
The effect of barium sulfate-doped lead oxide as a positive active material on the performance of lead acid batteries†
Xiqing Yuan‡
,
Jingping Hu‡,
Jingyi Xu,
Yucheng Hu,
Wei Zhang,
Jinxin Dong,
Sha Liang,
Huijie Hou,
Xu Wu and
Jiakuan Yang*
School of Environmental Science and Engineering, Huazhong University of Science and Technology (HUST), Wuhan, 430074, P. R. China. E-mail: jkyang@hust.edu.cn; yjiakuan@hotmail.com; Fax: +86-27-87792101; Tel: +86-27-87792207
First published on 7th March 2016
Abstract
Barium sulfate (BaSO4) is a common impurity in recycled lead paste that is challenging to eliminate completely during hydrometallurgical recycling of spent lead acid batteries, so the effect of this impurity in positive active materials on the performance of recycled lead acid batteries was investigated. The BaSO4 doped lead oxide composite was used as a positive active material in positive plates of lead acid batteries with theoretical capacities of 2.0 A h. BaSO4 was retained in the solid phase throughout the battery fabrication process. Different BaSO4 dosages affected the phase of the positive plates during the curing process, with the highest content of metallic lead obtained at a BaSO4 dosage of 0.06 wt%. Morphology analysis indicated that aggregates were formed in the positive plates and the particles became rougher with increasing addition of BaSO4 during the formation process. BaSO4 also demonstrated a large impact on charge/discharge cycles with 100% DOD in battery testing. Analysis of disassembled failed batteries indicated that the expansion and shedding-off of the positive active material were mainly responsible for the failure of these batteries, and this could be attributed to the non-uniform growth of lead oxide on the BaSO4 nucleus, and the accumulation of internal stress.
1 Introduction
Lead–acid batteries have been extensively utilized in electric bicycles,1 energy storage2–5 and many other applications6 over the last few decades. They account for about fifty percent of the battery market.7,8 However, the lead–acid battery industry faces many serious challenges with the development of the energy industry.9 Besides the issue with relatively low theoretical mass capacity and limited utilization of the active mass of lead–acid batteries, the green recovery of spent lead paste is also a big challenge during the recovery of waste lead acid batteries.Two recovery methods of spent lead paste have been widely studied,10 including traditional pyrometallurgical and alternative hydrometallurgical processes. Traditional pyrometallurgical process could have barium sulfate (BaSO4) easily removed from final lead ingot product, however, it is largely criticized due to serious emission of SO2 and release of lead dust to the environment.11 In contrast, hydrometallurgical process has been studied recently as an alternative method for spent lead paste recovery without emission of hazard gases and lead particulates,12 but it couldn't eliminate BaSO4 component completely. Pan et al. proposed a green lead hydrometallurgical process based on a hydrogen–lead oxide fuel cell,13 and demonstrated a new green hydrometallurgical process to recover lead based on this fuel cell, and lead and electricity were produced together with water as the only by-product. Nano-structured lead oxide product was synthesized by calcination of lead citrate precursor recovered from spent lead paste via novel hydrometallurgical process.14 Removal of impurities from the recovered product is a common challenge in the hydrometallurgical process. When lead oxide products recovered from spent lead paste was re-used as active materials for new lead–acid battery fabrication, BaSO4 inevitably existed in both positive active materials (PAM) and negative active materials (NAM). Studies on synthesized leady oxide for PAM of lead acid battery showed low capacity retention ratio15,16 and the existence of BaSO4 were mainly responsible for the poor capacity retention ratio. So, it is inspiring to investigate the effect of BaSO4 impurities in lead oxide as PAM on the performance of lead–acid battery.
Many additives, such as BaSO4, acetylene black, lignosulfonates, have been used in NAM on the negative plate in order to improve the utilization of active mass and cycle stability in lead–acid battery. BaSO4 can be used as bulking agent and has been intensively studied as a common additive of negative plate in the lead–acid battery. It has been shown that BaSO4 lowered the overpotential of PbSO4 nucleation,17 or served as seed crystals for the precipitation of PbSO4.18 However, when lignosulfonates and BaSO4 were utilized as negative plate additives, they have demonstrated to improve the reversibility during the charge–discharge process in the high-rate partial-state-of-charge (HRPSoC) mode and prolong the cycle life time.17 The effect of particles size variation of BaSO4 was also investigated when used as additive for the negative plate,19 and no influence on the cycle-life performance of cells under HRPSoC condition was observed. The effect of BaSO4 and SrSO4 was investigated as seed crystals by electrochemical atomic force microscopy (AFM).20 PbSO4 crystals were formed more rapidly on SrSO4 than on BaSO4 during the discharge process. Electrochemical reactions on negative plate were also investigated using electrochemical AFM to explore the mechanisms of oxidation and reduction on the lead/sulphuric acid interface when barium sulfate was added to the negative plate in the lead–acid battery,18 and the result indicated that BaSO4 provided nucleation seeds for the formation of PbSO4 crystallites. However, there is few reports on the effect of BaSO4 as additive for positive plate on the performance of battery. The effect of BaSO4 as additive in the electrolyte for the electrochemical deposition of leady dioxide was studied, and cyclic voltammetry and battery testing results showed that BaSO4 with concentration of 10−5 M could be used as suitable electrolyte additive to improve the performance of the battery,21 but the effect of BaSO4 as solid additive to the positive plate was still not clear when the plate was prepared through the commercial battery manufacturing process.
In this work, we studied the effect of BaSO4 impurity in PAM on the performance of lead acid battery and investigated the impact on the change of morphology and crystal structure of the plate during the whole battery production process.
2 Experimental
2.1 Materials and chemicals
Simulated spent lead paste (comprising PbSO4, PbO2, PbO, and metallic Pb) was prepared by mixing analytical reagents according to the chemical composition of spent lead paste in spent/discarded lead acid battery, as shown in Table S2.† Synthesized lead oxide was prepared following the method in the literature.14 The solution of sodium citrate and acetic acid were utilized as leaching reagents. Hydrogen peroxide was used as a reduction reagent to reduce lead dioxide to lead monoxide. The lead citrate precursor was firstly synthesized from simulated spent lead paste via hydrometallical process. And then novel ultrafine lead oxide products were prepared by the calcination of lead citrate precursor at 375 °C which was much lower than typical temperature used in pyrometallurgical process. Sodium citrate (Na3C6H5O7·2H2O, 99 wt%), acetic acid (CH3COOH, 99.5%), hydrogen peroxide (H2O2, 30%), BaSO4 (99 wt%) and concentrated sulfuric acid (98% purity) were obtained from Sino-pharmaceutical company of China.2.2 Barium sulfate addition and measurement
The solid phase powder was mixed with synthesized lead oxide (100 g) with different mass dosage of BaSO4 (0.02 wt%, 0.04 wt%, 0.06 wt%, 0.08 wt%, 0.1 wt%) by grinding in a mortar for 40 min. These concentrations were selected according to the typical BaSO4 concentration found in recycled spent lead paste (0.05–0.1 wt%). The concentration of barium ion in the solution was measured by Inductively Coupled Plasma (Optima 8300, PerkinElmer, US) after digesting 0.2 g sample with 15 mL boiled aqua regia.2.3 Electrochemical study of lead oxide
Detailed procedures for the preparation of working electrode (WE) for the electrochemical study are provided as follows. Firstly, lead oxide, carbon black and PVDF were mixed according to the mass ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the mortar, followed by grinding using pestle and mortar for about 30 minutes, and then mixed with NMP to prepare a homogeneous slurry. Then the resulting paste was transferred onto the surface of a glassy carbon electrode, followed by drying in an oven at 60 °C for 24 hours. Cyclic voltammetry (CV) was carried out using a CHI660E electrochemical workstation (Shanghai Chenhua Instruments Co. Ltd., China). The CV curves of lead oxide were acquired at a scan rate of 10 mV s−1 in 3 mol L−1 sulfuric acid in a typical three-electrode system, with a lead oxide sample loaded glassy carbon electrode (GCE) as working electrode (WE), a platinum wire as counter electrode (CE), and a Hg/Hg2SO4/K2SO4(sat.) reference electrode (RE).
:1:1 in the mortar, followed by grinding using pestle and mortar for about 30 minutes, and then mixed with NMP to prepare a homogeneous slurry. Then the resulting paste was transferred onto the surface of a glassy carbon electrode, followed by drying in an oven at 60 °C for 24 hours. Cyclic voltammetry (CV) was carried out using a CHI660E electrochemical workstation (Shanghai Chenhua Instruments Co. Ltd., China). The CV curves of lead oxide were acquired at a scan rate of 10 mV s−1 in 3 mol L−1 sulfuric acid in a typical three-electrode system, with a lead oxide sample loaded glassy carbon electrode (GCE) as working electrode (WE), a platinum wire as counter electrode (CE), and a Hg/Hg2SO4/K2SO4(sat.) reference electrode (RE).
2.4 Battery assembly, testing and dismantling
The preparation procedure of the positive plate, assembly and testing of the battery have been described previously in the literature.15 The positive plates (39.5 mm × 65.6 mm) for designed 2 A h testing battery were produced by standard battery assembly process including paste mixing, curing, formation, washing and drying treatment. The nominal capacity of the assembled battery was determined by the depth of discharge, DOD = 100%. After about 47 charge/discharge cycles, the batteries were dismantled after the final charging procedure. The dismantled positive plate was washed in distilled water to remove excess electrolyte on the surface and dried in an oven at 75 °C for 24 h.The positive paste was scrapped after dismantled, and the compositions of PbSO4 and PbO2 were measured: 0.2 g sample was screened by the 120 mesh sieve, mixed and sonicated with 10 mL 58.86 wt% HNO3 and 6 mL 0.83 wt% H2O2 separately, and mixed with potassium permanganate (C = 0.15 mol L−1) to titration the solution to pink color to determine the amount of residual H2O2. The electrolyte acid density was measured by the gravimetric method. The carbon and sulfur composition were measured by frequency infrared carbon and sulfur analyzer (HCS140, Shanghai).
2.5 XRD analysis and SEM analysis
The crystalline phases of lead oxide, plates after curing, plates after formation and dismantled pastes in positive plate of testing battery were identified by X-ray diffraction (XRD) analysis (D/MAX 2550, Rigaku, Japan) using Cu Kα radiation (λ = 1.54 Å), with the operation voltage and current of 40 kV and 300 mA, respectively.The morphologies of lead oxide, and the positive plates of testing battery were investigated using scanning electron microscopy (SEM, Sirion 200, FEI, Netherland and JEM-2100F, JEOL, Japan) operated at 10 kV after coating the samples with a thin layer of gold to eliminate charging effect.
3 Results and discussion
3.1 Characterization of lead oxides
The crystal structure and morphology of BaSO4-free synthesized lead oxides were investigated, and the XRD patterns of synthesized lead oxide specimens indicated that lead oxide power mainly comprised PbO with trace amount of metallic Pb. In contrary to the conventional lead oxide, the samples prepared from our novel hydrometallurgical method exhibited a porous structure with ultrafine particles which provides larger contact area to facilitate the electrochemical reaction of the active materials to improve the capacity.The physicochemical characteristics of leady oxides are summarized in Table S3.† The apparent density of synthesized lead oxide was slightly lower than the traditional ball-milled lead oxide, which was provided by Wuhan Changguang Power. Co. Ltd. The oxidizability of lead oxide was around 85%. Owing to the much smaller particle size and higher porosity, the synthesized lead oxide exhibited much higher water absorption value than traditional lead oxide which was beneficial for higher initial discharge capacity.
The electrochemical assessment was carried out in a three-electrode system by potential cycling for 30 times. It showed a decreased oxidation current attributable to oxygen evolution and a decreased reduction peak current with increasing potential cycles. Furthermore, the redox potential difference (0.25 V initially and 0.15 V after 30 cycles) also became smaller with the potential cycles.
3.2 The effect of BaSO4 on the curing of positive plate
The ultrafine lead oxide was used as PAM, and the prepared positive plate was cured at high temperature. The XRD patterns of PAM specimens after curing are presented in Fig. 1. In this figure, 4BS refers to the 4PbO·PbSO4 and 3BS refers to 3PbO·PbSO4, which were formed during curing process. 4BS and 3BS would transform to β-PbO2 and α-PbO2 respectively in the subsequent formation process, and the former contributed to the initial capacity and the latter contributed to the cycle stability for the batteries. The weak peaks presented in Fig. 1 were attributable to unlabeled peaks from the lead paste. The peak intensity of 4BS at 27.7° was 350 counts in the absence of BaSO4, and increased to 1521 with increasing dosage of BaSO4 to 0.1%. This indicated that the portion of 4BS phase increased with the increasing dosage of BaSO4. The increased ratio of 4BS phase with the addition of BaSO4 can be attributed to the high nucleation density of 4BS on the BaSO4 crystal nucleus.13 Nevertheless, the content of 4BS did not increase much when the dosage of BaSO4 is 0.1 wt% in contrast to the case of 0.08 wt%. The peaks of BaSO4 became evident for samples with the dosage of BaSO4 of 0.08 wt% and 0.1 wt%.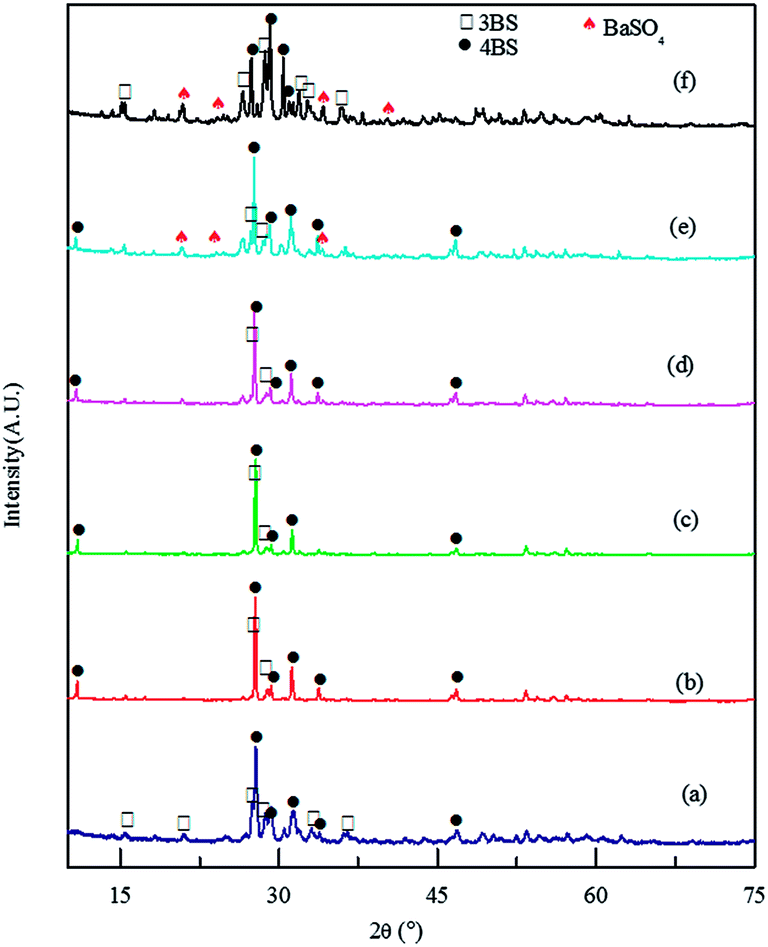 | ||
| Fig. 1 XRD patterns of the paste after curing with BaSO4 dosages of (a) 0 wt%, (b) 0.02 wt%, (c) 0.04 wt%, (d) 0.06 wt%, (e) 0.08 wt%, and (f) 0.1 wt%. | ||
The mass content of metallic lead in the plate after curing is shown in Table 1. The average content of metal lead was 14.5 ± 1.9 wt% with the addition of BaSO4. It can be seen that the content of metallic lead was higher in comparison with the plate without BaSO4. Hence, it indicated that BaSO4 had a significant influence on the curing of the positive plates.
| The dosage of BaSO4 (wt%) | 0 | 0 | 0 | 0.02 | 0.04 | 0.06 | 0.08 | 0.1 |
|---|---|---|---|---|---|---|---|---|
| The content of metallic Pb (wt%) | 7 | 7.2 | 15.0 | 11.4 | 16.7 | 15.2 | 14.4 |
Meanwhile, BaSO4 also had a significant impact on morphology of plate during the curing, as shown in Fig. 2. The particles aggregated together with the increasing dosage of BaSO4. The aggregation can be attributed to the promotion of the 4BS nucleation density on non-uniform distributed BaSO4, consistent with the XRD analysis.
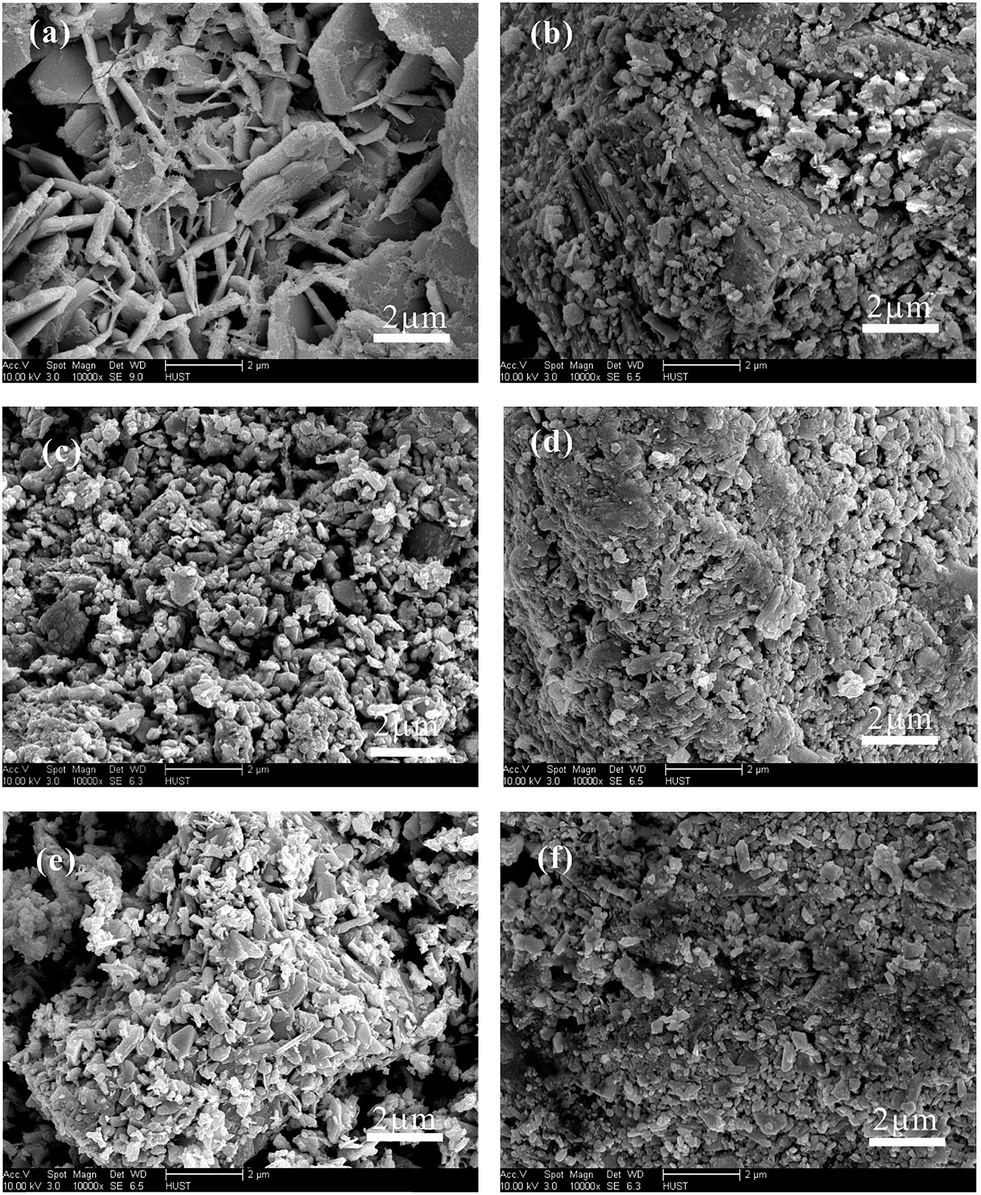 | ||
| Fig. 2 SEM images of cathode plates after curing with BaSO4 dosages of (a) 0 wt%, (b) 0.02 wt%, (c) 0.04 wt%, (d) 0.06 wt%, (e) 0.08 wt%, and (f) 0.1 wt%. The acceleration voltage was 10 kV and the spot size was 3 during SEM image acquisition. | ||
3.3 The effect of BaSO4 on the formation of positive plate
In contrast to the significant impact of BaSO4 doping on the curing of positive plate, it had different influence on the formation of positive plate, both in term of structure and morphology. The active mass consisted of β-PbO2 and PbSO4, and was dominated by β-PbO2, which can be seen from Fig. 3. The average content of lead dioxide was 92.1 ± 1.1 wt% in the presence of BaSO4, and 94.1 wt% in the absence of BaSO4, as summarized in Table 2, indicating that BaSO4 had negligible impact on the content of lead dioxide during formation. As can be seen from Fig. 3, the phase of PAM after formation procedure consisted of β-PbO2 and PbSO4, and the predominant crystal phase was β-PbO2.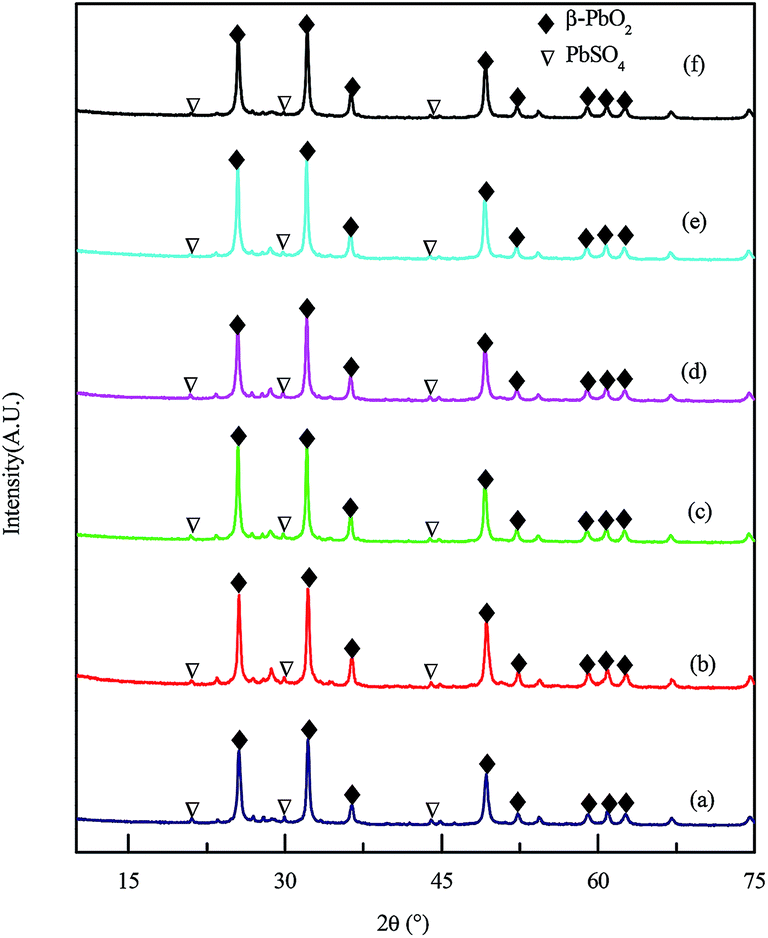 | ||
| Fig. 3 XRD patterns of the active mass with BaSO4 dosages of (a) 0 wt%, (b) 0.02 wt%, (c) 0.04 wt%, (d) 0.06 wt%, (e) 0.08 wt%, and (f) 0.1 wt%. | ||
| The dosage of BaSO4 (wt%) | 0 | 0.02 | 0.04 | 0.06 | 0.08 | 0.1 |
|---|---|---|---|---|---|---|
| The content of PbO2 (wt%) | 94.1 | 92.9 | 90.9 | 92.2 | 93.4 | 91.0 |
The morphology after formation is shown in Fig. 4. As can be seen from the photo of the plates in Fig. 4(a), there was no significant change on the surface of plates with varied dosage of BaSO4, with some white dots distributed on the surface of these plates. However, BaSO4 had a dramatic impact on the microstructure of the PAM in the formation process. The particles in PAM became more agglomerated with the increasing dosage of BaSO4, which was in line with the aggregation of 4BS after the curing process.
 | ||
| Fig. 4 (a) Photos of the active mass after formation with different dosages of BaSO4, where (I)–(V) denotes for the theoretical addition of 0.02 wt%, 0.04 wt%, 0.06 wt%, 0.08 wt%, and 0.1 wt% BaSO4; SEM images of the active mass with BaSO4 dosage of (b) 0 wt%, (c) 0.02 wt%, (d) 0.04 wt%, (e) 0.06 wt%, (f) 0.08 wt% and (g) 0.1 wt%. The acceleration voltage was 10 kV and the spot size was 3 during SEM image acquisition. | ||
3.4 The effect of BaSO4 on the battery performance
After curing and formation process, the plates were assembled to a battery to evaluate the effect of BaSO4 on the performance of lead acid battery. The 1C discharge time is shown in Fig. 5 for the testing batteries with different dosage of BaSO4. The 1C discharge time was 47 min in the absent of BaSO4, which was quite similar with that in the presence of BaSO4, and the average discharging time was 43.6 ± 0.7 min. This indicated that BaSO4 did not exhibit evident negative impact on the large current discharge performance of the battery.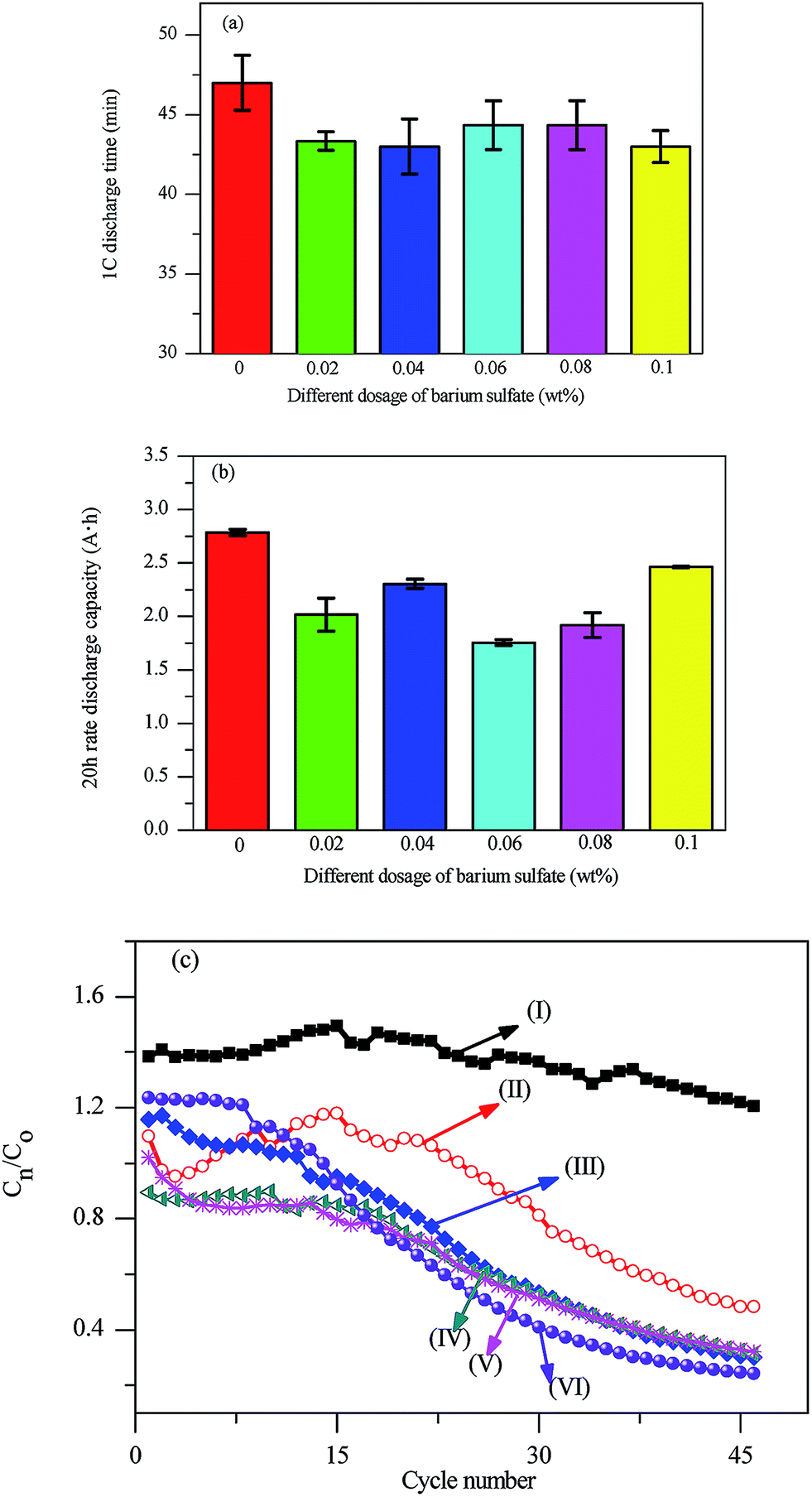 | ||
| Fig. 5 The 1C discharge time in the discharge current of 2 A (a) and 20 h rate of the battery with different dosage of BaSO4 (b); the capacity retention ratio of battery with various dosage of BaSO4 (c), where (I)–(VI) denotes for the theoretical addition of 0 wt%, 0.02 wt%, 0.04 wt%, 0.06 wt%, 0.08 wt%, and 0.1 wt% BaSO4. | ||
The 20 h rate of the testing batteries was evaluated with different dosage of BaSO4 added in the plate, as can be seen from Fig. 5. The 20 h rate of all batteries remained at about 2 A h with the increasing dosage of BaSO4, and it could be found that the 20 h rate achieved 2.46 A h in the presence of 0.1% BaSO4, and the average mean capacity was 2.1 ± 0.3 A h. It was clear the 20 h rates of the batteries in the presence of BaSO4 were lower than the batteries in the absent of BaSO4 (2.78 A h). Hence, the addition of BaSO4 had remarkable influence on the 20 h rate of all the batteries.
The change of the 10 h capacity retention rate of the testing batteries with different dosages of BaSO4 added in the plate is presented in Fig. 5, where C0 is the rated 10 h rate capacity of the battery, and Cn is the n times of 10 h rate capacity of the battery. The capacity retention ratios of all batteries with BaSO4 remained at about 80% before 25 cycles, and drops to below 80% in the subsequent cycles. In contrast, the battery in the absence of BaSO4 worked very well during all of 46 cycles. When the capacity retention ratio decreased from 0.8 to 0.6, the cycle stability of batteries deteriorated with the increasing dosage of BaSO4, which indicated that BaSO4 had a negative effect on the cycle stability of the battery. When the capacity retention ratio decreased further from 0.6 to 0.3, the capacity decreased with the increasing dosage of BaSO4, but the cycle stability curve stayed the same when the dosage varies from 0.04 wt% to 0.08 wt%. The battery stability curve indicated that BaSO4 had a severe negative effect on the performance of the battery.
The battery testing with BaSO4 demonstrated an overall tendency that the capacity retention ratio decreased with the increasing dosage of BaSO4. This was different from the previous study that the addition of BaSO4 as electrolyte additive can improve the performance of the battery.21 As discussed previously, BaSO4 acted as nucleuses to promote nucleation density of lead oxide on top and consequently led to severe aggregation of particles on non-uniform distributed BaSO4 particles. So severe non-uniformity was developed during charging and discharging cycles, and the growing internal stress finally undermined the mechanical integrity of positive plate and causes expansion and shedding-off of PAM on the positive plate, which eventually led to the failure of these batteries.
3.5 Chemical compositions of positive plates of dismantled batteries
After charging and discharging for 46 cycles, the batteries were dismantled after the final charging procedure. The content of PbSO4 and PbO2 in the positive plate is listed in Table 3. The average content of PbO2 was 60.5 ± 4.5 wt%, with the highest level found in the sample with 0.1 wt% of BaSO4, which indicated that sulfation was not the primary reason for the failure of batteries. This was supported by the low S content after formation (in Table 4). The XRD analysis also confirmed that the dominant crystalline phase in the dismantled paste was PbO2, and a small amount of PbSO4 was also identified at the end of charging procedure (in Fig. 6). Considering that PbO2 was the dominant phase in dismantled batteries and the content of S was not high after failure, the sulfation was not the dominant reason for the failure of these batteries. This is different from the widely accepted interpretation on the failure of conventional lead acid batteries.7,22–24| Symbol | Content of PbO2 (wt%) | Content of PbSO4 (wt%) | Acid density (g cm−3) |
|---|---|---|---|
| a Ba-x stands for the theoretical addition of BaSO4 is x% in weight percent. | |||
| Ba-0.02 | 71.0 | 39.0 | 1.23 |
| Ba-0.04 | 60.3 | 31.2 | 1.27 |
| Ba-0.06 | 63.2 | 37.8 | 1.32 |
| Ba-0.08 | 52.7 | 42.3 | 1.29 |
| Ba-0.10 | 63.6 | 27.5 | 1.21 |
| The dosage of BaSO4 (wt%) | 0.02 | 0.04 | 0.06 | 0.08 | 0.1 |
|---|---|---|---|---|---|
| The content of S after formation (wt%) | 0.75 | 0.96 | 0.83 | 0.70 | 0.95 |
| The content of S after failure (wt%) | 3.06 | 4.19 | 3.89 | 5.00 | 3.85 |
 | ||
| Fig. 6 XRD patterns of the active mass after failure with the dosage of BaSO4: (a) 0.02 wt%, (b) 0.04 wt%, (c) 0.06 wt%, (d) 0.08 wt%, and (e) 0.1 wt%. | ||
To examine the failure of lead acid batteries using BaSO4 doped lead oxide as PAM, optical photos and SEM images of the dismantled paste at the 47th battery cycles were acquired. From the optical photos of dismantled positive plates after failure as presented in Fig. 7(a), significant fracture and shedding of PAM were observed. This was believed to be the primary reason for the failure of these batteries in the present of BaSO4. As shown in the microstructure images in Fig. 7(b), the aggregation of active material particles appeared more and more evident with the increase of BaSO4 content. This observation confirmed the hypothesis that the failure of battery was mostly due to the accumulation of internal stress because of the inhomogeneous growth of lead oxide on BaSO4 nucleus during cycled charge and discharge processes.
 | ||
| Fig. 7 (a) Photos of the active mass after failure with different dosage of BaSO4, where (I)–(V) denotes for the theoretical addition of 0.02 wt%, 0.04 wt%, 0.06 wt%, 0.08 wt%, and 0.1 wt% BaSO4; SEM images of the active mass after failure with BaSO4 dosage of (b) 0.02 wt%, (c) 0.04 wt%, (d) 0.06 wt%, (e) 0.08 wt% and (f) 0.1 wt%. The acceleration voltage was 10 kV and the spot size was 3 during SEM image acquisition. | ||
The content of BaSO4 during the whole process is shown in Fig. 8. The dosage of BaSO4 in lead oxide was measured by ICP and the result was close to the theoretical content, which clearly implied that the quantification method was consistent. The relative portion of barium sulfate versus the mass of battery plate appeared to vary significantly at various fabrication stages in Fig. 8 and this was solely attributed to the mass variation of lead containing compound, dominated by PbSO4 during curing, PbO2 during formation, and mixture of PbSO4 and PbO2 after failure. In fact the absolute mass of barium sulfate stayed in the plate at various stages and the loss due to its dissolution in the electrolyte was negligible. This indicated that BaSO4 remained in PAM of the plates during the whole process since BaSO4 was insoluble in the sulfuric acid electrolyte. The content of BaSO4 after battery failure was lower than after formation of positive plate as a result of mass increase due to partial transformation of lead dioxide to lead sulfate.
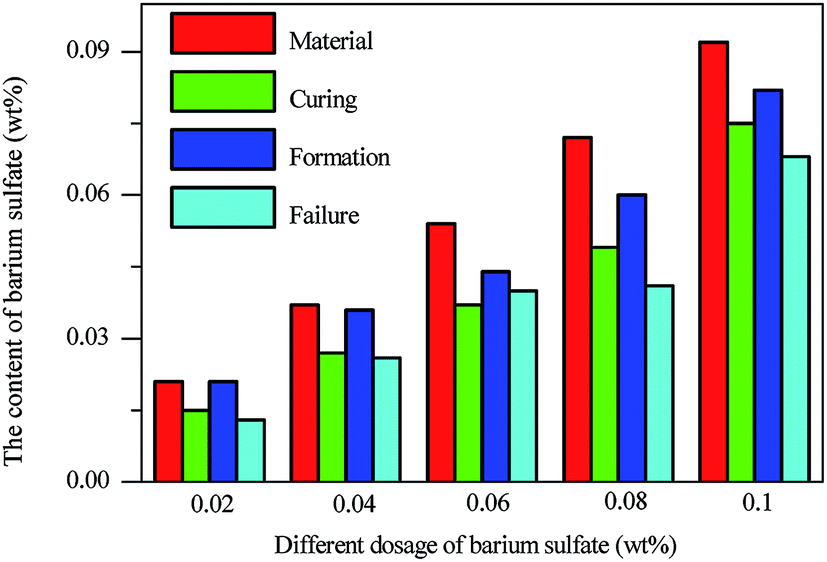 | ||
| Fig. 8 The content of BaSO4 in the battery during the whole procedure. Material refers to the content of BaSO4 in raw lead oxide; curing, formation, failure refer to the samples after curing, formation and failure of battery. | ||
4 Conclusions
The effect of BaSO4 impurities on the microstructural characteristics of ultrafine lead oxide and the performance of lead acid battery made from these lead oxide were investigated. Ultrafine lead oxide was prepared via low temperature calcination of lead citrate, and novel lead acid batteries were assembled using this material.The presence of BaSO4 impurities demonstrated dramatic influence on the crystalline phase of the positive plates during curing, of which the highest content of metallic lead was 16.7% with the BaSO4 dosage of 0.06%. It also showed a drastic effect on the morphology, resulting in severe particles aggregation with the increasing dosage of BaSO4. The presence of BaSO4 had negligible effect on the crystalline phase of the positive plates during formation, but the morphology analysis indicated that the active mass particle became strongly aggregated with the increasing dosage of BaSO4. BaSO4 also had a significant negative effect on charge/discharge cycles with 100% DOD, which can be attributed to the expansion and shedding of active mass. The non-uniform growth of lead oxide on BaSO4 nucleus and accumulation of internal stress were proposed to be the primary reasons for the failure of batteries through the analysis of the dismantled spent batteries. This study also provides useful guidance on impurities control during the recovery of spent lead paste through hydrometallurgical process.
Acknowledgements
The authors thank for the financial supports from the National Science-technology Support Plan Projects (2014BAC03B02), key project of Hubei Provincial Natural Science Foundation (2014CFA109, 2015CFA133), the Wuhan Planning Project of Science and Technology, China (2013060501010168, 2013011801010593, 2014030709020313 and 2015070404010200), and Innovative and Interdisciplinary Team at HUST (0118261077). The authors would like to thank the Analytical and Testing Center of Huazhong University of Science and Technology for providing the facilities to fulfill the experimental measurements. The technical supports from Wuhan Changguang Power Sources. Co. Ltd., is also gratefully acknowledged. The assistance from Mr Anxu Sheng during electrode preparation and battery assembly, and Dr Bingchuan Liu during manuscript preparation are acknowledged.References
- C. R. Cherry, J. X. Weinert and Y. Xinmiao, Transport. Res. Transport. Environ., 2009, 14, 281–290 CrossRef.
- H. Y. Chen, L. Wu, C. Ren, Q. Z. Luo, Z. H. Xie, X. Jiang, S. P. Zhu, Y. K. Xia and Y. R. Luo, J. Power Sources, 2001, 95, 108–118 CrossRef CAS.
- Y. Chang, X. X. Mao, Y. F. Zhao, S. L. Feng, H. Y. Chen and D. Finlow, J. Power Sources, 2009, 191, 176–183 CrossRef CAS.
- T. W. Ellis and A. H. Mirza, J. Power Sources, 2010, 195, 4525–4529 CrossRef CAS.
- A. Poullikkas, Renewable Sustainable Energy Rev., 2013, 27, 778–788 CrossRef.
- X. Tian, Y. Gong, Y. F. Wu, A. Agyeiwaa and T. Y. Zuo, Resour., Conserv. Recycl., 2014, 93, 75–84 CrossRef.
- L. T. Lam, N. P. Haigh, C. G. Phyland and A. J. Urban, J. Power Sources, 2004, 133, 126–134 CrossRef CAS.
- M. A. Kreusch, M. J. J. S. Ponte, H. A. Ponte, N. M. S. Kaminari, C. E. B. Marino and V. Mymrin, Resour., Conserv. Recycl., 2007, 52, 368–380 CrossRef.
- A. Zabaniotou, E. Kouskoumvekaki and D. Sanopoulos, Resour., Conserv. Recycl., 1999, 25, 301–317 CrossRef.
- E. Sayilgan, T. Kukrer, G. Civelekoglu, F. Ferella, A. Akcil, F. Veglio and M. Kitis, Hydrometallurgy, 2009, 97, 158–166 CrossRef CAS.
- N. K. Lyakov, D. A. Atanasova, V. S. Vassilev and G. A. Haralampiev, J. Power Sources, 2007, 171, 960–965 CrossRef CAS.
- X. F. Zhu, X. He, J. K. Yang, L. X. Gao, J. W. Liu, D. N. Yang, X. J. Sun, W. Zhang, Q. Wang and R. V. Kumar, J. Hazard. Mater., 2013, 250, 387–396 CrossRef PubMed.
- J. Q. Pan, Y. Z. Sun, W. Li, J. Knight and A. Manthiram, Nat. Commun., 2013, 4, 2178 Search PubMed.
- L. Li, X. F. Zhu, D. N. Yang, L. X. Gao, J. W. Liu, R. V. Kumar and J. K. Yang, J. Hazard. Mater., 2012, 203, 274–282 CrossRef PubMed.
- D. N. Yang, J. W. Liu, Q. Wang, X. Q. Yuan, X. F. Zhu, L. Li, W. Zhang, Y. C. Hu, X. J. Sun, R. V. Kumar and J. K. Yang, J. Power Sources, 2014, 257, 27–36 CrossRef CAS.
- X. J. Sun, J. K. Yang, W. Zhang, X. F. Zhu, Y. C. Hu, D. N. Yang, X. Q. Yuan, W. H. Yu, J. X. Dong, H. F. Wang, L. Li, R. V. Kumar and S. Liang, J. Power Sources, 2014, 269, 565–576 CrossRef CAS.
- D. Pavlov, P. Nikolov and T. Rogachev, J. Power Sources, 2010, 195, 4435–4443 CrossRef CAS.
- H. Vermesan and N. Hirai, Rev. Chim., 2007, 58, 1221–1225 CAS.
- K. Sawai, T. Funato, M. Watanabe, H. Wada, K. Nakamura, M. Shiomi and S. Osumi, J. Power Sources, 2006, 158, 1084–1090 CrossRef CAS.
- H. Vermesan, N. Hirai, M. Shiota and T. Tanaka, J. Power Sources, 2004, 133, 52–58 CrossRef CAS.
- H. Karami and A. Yaghoobi, J. Cluster Sci., 2010, 21, 725–737 CrossRef CAS.
- J. H. Yan, W. S. Li and Q. Y. Zhan, J. Power Sources, 2004, 133, 135–140 CrossRef CAS.
- M. Saravanan and S. Ambalavanan, Eng. Failure Anal., 2011, 18, 2240–2249 CrossRef CAS.
- Y. L. Guo, S. Q. Tang, G. Meng and S. J. Yang, J. Power Sources, 2009, 191, 127–133 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: see DOI: 10.1039/c6ra01873d |
| ‡ These two authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2016 |