands jointly demonstrated the formation of H-type aggregates during the self-assembly of compounds 1.
 | ||
| Fig. 2 UV-Vis spectra of 1c in CCl4 (10−5 M) at different temperatures. | ||
FT-IR spectra were measured to get more detailed information on the intermolecular interactions in self-assembled structures. The FT-IR spectra of all gels of 1a–c showed N–H stretching bands at 3254 cm−1, amide I bands at 1644 cm−1, and amide II bands at 1519 cm−1 (Fig. 3). Compared with those of free amide groups (N–H stretching bands at 3420 cm−1 and amide I bands at 1683 cm−1) measured in dilute solution (Fig. S7, see the ESI†), the relatively low wavenumbers in the aggregation state clearly proved the presence of strong intermolecular hydrogen bonds between amide groups.27,28 In addition, the characteristic absorption bands of saturated C–H stretching vibrations appeared at lower frequencies (νanti: 2922 cm−1; νsym: 2855 cm−1).24,29 This could be ascribed to the close packing of alkyl chains,29,30 indicative of the existence of van der Waals interactions in the gel-state. The FT-IR spectra of compound 2 and 3 aggregates are quite similar to those of compounds 1a–c (Fig. S8, see the ESI†), reflecting that both hydrogen bonds and van der Waals forces played important roles. On the basis of these results, we deduced that the π–π stacking of rigid cores predominantly drives the formation of supramolecular gels, while hydrogen bonding and van der Waals interactions also facilitate the self-assembly process.
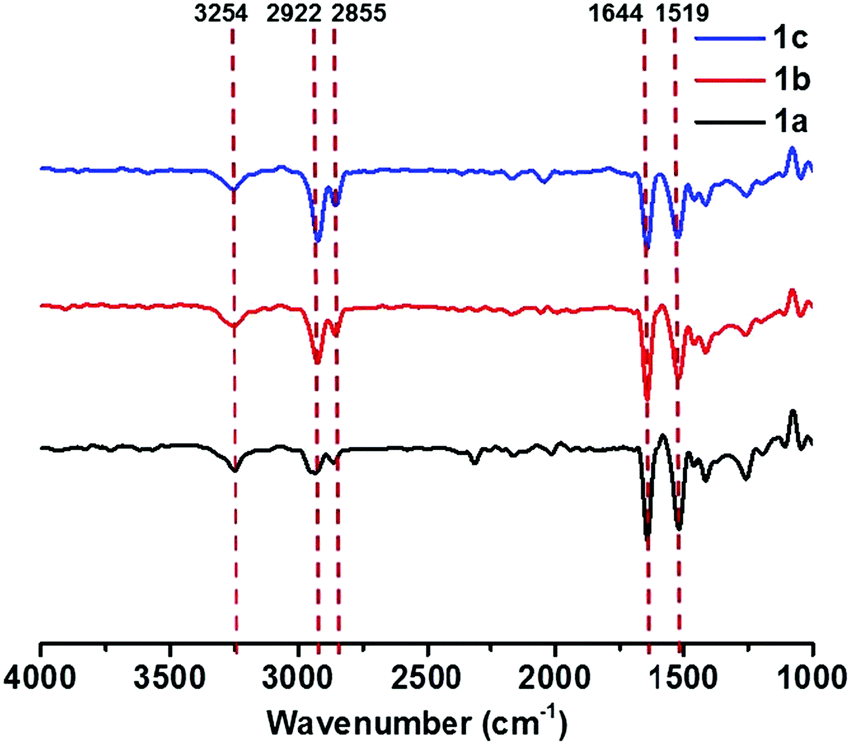 | ||
| Fig. 3 FT-IR spectra of 1a partial gel, 1b gel and 1c gel (gelation in chlorobenzene). | ||
The concentration-dependent 1H NMR spectra were then acquired to prove the above inferences, and are shown in Fig. 4. An up-field shift of the N–H signal was detected from 7.65 ppm to 7.57 ppm, upon decreasing the concentration of 1c from 10 mM to 1 mM, proving the formation of the intermolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds at high concentrations.31 Besides, the signals of triphenylene protons showed slight down-field shifts simultaneously, which could be ascribed to the π–π stacking between the triphenylene cores.1 Temperature-dependent 1H NMR measurements were also measured and are shown in Fig. S9 (see the ESI†). The resonance signals in toluene-d8 corresponding to the aromatic protons at 8.28, 8.08, and 7.23 ppm could be observed at 80 °C. Upon cooling, the signals became significantly broad and weak, owing to the efficient π–π stacking upon aggregation. Accordingly, we can conclude that supramolecular self-assembly of the gelators relies mainly on π–π stacking interactions and intermolecular hydrogen bonds, together with van der Waals forces.
C hydrogen bonds at high concentrations.31 Besides, the signals of triphenylene protons showed slight down-field shifts simultaneously, which could be ascribed to the π–π stacking between the triphenylene cores.1 Temperature-dependent 1H NMR measurements were also measured and are shown in Fig. S9 (see the ESI†). The resonance signals in toluene-d8 corresponding to the aromatic protons at 8.28, 8.08, and 7.23 ppm could be observed at 80 °C. Upon cooling, the signals became significantly broad and weak, owing to the efficient π–π stacking upon aggregation. Accordingly, we can conclude that supramolecular self-assembly of the gelators relies mainly on π–π stacking interactions and intermolecular hydrogen bonds, together with van der Waals forces.
 | ||
| Fig. 4 Concentration-dependent 1H NMR spectra of the aromatic protons of 1c in CDCl3. | ||
In order to investigate the morphology of the supramolecular assemblies, the partial gel of 1a, and the gels of 1b and 1c were subjected to field emission scanning electron microscopy (FE-SEM) analysis. Interwoven network structures were observed for both 1b and 1c, composed of microscale fibers with high aspect ratios (Fig. 5a and d). In contrast, microfibers in the partial gel of 1a were much shorter and straighter (Fig. S10, see the ESI†). It could be rationally explained that the longer alkyl chains of gelators, especially for compound 1c, greatly improved the flexibility in bulk, thus facilitating the formation of interconnected networks, i.e., supramolecular gelation. On the other hand, nanospheres were observed for the aggregates of compounds 2 and 3 (Fig. S13, see the ESI†), suggesting that the additional phenyl groups induced the molecules to tend to be agglomerated instead of undergoing anisotropic stacking to generate nanofibers, which are typically observed in supramolecular gels.
 | ||
| Fig. 5 (a and d) SEM, (b and e) AFM, and (c and f) TEM images of (a–c) 1b gel, and (d–f) 1c gel, respectively. | ||
A more detailed investigation on the morphology of the formed gels was performed by atomic force microscopy (AFM) and transmission electron microscopy (TEM). Fibrous networks (Fig. 5b, c, e and f) were observed for gels of both 1b and 1c, which should be the aggregation of the microfibers. A more rigid fibrous morphology was observed again by AFM for the 1a partial gel (Fig. S14, see the ESI†). The intermolecular packing modes of the two gelators were investigated by X-ray diffraction (XRD) measurements.32 As shown in Fig. S15 (see the ESI†), the d-spacing of 3.7 Å and 3.9 Å could be assigned to the π–π stacking distances between the triphenylene cores in the 1b and 1c assemblies respectively, and the d-spacing of 4.1 Å was ascribed to the packing of alkyl chains for both compounds. In addition, several sharp peaks in the range of 3–20° for 1c assembly suggested its good crystallinity, while the weak peaks for 1b assembly implied a low degree of ordering. The d-spacing of 34 Å for 1c could be assigned to the horizontal distance perpendicular to the π–π stacking direction, which agrees well with the molecular diameter calculated by DFT calculations (Fig. S16, see the ESI†), implying that the fibrous aggregates consisted of bundles of 1D molecular assemblies. In light of the above results, it could be concluded that supramolecular gelation started from linear self-assembly, which hierarchically aggregated to form microscale fibers and further entangled to entrap a large number of solvent molecules.
Triphenylene derivatives have been well known as promising liquid-crystalline materials and attracted great research interest.33 Thus, the thermodynamic transitions of the trisubstituted triphenylenes were measured by differential scanning calorimetry (DSC) and polarizing optical microscopy (POM). According to the thermal gravimetric analysis (TGA) results, compounds 1a–c were stable below 300 °C (Fig. S17, see the ESI†). DSC data showed that the melting point decreased with the increase of the length of the alkyl chains (Table S1 and Fig. S19a–c, see the ESI†), revealing a great effect of the flexibility of the terminal chains on the assembly phase transitions. On the other hand, in comparison to 1a–c, compounds 2 and 3 were stable up to 250 °C. But no obvious exothermic peaks on the cooling scan in the DSC trace were observed (Fig. S18 and S19d, e, see the ESI†), suggesting that no characteristic phase transitions occurred during the heating and cooling cycles.
As for 1b and 1c, two endothermic peaks were observed before melting at 121 and 110 °C respectively, along with an additional peak only observed for 1c at 199 °C. Moreover, the lower transition temperature and the broader signal of 1c indicated a less cooperative transition. The phase transitions were further studied by polarizing optical microscopy (POM). Birefringent droplets of 1c with branched texture were observed at 255 °C during the cooling process from the isotropic liquid (Fig. 6b, also see Fig. S20 ESI†), signifying the presence of a mesophase.34 However, the POM image of 1b resembled that of a crystallization phase (Fig. 6a). A variable-temperature XRD experiment was used to identify the corresponding structural evolution upon heating (Fig. 7). With the increase of temperature, diffraction peaks at q of ca. 16 nm−1 (4.1 Å, assigned to the packing of alkyl chains35a) became ambiguous for both 1b and 1c, indicative of the appearance of a new phase in which the arrangement of the alkyl chains was disordered. It should be noted that the new peak appeared in the low-q region (2–3 nm−1) for 1c observed at 220 °C (Fig. 7b, blue line), which is between the two endothermic peaks (199 and 260 °C, Fig. S19c, ESI†), featuring the formation of a new ordered mesophase.35b In contrast, 1b did not exhibit such transition (Fig. 7a). This phenomenon was in nice agreement with the DSC result. Therefore, we concluded that only 1c exhibited the nature of liquid crystal, probably owing to the longer alkyl chains which endowed the columnar stacking with better flexibility.36 The liquid crystalline phase of 1c is assigned by the standard PXRD refinement procedure. The column arrangement of 1c supramolecular columns is determined to be a monoclinic liquid crystalline superlattice with the dimensions of a = 3.02 nm, b = 2.44 nm, γ = 94.96°. To further investigate the molecular orientation in the column packing of these C3-symmetric 1,5,9-trisubstituted triphenylenes, we carried out two dimensional wide angle X-ray diffraction (2D WAXD) experiments of 1b (Fig. S15c, ESI†). In principle, it should have three possible packing modes with rotational directionalities from head and tail (Fig. S15d, ESI†). Based on the 2D WAXD results, it is speculated that the directionalities of the C3-symmetric 1,5,9-trisubstituted triphenylene molecules might be aligned randomly in the supramolecular column.
 | ||
| Fig. 6 Polarizing optical micrographs of (a) 1b (recorded at 260 °C), and (b) 1c (recorded at 255 °C), respectively, in the first cooling cycle. | ||
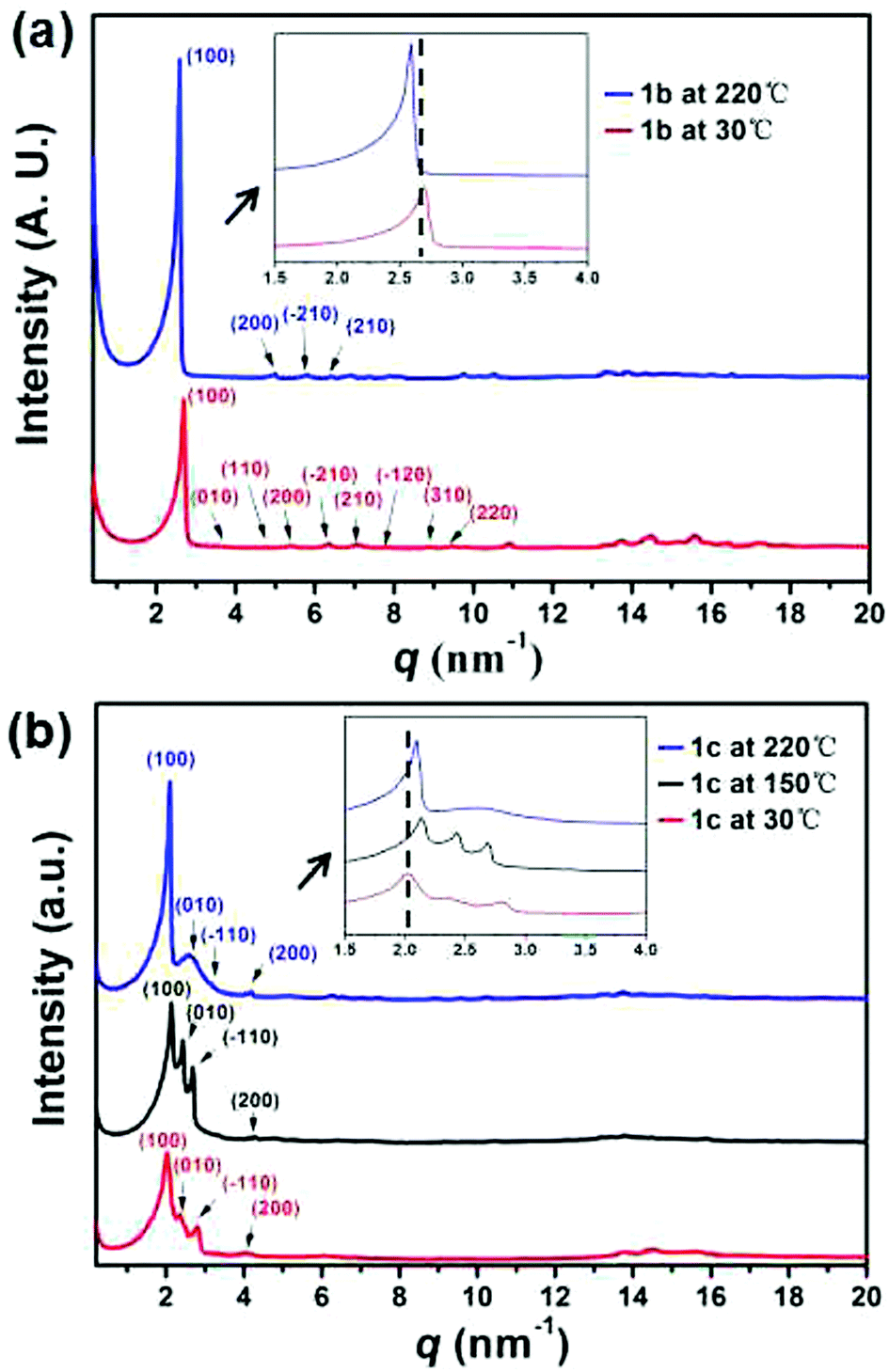 | ||
| Fig. 7 Variable-temperature XRD profiles recorded in the second heating process of (a) 1b and (b) 1c. | ||
Conclusions
In summary, we have designed and synthesized a series of C3-symmetrical trisubstituted triphenylenes, in which three amide groups were directly anchored at the 1-, 5-, and 9-positions. Their self-assembly behaviours and gelation properties were systematically investigated by UV-Vis, PL, FT-IR, 1H NMR, SEM, AFM, TEM, DSC, XRD, and POM measurements. Compared with 1a, triphenylene derivatives 1b and 1c bearing longer alkyl chains could form supramolecular gels in appropriate apolar organic solvents driven by π–π stacking, intermolecular hydrogen bonding, and van der Waals forces. Moreover, 1c with the longest alkyl chains exhibited liquid crystallinity, suggesting the key role of van der Waals force in regulating the phase behaviour. The current research provides important supplements for the structural investigation of triphenylene-based supramolecular self-assembly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51522303 and 21602154) and the National Key R&D Program of China (2017YFA0207500).Notes and references
- L. Chen, X. Dou, W. Pisula, X. Yang, D. Wu, G. Floudas, X. Feng and K. Müllen, Chem. Commun., 2012, 48, 702–704 RSC.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- D. Bardelang, F. Camerel, J. C. Margeson, D. M. Leek, M. Schmutz, M. B. Zaman, K. Yu, D. V. Soldatov, R. Ziessel, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem. Soc., 2008, 130, 3313–3315 CrossRef CAS PubMed.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- Y. Zhou, M. Xu, T. Yi, S. Xiao, Z. Zhou, F. Li and C. Huang, Langmuir, 2007, 23, 202–208 CrossRef CAS PubMed.
- N. Koumura, M. Kudo and N. Tamaoki, Langmuir, 2004, 20, 9897–9900 CrossRef CAS PubMed.
- X. Zhou, Q. Jin, L. Zhang, Z. Shen, L. Jiang and M. Liu, Small, 2016, 12, 4743–4752 CrossRef CAS PubMed.
- P. Yadav and A. Ballabh, New J. Chem., 2015, 39, 721–730 RSC.
- S. Yagai, T. Karatsu and A. Kitamura, Langmuir, 2005, 21, 11048–11052 CrossRef CAS PubMed.
- D. J. Lunn, O. E. C. Gould, G. R. Whittell, D. P. Armstrong, K. P. Mineart, M. A. Winnik, R. J. Spontak, P. G. Pringle and I. Manners, Nat. Commun., 2016, 7, 12371 CrossRef PubMed.
- (a) Y.-J. Choi, D.-Y. Kim, M. Park, W.-J. Yoon, Y. Lee, J.-K. Hwang, Y.-W. Chiang, S.-W. Kuo, C.-H. Hsu and K.-U. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 9490–9498 CrossRef CAS PubMed; (b) N. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2012, 48, 5007–5009 RSC.
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS PubMed.
- T. Sagawa, S. Fukugawa, T. Yamada and H. Ihara, Langmuir, 2002, 18, 7223–7728 CrossRef CAS.
- S. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS PubMed.
- L. Chen, K. S. Mali, S. R. Puniredd, M. Baumgarten, K. Rarvez, W. Pisula, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2013, 135, 13531–13537 CrossRef CAS PubMed.
- M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2003, 1354–1355 RSC.
- N. V. Ghule, D. D. La, R. S. Bhosale, M. A. Kobaisi, A. M. Raynor, S. V. Bhosale and S. V. Bhosale, ChemistryOpen, 2016, 5, 157–163 CrossRef CAS PubMed.
- L. Frkanec, M. Jokic, J. Makarevic, K. Wolsperger and M. Zinic, J. Am. Chem. Soc., 2002, 124, 9716–9717 CrossRef CAS PubMed.
- K. Kobayashi, K. Ishii, S. Sakamoto, T. Shirasaka and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 10615–10624 CrossRef CAS PubMed.
- S. Kumar, Liq. Cryst., 2004, 31, 1037–1059 CrossRef CAS.
- Q. Tan, H. Chen, H. Xia, B. Liu and B. Xu, Chem. Commun., 2016, 52, 537–540 RSC.
- K. R. Reddy, E. Varathan, N. Lobo, S. Easwaramoorthi and T. Narasimhaswamy, J. Phys. Chem. C, 2016, 120, 22257–22269 CAS.
- Y. Chen, F. Zhang, B. Zhu, Y. Han and Z. Bo, Chem. − Asian J., 2011, 6, 226–233 CrossRef CAS PubMed.
- Y. Setoguchi, H. Monobe, W. Wan, N. Terasawa, K. Kiyohara, N. Nakamura and Y. Shimizu, Thin Solid Films, 2003, 438, 407–413 CrossRef.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- T. Shimizu and M. Masuda, J. Am. Chem. Soc., 1997, 119, 2812–2818 CrossRef CAS.
- A. A. Maskevich, A. V. Lavysh, I. M. Kuznetsova, A. I. Sulatskaya and K. K. Turoverov, J. Appl. Spectrosc., 2015, 82, 33–39 CrossRef CAS.
- Y. Chen, Y. Lv, Y. Han, B. Zhu, F. Zhang, Z. Bo and C.-Y. Liu, Langmuir, 2009, 25, 8548–8555 CrossRef CAS PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- Y. Sun, Y.-X. Wang, M. Wu, W. Yuan and Y. Chen, Chem. − Asian J., 2017, 12, 52–59 CrossRef CAS PubMed.
- X.-D. Xu, J. Zhang, L.-J. Chen, X.-L. Zhao, D.-X. Wang and H.-B. Yang, Chem. − Eur. J., 2012, 18, 1659–1667 CrossRef CAS PubMed.
- L. A. Estroff, L. Leiserowitz, L. Addadi, S. Weiner and A. D. Hamilton, Adv. Mater., 2003, 15, 38–42 CrossRef CAS.
- M. Gupta, S. P. Gupta, M. V. Rasna, D. Adhikari, S. Dharab and S. K. Pal, Chem. Commun., 2017, 53, 3014–3017 RSC.
- I. Niezgoda, D. Pociecha and Z. Galewski, Thermochim. Acta, 2014, 587, 59–66 CrossRef CAS.
- (a) M. Li, J.-R. Xu, Y. Zeng, H.-J. Ben, F.-L. Yao, S. Yang, E.-Q. Chen and X.-K. Ren, Dyes Pigm., 2017, 139, 79–86 CrossRef CAS; (b) H. Jing, L. Lu, Y. Feng, J.-F. Zheng, L. Deng, E.-Q. Chen and X.-K. Ren, J. Phys. Chem. C, 2016, 120, 27577–27586 CrossRef CAS.
- Y. F. Bai, K. Q. Zhao, P. Hu, B. Q. Wang and Y. Shimizu, Mol. Cryst. Liq. Cryst., 2009, 509, 60–76 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, synthesis and characterization, and supplementary data. See DOI: 10.1039/c7qm00361g |
| This journal is © the Partner Organisations 2017 |