DOI:
10.1039/C6RA20765K
(Paper)
RSC Adv., 2016,
6, 113826-113833
Microporous organic polymers based on tetraethynyl building blocks with N-functionalized pore surfaces: synthesis, porosity and carbon dioxide sorption†
Received
17th August 2016
, Accepted 15th November 2016
First published on 30th November 2016
Abstract
A series of microporous organic polymers (MOPs) based on tetraethynyl monomers such as tetrakis(4-ethynylphenyl)methane and tetrakis(4-ethynylphenyl)silane was synthesized via conventional Sonogashira–Hagihara coupling reaction. The resulting MOPs were characterized by thermogravimetric analyses, IR-spectra, scanning electron microscopies, and the Brunauer–Emmett–Teller (BET) method. The incorporation of triphenylamine or azobenzene moieties into the polymer skeleton increases the number of electron donating basic nitrogen sites in the porous frameworks. Thus, these MOPs could exhibit efficient adsorption of Lewis acidic CO2 molecules and display good CO2-over-N2 selectivity. The triphenylamine-based polymer, TEPM-TPA, shows a high BET specific surface area up to 1072 m2 g−1 with a moderate CO2 uptake capacity of 2.41 mmol g−1 at 273 K and 1.13 bar. As for separation of CO2, both TEPM-Azo and TEPS-Azo exhibit relatively high CO2-over-N2 selectivities of 70.8 and 64.7 at 273 K, respectively, due to the N2-phobic feature of azo-based polymers.
Introduction
In recent years, global warming has become a major issue in climate change.1 Overwhelming scientific consensus has correlated global warming to anthropogenic CO2 emission. Thus, one of the promising technologies to protect the environment is to develop new efficient adsorbents for CO2 capture and storage (CCS).2 Traditional CO2 capture is based on the chemical adsorption of CO2 using amine-containing solvents.3 However, because of high energy consumption, solvent regeneration, and the corrosion of the equipment, scientists are seeking new materials for CCS.
Microporous organic polymers (MOPs) have drawn much attention due to their promising applications such as gas separation matrices,4–6 heterogeneous catalysis,7–10 supercapacitors,11–13 and sensing materials.14–16 MOPs generally possess large surface area, excellent chemical and thermal stability, tunable pore properties, low skeleton density, and synthetic diversity.6 These features could make MOPs became strong candidates for post-combustion CCS, and they have been the subject of recent research interests.4,17–29 The surface modification of MOPs is an effective approach to modulate framework–CO2 interactions. It has been proved that the introduction of nitrogen functionalities into the backbone of MOPs could enhance CO2 and energy storage properties owing to Lewis-acid–Lewis-base electrostatic interactions of the nitrogen atoms with CO2 molecules.17,20–23,30–34 Therefore, synthesis of new MOPs using nitrogen-rich monomers has attracted considerable scientific interest in recent years. A range of MOPs have been successfully developed for CCS applications based on various nitrogen-containing building blocks, such as covalent triazine frameworks,30–32 benzimidazole-linked polymers,33,34 carbazole-based MOPs,20,23 and triphenylamine-containing polymers.21 A recent study by Patel and coworkers has shown that nitrogen–nitrogen double bonds (azo-bond) in the porous polymer main chain can play important roles in CO2 storage and separation application.35 These new azo-functionalized MOPs demonstrated that the π-conjugated azo-bond could act as an electron donor, and thus enhance host–guest interactions.35–37
Cooper and coworkers applied Sonogashira–Hagihara coupling chemistry to prepare a series of MOPs with controllable pore dimensions and surface areas by varying building blocks.38 Impressively, the use of tetraethynyl monomers such as tetrakis(4-ethynylphenyl)methane28 and tetrakis(4-ethynylphenyl)silane29 is prominent for the generation of 3D-porous assemblies. Their strong covalent linkage leads to high chemical and thermal stability. In this work, we have rationally designed and synthesized a series of nitrogen-rich MOPs by copolymerization of tetraethynyl monomers with tris(4-iodophenyl)amine or 4,4′-diiodoazobenzene via Sonogashira–Hagihara coupling reaction. The highly cross-linked porous structure can substantially increase the surface area of the resulting polymer networks and, on the other hand, the incorporation of azobenzene or triphenylamine groups from the building block into the polymer skeleton could enhance the binding affinity between the adsorbent and CO2 molecules, and thus lead to the increase of CO2 capture capacity. As we expected, azo-based polymers exhibited excellent CO2 adsorption selectivity against N2. This unique property of TEPM-Azo and TEPS-Azo, along with their chemical and thermal stability, makes them ideal candidates for post-combustion CO2 separation.
Experimental
Materials
All the reagents, unless otherwise specified, were purchased from Sigma-Aldrich Co., Acros, and Tokyo Chemical Industry Co., Ltd. and were used without further purification. All the solvents were further purified under nitrogen flow. Tetrakis(4-ethynylphenyl)methane (1), tetrakis(4-ethynylphenyl)silane (2) and tris(4-iodophenyl)amine (3) were prepared according to the previously reported method.28,29,39
Measurements
The NMR spectra were recorded on an Agilent 300-MR NMR spectrometer. Thermogravimetric analysis (TGA) was performed by a differential thermal analysis instrument (Q1000DSC + LNCS + FACSQ600SDT) over the temperature range from 30 to 1000 °C with a heating and cooling rate of 10 °C min−1 under nitrogen. Solid state magic angle spinning 13C CP/MAS NMR measurements were carried out on a Varian Infinity-Plus 300 MHz NMR spectrometer at a MAS rate of 10 kHz. The FT-IR spectra were collected in transmission on a Tensor 27 FT-IR spectrometer (Bruker) using KBr disks. The polymer morphology was achieved using a field-emission scanning electron microscopy (SEM) (JSM-7001F, JEOL, Tokyo, Japan). Powder X-ray diffraction measurement (PXRD) was carried out on an X-ray diffractometer (D/Max-3c). The measurements of the surface area and pore-size distributions (PSDs) of the samples were performed on an ASAP 2420-4 (Micromeritics) volumetric adsorption analyzer by means of nitrogen adsorption and desorption at 77.3 K. Samples were degassed at 120 °C for 15 h under vacuum (10−5 bar) before analysis. The surface areas were calculated in the relative pressure (P/P0) range from 0.05 to 0.20. PSDs and pore volumes were derived from the adsorption branches of the isotherms using the nonlocal density functional theory (NL-DFT). Gas sorption isotherms were measured on an ASAP 2420-4 as well.
Synthesis
4,4′-Diiodoazobenzene (4). 4-Iodoaniline (6 g, 27.4 mmol), KMnO4 (10.5 g, 6.26 mmol) and CuSO4·5H2O were added to a degassed solution of dichloromethane (160 mL) and stirred at room temperature for 3 days. The reaction mixture was filtered through Celite and the solvent was evaporated under reduced pressure. The crude product was purified on a silica gel column using a petroleum ether/dichloromethane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) mixture as eluent. An orange solid of 4 was isolated in 15% yield (0.89 g). 1H NMR (300 MHz, CDCl3), δ (TMS, ppm): 7.84 (d, 4H), 7.61 (d, 4H).
:1 by volume) mixture as eluent. An orange solid of 4 was isolated in 15% yield (0.89 g). 1H NMR (300 MHz, CDCl3), δ (TMS, ppm): 7.84 (d, 4H), 7.61 (d, 4H).
Polymerization
All of the polymer networks were synthesized by palladium-catalyzed Sonogashira–Hagihara cross-coupling polycondensation of arylethynylenes and aryl halides. The molar ratio of ethynyl to halogen functionalities in the monomer feed was set at 1.5:1 according to the reported procedure.38 The purification of the polymers was conducted in air with yield of 80–90%. A typical procedure for TEPM-Azo is given below:
Tetrakis(4-ethynylphenyl)methane (81 mg, 0.195 mmol), 4,4′-diiodoazobenzene (113 mg, 0.26 mmol), tetrakis(triphenylphosphine)palladium(0) (15 mg), and copper(I) iodide (10 mg) were dissolved in a mixture of anhydrous DMF (5 mL) and triethylamine (5 mL). The reaction mixture was heated to 90 °C and stirred for 72 h under nitrogen atmosphere. The mixture was cooled to room temperature, and the precipitated polymer networks was filtered and washed four times with dichloromethane, water, methanol, and acetone to remove any unreacted monomer or catalyst residues. The product was dried in vacuum for 24 h at 100 °C. Yield: 80.1%. Apparent BET surface area: 564 cm3 g−1.
Result and discussion
Synthesis and characterization
The synthesis of 4,4′-diiodoazobenzene (monomer 4) is shown in Scheme 1. Starting from the commercially available 4-iodoaniline, 4,4′-diiodoazobenzene was prepared by the oxidative coupling of two equivalents of 4-iodoaniline in the presence of CuSO4·5H2O and KMnO4 under nitrogen atmosphere in dichloromethane.40 The polymer networks in this study were synthesized by palladium-catalyzed Sonogashira–Hagihara coupling polycondensation. The polymerization reactions were performed at a fixed reaction temperature (90 °C) and the reaction time (72 h). The molar ratio of ethynyl to iodine functionalities in the monomer feed was fixed at 1.5:1 since maximum surface areas was observed at this ratio based on the previous reports.38 The general synthetic routes to the polymer networks are shown in Scheme 1. The polymer networks are insoluble in common organic solvents due to their highly cross-linked structures and the rigid skeleton. The FT-IR spectra revealed no residual trace of the specific vibration bands related to terminal alkynes at around 3280 cm−1,28 therefore suggesting high conversion of the starting monomers (Fig. S1†). The 13C solid-state NMR (CP/MAS) measurements were carried out to study the local structures of these polymers (Fig. S2†). All of the carbon signals with a chemical shift exceeding 110 ppm are related to aromatic carbon atoms of the building phenylene groups in the framework. The peak at around 91 ppm in the NMR spectra is assigned to the sp carbons in alkyne bonds, thus suggesting that this cross-coupling reaction is effective for the synthesis of these polymer networks.41,42 A well-resolved peak is detected at 65 ppm associated with the quaternary carbon atom in tetrakis(4-ethynylphenyl)methane, indicating that tetrakis(4-ethynylphenyl)methane is the main building units in the polymer networks TEPM-TPA and TEPM-Azo.41,42 Additionally, the azo-linkage formation was further confirmed by the presence of characteristic signals for the –C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C– bond at around 152 ppm in NMR spectra of azo-based polymers.35 TGA measurements were carried out to evaluate the thermal stability of these polymers (Fig. S3†). All the polymer networks exhibit good stability, showing less than 5% weight loss at around 480 °C under nitrogen flow. Powder X-ray diffraction measurements suggested that all the polymer networks are amorphous solid in nature (Fig. S4†). The SEM images of the obtained polymer networks are shown in Fig. 1. For the polymer networks, similar morphology can be observed to be solid submicrometer particles with different size range from 10 to 100 nm.
N–C– bond at around 152 ppm in NMR spectra of azo-based polymers.35 TGA measurements were carried out to evaluate the thermal stability of these polymers (Fig. S3†). All the polymer networks exhibit good stability, showing less than 5% weight loss at around 480 °C under nitrogen flow. Powder X-ray diffraction measurements suggested that all the polymer networks are amorphous solid in nature (Fig. S4†). The SEM images of the obtained polymer networks are shown in Fig. 1. For the polymer networks, similar morphology can be observed to be solid submicrometer particles with different size range from 10 to 100 nm.
 |
| | Scheme 1 Synthetic routes to 4,4′-diiodoazobenzene (4) and the polymer networks. | |
 |
| | Fig. 1 SEM images of TEPM-TPA (a), TEPM-Azo (b), TEPS-TPA (c), TEPS-Azo (d), scale bars: 1 μm. | |
Porosity measurements
In order to study the porosity of these polymer networks, sorption analysis using nitrogen as the sorbent molecules was carried out. Nitrogen adsorption/desorption isotherms of these materials measured at 77.3 K are shown in Fig. 2a. The polymer networks exhibited Type I isotherms with a steep nitrogen gas uptake at low pressure according to the International Union of Pure and Applied Chemistry (IUPAC) classification, suggesting that micropores are dominant in these polymer networks.18 The gradual increase in N2 uptake (P/P0 = 0.05–0.9) at 77.3 K is due to the presence of mesopores in the polymers. The significant hysteresis between adsorption and desorption was observed in the isotherms for all the polymer networks in the whole range of relative pressure, which is in agreement with the fact that all the MOPs contain both meso- and microporosity. As shown in Fig. 2b, similar PSD profiles based on the adsorption branch of the isotherm using nonlocal density functional theory (NL-DFT) were found for the obtained polymer TEPM-TPA and TEPS-TPA. Both polymers show abundant micropore structures with micropore diameters centered at around 1.1 nm. As for the polymer TEPM-Azo and TEPS-Azo, the PSDs are relative broader in the range of 1.1–2.1 nm compared with those of TEPM-TPA and TEPS-TPA, indicating that much more mesopores and/or macropores in these two polymers. Besides, TEPM-Azo and TEPS-Azo show a spot mesopores peaks at around 5 nm. The PSD curves of the polymer networks are in good agreement with the shape of the nitrogen isotherms (Fig. 2a) and suggest that the pores of the polymer networks are dominated by micropores. The contribution of microporosity to the networks can be calculated as the ratio of the micropore volume over the total pore volume. As shown in Table 1, the ratios indicated that polymer networks TEPM-TPA, TEPS-TPA, TEPM-Azo, and TEPS-Azo possess a ratio of 0.63, 0.74, 0.79, and 0.90, respectively. The BET specific surface areas of TEPM-TPA and TEPS-TPA are 1072 and 937 m2 g−1, respectively, which are higher than those of TEPS-Azo (564 m2 g−1) and TEPM-Azo (598 m2 g−1) containing the azobenzene building block. It should be noted that TEPM-TPA and TEPS-TPA exhibit comparable surface area to the most of reported conjugated microporous poly(aryleneethynylene) (PAE) networks by Sonogashira–Hagihara cross-coupling chemistry.26
 |
| | Fig. 2 (a) Nitrogen adsorption/desorption isotherms (the adsorption branch is labeled with filled symbols and desorption branch is labeled with open symbols); (b) pore size distribution curves calculated by NL-DFT for the polymer networks. | |
Table 1 Summary of pore properties for the polymer networks
| Polymer |
SBETa (m2 g−1) |
SMicrob (m2 g−1) |
VMicroc (cm3 g−1) |
VTotald (cm3 g−1) |
SMicro/SBET (%) |
VMicro/VTotal (%) |
| Surface area calculated from N2 adsorption isotherm in the relative pressure (P/P0) range from 0.05 to 0.20. Micropore surface area calculated from the N2 adsorption isotherm using the t-plot method based on the Harkins–Jura equation. The micropore volume derived from the t-plot method. Total pore volume at P/P0 = 0.988. |
| TEPM-TPA |
1072 |
901 |
0.446 |
0.708 |
84.05 |
62.99 |
| TEPM-Azo |
598 |
552 |
0.267 |
0.338 |
92.3 |
78.99 |
| TEPS-TPA |
937 |
787 |
0.396 |
0.536 |
83.99 |
73.88 |
| TEPS-Azo |
564 |
489 |
0.236 |
0.261 |
86.7 |
90.42 |
Gas uptake of CO2, H2, and CH4
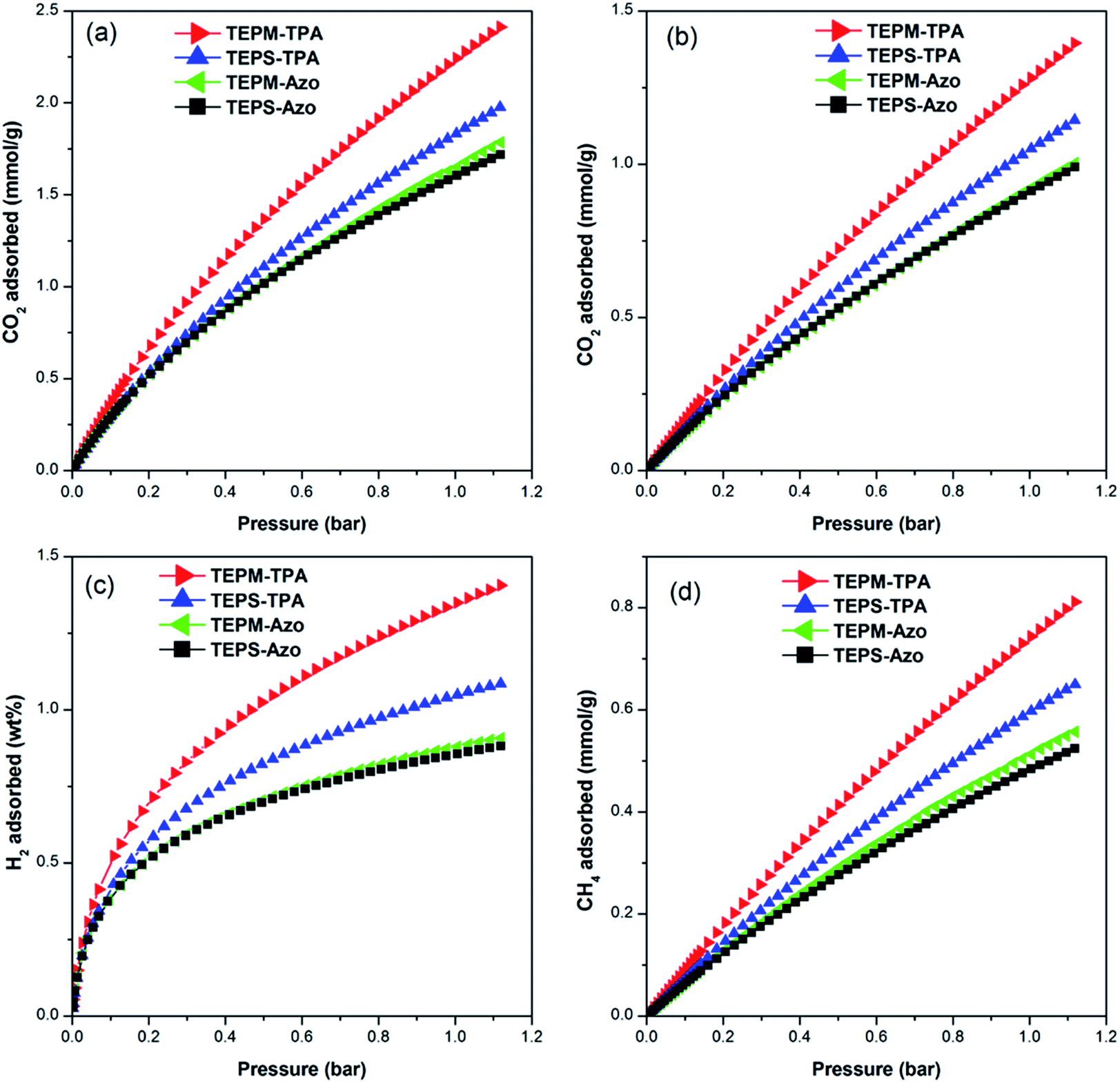
Because of the considerable porosity and nitrogen-rich features, we were interested in assessing their performance in gas uptake. The CO2 uptake of the polymer networks were measured up to 1.13 bar at 273, 283, and 298 K, respectively (Fig. 3a and b, S5,† and Table 2). It can be seen that the CO2 uptake capacity enhances monotonically with increasing CO2 pressure. TEPM-TPA exhibited the highest CO2 uptake at both 273 and 298 K, adsorbing 2.41 and 1.39 mmol g−1 at 1.13 bar among the resulting four polymers. When azobenzene is copolymerized with tetrakis(4-ethynylphenyl)methane, the absolute CO2 uptake capacity of the polymer decreases from 2.41 mmol g−1 for TEPM-TPA to 1.78 mmol g−1 for TEPM-Azo. This could be explained by the decrease of surface area and pore volume for the polymer networks as mentioned above (Table 1). The CO2 uptake of 2.41 mmol g−1 for TEPM-TPA is comparable to that of many reported MOPs produced by Sonogashira–Hagihara coupling reaction at the same conditions, such as the conjugated microporous PAE networks without nitrogen-containing building blocks and their metalized derivatives,41,43–47 tetraphenylmethane-based hypercrosslinked porous aromatic frameworks (2.27 mmol g−1 for PAF-32-OH),48 the tri(4-ethynylphenyl)amine-based porous aromatic frameworks (1.19–2.50 mmol g−1),42 the triazine-functionalized CMPs (2.62 mmol g−1 for TNCMP-2),49 the phenolsulfonephthalein-containing CMPs (2.77 mmol g−1 for BFCMP-2),50 but still lower than that of other tetrakis(4-ethynylphenyl)methane-based MOPs (3.77 mmol g−1, 5.07 mmol g−1, and 4.28 mmol g−1 for F-MOP-1, F-MOP-2, and HEX-POP-3, respectively),51,52 the imine-linked porous polymer frameworks (e.g. 6.1 mmol g−1 for PPF-1),53 the benzimidazole-incorporated porous polymer network of PPN-101 (5.1 mmol g−1),54 the microporous polycarbazole of CPOP-1 (4.8 mmol g−1),55 and the nanoporous azo-linked polymer of ALP-1 (5.36 mmol g−1).36 To determine the binding affinity between the polymer network and CO2 molecules, the isosteric heats of adsorption (Qst) were calculated from the CO2 adsorption isotherms at 273, 283, and 298 K by using the Clausius–Clapeyron equation. As illustrated in Fig. 4, the Qst values of the obtained MOPs are in the range from 22 to 33 kJ mol−1 at the near zero coverage. TEPM-Azo shows the highest Qst of 33 kJ mol−1 among the polymers, which could be attributed to the high nitrogen content from azo groups in the skeleton of MOPs leading to a strong induce-dipole force with CO2 molecules.35–37 TEPS-Azo shows somewhat lower Qst values than TEPM-Azo. We suspect that even though the azo framework is same in both, TEPS-Azo with tetrakis(4-ethynylphenyl)silane units in the structure generated diminish basicity in the nitrogen species compared with those in TEPM-Azo with no Si (silicon is acidic in nature).56 As for triphenylamine-containing polymers, the Qst value of TEPS-TPA is lower than that of TEPM-TPA, which is further support the above results in the case of azo-based polymers.
 |
| | Fig. 3 (a) CO2 adsorption isotherms collected at 273 K up to 1.13 bar for the polymer networks; (b) CO2 adsorption isotherms collected at 298 K; (c) volumetric H2 sorption curves collected at 77.3 K; (d) CH4 adsorption isotherms collected at 273 K. | |
 |
| | Fig. 4 Isosteric heats of adsorption for CO2 calculated from the adsorption isotherms collected at 273, 283, and 298 K. | |
H2 and CH4 are important potential clean fuels for automotive technology; therefore, we also investigated the uptake capacities of these two gases by volumetric methods. The hydrogen sorption performance for the polymers networks was measured at 77.3 K up to a pressure of 1.13 bar. All adsorption isotherms show a gradual rise and reached a maximum of 0.88–1.4 wt% for hydrogen uptake (Fig. 3c). The H2 uptake sorption properties of polymer networks can be correlated with the micropore volume and micropore surface area, which was also observed in other MOPs. Triphenylamine-based polymer networks exhibit higher H2 adsorption capacity than azo-based polymer networks under the same conditions because of their lager surface areas and narrower pore sizes. The CH4 uptakes at 273 K up to 1.13 bar (Fig. 3d) were also conducted. As expected, TEPM-TPA having the highest micropore volume and highest surface area shows the largest CH4 uptake of 0.81 mmol g−1.
Selective CO2 capture over N2 and CH4
We also investigated the CO2 sorption selectivity over N2 and CH4 to assess their potential application in gas separation. The CO2, N2, and CH4 adsorption isotherms were measured at 273 K up to 1.13 bar (Fig. S6†). The selectivity of the polymer networks was estimated using the ratios of the Henry's law constant calculated from the initial slopes of the single-component gas adsorption isotherms collected at 273 K at low pressure coverage less than 0.15 bar, which is typical partial pressure of CO2 in flue gas. As shown in Table 2, TEPM-Azo and TEPS-Azo exhibit relative high CO2/N2 selectivity ratios of 70.8 and 64.7 at 273 K, whereas TEPM-TPA and TEPS-TPA show poor selectivities of 26.3 and 20.5, respectively. These results suggested that the azo-based polymer is efficient for CO2/N2 separation as expected because of the “N2-phobicity” of the azo-linkage.35–37 The differences in CO2/N2 selectivity between azo- and triphenylamine-based polymer networks indicate that in addition to the function of the surface area and pore size, the linkage is very important factor that affect CO2 uptake and the CO2/N2 selectivity. The calculated CO2/N2 selectivities by both TEPM-Azo and TEPS-Azo are significantly higher than those of TEPM-TPA, TEPS-TPA, and some other MOPs such as tetraethynylspirobifluorene-based porous organic polymers POP (6.7–11.8 at 298 K),57 porous hypercrosslinked aromatic polymers (PHAPs) using tetrahedral precursors (29.3–34.2 at 273 K),56 tetraphenylsilane-containing porous covalent triazine polymer networks PCTP-2 (31.6 at 298 K),58 tetra(4-(N-carbazolyl)phenyl)silane-based polycarbazole microporous polymer P-TCzPhSi (24.8 at 273 K),59 the nanoporous azo-bridged polymers (30–43 at 273 K),36 the carbazolic porous organic frameworks (19–37 at 273 K),60 the imine-linked porous polymer frameworks (14.5–20.4 at 273 K),53 the triazine-functionalized CMPs (9.0–25.2 at 298 K),49 and the amide-functionalized CMPs (8.5–12 at 298 K) under the similar conditions.61 Moreover, they are also comparable to the best azo-polymers ever reported like azo-COPs,37 tri(4-ethynylphenyl)amine-containing polymers (72 and 63.9 for PAF-34 and PAF-34-OH, respectively),42 tetrakis(4-ethynylphenyl)methane-based porous aromatic frameworks PAF-18-OH, PAF-26-COOH, and their metalized derivatives (16–73 at 298 K).41,46 As for separation of CH4, the calculated CO2/CH4 adsorption selectivity was about from 3.9 to 4.6 for the four polymers at 273 K. The results are comparable to that of most other reported porous organic polymers, such as tetrakis(4-ethynylphenyl)methane-based porous materials PAF-26 (4–9 at 298 K),41 tetraethynylspirobifluorene-containing porous organic polymers POP (3.4–4.8 at 298 K),57 the carbazole-based microporous organic polymer of Cz-POF (4.4–7.1 at 273 K),60 the nanoporous covalent triazine-based frameworks (5–7 at 273 K),62 the tetra-armed triphenylamine-containing MOPs (3.5–4.3 at 273 K),63 and the azo-containing polymer networks ALP (8 at 273 K)36 under the same conditions. The CO2/CH4 selectivity of the resultant polymers is considerably lower than that of CO2/N2 because of the higher polarizability of CH4 than that of N2.60
Table 2 Summary of gas uptakes values for the polymer networks
| Polymer |
H2 uptakea (wt%) |
CH4 uptakeb (mmol g−1) |
CO2 uptakeb (mmol g−1) |
CO2 uptakec (mmol g−1) |
Henry law selectivityd |
IAST selectivitye |
| CO2/N2 |
CO2/CH4 |
CO2/N2 |
CO2/CH4 |
| Data collected by volumetric H2 sorption method at 77.3 K and 1.13 bar. Data collected at 273 K and 1.13 bar. Data collected at 298 K and 1.13 bar. Adsorption selectivity based on the Henry's law. Calculated from ideal adsorbed solution theory (IAST) by using the CO2, N2, and CH4 adsorption isotherms collected at 273 K. |
| TEPM-TPA |
1.4 |
0.81 |
2.41 |
1.39 |
26.3 |
4.6 |
36.9 |
5.1 |
| TEPM-Azo |
0.91 |
0.55 |
1.78 |
1 |
70.8 |
4.6 |
121.1 |
6.3 |
| TEPS-TPA |
1.08 |
0.65 |
1.97 |
1.14 |
20.5 |
3.9 |
30.5 |
4.9 |
| TEPS-Azo |
0.88 |
0.52 |
1.71 |
0.99 |
64.7 |
4.3 |
111.9 |
6.5 |
In addition, the gas adsorption selectivity for mixtures of CO2:N2 (0.15:0.85) and CO2:CH4 (0.05:0.95) were predicted by using ideal adsorbed solution theory (IAST) at a standardized temperature of 273 K. The method allows for the determination of the selectivities as a function of pressure, and has been widely used to predict binary gas mixture adsorption behaviors in many MOPs.64,65 The adsorption data in Table 2 exhibit that the calculated IAST selectivities are higher than the values obtained from Henry's constant ratios, as observed for most reported porous materials.17,30,31
Conclusions
In summary, we have synthesized a series of microporous organic polymer networks by using Sonogashira–Hagihara cross-coupling chemistry from two kinds of tetrahedral-shaped monomers of tetrakis(4-ethynylphenyl)methane and tetrakis(4-ethynylphenyl)silane. The resulting polymer networks incorporated nitrogen-rich triphenylamine or azobenzene groups present a high affinity to CO2 molecules, which has been significantly verified by gas adsorption measurements. TEPM-TPA shows high BET specific surface area up to 1072 m2 g−1 with a moderate CO2 uptake capacity of 2.41 mmol g−1 at 273 K and 1.13 bar, which reinforce the benefits of tetrahedral monomers for the construction of high surface area materials. In addition, TEPM-Azo and TEPS-Azo exhibit relative high CO2/N2 selectivity of 70.8 and 64.7 at 273 K, respectively, which compared to that of some other reported polymer networks.
Acknowledgements
This work was supported by Natural Science Foundation of China (Grant No. 51103111, 21304055 and 21574077), Education Ministry of China (Program for NCET-12-0714), Shaanxi Innovative Team of Key Science and Technology (2013KCT-17), the Fundamental Research Funds for the Central Universities (GK201501002), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (2014-skllmd-11), and Graduate Innovative Fund of Wuhan Institute of Technology (Grant No. CX2014017, CX2014026).
Notes and references
- J. A. Logan, J. Regniere and J. A. Powell, Front. Ecol. Environ., 2003, 1, 130–137 CrossRef.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- F. Li and L. S. Fan, Energy Environ. Sci., 2008, 1, 248–267 CAS.
- S. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- J. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- F. Xu, X. Chen, Z. Tang, D. Wu, R. Fu and D. Jiang, Chem. Commun., 2014, 50, 4788–4790 RSC.
- C. Zhang, X. Yang, W. Ren, Y. Wang, F. Su and J. Jiang, J. Power Sources, 2016, 317, 49–56 CrossRef CAS.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- Y. Zhang, A. Sigen, Y. Zou, X. Luo, Z. Li, H. Xia, X. Liu and Y. Mu, J. Mater. Chem. A, 2014, 2, 13422–13430 CAS.
- J. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- X. Wang, Y. Zhao, L. Wei, C. Zhang and J. Jiang, J. Mater. Chem. A, 2015, 3, 21185–21193 CAS.
- Y. Zhang, Y. Li, F. Wang, Y. Zhao, C. Zhang, X. Wang and J. Jiang, Polymer, 2014, 55, 5746–5750 CrossRef CAS.
- S. Qiao, Z. Du, C. Yang, Y. Zhou, D. Zhu, J. Wang, X. Chen and R. Yang, Polymer, 2014, 55, 1177–1182 CrossRef CAS.
- S. Qiao, Z. Du and R. Yang, J. Mater. Chem. A, 2014, 2, 1877–1885 CAS.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J. Jiang, J. Mater. Chem. A, 2014, 2, 15139–15145 CAS.
- C. Gu, D. Liu, W. Huang, J. Liu and R. Yang, Polym. Chem., 2015, 6, 7410–7417 RSC.
- L. Pan, Q. Chen, J. Zhu, J. Yu, Y. He and B. Han, Polym. Chem., 2015, 6, 2478–2487 RSC.
- W. Huang, C. Gu, T. Wang, C. Gu, S. Qiao and R. Yang, RSC Adv., 2014, 4, 62525–62531 RSC.
- J. Huang, X. Zhou, A. Lamprou, F. Maya, F. Svec and S. R. Turner, Chem. Mater., 2015, 27, 7388–7394 CrossRef CAS.
- U. H. F. Bunz, K. Seehafer, F. L. Geyer, M. Bender, I. Braun, E. Smarsly and J. Freudenberg, Macromol. Rapid Commun., 2014, 35, 1466–1496 CrossRef CAS PubMed.
- W. Lu, Z. Wei, D. Yuan, J. Tian, S. Fordham and H. Zhou, Chem. Mater., 2014, 26, 4589–4597 CrossRef CAS.
- J. Chun, J. H. Park, J. Kim, S. M. Lee, H. J. Kim and S. U. Son, Chem. Mater., 2012, 24, 3458–3463 CrossRef CAS.
- L. Monnereau, T. Muller, M. Lang and S. Brase, Chem. Commun., 2016, 52, 571–574 RSC.
- W. Song, X. Xu, Q. Chen, Z. Zhuang and X. Bu, Polym. Chem., 2013, 4, 4690–4696 RSC.
- H. A. Patel, F. Karadas, J. Byun, J. Park, E. Deniz, A. Canlier, Y. Jung, M. Atilhan and C. T. Yavuz, Adv. Funct. Mater., 2013, 23, 2270–2276 CrossRef CAS.
- A. Karmakar, A. Kumar, A. K. Chaudhari, P. Samanta, A. V. Desai, R. Krishna and S. K. Ghosh, Chem.–Eur. J., 2016, 22, 4931–4937 CrossRef CAS PubMed.
- S. Altarawneh, T. Islamoglu, A. K. Sekizkardes and H. M. El-Kaderi, Environ. Sci. Technol., 2015, 49, 4715–4723 CrossRef CAS PubMed.
- V. Neti, J. Wang, S. Deng and L. Echegoyen, RSC Adv., 2015, 5, 10960–10963 RSC.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- P. Arab, M. G. Rabbani, A. K. Sekizkardes, T. Islamoglu and H. M. El-Kaderi, Chem. Mater., 2014, 26, 1385–1392 CrossRef CAS.
- H. A. Patel, S. H. Je, J. Park, Y. Jung, A. Coskun and C. T. Yavuz, Chem.–Eur. J., 2014, 20, 772–780 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- M. Grigoras and L. Stafie, High Perform. Polym., 2009, 21, 304–314 CrossRef CAS.
- J. Zeitouny, C. Aurisicchio, D. Bonifazi, R. De Zorzi, S. Geremia, M. Bonini, C. A. Palma, P. Samori, A. Listorti, A. Belbakra and N. Armaroli, J. Mater. Chem., 2009, 19, 4715–4724 RSC.
- H. Ma, H. Ren, X. Zou, S. Meng, F. Sun and G. Zhu, Polym.
Chem., 2014, 5, 144–152 RSC.
- R. Yuan, H. Ren, Z. Yan, A. Wang and G. Zhu, Polym. Chem., 2014, 5, 2266–2272 RSC.
- Z. Yan, H. Ren, H. Ma, R. Yuan, Y. Yuan, X. Zou, F. Sun and G. Zhu, Microporous Mesoporous Mater., 2013, 173, 92–98 CrossRef CAS.
- M. Cha, Y. Lim and J. Chang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2336–2342 CrossRef CAS.
- M. Trunk, A. Herrmann, H. Bildirir, A. Yassin, J. Schmidt and A. Thomas, Chem.–Eur. J., 2016, 22, 7179–7183 CrossRef CAS PubMed.
- H. Ma, H. Ren, X. Zou, F. Sun, Z. Yan, K. Cai, D. Wang and G. Zhu, J. Mater. Chem. A, 2013, 1, 752–758 CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Brase, J. Guenther, J. Blumel, R. Krishna, Z. Li and H. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- X. Jing, D. Zou, P. Cui, H. Ren and G. Zhu, J. Mater. Chem. A, 2013, 1, 13926–13931 CAS.
- S. Ren, R. Dawson, A. Laybourn, J. Jiang, Y. Khimyak, D. J. Adams and A. I. Cooper, Polym. Chem., 2012, 3, 928–934 RSC.
- C. Zhang, X. Yang, Y. Zhao, X. Wang, M. Yu and J. Jiang, Polymer, 2015, 61, 36–41 CrossRef CAS.
- Z. Yang, Y. Zhao, H. Zhang, B. Yu, Z. Ma, G. Ji and Z. Liu, Chem. Commun., 2014, 50, 13910–13913 RSC.
- C. M. Thompson, G. T. McCandless, S. N. Wijenayake, O. Alfarawati, M. Jahangiri, A. Kokash, Z. Tran and R. A. Smaldone, Macromolecules, 2014, 47, 8645–8652 CrossRef CAS.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- M. Zhang, Z. Perry, J. Park and H. Zhou, Polymer, 2014, 55, 335–339 CrossRef CAS.
- Q. Chen, M. Luo, P. Hammershoj, D. Zhou, Y. Han, B. W. Laursen, C. Yan and B. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- P. Puthiaraj and W. Ahn, Ind. Eng. Chem. Res., 2016, 55, 7917–7923 CrossRef CAS.
- Q. Ma, B. Yang and J. Li, RSC Adv., 2015, 5, 64163–64169 RSC.
- P. Puthiaraj, S. Kim and W. Ahn, Chem. Eng. J., 2016, 283, 184–192 CrossRef CAS.
- F. Jiang, J. Sun, R. Yang, S. Qiao, Z. An, J. Huang, H. Mao, G. Chen and Y. Ren, New J. Chem., 2016, 40, 4969–4973 RSC.
- X. Zhang, J. Lu and J. Zhang, Chem. Mater., 2014, 26, 4023–4029 CrossRef CAS.
- T. Ratvijitvech, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Polymer, 2014, 55, 321–325 CrossRef CAS.
- A. Bhunia, I. Boldog, A. Moller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990–14999 CAS.
- X. Yang, S. Yao, M. Yu and J. Jiang, Macromol. Rapid Commun., 2014, 35, 834–839 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- C. Xu and N. Hedin, J. Mater. Chem. A, 2013, 1, 3406–3414 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20765k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.