
Fe–N-doped carbon-based composite as an efficient and durable electrocatalyst for the oxygen reduction reaction†
Gasidit Panomsuwan*ab,
Nagahiro Saitobcd and
Takahiro Ishizaki*de
aDepartment of Materials Engineering, Faculty of Engineering, Kasetsart University, Bangkok 10900, Thailand. E-mail: g.panomsuwan@gmail.com
bNU-PPC Plasma Chemical Technology Laboratory, The Petroleum and Petrochemical College, Chulalongkorn University, Bangkok 10330, Thailand
cDepartment of Materials, Physics and Energy Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
dCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST), Saitama 333-0012, Japan
eDepartment of Materials Science and Engineering, Faculty of Engineering, Shibaura Institute of Technology, Tokyo 135-8548, Japan. E-mail: ishizaki@sic.shibaura-it.ac.jp
First published on 28th November 2016
Abstract
We herein report the preparation of an Fe–N-doped carbon nanoparticle–carbon nanofiber (Fe–N-CNP–CNF) composite using a solution plasma process followed by heat treatment. The resulting Fe–N-CNP–CNF exhibits excellent catalytic activity, durability, and methanol tolerance for the oxygen reduction reaction (ORR) in an alkaline solution. The enhanced ORR activity of Fe–N-CNP–CNF can reasonably be attributed to the synergistic contributions provided by a high degree of graphitization of CNF, meso/macroporosity of CNP, presence of catalytically active sites for ORR (i.e., graphitic N and Fe–N bond), and existence of carbon-encapsulated Fe/Fe3C particles.
The electrochemical oxygen reduction reaction (ORR) plays the most vital role in determining the performance and reliability of fuel cells.1 Until now, Pt-supported carbon materials (Pt/C) have been regarded as the most effective catalysts for catalyzing ORR at the cathode owing to their superb intrinsic ORR activity through an efficient four-electron reduction pathway in both alkaline and acidic media.2 Nevertheless, the sluggish ORR kinetics, limited availability, high price, and poor durability of Pt are major barriers for the next steps towards further development and manufacturing scale-up of fuel cells.3 The key technical challenge facing this technology is to develop inexpensive and sustainable cathode catalysts with high ORR performance in terms of both catalytic activity and durability.4
Over recent years, considerable research efforts have been devoted to developing non-Pt based catalysts, such as heteroatom-doped carbons,5 metal oxides,6 transition metal chalcogenides,7 and transition metal–nitrogen (Me–N) doped carbons.8 Among the various alternative catalysts pursued, a family of Me–N-doped carbons, especially Fe–N-doped carbon, has demonstrated the most promising prospects as an ideal alternative ORR catalyst for ultimately replacing a commercial Pt/C catalyst because of its excellent balance between price, catalytic activity, and durability. Although the exact nature of the Fe–N active sites in these catalysts is still under intense debate in the catalysis community, both experimental studies and quantum mechanical calculations have reported the same conclusion that the Fe–N coordination sites play a crucial role in promoting the ORR activity with high selectivity towards a direct four-electron pathway.9
With a continuous endeavor to further improve ORR activity, an integration of carbon materials into binary or ternary composite systems with the presence of Fe–N active sites (e.g., carbon nanotube–graphene,10 carbon nanotube–carbon nanoparticle,11 carbon fiber–graphene,12 carbon nanosheet–carbon nanotube,13 carbon nanosphere–carbon nanotube–graphene14) has recently been considered as an effective strategy to boost ORR activity beyond the use of individual constituent carbon materials. A remarkable enhancement in the ORR activity of such composite systems could be attributed to the unique architectural morphology as well as the combined advantages of each constituent carbon material. Specifically, the ORR performance of Fe–N-doped carbon catalysts as reported in the literature was greatly varied, depending on the type of carbon allotrope, carbon/nitrogen source precursor, heat-treatment temperature, and synthesis method. Despite significant advancements in this field, the rational design and synthesis of Fe–N-doped carbon-based composite through an innovative synthesis method are still an ongoing challenge in ORR catalyst research.
Recently, plasma in liquid phase, termed solution plasma, has emerged as one of the simple and effective methods for the synthesis of Pt-free carbon-based ORR catalysts, such as heteroatom-doped carbons (single15,16 or multiple doping17) and Fe–N-doped carbons.18 The main advantages of solution plasma in the synthesis of Pt-free carbon-based ORR catalysts are its simplicity, a fast synthesis rate, and flexible use of a wide variety of precursors (both liquid and solid compounds). Moreover, synthesis under unusual conditions in the plasma reaction may lead to exploration into a new class of catalysts for ORR, and groundbreaking findings could be expected. Thus, solution plasma is a fertile field for a brighter research future in ORR catalysts.
Herein, we present the synthesis of Fe–N-doped carbon nanoparticle–carbon nanofiber (Fe–N-CNP–CNF) composite using a solution plasma process. In brief, the plasma was generated by applying a bipolar high-voltage pulse through a symmetric pair of metallic Fe electrodes, which were submerged in a homogeneous mixture of CNFs and liquid 2-cyanopyridine (C4H4N2) (Fig. S1†). The carbon particles with in situ N doping were produced directly from 2-cyanopyridine through dissociation and recombination processes, while Fe atoms were sputtered simultaneously from the electrode surface. In the plasma zone, the Fe atoms could react with highly energetic C2 and CN radicals generated from the dissociation of 2-cyanopyridine molecules (Fig. S2†), giving rise to the formation of Fe–N coordination. The N-CNP containing Fe–N sites formed in the plasma zone diffused to the suspension and attached to the CNF surface, forming a composite structure. After synthesis, the resulting Fe–N-CNP–CNF composite was collected and subsequently heated to 900 °C for 1 h under Ar flow. The synthesis procedure of Fe–N-CNP–CNF is schematically depicted in Fig. 1. For comparison, two control catalysts, including N-CNP and N-CNP–CNF composite, were prepared concurrently using the same procedure and conditions, except for the use of tungsten as the electrodes. More detailed information on the synthesis procedure of all the catalysts is given in ESI.† Various physicochemical and electrochemical characterizations were conducted thoroughly to elucidate the possible factors that play roles in the enhancement of ORR activity for Fe–N-CNP–CNF.
 | ||
| Fig. 1 A schematic representation for the preparation of Fe–N-CNP–CNF composite using a solution plasma process followed by a subsequent heat treatment at 900 °C in Ar atmosphere. | ||
The morphology and microstructure of Fe–N-CNP–CNF were investigated by means of field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM). The FE-SEM image in Fig. 2a shows the presence of CNFs interspaced with aggregated carbon particles, forming an interconnected three-dimensional pore structure. In the TEM image in Fig. 2b, CNF exhibits a hollow-core structure and carbon particles are aggregated with each other. Moreover, it can be seen that the Fe particles are embedded in the carbon particle matrix, as indicated by the dark contrast in the TEM image. The average diameter of the Fe particles was found to be 22.0 ± 6.9 nm. The selected area electron diffraction (SAED) pattern of Fe–N-CNP–CNF displays both diffraction rings and dots corresponding to the carbon and Fe phases, respectively (inset of Fig. 2b). Fig. 2c and S4† present high-resolution TEM (HR-TEM) images at the area where the single Fe particle exists. It can be seen that the outer surface of Fe particles is surrounded by the graphitic layers. The formation of Fe particles is possibly due to the reduction of Fe ions during heat treatment at high temperature. These Fe particles can accelerate the graphitization of carbon around them, resulting in encapsulation inside the graphitic carbon shells. HR-TEM images were also taken at the areas where the carbon particles and wall of CNF exist (Fig. S5†). The presence of short and wrinkled graphitic planes was observed for the carbon particles, indicating its amorphous nature. On the other hand, a long-range order of graphitic layers was clearly visible along the wall of CNF, reflecting its high degree of graphitization. The individual distributions of C, N, and Fe elements in Fe–N-CNP–CNF were acquired by energy dispersive X-ray (EDX) spectroscopy. The elemental mapping images of C, N, and Fe are illustrated in Fig. 2d. As can be seen, both C and N elements are uniformly distributed throughout the whole area of Fe–N-CNP–CNF, while the Fe elements are highly concentrated at the areas where Fe particles exist, and also slightly distributed in the carbon particle matrix. Interestingly, the Fe signal seems to be adjacent or overlaid with the N signal at the atomic level, which roughly implies the existence of Fe–N sites in Fe–N-CNP–CNF. This result may suggest that Fe–N-CNP–CNF was composed of not only carbon-encapsulated Fe particles, but also that the Fe–N sites were dispersed in carbon particles.
 | ||
| Fig. 2 Electron microscopy characterizations of Fe–N-CNP–CNF: (a) FE-SEM, (b) TEM, and (c) HR-TEM images. The corresponding SAED pattern is shown in the inset of (b). (d) STEM and the corresponding elemental mapping images: C, N, and Fe are given in blue, red, and yellow colors, respectively. | ||
The phase structure of all the catalysts was examined with X-ray diffraction (XRD). The XRD pattern of Fe–N-CNP–CNF (Fig. 3a) is comprised of the broad and narrow 002 diffraction peaks arising from amorphous N-CNP (dashed line) and highly graphitic CNF (dotted line), respectively. The diffraction peaks detected at 44.6° and 64.9° (denoted as *) are associated with the crystalline phase of metallic Fe (ICDD: no. 00-001-1262). Additionally, a set of weak diffraction peaks is also detected beside the Fe 110 peak (denoted as #), which corresponds to the Fe3C phase (ICDD: no. 00-006-0688). Taking into account the TEM results, the metal particles encapsulated in graphitic shells should be in the form of the Fe/Fe3C phase. The absence of the diffraction peaks from oxidized Fe species is likely due to the encapsulation of Fe/Fe3C particles inside the graphitic shells, which can protect them from direct contact with oxygen. The XRD data gives us the evidence that Fe–N-CNP–CNF contains a mixture of highly graphitized CNF, amorphous carbon particles, and crystalline Fe/Fe3C phases. More structural information was further obtained using Raman spectroscopy measurement. Two evident characteristic peaks of carbon were detected at 1350 and 1590 cm−1 for all catalysts, which are attributed to the D band (disordered carbon) and G band (sp2 carbon), respectively (Fig. S6b†).19 The intensity ratio of the D band to the G band (ID/IG) was estimated to be 0.83, 1.05, 0.97, and 0.94 for CNF, N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF, respectively. The result is in accordance with the aforementioned HR-TEM and XRD data that CNF has a high degree of graphitization, while N-CNP exhibits a relatively lower degree of graphitization (more disorder structure).
 | ||
| Fig. 3 (a) XRD patterns of Fe–N-CNP–CNF. The dashed and dotted lines indicate the position of amorphous carbon (N-CNP) and highly graphitic carbon (CNF), respectively. (b) N2 adsorption–desorption isotherm and the corresponding pore-size distribution of Fe–N-CNP–CNF. The solid (●) and open circles (○) represent adsorption and desorption data, respectively. High-resolution XPS spectrum of (c) Fe 2p and (d) N 1s regions for Fe–N-CNP–CNF. | ||
The N2 adsorption–desorption isotherms of all catalysts were measured to investigate their specific surface area, pore volume, and pore-size distribution. A typical N2 adsorption–desorption isotherm of Fe–N-CNP–CNF is shown in Fig. 3b. A low adsorbed volume at very low relative pressure (P/P0 < 0.01) indicates the low amount of micropores. At high relative pressures (P/P0 = 0.85–0.99) a steep increase in the isotherm curve with a narrow hysteresis loop was observed, indicating the presence of meso- and macropores. A similar feature of N2 adsorption–desorption isotherm was also observed for N-CNP and N-CNP–CNF (Fig. S7†). The pore-size distributions of N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF clearly reveal a broad peak covering the range of 10–100 nm (Fig. S8†), confirming the presence of multiple porous structures ranging from mesopore to macropore size. The specific surface area of Fe–N-CNP–CNF determined by the Brunauer–Emmett–Teller (BET) method was found to be 156.5 m2 g−1, which is comparable with that of N-CNP (171.9 m2 g−1) and N-CNP–CNF (159.8 m2 g−1), but much higher than that of CNF (47.4 m2 g−1). The pore volumes of CNF, N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF were calculated to be 0.34, 0.94, 0.84, and 0.79 cm3 g−1, respectively (Table S1†). From pore structural analysis, it is clear that morphological features and pore structure of both N-CNP–CNF and Fe–N-CNP–CNF are governed by N-CNP rather than CNF. Hierarchical porous textures with meso- and macropores are known to be beneficial for enhancing accessible specific surface area and also facilitating the diffusion of the electrolyte and O2 to active sites.20
Furthermore, X-ray photoelectron spectroscopy (XPS) measurements were carried out to probe the elemental compositions and chemical states of the catalysts. The XPS survey spectrum of Fe–N-CNP–CNF (Fig. S9†) was composed of the signal peaks of C, N, O, and Fe elements without discernable signals from other elements. The surface atomic contents of C, O, N, and Fe were determined to be 90.10, 8.59, 1.16, and 0.15 at%, respectively (Table S2†). This result corroborates that N and Fe were successfully incorporated into the carbon composite structure; however, the Fe content was very low, which was as a result of the encapsulation of Fe inside the graphitic shells. High-resolution XPS C 1s spectra of all catalysts revealed an asymmetric peak shape with the main component at 284.5 eV corresponding to the sp2 carbon. An extended tail at higher binding energies was composed of several peaks assigned to the C atoms bonded to N and/or O atoms (Fig. S10†). The emergence of an O 1s peak is likely due to the adsorbed oxygen and partial oxidation on the catalyst surface (Fig. S11†). The high-resolution XPS Fe 2p spectrum of Fe–N-CNP–CNF reveals a double-peak feature, which can be deconvoluted into four peaks (Fig. 3c).21 The peaks at 710.8 and 712.7 eV represent the binding energies of the Fe 2p3/2 band of Fe2+ and Fe3+ species, respectively. For the Fe 2p1/2 band, the peak at 724.2 eV is assigned to the binding energy of Fe2+, while that at 725.8 eV corresponds to Fe3+. The absence of a peak assignable to metallic Fe0 peaks is due to the encapsulation of Fe inside the graphitic carbon shell, as demonstrated in the HR-TEM result. This result suggests that Fe species on the catalyst surface dominantly exist in the ionic states (i.e., Fe2+ and Fe3+) and can potentially be bound by N atoms, which can act as active sites for the ORR.
High-resolution XPS N 1s spectra of N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF can be deconvoluted into three peaks associated with pyridinic N and/or Fe–N (398.4 ± 0.1 eV), graphitic N (400.9 ± 0.1 eV), and pyridinic N-oxide (403.5 ± 0.2 eV) (Fig. 3d and S12†).15,22 It should be noted that the peaks of pyridinic N and Fe–N are assumed to locate at the same position owing to the small difference in their binding energies.23 The N contents of N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF were estimated to be 1.35, 1.33, and 1.16 at%, respectively. The proportion of various N states for N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF reveal a similar trend, of which graphitic N is a major component, while pyridinic N and pyridinic N-oxide are minor components (Table S3†). This result implies that the surface of N-CNP–CNF and Fe–N-CNP–CNF composites is dominantly governed by N-CNP. In the case of Fe–N-CNP–CNF, the N content was relatively lower (1.16 at%) and the proportion of graphitic N was as high as 73%, which is higher than that of N-CNP (68%) and N-CNP–CNF (64%). The most abundant proportion of graphitic N suggests that N atoms were preferentially doped into the graphitic basal plane rather than at the edge for Fe–N-CNP–CNF. Our finding is in agreement with previous report by Jiang et al. in which the addition of Fe into N-doped carbon materials resulted in a slight decrease of N content and also played a role in the favorable formation of graphitic N.24 Although the exact role of active sites in Fe–N-based carbon catalysts is still debatable, the consensus in the literature is that Fe–N, graphitic N and pyridinic N are considered to be electrocatalytically active sites for ORR,9,25 while pyridinic N-oxide has a negligible effect on ORR.
The electrocatalytic activity for ORR of all catalysts was first evaluated using cyclic voltammetry (CV) in 0.1 M KOH solution saturated with N2 and O2 at room temperature (Fig. S13†). For comparison, a commercial 20 wt% Pt supported on Vulcan XC-72 (denoted as 20% Pt/C) was also tested under the same conditions as a reference. As shown in Fig. 4a, the CV curve of Fe–N-CNP–CNF reveals a quasi-rectangular capacitive behavior in the N2-saturated solution (dashed line), while a pronounced ORR peak emerges at −0.24 V in the O2-saturated solution (solid line). To gain better insight into the ORR activity, the polarization curves of the catalysts were measured with linear sweep voltammetry (LSV) in an O2-saturated 0.1 M KOH solution at a rotation speed of 1600 rpm and a scan rate of 10 mV s−1 (Fig. 4b). The polarization curve of CNF exhibited a two-step reduction process with the onset potentials at −0.21 and −0.64 V. This curve feature suggests that the CNF possesses poor ORR activity via two consecutive steps of a two-electron pathway with the generation of peroxide as intermediates. In contrast, a single-step plateau of current density was clearly visible for N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF. The polarization curve of Fe–N-CNP–CNF had an onset potential at −0.10 V, which is much more positive than that of N-CNP (−0.17 V) and N-CNP–CNF (−0.14 V), but still more negative than that of 20% Pt/C (−0.08 V). The half-wave potential (E1/2) of Fe–N-CNP–CNF and 20% Pt/C was −0.19 and −0.15 V, respectively. It can be concluded that the onset potential and half-wave potential of Fe–N-CNP–CNF are only 20 mV and 40 mV more negative than those of 20% Pt/C, respectively. Moreover, the measured current density of Fe–N-CNP–CNF was larger than that of CNF, N-CNP, and N-CNP–CNF over the potential range investigated. This result demonstrates that although the total N content of Fe–N-CNP–CNF was relatively lower in comparison with that of N-CNP and N-CNP–CNF, the Fe–N-CNP–CNF exhibited the highest ORR activity in terms of both onset potential and current density. There was no straightforward relationship between the ORR activity and the total N content, and the coexistence of graphitic N and Fe–N coordination sites is believed to play a major role in significant improvements in ORR activity for Fe–N-CNP–CNF.
 | ||
Fig. 4 Electrocatalytic activity for ORR: (a) CV curves of Fe–N-CNP–CNF in N2 and O2-saturated 0.1 M KOH solutions at a scan rate of 50 mV s−1 (N2: dashed line, O2: solid line). (b) Polarization curves of various catalysts measured on an RRDE in an O2-saturated 0.1 M KOH solution at a rotation speed of 1600 rpm and a scan rate of 10 mV s−1. A constant potential of 0.5 V was applied to the Pt ring electrode to detect peroxide species generated at the disk electrode during ORR. (c) HO2− yield and electron transfer number (n) of various catalysts obtained from the RRDE measurement at the potential range from −1.0 to −0.3 V. (d) The K–L plots of various catalysts at −0.4 V obtained from polarization curves at different rotation speeds from 225 to 2500 rpm (Fig. S14†). (e) Chronoamperometric response of Fe–N-CNP–CNF and 20% Pt/C at −0.35 V for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (1600 rpm). (f) Methanol tolerance test of Fe–N-CNP–CNF and 20% Pt/C at −0.35 V (1600 rpm). The 3.0 M methanol was introduced at 1000 s (indicated as a dashed line). The catalyst loading was 0.4 mgcat cm−2 for CNF, N-CNP, N-CNP–CNF and Fe–N-CNP–CNF, and 40 μgPt cm−2 for commercial 20% Pt/C. 000 s (1600 rpm). (f) Methanol tolerance test of Fe–N-CNP–CNF and 20% Pt/C at −0.35 V (1600 rpm). The 3.0 M methanol was introduced at 1000 s (indicated as a dashed line). The catalyst loading was 0.4 mgcat cm−2 for CNF, N-CNP, N-CNP–CNF and Fe–N-CNP–CNF, and 40 μgPt cm−2 for commercial 20% Pt/C. | ||
To better understand and elucidate the ORR catalytic pathway on the catalysts, the generation of peroxide (HO2−) and the corresponding electron transfer number (n) per O2 molecule during the ORR process can be determined on the basis of the measured disk and ring currents using the following equations:26
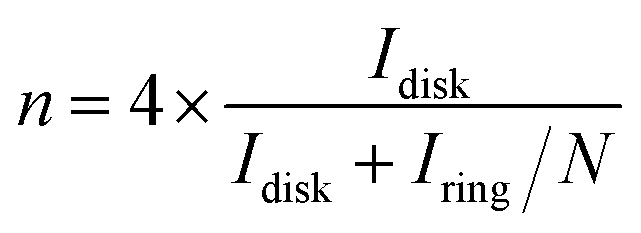 | (1) |
 | (2) |
To further verify the ORR catalytic pathway evaluated by the RRDE measurement, rotating disk electrode (RDE) measurements were carried out. The polarization curves of the catalyst were recorded at different rotation speeds, from 225 to 2500 rpm, with a scan rate of 10 mV s−1 in O2-saturated 0.1 M KOH solution (Fig. S14†). Obviously, the current density of all catalysts increased with increasing rotation speed, which can be explained by faster oxygen diffusion through the electrode surface. On the basis of these polarization curves recorded at different rotation speeds, the n values per O2 molecule can be determined from the slope of the plot of J−1 versus ω−1/2 according to the Koutecky–Levich (K–L) equation (see ESI† for more details). As shown in Fig. S15,† the slopes of the corresponding K–L plots for N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF exhibit parallelism of straight lines in the potentials between −1.0 and −0.4 V. This feature suggests that the electron transfer number and active surface area for the ORR did not change significantly within the potential range investigated. The linearity and parallelism of the K–L plots were considered to be typical of first-order reaction kinetics with respect to the concentration of dissolved O2. The n values calculated from the slope of the K–L plots at −0.4 V were 2.16, 3.07, 3.54, and 3.91, for CNF, N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF, respectively (Fig. 4d). The n values of all catalysts as a function of potential are shown in Fig. S15f.† Although the n values obtained from the K–L plots are not completely equal to those obtained from the RRDE measurement, they are close with each other and also show a similar changing trend within the potential range investigated.
An important investigation from the K–L plot that should be discussed here is regarding a non-zero intercept of the extrapolated K–L lines for all catalysts (Fig. 4d and S15†). This phenomenon is normally found when an ink-type electrode is used in RDE measurements because of an incomplete diffusion control. The non-zero intercept is possibly caused by the effects of an oxygen reduction kinetic current and oxygen diffusion in the Nafion layer.30 In this work, Nafion content in the catalyst layer seems to be the dominant effect because of its high content of 28% for CNF, N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF, and 56% for 20% Pt/C. High Nafion content results in coverage of the catalyst surface by a Nafion layer, which acts as a barrier for O2 diffusion and also blocks the active sites for catalyzing ORR. This undesirable effect can thus lead to a significant ORR inhibition and underestimated catalyst activities, as reflected by its limiting current density of 20% Pt/C being lower than the theoretical value (∼22%). Note that the limiting current density of a four-electron transfer catalyst (Pt-based catalyst) at a rotation speed of 1600 rpm should reach about 5.8 mA cm−2 according to the Levich equation. On the other hand, small deviations from theoretical values (<10%) were found for N-CNP, N-CNP–CNF, and Fe–N-CNP–CNF. Another possible Nafion effect that may affect the ORR evaluation of the catalysts is Nafion self-assembly, which tends to occur through the attractive interactions between the hydrophobic backbone of the Nafion and the graphitic surface of the catalysts. This phenomenon can occur even with small Nafion content in the catalyst ink. Spillover of self-assembled Nafion onto the catalytically active sites of the catalysts can impair their ORR activity.31 Therefore, the Nafion content exhibited a significant influence on the correct evaluation of ORR catalytic activity, which should be further emphasized in our next work.
The above electrochemical results can allow us to conclude that the Fe–N-CNP–CNF exhibits excellent ORR activity with high selectivity towards a four-electron reduction pathway in alkaline solution, which is nearly comparable to a commercial 20% Pt/C and other Fe–N-doped carbons reported in the literature (Table S4†). Based on the association of physicochemical and electrochemical results discussed above, the possible reasons that lead to the enhanced ORR activity of Fe–N-CNP–CNF can reasonably be explained as follows: (i) the existence of meso/macropores results in more accessible active sites for catalyzing ORR and also facilitates electrolyte/reactant transport. From the K–L analysis in the kinetic-controlled region (slow kinetics), the intercept is much greater than zero and the slope is quite low at E ≥ −0.2 V (Fig. S16†), indicating a high active surface area because of reduction within the catalyst pores. With lowering potential (E < −0.2 V), the slopes become lower and remain constant, indicating the surface area and other parameters are unchanged in this potential range.32 (ii) Highly graphitized CNF can provide an electrically conductive path, which thus promotes fast electron transport in the composite system.33 (iii) The existences of graphitic N and Fe–N coordination sites in Fe–N-CNP–CNF are considered as highly active sites for catalyzing ORR through a four-electron pathway. (iv) The existence of Fe/Fe3C particles encapsulated within graphitic shells in Fe–N-CNP–CNF may help to partially promote the ORR activity of neighboring Fe–N active sites.24
Apart from the catalytic activity and reduction pathway for ORR, durability and methanol-tolerant ability are also some of the most important concerns in practical fuel-cell applications. A durability test was performed by measuring the loss in current density against time with current–time chronoamperometry at a constant potential of −0.35 V in an O2-saturated 0.1 M KOH solution for 20000 s (Fig. 4e). After 20000 s, the relative current density (J/J0) of Fe–N-CNP–CNF remained as high as 87%, while that of 20% Pt/C diminished to 71%. This result confirms that Fe–N-CNP–CNF exhibits better durability in alkaline solution than 20% Pt/C. However, a slight reduction in current density of Fe–N-CNP–CNF was noticed. This was possibly caused by the fact that peroxide species (HO2−) formed during ORR reacted with active sites, which would cause the loss of the active sites and eventually result in the reduction in the ORR activity. Methanol tolerant ability was further tested by introducing 3.0 M methanol during the measurement of chronoamperometry (Fig. 4f). Upon the introduction of 3.0 M methanol at 1000 s (indicated as a vertical dashed line), the J/J0 of Fe–N-CNP–CNF was slightly disturbed and remained almost unchanged, whereas an abrupt drop in J/J0 was obvious for 20% Pt/C because of methanol oxidation over Pt. This result clearly indicates that the introduction of methanol has no significant influence on the ORR of Fe–N-CNP–CNF, but seriously deteriorates the ORR of 20% Pt/C. High durability and the methanol tolerant ability of Fe–N-CNP–CNF makes it a promising cathode catalyst for a direct methanol fuel cell.
In summary, we have developed a novel approach to prepare Fe–N-CNP–CNF catalyst through a solution plasma process followed by subsequent heat treatment. The Fe–N-CNP–CNF exhibits excellent ORR activity through a four-electron reduction pathway with low HO2− yield in alkaline solution, which closely approaches that of 20% Pt/C. Moreover, the electrochemical durability and methanol tolerant ability of Fe–N-CNP–CNF are superior to those of 20% Pt/C. The high ORR performance of Fe–N-CNP–CNF can be attributed to the synergistic contributions provided by a large specific surface with a meso/macropore structure of carbon particles, high degree of graphitization of CNF, presence of catalytically active sites for ORR (i.e., graphitic N and Fe–N coordination sites), and existence of Fe/Fe3C particles encapsulated within graphitic shells. The Fe–N-CNP–CNF synthesized in this study can be one of the cathode catalyst candidates in a next-generation fuel cell system. We also expect that this synthesis approach has shed new light on electrocatalysis research for the further design and synthesis of advanced Fe–N-doped carbons for ORR.
Acknowledgements
This work was financially supported by Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency (JST) and NU-PPC Plasma Chemical Technology Laboratory.Notes and references
- (a) S. B. Adler, Chem. Rev., 2004, 104, 4791 CrossRef CAS PubMed; (b) K. Shimizu, L. Sepunaru and R. G. Compton, Chem. Sci., 2016, 7, 3364 RSC.
- (a) Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184 RSC; (b) S. Guo, S. Zheng and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526 CrossRef CAS PubMed.
- (a) F. J. Berger, Science, 1999, 286, 49 CrossRef; (b) Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558 CrossRef CAS; (c) S. Sheng, X.-Z. Yuan, J. N. C. Hin, H. Wang, K. A. Friedrich and M. Schulze, J. Power Sources, 2009, 194, 588 CrossRef.
- (a) A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238 RSC; (b) H. Shi, Y. Shen, F. He, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Mater. Chem. A, 2014, 2, 15704 RSC; (c) M. Zhou, H.-L. Wang and S. Guo, Chem. Soc. Rev., 2016, 45, 1273 RSC; (d) R. Raj, A. Samanta, S. H. Noh, S. Mondai, T. Okajima and T. Ohsaka, J. Mater. Chem. A, 2016, 4, 11156 RSC; (e) M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594 CrossRef CAS PubMed.
- (a) J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC; (b) J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244 CrossRef CAS; (c) L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823 CrossRef CAS PubMed.
- (a) R. L. Toh, Z. Sofer and M. Pumera, ChemPhysChem, 2015, 16, 3527 CrossRef CAS PubMed; (b) J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS PubMed.
- (a) Y. Feng, A. Gago, L. Timperman and N. Alonso-Vate, Electrochim. Acta, 2011, 56, 1009 CrossRef CAS; (b) M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986 RSC.
- (a) G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed; (b) F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnsto and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 RSC; (c) Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167 RSC.
- (a) M. Lefèvre, E. Proietti, F. Jaouen and P.-P. Dodelet, Science, 2009, 324, 71 CrossRef PubMed; (b) L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027 CrossRef CAS PubMed; (c) A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed; (d) Y. Qian, P. Du, P. Wu, C. Cai and D. F. Gervasio, J. Phys. Chem. C, 2016, 120, 9884 CrossRef CAS; (e) J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2016, 9, 2418 RSC; (f) C. E. Szakacs, M. Lefèvre, U. I. Kramm, J.-P. Dodelet and F. Vidala, Phys. Chem. Chem. Phys., 2014, 16, 13654 RSC.
- S. Zhang, H. Zhang, Q. Liu and S. Chen, J. Mater. Chem. A, 2013, 1, 3302 CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- Y.-W. Ju, S. Yoo, C. Min, S. Kim, I.-Y. Jeon, J. Shin, J.-B. Baek and G. Kim, Adv. Sci., 2016, 3, 1500205 CrossRef PubMed.
- Y. Zhang, L.-B. Huang, W.-J. Jiang, X. Zhang, Y.-Y. Chen, Z. Wei, L.-J. Wan and J.-S. Hu, J. Mater. Chem. A, 2016, 4, 7781 CAS.
- S. Zhang, B. Liu and S. Chen, Phys. Chem. Chem. Phys., 2013, 15, 18482 RSC.
- (a) G. Panomsuwan, S. Chiba, Y. Kaneko, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2014, 2, 18677 RSC; (b) G. Panomsuwan, N. Saito and T. Ishizaki, Phys. Chem. Chem. Phys., 2015, 17, 6227 RSC; (c) G. Panomsuwan, N. Saito and T. Ishizaki, Carbon, 2016, 98, 411 CrossRef CAS; (d) G. Panomsuwan, N. Saito and T. Ishizaki, ACS Appl. Mater. Interfaces, 2016, 8, 6962 CrossRef CAS PubMed.
- (a) G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972 RSC; (b) G. Panomsuwan, N. Saito and T. Ishizaki, Electrochem. Commun., 2015, 59, 81 CrossRef CAS.
- D.-W. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2015, 17, 407 RSC.
- (a) D.-W. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2014, 16, 14905 RSC; (b) K. Hyun, T. Ueno, G. Panomsuwan, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2016, 18, 10856 RSC.
- (a) Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557 CrossRef CAS; (b) A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477 CrossRef CAS PubMed.
- (a) H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS PubMed; (b) C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517 RSC; (c) G. Tao, L. Zhang, L. Chen, X. Cui, Z. Hua, M. Wang, J. Wang, Y. Chen and J. Shi, Carbon, 2015, 86, 108 CrossRef CAS.
- L. Lin, Q. Zhu and Z.-X. Hu, J. Am. Chem. Soc., 2014, 136, 11027 CrossRef CAS PubMed.
- (a) G. Liu, X. Li, J.-W. Lee and B. N. Popov, Catal. Sci. Technol., 2011, 1, 207 RSC; (b) J. R. Pels, F. Kaptejin, J. A. Moulihin, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641 CrossRef CAS.
- (a) G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More, D. J. Myers and P. Zelenay, J. Mater. Chem., 2011, 21, 11392 RSC; (b) Y. Zhu, B. Zhang, X. Liu, D.-W. Wang and D. S. Su, Angew. Chem., Int. Ed., 2014, 53, 10673 CrossRef CAS PubMed.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 3570 CrossRef CAS PubMed.
- (a) W. He, C. Jiang, J. Wang and L. Lu, Angew. Chem., It. Ed., 2014, 53, 9503 CrossRef CAS PubMed; (b) R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed; (c) Y. Su, H. Jiang, Y. Zhu, X. Yang, J. Shen, W. Zou, J. Chen and C. Li, J. Mater. Chem. A, 2014, 2, 7281 RSC; (d) H. J. Cui, H. M. Yu, J. F. Zheng, Z. J. Wang, Y. Y. Zhu, S. P. Jia, J. Jia and Z. P. Zhu, Nanoscale, 2016, 8, 2795 RSC; (e) S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed; (f) L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 RSC.
- E. Claude, T. Addou, J.-M. Latour and P. Aldebert, J. Appl. Electrochem., 1998, 28, 57 CrossRef CAS.
- (a) A. Bonakdarpour, M. Lefevre, R. Yang, F. Jaouen, T. Dahn, J.-P. Dodele and R. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B105 CrossRef CAS; (b) X. Li, H.-J. Zhang, H. Li, C. Deng and J. Yang, Electrochem. Solid-State Lett., 2014, 3, H33 CrossRef CAS.
- M. Inaba, H. Yamada, J. Tokunaga and A. Tasaka, Electrochem. Solid-State Lett., 2004, 7, A474 CrossRef CAS.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720 CrossRef CAS.
- (a) S. L. Gojković, S. Gupta and R. F. Savinell, J. Electroanal. Chem., 1999, 462, 63 CrossRef; (b) U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134 CrossRef CAS; (c) S. Ye and A. K. Vijh, J. Solid State Electrochem., 2005, 9, 146 CrossRef CASS. Litster and G. Mclean, J. Power Sources, 2004, 130, 61 CrossRef CAS.
- J. Chlistunoff and J.-M. Sansiñena, J. Electroanal. Chem., 2016, 780, 134 CrossRef CAS.
- (a) P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83 CrossRef CAS; (b) L. Xu, G. Pan and X. Laing, RSC Adv., 2014, 4, 19756 RSC.
- (a) G.-L. Tian, M.-Q. Zhao, D. Yu, X.-Y. Kong, J.-Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251 CrossRef CAS PubMed; (b) S. Rasto, I. Kruusenberg, M. Vikkisk, U. Joost, E. Shulga, I. Kink, T. Kallio and K. Tammevski, Carbon, 2014, 73, 361 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterizations, electrochemical measurements, optical emission spectrum of plasma generated in cyanopyridine-2, electron microscopy images (FE-SEM, TEM, and HR-TEM images), XRD patterns, Raman spectra, N2 adsorption–desorption isotherms, BJH pore-size distributions, XPS measurements (survey spectra and high-resolution C 1s, O 1s, and N 1s spectra), CV curves in N2- and O2-saturated electrolytes, RDE measurements, K–L plots, n derived from the K–L plots as a function of potential, comparison table of ORR activity between the catalyst in this work and Fe–N-doped carbons in recent literature. See DOI: 10.1039/c6ra24214f |
| This journal is © The Royal Society of Chemistry 2016 |