
Dependence on size of supported Rh nanoclusters for CO adsorption†
Ting-Chieh Hunga,
Ting-Wei Liaoa,
Zhen-He Liaoa,
Po-Wei Hsua,
Pei-Yang Caia,
Wen-Hua Lub,
Jeng-Han Wang*b and
Meng-Fan Luo*a
aDepartment of Physics, National Central University, 300 Jhongda Road, Jhongli 32001, Taiwan. E-mail: mfl28@phy.ncu.edu.tw; Fax: +886-3-4251175; Tel: +886-3-42271751 ext. 65349
bDepartment of Chemistry, National Taiwan Normal University, No. 88, Sec. 4, Ting-Chow Rd., Taipei 11677, Taiwan. E-mail: jenghan@ntnu.edu.tw
First published on 22nd December 2015
Abstract
We studied the adsorption and lateral interactions of CO molecules on Rh nanoclusters supported on an ordered thin film of Al2O3/NiAl(100) with varied surface probe techniques under ultra-high vacuum conditions and with density-functional-theory (DFT) calculations. The Rh clusters were grown with vapor deposition onto the Al2O3/NiAl(100) surface at 300 K; with increasing deposition, their mean diameter evolved from 1.0 to 3.5 nm and their height from 0.4 to 0.8 nm. The initial adsorption energy (for sparse CO coverage) and the number of adsorbed CO per surface site on the Rh clusters increased with decreasing cluster size. The former effect results from the surface structure and expanded lattice parameter of small Rh clusters, whereas the latter effect involves not only the initial adsorption energy but also altered lateral interactions among CO molecules. In contrast with CO on Rh single crystals, CO on small clusters adsorbed with their axes tilted from the surface normal, weakening the CO–CO repulsive interactions for CO coverage over a wide range. The saturated density of CO on clusters of diameter near 1.0 nm and height near 0.4 nm is 2–3 times that of large clusters (diameter ≥ 3.5 nm) or a Rh(100) surface. The CO–CO repulsive interactions on small clusters became effective at large CO densities, given that the onset of desorption of CO at saturation was 100 K lower than that of large clusters.
1. Introduction
Adsorption and lateral interactions between adsorbates are important for catalytic reactions, as they might alter the reaction kinetics in a substantial manner. CO is involved in numerous important reactions, such as dehydrogenation of methanol and the Fischer–Tropsch process,1–7 and also it is used commonly as a probe molecule to investigate lateral interactions.8–21 Adsorption of CO on single-crystal transition-metal surfaces has thus been studied widely to model its behavior in catalytic reactions.8–31 The results indicated that CO, similar to other electronegative adspecies, such as oxygen and halogens, undergoes a significant charge re-distribution upon adsorption and exhibits non-negligible lateral interactions to weaken the bonding of neighboring adspecies.32,33 To bridge the material gap between single-crystal model systems and real-world catalysts, numerous researchers have turned toward metal nanoclusters supported on well-defined oxides, as realistic model systems.7,34–44 A disparity in the adsorption and lateral interactions of CO between single-crystal and supported-cluster models is expected. The present investigation is aimed to reveal the contrasting behavior on supported clusters.We investigated experimentally and computationally the adsorption of CO on Rh nanoclusters formed upon deposition of a vapor onto an ordered thin film of Al2O3/NiAl(100). Al2O3/NiAl(100) was chosen as a support because it was a prototype to model the properties of real supporting alumina in catalysts.45–48 Under ultrahigh vacuum (UHV) conditions, the morphology and structure of the Rh clusters were characterized with scanning tunneling microscopy (STM) and reflection high-energy electron diffraction (RHEED), and the adsorption and desorption of CO with infrared reflection absorption spectroscopy (IRAS) and temperature-programmed desorption (TPD). To rationalize the adsorption behavior observed from the experiments, we undertook calculations based on density-functional theory (DFT) to examine the optimized structures, adsorption energies and the corresponding electronic structures.
Preceding work on extended surfaces of Rh showed that the adsorption energy and sticking coefficient of CO depend on the CO coverage; the adsorption sites and ordered CO superstructures also varied with the CO coverage.11,12,23–25,27,28,30 The present study in contrast shows an effect of cluster size on the adsorption and lateral interactions. With decreasing cluster size, the initial adsorption energy (for sparse CO coverage) was enhanced, in contrast with CO on Pd clusters/Fe2O3/Pt(111),34,40,42 and the lateral interactions became attenuated. The origin of these effects is discussed with the structures of the Rh clusters and the adsorption configuration of CO on the Rh clusters. Both effects led to greater accommodation of CO. The density of absorbed CO on the Rh clusters, related to the number of adsorbed CO per Rh surface site, thus increased with the decreasing cluster size. At a cluster diameter near 1.0 nm and height near 0.4 nm, the saturated density of CO was 2–3 times of that on large clusters (diameter ≥ 3.5 nm) or a Rh(100) surface, but CO on Rh clusters remains on atop sites—with increasing CO density, no CO migrated to bridge or hollow sites like CO on extended Rh surfaces.
2. Experimental and computational methods
Our experiments were performed in UHV chambers with a base pressure in the 10−10 Torr regime. A NiAl(100) sample (MaTeck GmbH) was polished to a roughness less than 30 nm and an orientation accuracy better than 0.1°; a Rh(100) sample (MaTeck GmbH) was polished to an orientation accuracy better than 1°. To obtain a clean surface, the sample underwent alternative cycles of sputtering and subsequent annealing before each experiment. The cleanliness of the sample was monitored with Auger electron spectroscopy, low-energy electron diffraction and STM. An ultra-thin θ-Al2O3 film was formed upon oxidation of a NiAl(100) alloy surface at 1000 K; the formation of Al2O3 thin films is described elsewhere.45–48 To achieve a homogeneous crystalline Al2O3 surface with no NiAl facets,49,50 we refrained from protracted post-oxidation annealing of the oxide films. The amorphous oxide surface was also negligible. The grown θ-Al2O3 thin film had 0.5–1.0 nm thickness.47,48 The sample was then quenched to 300 K for vapor deposition of Rh from an ultra-pure Rh rod heated by electron bombardment in a commercial evaporator (Omicron EFM 3). The rate of deposition of Rh was fixed at about 0.15 ML per min, calculated according to the coverage prepared at 300 K. The coverage was estimated from the volume of the Rh clusters observed with STM; 1 ML corresponded to a density of 1.39 × 1015 atoms per cm2 of fcc Rh(100) surface atoms. After the deposition, the sample was cooled to the desired adsorption temperature (100 K, unless specified). CO gas was dosed by a doser pointing to the sample, with a 2–5 × 10−9 Torr background pressure. We report CO exposures in Langmuir units (1 L = 10−6 Torr s).STM images (recorded with a RHK UHV 300 unit), constant-current topographies, were obtained at 90 K with a sample bias voltage typically between 2.4–2.8 V and a 0.8–1.2 nA tunneling current. The STM tip consisted of an electrochemically etched tungsten wire. RHEED was performed with a 40 keV incident electron beam of energy at a 2–3° grazing angle to the surface. TPD spectra were obtained by ramping the sample at 3 K s−1 and monitoring the various masses on a quadruple mass spectrometer (Hiden), which was shielded and placed close (about 2 mm) to the sample. IRAS spectra were obtained using a Fourier transform infrared spectrometer (FTLA 2000) with external optics aligned for a 75° incident angle from the sample normal and a liquid nitrogen-cooled MCT detector. The IRAS spectra are presented as the ratio of sample and oxide surface (or Rh clusters) data measured at the same surface temperature (100 K) and are typically the average of 256 scans at 4 cm−1 resolution.
The calculations were performed using the Vienna Ab initio Simulation Package (VASP)51–53 at the density functional theory (DFT) level with a 3D periodic boundary condition. The exchange-correlation function was treated by the generalized gradient approximation54 with the Perdew–Wang 1991 formulation (GGA-PW91).55 The electron–ion interaction was modeled by the projector-augmented wave method (PAW),56,57 combining the accuracy of augmented plane waves with the cost-effective pseudopotentials. The kinetic cutoff energy of the plane-wave basis was set at 600 eV. The Brillouin-Zone (BZ) integration was sampled by a Monkhorst–Pack scheme58 at a 0.05 × 2 (Å−1) interval in the reciprocal space. All the modeled clusters, surfaces and the related adsorptions were optimized by a quasi-Newton method with an energetic convergence of 1 × 10−4 eV and a gradient convergence of 1 × 10−2 eV.
3. Results and discussion
3.1 Morphology and structure of supported Rh clusters
The Rh clusters were grown by deposition of Rh vapor onto Al2O3/NiAl(100) at 300 K and were characterized structurally with STM and RHEED. Fig. 1(a)–(d) exemplify the morphologies and sizes of the Rh clusters at various Rh coverages on Al2O3/NiAl(100); Fig. 1(e) plots the evolution of the mean diameter and height of the Rh clusters with the coverage. The insets in Fig. 1(a)–(d) show characteristic histograms of the height and diameter for each coverage; the curve in each histogram is a Gaussian fit to the size distribution. At the least coverage, 0.05 ML (Fig. 1(a)), the clusters had about a 1.0 nm mean diameter and about 0.4 nm height; with increasing coverage, both diameter and height of the clusters increased: at 0.25 (0.8) ML, the mean diameter and height increased to 1.3 (1.7) and 0.5 (0.7) nm, respectively (Fig. 1(b) and (c)). The clusters enlarged with increasing coverage as plotted in Fig. 1(e); the error bars in Fig. 1(e) indicate the full width at half maximum (FWHM) of the best Gaussian fits to the distributions of diameter and height of the clusters for each coverage. At coverages greater than 2.0 ML, the clusters generally coalesced (ESI†). At 4.0 ML (Fig. 1(d)), the diameter of the Rh clusters increased to 3.5 nm but the height was slightly altered. As the lower portions of the clusters merged at this stage, the measured height reflected only the upper portions of the clusters. The present study concentrated on the coverage regime for which coalescence scarcely occurs (≤1.6 ML). In this regime, the clusters grew through nucleation, rather than coalescence, so their properties were readily correlated to well-defined cluster sizes. The images also show that most of the Rh clusters grew along stripe protrusions of the crystalline Al2O3/NiAl(100) (Fig. 1(a)–(c)), indicating the protrusions were preferential nucleation sites.49,59 The formation of the protrusions is due to the lattice mismatch between the Al2O3 film and NiAl(100).45,46,49 Because of the rich protrusions on Al2O3/NiAl(100),49,59 the grown Rh clusters were smaller and the cluster density was greater than those on the surfaces with less defects such as graphene.60 The few clusters grown on the region other than the protrusions on Al2O3/NiAl(100) indicates that diffusion of Rh atoms was efficient at 300 K. This feature is consistent with that previously observed for Rh clusters on Al2O3/NiAl(110).35,61 | ||
| Fig. 1 (a)–(d) STM images for 0.05, 0.25, 0.8 and 4.0 ML Rh deposited on a thin film of Al2O3/NiAl(100) at 300 K; the insets in (a)–(d) show characteristic histograms of height and diameter for each coverage; the curves are the best Gaussian fits to the distributions. (e) Plots of the evolution of the mean diameter (red circles) and height (black squares) of Rh clusters with coverage. The error bars in (e) indicate the full width at half maximum (FWHM) of the best Gaussian fits to the histograms of diameter and height of Rh clusters at each coverage. | ||
The structural ordering of the Rh clusters is reflected in the RHEED patterns, as shown in Fig. 2(a)–(d). The reflection rods of the RHEED patterns in the figures are ascribed to the oxide and the NiAl(100) substrate; upon deposition of Rh, the half-order reflections from the (2 × 1) structure of ordered θ-Al2O3(100)45–48 either disappeared (Fig. 2(a)) or became vague (Fig. 2(c)). Additional patterns superimposed on the reflection rods at both azimuths [0−10] and [0−11] resulted from structurally ordered Rh clusters. The patterns were evident at 1.0 ML (Fig. 2(a) and (b)), and became even sharper with increasing coverage (Fig. 2(c) and (d)). The trend implies that the content of the ordered structures increased in larger clusters. The structurally ordered clusters had an fcc phase and grew preferentially with their (100) facets parallel to the θ-Al2O3(100) surface; their [110] axes lay along the [010] direction of the oxide surface, denoted as Rh(100)[110]//Al2O3(100)[010]. The blue circles in Fig. 2(a)–(d) are the corresponding points of the reciprocal lattice at the two azimuths. This orientation is preferred because the (100) facets of the Rh clusters match structurally better the square oxygen lattice of the θ-Al2O3(100) surface.50,62,63 The lattice parameter of the Rh clusters also increased to match the oxide surface. The Rh clusters at 1.0 ML (diameter near 1.8 nm and height near 0.7 nm) had about a 4.04 Å mean lattice parameter, which was increased by about 6% with respect to bulk Rh (3.80 Å). The expansion decreased with increasing size or coverage, as the substrate effect on the upper layers of the growing clusters attenuated. The mean lattice parameter of the Rh clusters at 4.0 ML increased to 3.92 Å, corresponding to a 3% expansion. For a coverage at, or smaller than, 0.5 ML, no clear diffraction pattern was observed for the clusters (diameter ≤ 1.5 nm and height ≤ 0.6 nm). The (100) orientation is expected to be retained for small clusters. No clear diffraction pattern was shown as the content of the ordered structures decreased.
 | ||
| Fig. 2 RHEED patterns for 1.0 and 4.0 ML Rh deposited on Al2O3/NiAl(100) at 300 K. (a) and (b) show patterns obtained from 1.0 ML Rh clusters at azimuths [0−10] and [0−11], respectively, and (c) and (d) from 4.0 ML Rh clusters at azimuths [0−10] and [0−11], respectively. Blue circles in (a)–(d) denote reciprocal-lattice points for the clusters of Rh(001)[110]//Al2O3(100)[010]. The lattice parameters were determined on fitting the reciprocal-lattice nets to the brightest areas of the diffraction pattern (top 2% in intensity). | ||
3.2 Adsorption of CO on supported Rh clusters
The adsorption of CO on Rh clusters on Al2O3/NiAl(100) was monitored with TPD and IRAS. Fig. 3 shows the CO TPD spectra for 2.0 L CO adsorbed on the Rh clusters at varied coverages (0.13–4.0 ML) at 100 K. As CO does not adsorb on Al2O3/NiAl(100) at 100 K, no CO signals were observed for Al2O3/NiAl(100) exposed to CO at 100 K, the desorption signals originate from the Rh clusters. As increasing CO exposure to 5.0 L gave signals similar to those of 2.0 L, 2.0 L CO already saturated the surface of the clusters to a great extent. On 4.0 ML Rh clusters (top in Fig. 3), the desorption of CO began at about 400 K and was complete at about 530 K, resembling that of 0.50–0.70 ML CO on a Rh(100) single crystal.11,12 With deceasing Rh coverage (cluster size), the desorption signal broadened and shifted. Not only the maximum but also the onset of CO desorption shifted towards lower temperature; concomitantly, the desorption also extended to higher temperature. For 0.13 ML Rh clusters, the desorption began at temperature <300 K and was complete at about 600 K. No comparable desorption features were observed on Rh single-crystal surfaces.11,12,23,25,27,28,31 This systematic alteration indicates that CO adsorbs in a manner depending on the cluster size. The concomitantly decreased and increased desorption temperatures, and broadening of desorption features, from small clusters reflect new adsorption states; they arise because both the density of adsorbed CO (number of CO per surface site) and the surface structures varied with the cluster size. These deductions are supported by the following results. | ||
| Fig. 3 CO TPD spectra for 2.0 L CO adsorbed on Rh clusters/Al2O3/NiAl(100) at varied coverages (0.13–4.0 ML) and 100 K. Rh was deposited at 300 K. | ||
Fig. 4 shows the integrated intensities of CO TPD spectra (black) as a function of Rh coverage compared with the estimated ones for total adsorbed CO (red) that contains both desorbing and dissociating CO.7,35,43 Our experiments with synchrotron-based photoelectron spectra (not shown) confirmed that the rate of CO dissociation depends on the size of the Rh clusters; this dependence resembles previous observations.7 The plot shows that adsorbed CO increases with the Rh coverage in a nonlinear manner; the rate of increase below 0.5 ML is significantly greater than that above 0.5 ML. The surface area of the Rh clusters, measured with STM, increases almost linearly with the coverage <1.6 ML (Fig. 4(b)), the result implies that the number of CO adsorbed on the clusters is not simply proportional to the number of surface sites. The density of adsorbed CO or the number of CO per surface site, must vary with the cluster size. Fig. 4(c) plots the ratio of the CO TPD intensity to the Rh surface area (measured with STM) as a function of cluster diameter. Either the measured desorbing CO (black) or the estimated total CO (red) signals showed that the ratio decreased with increasing cluster diameter. As the ratio reflects the number of CO per Rh surface site, this result indicates an enhanced density of CO on small clusters. This enhancement might produce a decreased adsorption energy, corresponding to the decreased onset temperature of desorption (Fig. 3). The adsorption energy of CO on supported Pd clusters was previously shown to decrease with increased CO density.40,42 We argue below that the enhancement resulted from the surface structures and altered lateral interactions of CO on small clusters.
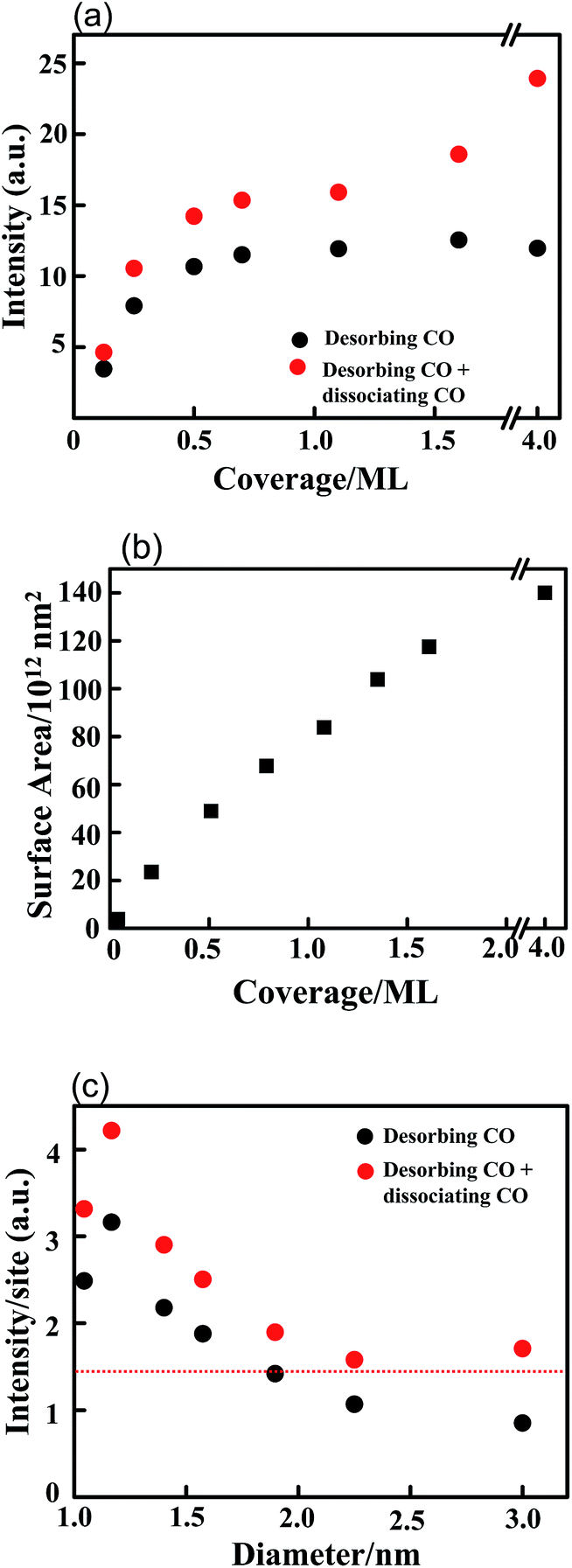 | ||
| Fig. 4 (a) The integrated intensity of CO TPD spectra (black) from 2.0 L CO adsorbed on Rh clusters/Al2O3/NiAl(100) at varied coverages and 100 K; (b) surface areas of Rh clusters, measured with STM, as a function of Rh coverage; (c) number of adsorbed CO per surface Rh site (from 2.0 L CO) as a function of cluster diameter. The red circles in (a) indicate the estimated quantities of total adsorbed CO, based on the integrated intensity of CO TPD spectra and the known rate of dissociation of adsorbed CO.7 In (c), the data are the ratios of the estimated quantities of total adsorbed CO to the surface area of the Rh clusters measured with STM. The sizes in (c) correspond to Rh clusters at 0.13, 0.25, 0.5, 0.7, 1.1, 1.6, 4.0 ML. Rh was deposited at 300 K. | ||
The CO lateral interactions altered when the Rh cluster size was decreased. Earlier studies of CO on a Rh(100) surface showed the desorption of 0.5–0.7 ML CO in the 400–550 K temperature range,11,12 resembling that of CO on large Rh clusters (Fig. 3); 0.5 ML CO on Rh(100) occupied only atop sites.11,27 The resembling desorption features imply similar adsorption and lateral interactions of CO. Our desorption experiments for 2.0 L CO on a Rh(100) surface confirmed comparable densities of CO on large clusters and single crystals (dashed line in Fig. 4(c)). Accordingly, the CO coverage on Rh clusters of diameter < 1.5 nm was greater than 1.0 ML (Fig. 4(c)) and exceeds the saturation coverage (0.83 ML) of CO on a Rh(100) surface.11,12 For 0.72–0.83 ML CO on a Rh(100) surface, the desorption feature became asymmetric and a shoulder extended below 400 K;11,12 a fraction of CO moved from atop to bridge sites (reflected in the CO IRAS spectra).11 Similar coverage-dependent adsorption sites were observed on other Rh facets.23,25 The change in adsorption sites with the CO coverage resulted largely from repulsive CO–CO interactions;11,32,33 the desorption below 400 K was associated with the enhanced CO lateral interactions and CO adsorbed on the less preferred sites. In contrast, CO adsorbed on Rh clusters remained on atop sites, despite the cluster size and the CO density. Even for CO on Rh clusters of diameter < 1.5 nm (CO coverage >1.0 ML), the CO lateral interactions induced no migration of CO.
Fig. 5(a) shows the IRAS spectra for CO on Rh clusters at varied coverages (sizes). The wavenumber of the CO absorption signal (C–O stretching) was about 2051 cm−1 at 0.3 ML Rh, and shifted positively with Rh coverage to about 2061 cm−1 at 1.6 ML. The absorption features are attributed to CO on top of Rh (atop sites) on the clusters; the signals for CO on bridge or hollow sites are expected in the 1770–1950 cm−1 regime.11,23,25,64 No signal for CO on the bridge or hollow sites was observed at any elevated temperature (see the ESI†). The CO desorption below 400 K from small clusters (Fig. 3) is attributed to CO on neither bridge nor hollow sites. The IRAS spectra showed no signal for carbonyl species, expected to be >2100 cm−1,65 indicating that the great CO density on small clusters was not from carbonyl species. The positive shift of the CO absorption signal with Rh coverage (2051 cm−1 to 2061 cm−1) is due to the increased long-range CO–CO interactions resulting from increased CO on the entire surface. Upon decreasing CO with stepwise annealing the sample, the wavenumber shifted negatively (ESI†). Similar shifts were previously observed for CO on supported Rh and Pt clusters,38,64 and also on Rh single crystals.11,23,25 The variation of the integrated intensity of CO IRAS spectra (black squares in Fig. 5(b)) with the Rh coverage (size) reveals the alteration of the CO adsorption configuration. The intensity increased with the coverage linearly below 1.0 ML, whereas it became almost saturated above 1.0 ML. The trend differed for that of the CO TPD spectra (Fig. 4(a)), for which the saturation occurred at about 0.5 ML. The ratio of the CO IRAS to the CO TPD intensities (red circles in Fig. 5(b)) increased notably with the Rh coverage below 1.0 ML. As the ratio reflects the absorption signals contributed by each adsorbed CO, the trend implies that CO on small clusters yielded fewer IRAS signals, which are governed by a dipole-selection rule.66 A great fraction of CO molecules on small clusters that cannot be detected with IRAS must thus have their C–O bond axes tilted from the surface normal, in contrast with those on Rh single crystals or large clusters. Such a tilted configuration is associated with the geometry of small clusters and the great density of CO. The result agrees with our DFT simulation mentioned below.
 | ||
| Fig. 5 (a) CO IRAS spectra from 2.0 L CO adsorbed on Rh clusters/Al2O3/NiAl(100) at varied coverages, as indicated, and 100 K. (b) Integrated intensities of CO IRAS spectra (black squares) and ratios of CO IRAS intensities to the corresponding CO TPD intensities (red circles), as a function of Rh coverage. Rh was deposited at 300 K. | ||
The origin of the decreased adsorption energy of CO on small Rh clusters differs from that on supported Pd clusters/Fe2O3/Pt(111). Published work on the supported Pd clusters showed that the adsorption energy of CO decreased with the cluster size.34,40,42 The size effect was attributed to a decreased length of the Pd–Pd bond in small clusters and a weakened van der Waals interaction on small clusters.34 As the present Rh clusters exhibit expanded lattice parameters and an increased Rh–Rh bond, a decreased adsorption energy is unexpected. Our DFT calculations confirmed this concept and show an increased initial adsorption energy (for small CO coverage) primarily due to the expanded lattice and surface structures on small clusters, which accounts largely for the extension of desorption to 600 K (Fig. 3). The increased initial adsorption energy is not simply attributed to defect sites, such as steps, on small clusters, indicated by previous TPD experiments on stepped Rh surfaces.25 Our experiments confirmed also that the defect sites introduced on a single crystal altered little the adsorption energy (Fig. 6). The ion-sputtered Rh(100) surface is expected to have more surface defects, whereas it shows a desorption temperature, 400–530 K, and a peak position similar to those of pristine Rh(100). The desorption features observed on small Rh clusters (Fig. 3) were absent. Surface defects such as those on the ion-sputtered Rh(100) surface are not responsible for the increased initial adsorption energy on small Rh clusters.
 | ||
| Fig. 6 CO TPD spectra for 2.0 L CO adsorbed on Rh(100) (lower panel) and Ar ion-sputtered Rh(100) (upper panel) single-crystal surfaces at 100 K. The sputtering had about a 4.0 μA sample current and was performed for 40 min. | ||
3.3 DFT calculations for CO adsorbed on supported Rh clusters
To examine the adsorption behaviors of CO on Rh clusters of varied sizes, we computed systematically the adsorption energies (Eads) for CO on Rh clusters (RhC) and surfaces (Rh(100) and Rh(100)E). Eads is defined as E(CO) − E(surface) − E(CO(g)), in which E(CO), E(surface) and E(CO(g)) denote the total energies of the surface with adsorbed CO, a clean surface and gas-phase CO, respectively. RhC, constructed by 38 Rh atoms forming a spherically shaped nanocluster of ca. 1.0 nm diameter (Fig. 7) in a 25 × 25 Å2 supercell, was applied to model small Rh clusters (∼0.05 ML). The large Rh clusters (≥1.0 ML) had an ordered structure of orientation (100) and expanded lattice parameters. Accordingly, Rh(100) surfaces with 6% expanded and optimized lattice parameters (denoted as Rh(100)E and Rh(100), respectively), comprising a 3 × 3 slab of five metal layers (bottom two layers fixed and top three layers allowed to relax) and an equivalent five-metal-layer vacuum space, were applied to model the surfaces for 1.0 and 4.0 ML Rh clusters, respectively. The simulation did not contain the Al2O3/NiAl(100) support because the metal-oxide interfacial interaction altered little the electronic structures of supported Rh clusters. Preceding PES spectra showed no dramatic BE shifts of Rh 3d, Al 2p and O 1s levels due to a strong interface interaction.67,68 A blue shift in the CO IRAS spectra, reflecting oxidized Rh, was also not observed for adsorbed CO.69–71 | ||
| Fig. 7 Top views of a Rh(100) surface and a RhC cluster. The structure of Rh(100)E is similar to that of Rh(100) and not shown. The adsorption sites on RhC include top (T), bridge (B), 4-fold-hollow (4FH) and 3-fold-hollow (3FH) sites. The adsorption sites on Rh(100) and Rh(100)E include T, B and 4FH sites. The yellow spheres represent the surface Rh atoms in the first layer. | ||
CO adsorbed on top (T), bridge (B), three-fold-hollow (3FH) and four-fold-hollow (4FH) sites (Fig. 7) were all examined. The results show that Eads for one CO adsorbed on these sites (for sparse CO coverage) are −2.45 to −2.24 eV on RhC, −2.17 to −2.02 eV on Rh(100)E and −1.99 to −1.84 eV on Rh(100). The most stable CO adsorption sites were the 4FH site on RhC, and the B sites on Rh(100)E and Rh(100). The computational method favored adsorption sites with increased coordination, which differs from the experimental observation.11,23,25,27–29,64 The difference is attributable to the CO adsorption puzzle,72 for which GGA functional favors hollow sites. An agreement with the observation can be achieved through van der Waals or nonlocal correlation calculations (Table S1 in the ESI†).73–75 Nevertheless, the discrepancy does not alter the general trends; the computed energetics with GGA-PW91 could be utilized to understand the adsorption behaviors in the experiments. Eads on Rh(100)E is greater (more negative) than that on Rh(100) as the expanded lattice narrows the metal d band and shifts the anti-bonding band upward to strengthen the adsorption.76 The greatest Eads (most negative) on RhC corresponds to CO bound to the under-coordinated Rh atoms on top and at the edge of a cluster.22,77 The four Rh atoms coordinated to CO resemble corner sites on the clusters. The calculated energetics agreed satisfactorily with the TPD experiments for sparse CO coverage (Fig. 3): the greatest Eads on RhC reflects the desorption at 600 K for small Rh clusters (0.13 ML), whereas the smallest Eads on Rh(100) is that at 530 K for large clusters (4.0 ML). The effect of the expanded lattice is also reflected in the desorption generally extending to higher temperature for smaller clusters (0.25–1.6 ML), confirming the above mentioned speculation.
Fig. 8(a) shows the coverage-dependent Eads, which were calculated with CO uniformly distributed on RhC, Rh(100)E and Rh(100). Eads is a mean adsorption energy for all CO: Eads = [E(nCO) − E(surface) − n × E(CO(g))]/n, in which n is the number of adsorbed CO. The computed results show that Eads decreased (becomes less negative) as the coverage (θ, defined as the ratio of the numbers of CO and surface Rh) increased for all surfaces. The result is attributed to an enhanced interaction among neighboring CO and is confirmed by the charge distribution such that the ionic character of the C–Rh bond attenuated as θ increased, and the density of states for CO at a large coverage showing a greater anti-bonding distribution above the Fermi level to weaken Eads (see Fig. S5 in the ESI†). On Rh(100), Eads is in the range from −1.99 to −1.84 eV at the smallest θ = 0.06, as one CO adsorbed on the surface of 18 Rh atoms; increasing θ to 6 CO on the surface, the change of Eads was limited (Eads = −1.98 eV at θ = 0.33). Upon adsorbing another six CO, Eads showed a steady decrease to −1.85 eV (θ = 0.67). The decrease was doubled upon adsorbing a further six CO, Eads = −1.60 at θ = 1.00 (18 surface CO). The trend of Eads on Rh(100)E was similar, but Eads was greater (Fig. 8(a)): a small decrease for the first six CO adsorptions (Eads = −2.17 to −2.13 eV as θ = 0.06 to 0.33) and moderate decreases for the second and third six CO adsorptions (Eads = −1.98 eV at θ = 0.67 and Eads = −1.81 eV at θ = 1.00). CO on RhC behaves similarly to that on Rh(100)E at θ < 0.75, indicated by the overlapping points of Rh(100)E and RhC in Fig. 8(a), whereas Eads on RhC decreased slowly at greater CO coverage. The adsorption of a single CO on RhC (θ = 0.03) has a wider range of Eads (−2.45 to −2.24 eV), because of distinct adsorption sites. Ten CO evenly distributed on RhC (θ = 0.31) yielded a slightly decreased Eads (−2.15 eV); further increasing the number of CO to 26 (θ = 0.80) maintained Eads near −2.0 eV; increasing CO to 36 (θ = 1.13) still maintained Eads at −1.90 eV, which is similar to values on Rh(100) at θ = 0.5 and Rh(100)E at θ = 0.83. Increasing θ above 1.31 on RhC, Eads began to decrease significantly, implying an evidently decreased onset desorption temperature.
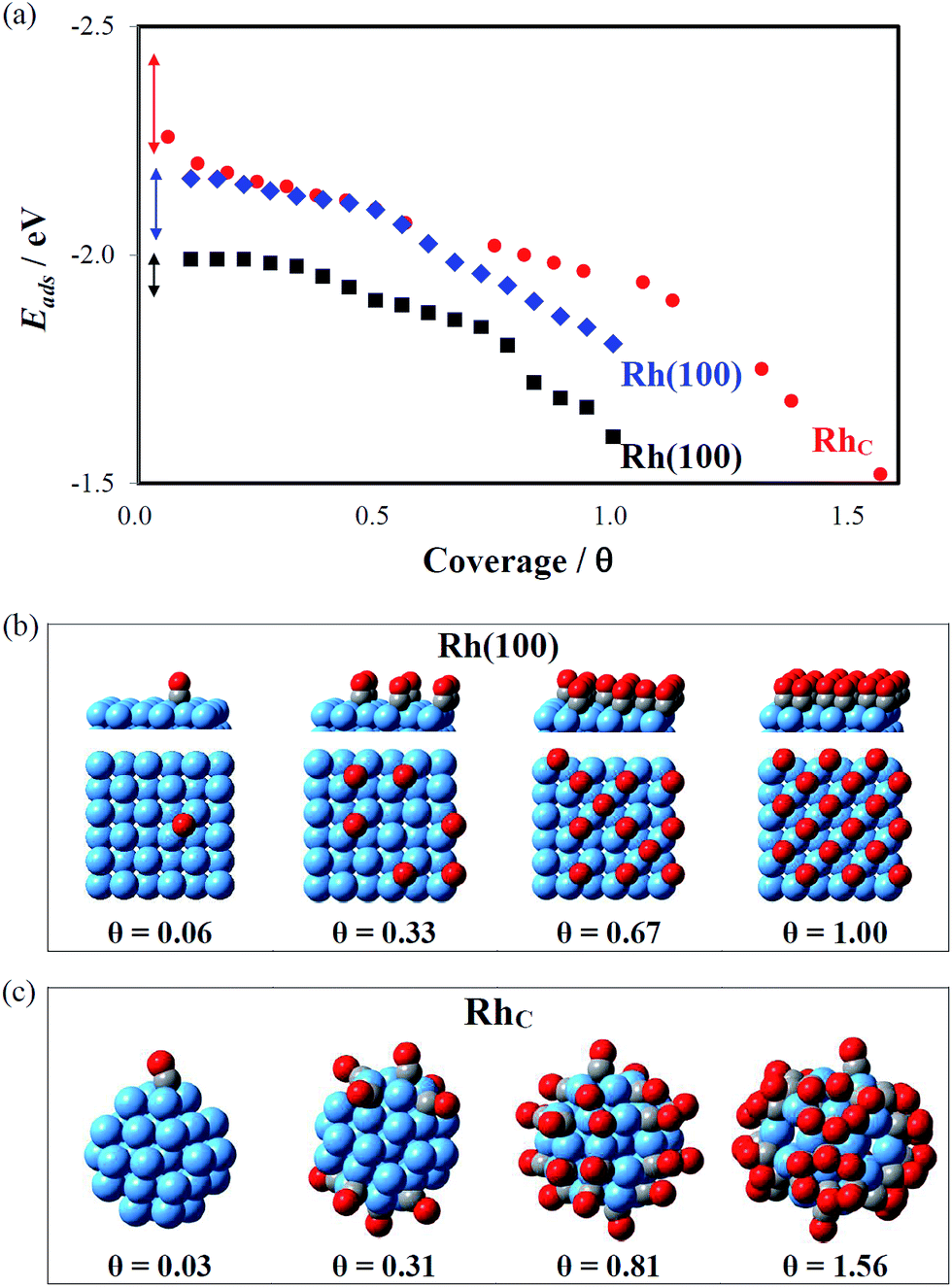 | ||
| Fig. 8 (a) Eads as a function of surface coverage (θ) of CO on Rh(100) and Rh(100)E surfaces and a RhC cluster, in black, blue and red symbols, respectively. The arrows are ranges of Eads at the least coverages on the surfaces and a nanocluster. (b) Selected adsorption structures at θ = 0.06, 0.33, 0.67 and 1.00 on Rh(100); the upper panel is side view and the lower one top view. (c) Selected adsorption structures at θ = 0.03, 0.31, 0.81 and 1.56 on RhC. | ||
The energetic trends (Fig. 8(a)) show that at the same θ, both Rh(100)E and RhC have greater Eads than Rh(100). These results imply that these two Rh surfaces can accommodate readily more CO (at greater θ) than Rh(100). The effects of both expanded lattice and low coordination of Rh clusters contribute to the enhanced density of CO on small clusters. The expanded lattice on Rh(100)E results in a more localized d band of surface Rh to increase Eads. In addition, Eads on RhC decreased notably slowly with θ, particularly for θ > 0.75, which allowed the density of CO to increase further to θ > 1.0. The characteristic trend of Eads on RhC is attributed to weakened lateral interactions of CO. CO adsorbed perpendicularly on the planes of Rh(100) (Fig. 8(b)) but radially on the sphere of RhC (Fig. 8(c)). Such a radial configuration eases the interactions among CO. The distinct adsorption configuration on RhC was confirmed with our IRAS measurements (Fig. 5(b)), for which the axes of adsorbed CO on smaller clusters are much off from the surface normal.
These simulation results match well the TPD experiments shown in Fig. 3. With the same exposure to CO (2.0 L), small Rh clusters had a great θ of CO as small Rh clusters accommodated readily more CO (Fig. 4(c)): the number of adsorbed CO per Rh surface site (CO density) was greater on small clusters. For example, Eads = 1.90 eV at θ = 0.50 and 1.13 for Rh(100) and RhC, respectively, reflecting that the smallest Rh cluster can adsorb 2–3 times more CO than a Rh(100) surface. In addition, a greater θ of CO decreased Eads on small clusters, producing a decreased onset temperature of desorption (<300 K) on small Rh clusters (0.13 ML). In contrast to that on small clusters, Eads on a single-crystal surface significantly decreased with θ for θ > 0.67, which prevented a further increase of θ. This result explains that the same exposure to CO (2.0 L) gives θ = 0.50–0.73 ML on large clusters or single-crystal surfaces.
4. Conclusion
With STM, RHEED, TPD, IRAS and DFT calculations, we investigated the adsorption and lateral interactions of CO on Rh nanoclusters grown with vapor deposition onto a Al2O3/NiAl(100) surface at 300 K. The mean diameter of Rh clusters evolved from 1.0 to 3.5 nm and the mean height from 0.4 to 0.8 nm with increasing coverage. These clusters exhibited a fcc phase and grew with their facets (100) parallel to the θ-Al2O3(100) surface. Relative to bulk Rh, the lattice parameter of the structurally ordered clusters increased, up to 6% with decreasing cluster size. With that decreasing cluster size, the initial adsorption energy and the number of CO adsorbed per surface site on the Rh clusters (CO density) were both enhanced. The former is attributed to the increased corner sites and expanded lattice parameter of small Rh clusters, whereas the latter results largely from weakened lateral interactions of CO, because of the radial adsorption configuration of CO on small Rh clusters. On clusters of diameter near 1.0 nm and height near 0.4 nm, the saturated density of CO was 2–3 times that on large clusters (diameter ≥ 3.5 nm) or a Rh(100) surface. At large CO densities, the repulsive CO–CO interactions became enhanced, resulting in a decreased temperature of onset of CO desorption.Acknowledgements
National Science Council provided support (NSC-100-2112-M-008-010-MY3 and NSC 101-2113-M-003-006-MY3) for this study. CPU time at Taiwan's National Center for High-performance Computing (NCHC) and Department of Applied Chemistry in Private Chinese Culture University (PCCU) is greatly appreciated.References
- B. Beden, J.-M. Léger and C. Lamy, Electrocatalytic Oxidation of Oxygenated Aliphatic Organic Compounds at Noble Metal Electrodes, Plenum Publishers, New York, 1992 Search PubMed.
- G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 1994 Search PubMed.
- J. R. Rostrup-Nielsen, Catal. Rev., 2004, 46, 247–270 CAS.
- G. T. Burstein, C. J. Barnett, A. R. Kucernak and K. R. Williams, Catal. Today, 1997, 38, 425–437 CrossRef CAS.
- A. Hamnett, Catal. Today, 1997, 38, 445–457 CrossRef CAS.
- K. R. Williams and G. T. Burstein, Catal. Today, 1997, 38, 401–410 CrossRef CAS.
- S. Andersson, M. Frank, A. Sandell, A. Giertz, B. Brena, P. A. Brühwiler, N. Mårtensson, J. Libuda, M. Baümer and H.-J. Freund, J. Chem. Phys., 1998, 108, 2967 CrossRef CAS.
- H. P. Bonzel and G. Pirug, The Chemical Physics of Solid Surfaces, Elsevier, Amsterdam, 1st edn, 1993 Search PubMed.
- L. J. Whitman and J. W. Ho, Chem. Phys., 1989, 90, 6018 CAS.
- A. P. V. Bavel, M. J. P. Hopstaken, A. P. V. Bavel, M. J. P. Hopstaken, D. Curulla-Ferré, J. W. Niemantsverdriet, J. J. Lukkien and P. A. J. Hilbers, J. Chem. Phys., 2003, 119, 524 CrossRef.
- M. M. M. Jansen, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, Phys. Chem. Chem. Phys., 2009, 11, 10009–10016 RSC.
- D. L. S. Nieskens, M. M. M. Jansen, A. P. V. Bavel, D. Curulla-Ferré and J. W. Niemantsverdriet, Phys. Chem. Chem. Phys., 2006, 8, 624–632 RSC.
- W. Braun, H.-P. Steinruck and G. Held, Surf. Sci., 2005, 574, 193 CrossRef CAS.
- E. L. Hardegree, P. Ho and J. M. White, Surf. Sci., 1986, 165, 488 CrossRef CAS.
- D. Hoge, M. Tueshaus and A. M. Bradshaw, Surf. Sci., 1988, 207, L935–L942 CrossRef CAS.
- S. Johnson and R. J. Madix, Surf. Sci., 1981, 108, 77 CrossRef CAS.
- S. W. Jorgensen and R. J. Madix, Surf. Sci., 1985, 163, 19 CrossRef CAS.
- M. Kiskinova, Surf. Sci., 1987, 182, 150 CrossRef CAS.
- M. Kiskinova and D. W. Goodman, Surf. Sci., 1981, 108, 64 CrossRef CAS.
- T. Yamada, Z. Runsheng, Y. Iwasawa and K. Tamaru, Surf. Sci., 1988, 205, 82 CrossRef CAS.
- R. Zenobi, J. Xu and J. T. Yates Jr, Surf. Sci., 1992, 276, 241–248 CrossRef CAS.
- M. Mavrikakis, M. Bäumer, H. J. Freund and J. K. Nørskov, Catal. Lett., 2002, 82, 153–156 CrossRef.
- G. Krenn, I. Bako and R. Schennach, J. Chem. Phys., 2006, 124, 144703 CrossRef PubMed.
- R. K. Kose, W. A. Brown and D. A. King, J. Phys. Chem. B, 1999, 103, 8722–8725 CrossRef CAS.
- H. P. Koch, P. Singnurkar, R. Schennach, A. Stroppa and F. Mittendorfer, J. Phys. Chem. C, 2008, 112, 806–812 CAS.
- A. Stroppa, F. Mittendorfer, J. N. Andersen, G. Parteder, F. Allegretti, S. Surnev and F. P. Netzer, J. Phys. Chem. C, 2009, 113, 942–949 CAS.
- A. Baraldi, L. Gregoratti, G. Comelli, V. R. Dhanak, M. Kiskinova and R. Rosei, Appl. Surf. Sci., 1996, 99, 1–8 CrossRef CAS.
- J. D. Batteas, D. E. Gardin, M. A. V. Hove and G. A. Somorjai, Surf. Sci., 1993, 297, 11–18 CrossRef CAS.
- M. Rebholz, R. Prins and N. Kruse, Surf. Sci., 1991, 259, L797–L803 CrossRef CAS.
- F. Strisland, A. L. Ramstad, T. Ramsvik and A. C. Borg, Surf. Sci., 1998, 415, L1020–L1026 CrossRef CAS.
- P. Singnurkar and R. Schennach, Vacuum, 2011, 85, 761–767 CrossRef CAS PubMed.
- T. Zhang, Z.-P. Liu, S. M. Driver, S. J. Pratt, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 95, 266102 CrossRef PubMed.
- P. Rodriguez, Y. Kwon and M. T. M. Koper, Nat. Chem., 2012, 4, 177–182 CrossRef CAS PubMed.
- M. Peter, J. M. F. Camacho, S. Adamovski, L. K. Ono, K.-H. Dostert, C. P. O'Brien, B. R. Cuenya, S. Schauermann and H.-J. Freund, Angew. Chem., Int. Ed., 2013, 52, 5175–5179 CrossRef CAS PubMed.
- M. Frank, S. Andersson, J. Libuda, S. Stempel, A. Sandell, B. Brena, A. Giertz, P. A. Brühwiler, M. Bäumer, N. Mårtensson and H. J. Freund, Chem. Phys. Lett., 1997, 279, 92–99 CrossRef CAS.
- E. S. Putna, R. J. Gorte, J. M. Vohs and G. W. Graham, J. Catal., 1998, 178, 598–603 CrossRef CAS.
- J. Zhou and D. R. Mullins, J. Phys. Chem. B, 2006, 110, 15994–16002 CrossRef CAS PubMed.
- C.-S. Chao, Y.-D. Li, B.-W. Hsu, W.-R. Lin, H.-C. Hsu, T.-C. Hung, C.-C. Wang and M.-F. Luo, J. Phys. Chem. C, 2013, 117, 5667–5677 CAS.
- M. Bäumer, J. Libuda, K. M. Neyman, N. Rösch, G. Rupprechter and H. J. Freund, Phys. Chem. Chem. Phys., 2007, 9, 3541–3558 RSC.
- J. M. Flores-Camacho, J.-H. Fischer-Wolfarth, C. T. C. M. Peter, S. Schauermann and H.-J. Freund, Phys. Chem. Chem. Phys., 2011, 13, 16800–16810 RSC.
- S. Schauermann, J. Hoffmann, V. Johánek, J. Hartmann and J. Libuda, Phys. Chem. Chem. Phys., 2002, 4, 3909–3918 RSC.
- J.-H. Fischer-Wolfarth, J. A. Farmer, J. M. Flores-Camacho, A. Genest, I. V. Yudanov, N. Rösch, C. T. Campbell, S. Schauermann and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 241416 CrossRef.
- B. F. D. Mongeot, A. Toma, A. Molle, S. Lizzit, L. Petaccia and A. Baraldi, Phys. Rev. Lett., 2006, 97, 056103 CrossRef PubMed.
- V. Nehasil, I. Stará and V. Matolín, Surf. Sci., 1995, 331–333, 105–109 CrossRef CAS.
- N. Frémy, V. Maurice and P. Marcus, J. Am. Ceram. Soc., 2003, 86, 669–675 CrossRef.
- R.-P. Blum, D. Ahlbehrendt and H. Niehus, Surf. Sci., 1998, 396, 176–188 CrossRef CAS.
- P. Gassmann, R. Franchy and H. Ibach, Surf. Sci., 1994, 319, 95–109 CrossRef CAS.
- M. C. Zei, C.-S. Lin, W.-H. Wen, C.-I. Chiang and M.-F. Luo, Surf. Sci., 2006, 600, 1942–1951 CrossRef CAS.
- M.-F. Luo, C.-I. Chiang, H.-W. Shiu, S. D. Sartale and C.-C. Kuo, Nanotechnology, 2006, 17, 360–366 CrossRef CAS.
- M.-F. Luo, W.-H. Wen, C.-S. Lin, C.-I. Chiang, S. D. Sartale and M. C. Zei, Surf. Sci., 2007, 601, 2139–2146 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566–569 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- M.-F. Luo, H.-W. Shiu, M.-H. Tien, S. D. Sartale, C.-I. Chiang, Y.-C. Lin and Y.-J. Hsu, Surf. Sci., 2008, 602, 241–248 CrossRef CAS.
- A. Cavallin, M. Pozzo, C. Africh, A. Baraldi, E. Vesselli, C. Dri, G. Comelli, R. Larciprete, P. Lacovig, S. Lizzit and D. Alfè, ACS Nano, 2012, 6, 3034 CrossRef CAS PubMed.
- M. Bäumer, M. Frank, M. Heemeier, R. Kühnemuth, S. Stempel and H. J. Freund, Surf. Sci., 2000, 454–456, 957–962 CrossRef.
- M.-F. Luo, M.-H. Tien, C.-C. Wang, W.-R. Lin, J.-Y. Ho, B.-W. Chang, C.-T. Wang, Y.-C. Lin and Y.-J. Hsu, J. Phys. Chem. C, 2009, 113, 12419–12426 CAS.
- M.-F. Luo, C.-C. Wang, G.-R. Hu, W.-R. Lin, J.-Y. Ho, Y.-C. Lin and Y.-J. Hsu, J. Phys. Chem. C, 2009, 113, 21054–21062 CAS.
- M. Frank, R. Kühnemuth, M. Bäumer and H. J. Freund, Surf. Sci., 1999, 427–428, 288–293 CrossRef CAS.
- M. Frank, R. Kühnemuth, M. Bäumer and H. J. Freund, Surf. Sci., 2000, 454–456, 968–973 CrossRef CAS.
- A. M. Bradshaw and E. Schweizer, Infrared reflection-absorption spectroscopy of adsorbed molecules, in Advances in Spectroscopy: Spectroscopy of Surfaces, Wiley, New York, 1988 Search PubMed.
- T.-C. Hung, T.-W. Liao, Z.-H. Liao, P.-W. Hsu, P.-Y. Cai, H. Lee, Y.-L. Lai, Y.-J. Hsu, H.-Y. Chen, J.-H. Wang and M.-F. Luo, ACS Catal., 2015, 5, 4276 CrossRef CAS.
- S. Andersson, P. A. Brühwiler, A. Sandell, M. Frank, J. Libuda, A. Giertz, B. Brena, A. J. Maxwell, M. Bäumer, H.-J. Freund and N. Mårtensson, Surf. Sci., 1999, 442, L964–L970 CrossRef CAS.
- K. I. Hadjiivanov, J. Chem. Soc., Faraday Trans., 1998, 94, 1901–1904 RSC.
- M. Happel, J. Mysliveček, V. Johánek, F. Dvořák, O. Stetsovych, Y. Lykhach, V. Matolín and J. Libuda, J. Catal., 2012, 289, 118–126 CrossRef CAS.
- K. Chakarova, K. Hadjiivanov, G. Atanasova and K. Tenchev, J. Mol. Catal. A: Chem., 2007, 264, 270 CrossRef CAS.
- P. J. Feibelman, B. Hammer, J. K. Nørskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4018 CrossRef CAS.
- P. Lazić, M. Alaei, N. Atodiresei, V. Caciuc, R. Brako and S. Blügel, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 045401 CrossRef.
- X. Ren, P. Rinke and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 045402 CrossRef.
- Q.-M. Hu, K. Reuter and M. Scheffler, Phys. Rev. Lett., 2007, 98, 176103 CrossRef PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- R. Pushpa, P. Ghosh, S. Narasimhan and S. D. Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 165406 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20384h |
| This journal is © The Royal Society of Chemistry 2016 |