
Tellurium-promoted stereoselective hydrodebromination of 1,1-dibromoalkenes: synthesis of (E)-bromoalkenes†
Gelson Perin*a,
Angelita M. Barcellosa,
Thiago J. Peglowa,
Patrick C. Nobrea,
Roberta Cargneluttib,
Eder J. Lenardão a,
Francesca Marinic and
Claudio Santic
a,
Francesca Marinic and
Claudio Santic
aLaboratório de Síntese Orgânica Limpa – LASOL, CCQFA, Universidade Federal de Pelotas – UFPel, P. O. Box 354, 96010-900, Pelotas, RS, Brazil. E-mail: gelson_perin@ufpel.edu.br; Fax: +55 5332757533; Tel: +55 5332757533
bLMI, Departamento de Química, Universidade Federal de Santa Maria – UFSM, P. O. Box 97105900, Santa Maria, RS, Brazil
cDepartment of Pharmaceutical Sciences, Group of Catalysis and Organic Green Chemistry, University of Perugia, Via del Liceo 1, 06100, Perugia, Italy
First published on 26th October 2016
Abstract
We describe herein an efficient and simple method for the stereoselective hydrodebromination of 1,1-dibromoalkenes by using a catalytic amount of the nucleophilic species of tellurium, generated in situ by the reaction of elemental tellurium with NaBH4. By this methodology, (E)-bromoalkenes were obtained in moderate to excellent yields under mild reaction conditions, without the use of transition metals or base. Furthermore, a high stereoselectivity for the (E)-isomer was observed when 1,1-dibromoarylalkenes were used, thus indicating a promising alternative for future applications in organic synthesis.
Introduction
The 1,1-dibromoalkenes systems are extremely useful intermediates in organic synthesis, and this attractive functionality has been applied as a precursor of alkynes, heterocycles, carbocycles and other chemicals.1 In general, 1,1-dibromoalkenes are conveniently and easily prepared from aldehydes/CBr4/PPh3 (the Ramirez dibromolefination)2 or aldehydes and ketones/CBr4/P(OiPr)3 (the Lautens modification).3 1,1-Dibromoalkenes can be readily converted to (E)- and (Z)-bromoalkenes, via a hydrodebromination reaction. This synthetic strategy has been described as the key step in the total synthesis of many natural occurring compounds4 and to prepare compounds for application in therapeutics5 and materials sciences.6The number of different approaches reported for the hydrodebromination of 1,1-dibromoalkenes reflects their importance, particularly with respect to controlling the double-bond's geometry of the bromoalkene obtained. More specifically, the classic method to afford the Z isomer uses Bu3SnH (1.1 equiv.)/Pd(PPh3)4 (4.0 mol%),7 while the E one is typically prepared using 4.0 equiv. of diethylphosphite and 2.0 equiv. of trimethylamine at 90 °C for 5 h.8 In this sense, other methodologies were developed to prepare (E)-bromoalkenes, such as: (a) nBu3ZnLi (1.2 equiv.) at −85 °C for 3 h;9 (b) a solution of organolithium (1.0 equiv.) at −105 °C for few minutes;10 (c) Fe(acac)3 (5.0 mol%) and iPrMgCl (1.1 equiv.) at −10 °C for 0.5 h;11 (d) microwave irradiation using a diethyl phosphonate/EtONa/EtOH system;12 (e) 2.0 equiv. of LiAlH4 and EtOAc in THF at −40 °C for 2–13 h;13 (f) (MeO)2P(O)H (2.0 equiv.) and K2CO3 (1.0 equiv.) in dioxane at 80 °C for 13 h;14 (g) indium metal (1.1 equiv.) in [bmim]Br under ohmic heating15 at 95 °C and (h) In0/InCl3/Pd(PPh3)4 system at r.t. for 3 h.16 Furthermore, in a number of the described protocols there is no control over the geometry of the generated double bond at all.17
On the other hand, due their unique properties, a lot of applications of tellurium compounds in both organic and inorganic chemistry have been described.18 Consequently, a variety of tellurium reagents, such as sodium telluride (Na2Te) and sodium hydrogen telluride (NaTeH),19 has been employed by chemists in the synthesis of organotellurides,20 the cleavage of carboxylic esters or epoxides,21 cyclization reactions,22 among others.19 Generally, these species are formed in situ by reaction of elemental tellurium with sodium borohydride (NaBH4), in different solvents,23 stoichiometry ratios21a and reaction conditions.24 The nucleophilic species of tellurium is a valuable reagent widely employed for the reduction of different functional groups.25 For example, it can reduce the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of α,β-unsaturated carbonyl compounds,26 conjugated arylalkenes27 as well as nitroarenes.28 Further, nucleophilic tellurium is used in the desulphonation of vic-dimesylates/ditosylates,29 the debromination of vic-dibromoalkenes30 to afford alkenes and in the dehalogenation of polyhalogenated organic compounds.31
C bond of α,β-unsaturated carbonyl compounds,26 conjugated arylalkenes27 as well as nitroarenes.28 Further, nucleophilic tellurium is used in the desulphonation of vic-dimesylates/ditosylates,29 the debromination of vic-dibromoalkenes30 to afford alkenes and in the dehalogenation of polyhalogenated organic compounds.31
In this sense – and due to our interest on chalcogen chemistry,32 we describe herein an alternative method for preparation of (E)-bromoalkenes under mild reaction conditions without the use of transition metal or base. This method involves hydrodebromination of 1,1-dibromoalkenes 1 by using a catalytic amount of nucleophilic species of tellurium, which was generated in situ by the reaction of elemental tellurium with NaBH4 in EtOH (Scheme 1).
 | ||
| Scheme 1 General scheme of the reaction. | ||
Results and discussion
First of all, we choose 1,1-dibromoalkene 1a, tellurium and NaBH4 as the standard starting materials to establish the best reaction conditions under argon atmosphere (Table 1). We examined the amount of elemental tellurium, temperature and the nature of the solvent.
|
||||
|---|---|---|---|---|
| Entry | Te0 (mol%) | Solvent | E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratiob :Z ratiob |
Yieldc (%) |
| a Reaction was performed with 1,1-dibromoalkene 1a (0.5 mmol), tellurium and NaBH4 (0.7 mmol) in 1.0 mL of the solvent under argon.b E/Z ratio was determined by GC/MS from the crude.c Yields are given for isolated products.d Reaction was performed without NaBH4.e Reaction was performed using 10 mol% of (C6H5Te)2 at room temperature.f Second step was performed at 50 °C. | ||||
| 1 | 5 | EtOH | 96:4 |
73 |
| 2 | 10 | EtOH | 96:4 |
90 |
| 3 | 15 | EtOH | 96:4 |
86 |
| 4 | 10 | H2O | 94:6 |
Traces |
| 5 | 10 | PEG-400 | 95:5 |
76 |
| 6 | 10 | EtOH/THF | 96:4 |
83 |
| 7 | — | EtOH | 48:52 |
Traces |
| 8d | 10 | EtOH | — | n.r. |
| 9e | — | EtOH | 97:3 |
26 |
| 10f | 10 | EtOH | 94:6 |
71 |

In our preliminary experiment, a mixture of 0.025 mmol of tellurium powder and 0.7 mmol of NaBH4 in ethanol (1.0 mL) was stirred at 50 °C for 0.5 h under argon atmosphere to afford in situ a nucleophilic species of tellurium, which based on the literature in selenium, could be Na2Te[B(OEt)3]2.33 The reduction of tellurium metal was accompanied by the change in the color of the reaction mixture, which gradually altered from a gray suspension to a pale purple, and then a colorless solution. After that, the mixture was cooled to room temperature and 1,1-dibromoalkene 1a (0.5 mmol) was added in the reaction vessel and the stirring was maintained for one additional hour. Under these reaction conditions, the desired (E)-bromostyrene 2a was obtained in 73% yield and a E:Z ratio = 96:4 (Table 1, entry 1). This method is particularly advantageous compared to the conventional ones8 due to the high selectivity to the olefin with E geometry. Aiming to improve the yield, we performed reactions using larger amounts of tellurium and good results were obtained when 10 or 15 mol% of Te0 were used (Table 1, entries 2 and 3).
Regarding the influence of the solvent in the reaction, we explored the use of H2O, PEG-400 and a 1:1 mixture of EtOH/THF and we observed that EtOH is slightly superior to all the others giving bromostyrene 2a in 90% yield (Table 1, entries 2 vs. 4–6). The reactions performed without a tellurium source or reducing agent did not afford dehydrobrominated alkene 2a, as indicated by GC/MS analysis (Table 1, entries 7 and 8). We also checked if a different source of nucleophilic tellurium could be used in the reaction. When a solution of phenyl tellurolate (10 mol%), prepared by the reduction of the diphenyl ditelluride with NaBH4 in EtOH at 25 °C, was used in the place of Na2Te[B(OEt)3]2, bromostyrene 2a was obtained in only 26% yield (Table 1, entry 9). After 24 h GC analysis confirmed the presence of the starting material, even using 50 mol% of (C6H5Te)2. This condition is similar to that previously described by us to prepare (E)-1-bromo-1-seleno alkenes and ketene selenoacetals by a stoichiometric and temperature-controlled reaction.34 Finally, when the two steps were performed at 50 °C, after 1 h the product 2a was obtained in 71% yield due the parallel reactions (Table 1, entry 10).
From the results collected in Table 1, we defined the best reaction conditions: 10 mol% of tellurium powder and 0.7 mmol of NaBH4 in EtOH at 50 °C for 0.5 h under Ar (for the formation of Na2Te[B(OEt)3]2 in situ) followed by the dropwise addition of 1,1-dibromoalkene 1a (0.5 mmol) and stirring at room temperature (Table 1, entry 2). Furthermore, we observed that under the same experimental conditions, selenium was ineffective after several hours, proving that tellurium exhibit unique reactivity in this type of reaction.
Once the best reaction conditions were determined for 1a, the scope and limitations of the Te-promoted hydrodebromination for a number of 1,1-dibromoalkenes 1 was verified (Table 2). The results showed in Table 2 reveal that our protocol worked well for aromatic substrates containing electron-donating and electron-withdrawing groups to give products in high yields and a (E)-selectivity. The electronic effects on the aryl moiety of the 1,1-dibromoalkenes 1a–i seemed to have an influence on the time of consumption of the starting material. For example, 1,1-dibromoalkenes with chloro, bromo or electron–neutral substituents reacted faster than methyl or methoxy ones (Table 2, entries 1, 6–8 vs. 2–5). This effect was pronounced when disubstituted 1,1-dibromoarylalkenes were employed (Table 2, entries 3 vs. 4 and 5). The yield and selectivity decreased when a substituent was present at the ortho position of the aromatic ring, due to steric hindrance (Table 2, entries 4 vs. 5 and 7 vs. 8). The reduction of the functional group occurred when the nitroaryl derivative 1i was used28 affording the resulting aniline isolated in 51% yield after 5 h (Table 2, entry 9). Additionally, our protocol is not suitable to phenolic derivatives (2-HOC6H4; 2-HO-3-CH3OC6H3), probably due the acidity of the phenolic hydrogen, which competes with the hydride from the NaBH4 to give molecular hydrogen and the respective borates.
|
||||
|---|---|---|---|---|
| Entry | 2 (major isomers) | Time (h) | E:Z ratiob |
Yieldc (%) |
| a Reactions were performed in the presence of 1,1-dibromoalkenes 1a–n (0.5 mmol), 10 mol% of tellurium and NaBH4 (0.7 mmol) in 1.0 mL of the EtOH under argon.b E/Z ratio was determined by GC/MS for crude products.c Yields are given for isolated products.d Starting from 1-(2,2-dibromovinyl)-4-nitrobenzene.e The isomers were not separable by flash chromatography.f E/Z ratio was determined by 1H NMR for isolated products. | ||||
| 1 |  |
1 | 96:4 |
90 |
| 2 |  |
1.5 | 97:3 |
87 |
| 3 |  |
1.5 | 96:4 |
95 |
| 4 |  |
4 | 99:1 |
87 |
| 5 |  |
6 | 93:7 |
80 |
| 6 |  |
1 | 97:3 |
91 |
| 7 |  |
1 | 97:3 |
96 |
| 8 | 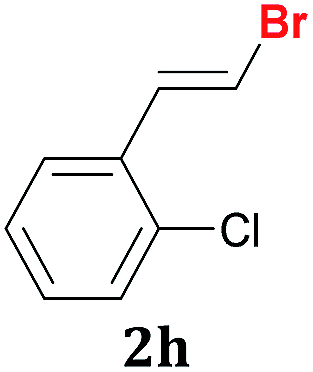 |
1 | 96:4 |
90 |
| 9 |  |
5 | 94:6d |
51 |
| 10 |  |
1 | 98:2 |
68 |
| 11 |  |
2 | 74:26e |
53 |
| 12 |  |
3 | 78:22e |
94 |
| 13 |  |
1 | 5:95f |
90 |
| 14 | 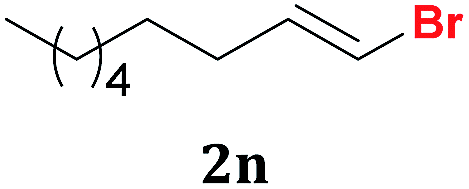 |
5 | 51:49e |
65 |

The possibility of performing the reaction with heteroaromatic 1,1-dibromoalkenes was also investigated. The reaction of Na2Te[B(OEt)3]2 with 2-(1,1-dibromovinyl)pyridine 1j furnished the respective product 2j in acceptable yield and high selectivity after 1 h. Analysis of the co-products of the reaction indicated that mono-debromination competes with di-debromination and formation of telluroalkene derivatives (Table 2, entry 10). The same parallel reactions were observed by using 1,1-dibromo-2-furylethene 1k, which afforded the expected product 2k in 53% yield after 2 h (Table 2, entry 11).
To our delight, this Te-promoted method showed to be chemoselective when conjugated 1,1-dibromo-butadiene 1l was used as starting material, affording exclusively product 2l in 94% yield after 3 h. It was expected that Na2Te[B(OEt)3]2 could reduce the conjugated double bond, as extensively described under similar conditions,26 but probably the low-loading of tellurium and the short reaction time contributed to the exclusive formation of a mixture E/Z isomers of the bromoalkene 2l (Table 2, entry 12). Similarly, the butenyne derivative 1m was exclusively reduced in the brominated double bond. In this case, after 1 h of reaction, the (Z)-isomer 2m was selectively isolated in 90% yield (Z:E ratio of 95:5; Table 2, entry 13). Finally, the reaction with alkyl substituted 1,1-dibromoalkene 1n afforded a mixture E/Z isomers of product 2n in 65% yield (Table 2, entry 14).
According to the mechanism proposed by Kuang and co-workers12 for the preparation of (E)-bromoalkenes using diethyl phosphonate two different pathways can be envisioned: a Michael type attack (Scheme 2, path I) and/or an halophilic attack (Scheme 2, path II). In the first case the nucleophilic tellurium species adds into the 1,1-dibromoalkene to give the intermediate A. After that, a formal protonation and elimination of Te0 should afford prevalently the (E)-isomer (Scheme 2). In the case of halophilic attack, a similar involvement of the borane should drive the formation of a mixture of isomers, most probably enriched with (Z)-isomer C. Based on these considerations, even if it is not possible to exclude one of the two mechanisms as well as the reasonable involvement of single electron transfer process and/or radicals, those depicted in Scheme 2 (path I) nicely explain our experimental results.
 | ||
| Scheme 2 Proposed mechanism. | ||
As an example of the synthetic utilities of (E)-bromoalkenes, 2a was converted into the corresponding (E)-phenylselenostyrene 4 by a previously reported procedure.34 The compound 2a (0.5 mmol) was reacted with the nucleophilic selenium species, generated in situ by reacting diphenyl diselenide 3 (0.3 mmol) and NaBH4 in PEG-400 at room temperature. After 4 h at 120 °C, (E)-phenylselenostyrene 4 was isolated in 72% yield with total retention of the stereochemistry of the double bond (Scheme 3). Furthermore some of us reported that 2a can react with PhSeZnCl in ‘on water’ conditions affording 4 in 2 h and 94% yield.35
 | ||
| Scheme 3 Synthetic utility of (E)-bromoalkene 2a. | ||
Conclusions
In summary, we have developed a simple and efficient method to hydrodebromination of 1,1-dibromoalkenes in the presence of 10 mol% of tellurium and NaBH4 in ethanol. This transition metal-free protocol is suitable to several 1,1-dibromoalkenes, affording the corresponding (E)-bromoalkenes in good yields and excellent stereoselectivity in short reaction times and under mild reaction conditions.Acknowledgements
The authors thank FAPERGS, CNPq and CAPES for financial support. CNPq is also acknowledged for the fellowship for G. P. and E. J. L. This work is part of the scientific collaborations carried out under the umbrella of the Network SeS-Redox and Catalysis.Notes and references
- G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS PubMed.
- F. Ramirez, N. B. Desal and N. McKelvie, J. Am. Chem. Soc., 1962, 84, 1745 CrossRef CAS.
- Y.-Q. Fang, O. Lifchits and M. Lautens, Synlett, 2008, 413 CAS.
- (a) A. Sorg and R. Brückner, Angew. Chem., Int. Ed., 2004, 43, 4523 CrossRef CAS PubMed; (b) L. F. Tietze, L. P. Lücke, F. Major and P. Müller, Aust. J. Chem., 2004, 57, 635 CrossRef CAS; (c) D. Xu, M. A. Drahl and L. J. Williams, Beilstein J. Org. Chem., 2011, 7, 937 CrossRef CAS PubMed; (d) T. Kim, W. Lee, K. H. Jeong, J. H. Song, S.-H. Park, P. Choi, S.-N. Kim, S. Lee and J. Ham, Bioorg. Med. Chem. Lett., 2012, 22, 4122 CrossRef CAS PubMed; (e) L. F. Tietze and W.-R. Krahnert, Chem.–Eur. J., 2002, 8, 2116 CrossRef CAS PubMed; (f) A. Iwata, S. Inuki, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 5506 CrossRef CAS PubMed; (g) S. López, F. Fernández-Trillo, L. Castedo and C. Saá, Org. Lett., 2003, 5, 3725 CrossRef PubMed; (h) I. Paterson, T. Paquet and S. M. Dalby, Org. Lett., 2011, 13, 4398 CrossRef CAS PubMed; (i) A. E. Pasqua, F. D. Ferrari, J. J. Crawford and R. Marquez, Tetrahedron Lett., 2014, 55, 6042 CrossRef CAS.
- (a) M. Braun, A. Holmann, J. Rahematpura, C. Bühne and S. Grimme, Chem.–Eur. J., 2004, 10, 4584 CrossRef CAS PubMed; (b) J. L. Burkemper, C. Huang, A. Li, L. Yuan, K. Rich, J. McConathy and S. E. Lapi, J. Med. Chem., 2015, 58, 8542 CrossRef CAS PubMed; (c) J. Alzeer, J. Chollet, I. Heinze-Krauss, C. Hubschwerlen, H. Matile and R. G. Ridley, J. Med. Chem., 2000, 43, 560 CrossRef CAS PubMed.
- (a) S. Tanaka, Y. Fukui, N. Nakagawa, K. Murakami, T. N. Murakami, N. Koumura and A. Mori, Org. Lett., 2016, 18, 650 CrossRef CAS PubMed; (b) S. M. A. Rahman, M. Sonoda, M. Ono, K. Miki and Y. Tobe, Org. Lett., 2006, 8, 1197 CrossRef CAS PubMed; (c) A. Hayford, J. J. Kaloko, S. EI-Kazaz, G. Bass, C. Harrison and T. Corprew, Org. Lett., 2005, 7, 2671 CrossRef CAS PubMed; (d) M. Nojima, Y. Ohta and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2643 CrossRef CAS.
- (a) J. Uenishi, R. Kawahama, Y. Shiga and O. Yonemitsu, Tetrahedron Lett., 1996, 37, 6759 CrossRef CAS; (b) J. Uenishi, R. Kawahama, Y. Shiga and O. Yonemitsu, J. Org. Chem., 1998, 63, 8965 CrossRef CAS.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, J. Org. Chem., 1981, 46, 3745 CrossRef CAS.
- (a) T. Harada, D. Hara, K. Hattori and A. Oku, Tetrahedron Lett., 1988, 29, 3821 CrossRef CAS; (b) T. Harada, T. Katsuhira, D. Hara, Y. Kotani, K. Maejima, R. Kaji and A. Oku, J. Org. Chem., 1993, 58, 4897 CrossRef CAS.
- D. Grandjean and P. Pale, Tetrahedron Lett., 1993, 34, 1155 CrossRef CAS.
- M. A. Fakhfakh, X. Franck, R. Hocquemiller and B. Figadére, J. Organomet. Chem., 2001, 624, 131 CrossRef CAS.
- C. Kuang, H. Senboku and M. Tokuda, Tetrahedron, 2002, 58, 1491 CrossRef CAS.
- H. Horibe, K. Kondo, H. Okuno and T. Aoyama, Synthesis, 2004, 986 CAS.
- Y. Zhao, T. Chen, X.-B. Wang and L.-B. Han, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1820 CrossRef CAS.
- R. G. Soengas, V. L. M. Silva, J. Pinto, H. Rodríguez-Solla and A. M. S. Silva, Eur. J. Org. Chem., 2016, 99 CrossRef CAS.
- R. G. Soengas, H. Rodríguez-Solla and A. M. S. Silva, Synlett, 2016, 1096 CrossRef CAS.
- (a) K. Miura, Y. Ichinose, K. Nozaki, K. Fugami, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1989, 62, 143 CrossRef CAS; (b) T. Harada, T. Katsuhira and A. Oku, J. Org. Chem., 1992, 57, 5805 CrossRef CAS; (c) Y. Taskesenligil, R. P. Kashyap, W. H. Watson and M. Balci, J. Org. Chem., 1993, 58, 3216 CrossRef CAS; (d) S. Abbas, C. J. Hayes and S. Worden, Tetrahedron Lett., 2000, 41, 3215 CrossRef CAS; (e) B. C. Ranu, S. Samanta and S. K. Guchhait, J. Org. Chem., 2001, 66, 4102 CrossRef CAS PubMed; (f) L. Wang, P. Li, Y. Xie and Y. Ding, Synlett, 2003, 1137 CrossRef CAS.
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725 RSC.
- (a) N. Petragnani and J. V. Comasseto, Synthesis, 1991, 793 CrossRef CAS; (b) N. Petragnani and J. V. Comasseto, Synthesis, 1991, 897 CrossRef CAS; (c) N. Petragnani and H. A. Stefani, Tetrahedron, 2005, 61, 1613 CrossRef CAS.
- E. L. Borges, T. J. Peglow, M. S. Silva, C. G. Jacoby, P. H. Schneider, E. J. Lenardão, R. G. Jacob and G. Perin, New J. Chem., 2016, 40, 2321 RSC.
- (a) J. Chen and X. J. Zhou, Synthesis, 1987, 586 CrossRef CAS; (b) D. H. R. Barton, A. Fekih and X. Lusinchi, Tetrahedron Lett., 1985, 26, 6197 CrossRef CAS; (c) A. S. Pepito and D. C. Dittmer, J. Org. Chem., 1997, 62, 7920 CrossRef CAS PubMed.
- C. A. Landis, M. M. Payne, D. L. Eaton and J. E. Anthony, J. Am. Chem. Soc., 2004, 126, 1338 CrossRef CAS PubMed.
- (a) X. Huang, J.-H. Pi and Z.-Z. Huang, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 67, 177 CrossRef CAS; (b) X. Huang and J.-H. Pi, Synth. Commun., 1990, 20, 2297 CrossRef CAS; (c) X. Huang and H.-Z. Zhang, Synth. Commun., 1989, 19, 97 CrossRef CAS.
- L. Engman, Phosphorus, Sulfur Silicon Relat. Elem., 1988, 38, 105 CrossRef.
- N. Petragnani and H. A. Stefani, Tellurium in Organic Synthesis: Second, Updated and Enlarged Edition, Elsevier, Amsterdam, 2nd edn, 2007 Search PubMed.
- M. Yamashita, Y. Tanaka, A. Arita and M. Nishida, J. Org. Chem., 1994, 59, 3500 CrossRef CAS.
- K. Ramasamy, S. K. Kalyanasundaram and P. Shanmugam, Synthesis, 1978, 545 CrossRef CAS.
- (a) S. Uchida, K. Yanada, H. Yamaguchi and H. Meguri, Chem. Lett., 1986, 1069 CrossRef CAS; (b) H. Suzuki, K. Takaoka and A. Osuka, Bull. Chem. Soc. Jpn., 1985, 58, 1067 CrossRef CAS.
- V. Bargues, G. Blay, J. Fernandez and J. R. Pedro, Synlett, 1996, 655 CrossRef CAS.
- K. Ramasamy, S. K. Kalyanasundaram and P. Shanmugam, Synthesis, 1978, 311 CrossRef CAS.
- (a) W. Mack, Angew. Chem., Int. Ed., 1967, 6, 1083 CrossRef CAS; (b) A. A. Vasilev and L. Engman, J. Org. Chem., 1998, 63, 3911 CrossRef CAS; (c) A. Osuka, K. Takechi and H. Suzuki, Bull. Chem. Soc. Jpn., 1984, 57, 303 CrossRef CAS.
- G. Perin, D. Alves, R. G. Jacob, A. M. Barcellos, L. K. Soares and E. J. Lenardão, ChemistrySelect, 2016, 2, 205 CrossRef.
- M. Miyashita, T. Suzuki, M. Hoshino and A. Yoshikoshi, Tetrahedron, 1997, 53, 12469 CrossRef CAS.
- R. Webber, T. J. Peglow, P. C. Nobre, A. M. Barcellos, J. A. Roehrs, R. F. Schumacher and G. Perin, Tetrahedron Lett., 2016, 57, 4128 CrossRef CAS.
- S. Santoro, B. Battistelli, L. Testaferri, M. Tiecco and C. Santi, Eur. J. Org. Chem., 2009, 4921 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures as well as spectral data of all synthesized compounds are presented. See DOI: 10.1039/c6ra24295b |
| This journal is © The Royal Society of Chemistry 2016 |