
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of unsymmetrical NCN′ and PCN pincer palladacycles and their catalytic evaluation compared with a related SCN pincer palladacycle†‡
Gavin W.
Roffe
a,
Graham J.
Tizzard
b,
Simon J.
Coles
 b,
Hazel
Cox
a and
John
Spencer
*a
b,
Hazel
Cox
a and
John
Spencer
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Falmer, Brighton, East Sussex BN1 9QJ, UK. E-mail: j.spencer@sussex.ac.uk
bUK National Crystallography Service, School of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 8th June 2016
Abstract
1-(3-(Pyridin-2-yl)phenyl)methanamine derivatives have been synthesized and underwent C–H bond activation to afford unsymmetrical NCN′ pincer palladacycles, which were characterised in the solid state. 2-Pyridinyl-phenol and -benzyl alcohols were then used as precursors to unsymmetrical PCN pincer palladacycles. Catalytic applications, where the palladacycle remains in the Pd(II) state, have been carried out and show good activity and selectivity.
Introduction
Palladacycles contain a covalent Pd–C bond intramolecularly stabilised by a coordinating group such as an amine, phosphine or thioether and have been extensively studied since their discovery in the mid-1960s.1 Pincer palladacycles, where a tridentate ligand can coordinate to palladium were first synthesized by Shaw and Moulton in 1976.2 However, the field of palladacycle-mediated catalysis only truly gathered momentum after the seminal discovery by Herrmann and Beller et al.3,4 that their eponymous palladacycle (Fig. 1) was highly active in catalytic C–C bond coupling reactions. Since, a number of reviews and books have been published covering the wide array of catalytic applications.5–14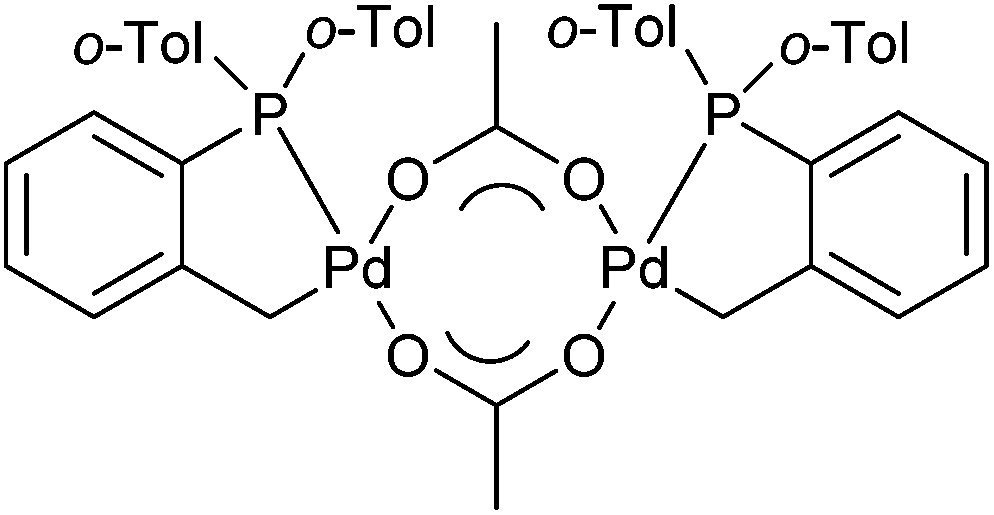 | ||
| Fig. 1 Herrmann–Beller palladacycle. | ||
The majority of pincer palladacycles are symmetrical, such as I (Fig. 2).15 However, unsymmetrical analogues such as II (Fig. 2),16 have been explored,17 and may provide a greater opportunity to fine tune catalysis due to the potential hemilability of the ligand18 and the ability to influence catalytic activity by altering the steric and electronic properties of the donor atoms.19,20
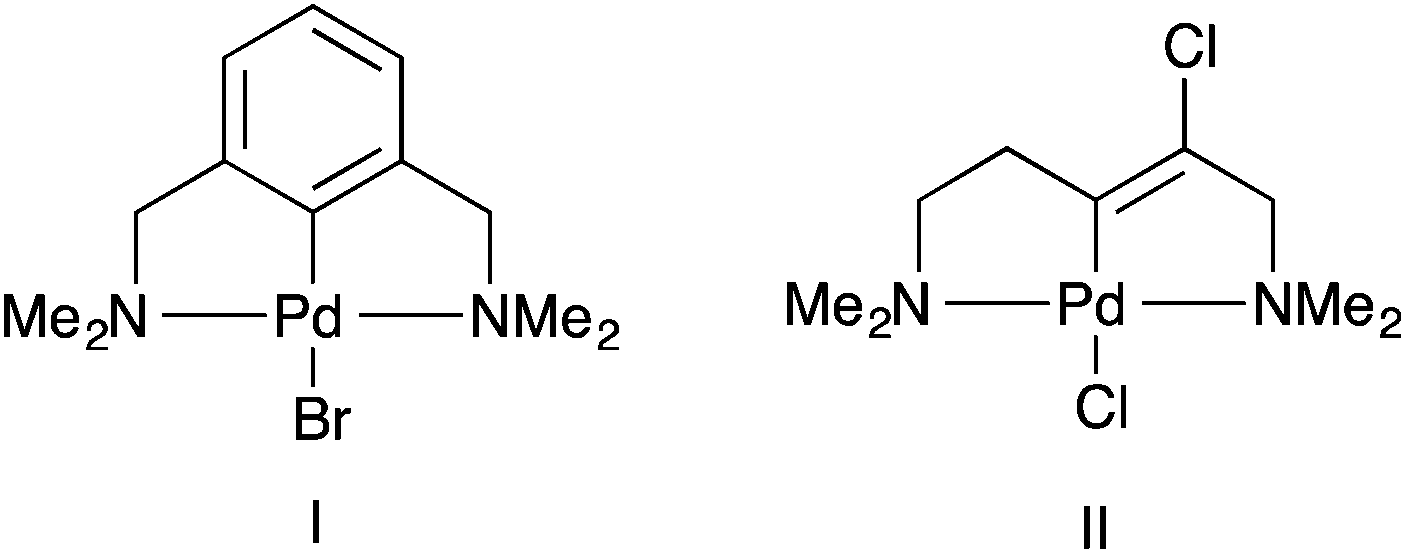 | ||
| Fig. 2 (I) symmetrical, NCN, and (II) unsymmetrical NCN′ pincer palladacycles. | ||
The synthesis of NCN pincers can be complicated compared with the analogous PCP and SCS pincers due to the hard-ligand (amine) soft-acid (metal) mismatch between palladium and nitrogen. This results in competition in forming the kinetic product III or the thermodynamic pincer product IV (Scheme 1).21 Hence, incorporating functional groups in the mutual ortho position such as SiMe3 or Br can be beneficial in their synthesis, compared with C–H activation routes.8 However, this is less attractive synthetically, since an additional step is required in making the functionalised ligand.
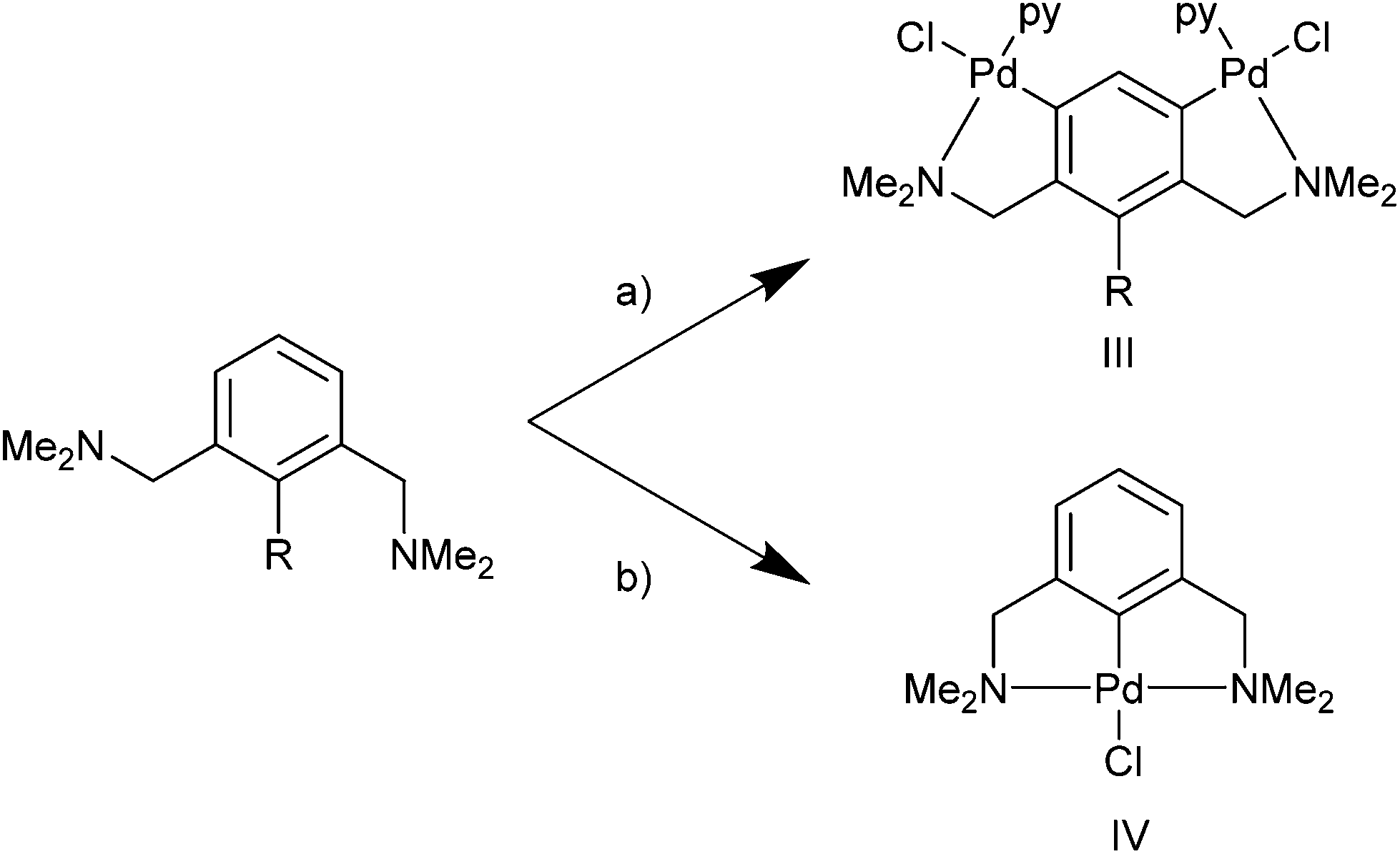 | ||
| Scheme 1 py = pyridine (III) kinetic product, (IV) thermodynamic product. (a) (i) R = H; Pd(OAc)2, MeOH, NEt3, (ii) LiCl, (iii) R = SiMe3. py. (b) Li2[PdCl4]. | ||
Examples of unsymmetrical PCN pincer palladacycles (Fig. 3) are synthesised by C–H bond activation or chloropalladation.16,22,23 A potential route to phosphorus containing palladacycles is via phenolic or benzylic alcohols, such as that by Eberhard et al.,24 in the formation of mixed 5-,6-membered PCP′, and 5-,5-pincer palladacycles (Fig. 3) containing both phosphine and phosphinite groups.
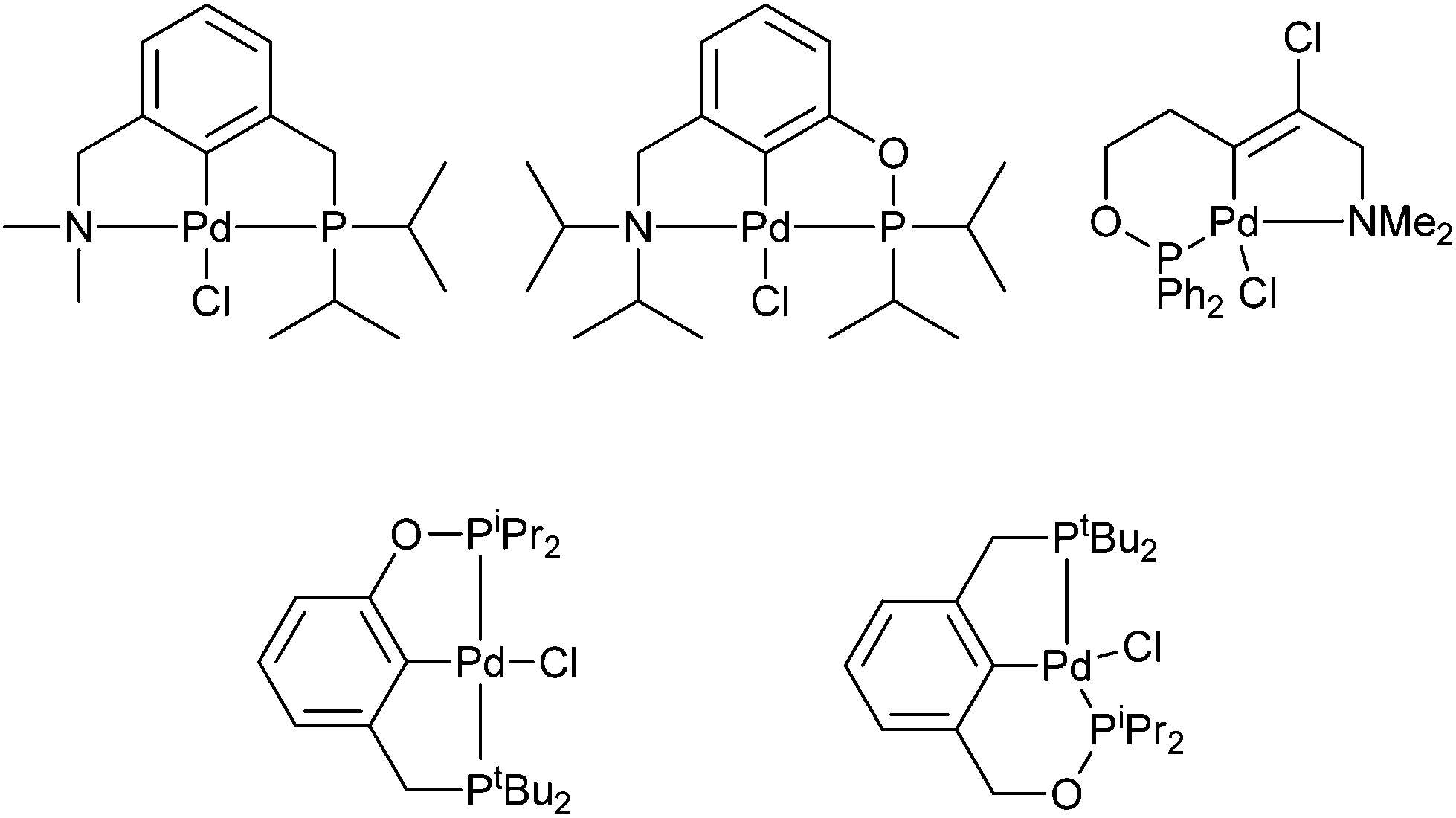 | ||
| Fig. 3 Unsymmetrical PCN, and P′CP pincer palladacycles. | ||
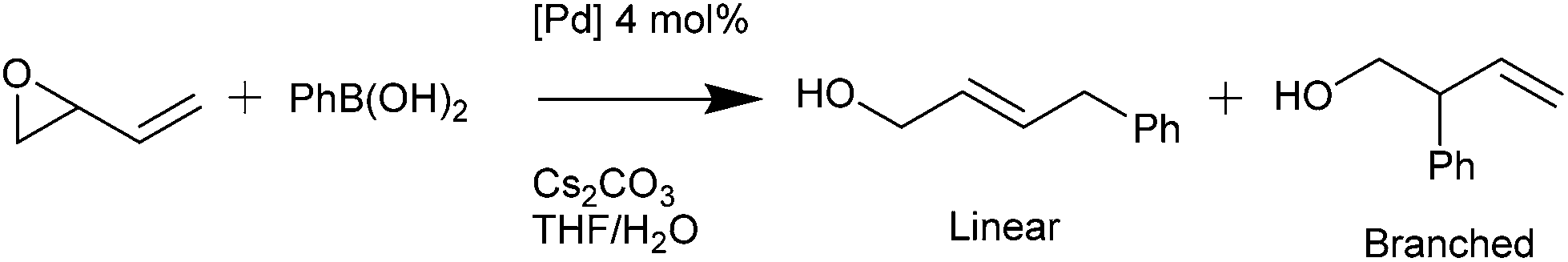
Palladacycles normally act as a reservoir of catalytically active Pd(0) species in applications such as the Suzuki–Miyaura/Heck reaction.25,26 Other catalytic applications of pincer palladacycles utilise the palladium in its Pd(II) oxidation state, retaining its ligand structure, and are more likely to take advantage of the tuning abilities of the unsymmetrical ligand design. The use of pincer palladacycles as a Lewis acid catalyst in an aldol condensation (Scheme 2) demonstrated the ability to change the stereochemical outcome of the reaction depending on the ligand donor atoms.27 Another application utilising pincer palladacycles in Pd(II) catalysis, is in the coupling of vinyl epoxides and boronic acids using a symmetrical SeCSe palladacycle (Scheme 3).28
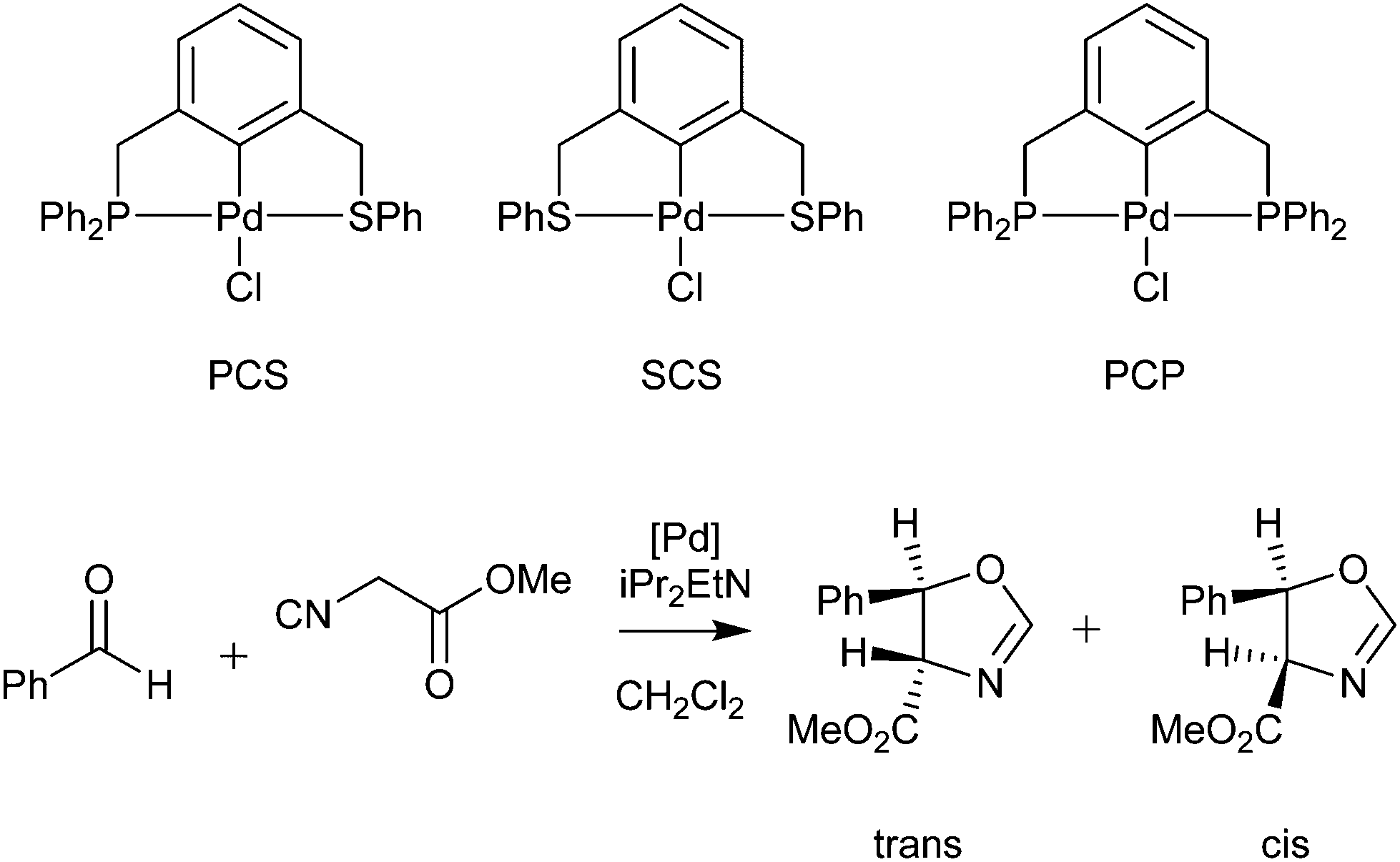 | ||
| Scheme 2 Aldol condensation catalysed by pincer palladacycles. | ||
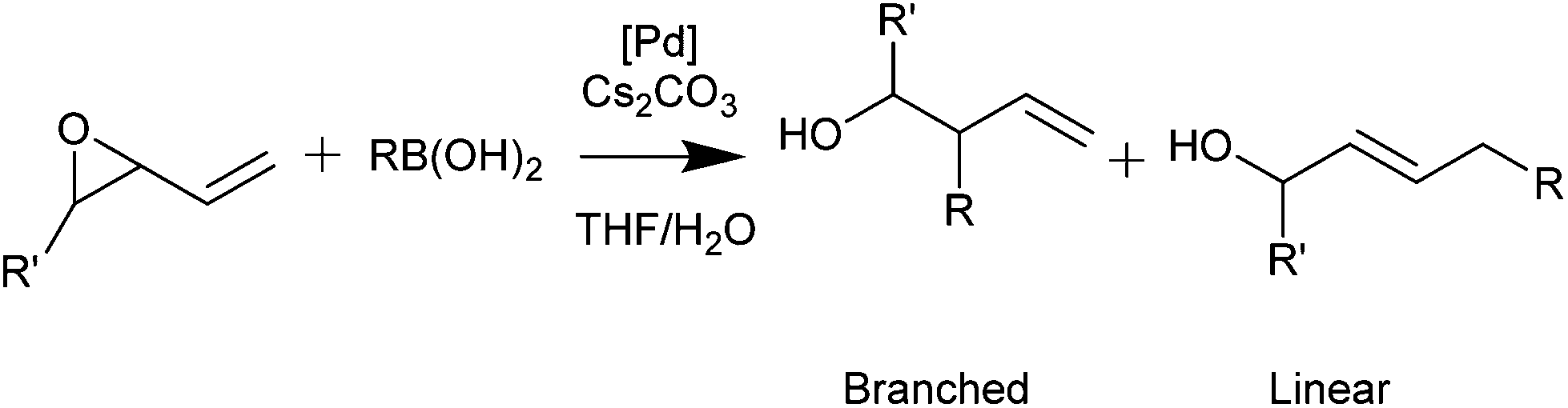 | ||
| Scheme 3 Vinyl epoxide and boronic acid coupling. | ||
Unsymmetrical pincer palladacycles are relatively rare and their synthesis and that of their respective ligands often poses a greater challenge than for their symmetrical counterparts. Given some distinct advantages of employing unsymmetrical pincer palladacycles in catalytic applications,17 such as the tandem catalysis reported by Szabó et al.27 recent work in our group has focused on a robust synthetic route to useful, modular unsymmetrical ligands and their pincer palladacycles.29 The present work is focused on the synthesis of several new unsymmetrical nitrogen-based pincer ligands, denoted NCN′, their respective palladacycles, phosphinite PCN pincer palladacycles, and catalytic evaluation in aldol condensation and vinyl epoxide coupling reactions in comparison to a related SCN pincer palladacycle.
Results and discussion
NCN′ ligand synthesis
The simple biaryl motif 1 was deemed to be a potentially useful precursor to NCN′ pincer ligands. 1 was formed via a Suzuki–Miyaura (SM) coupling and several catalysts, Pd(PPh3)4, Pd(dppf)Cl2, Pd(OAc)2 and Buchwald's XPhos Pd G2 catalyst30 were trialled in order to optimise its yield (Table 1). It was found that Pd(dppf)Cl2 (Pd-118) was the catalyst of choice utilising microwave-mediated (MW) synthesis (Table 1, entry 4).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Reaction conditions | Isolated yield/% |
A = thermal, 85 °C, 18 h. B = thermal, 85 °C, 48 h. C = MW, 150 °C, 20 min. Entries 1–5, 4 mol% catalyst, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 base:toluene:EtOH. Entry 6, 2 mol% catalyst 1:2 base:THF. :2:1 base:toluene:EtOH. Entry 6, 2 mol% catalyst 1:2 base:THF. |
||||
| 1 | Pd(PPh3)4 | 1 M Na2CO3 | A | 65 |
| 2 | Pd(PPh3)4 | 1 M Na2CO3 | B | 72 |
| 3 | Pd(PPh3)4 | 1 M Na2CO3 | C | 58 |
| 4 | Pd(dppf)Cl 2 | 1 M Na 2 CO 3 | C | 79 |
| 5 | Pd(OAc)2 | 1 M Na2CO3 | C | 37 |
| 6 | Buchwald XPhos Pd G2 | 0.5 M K2PO4 | C | 22 |
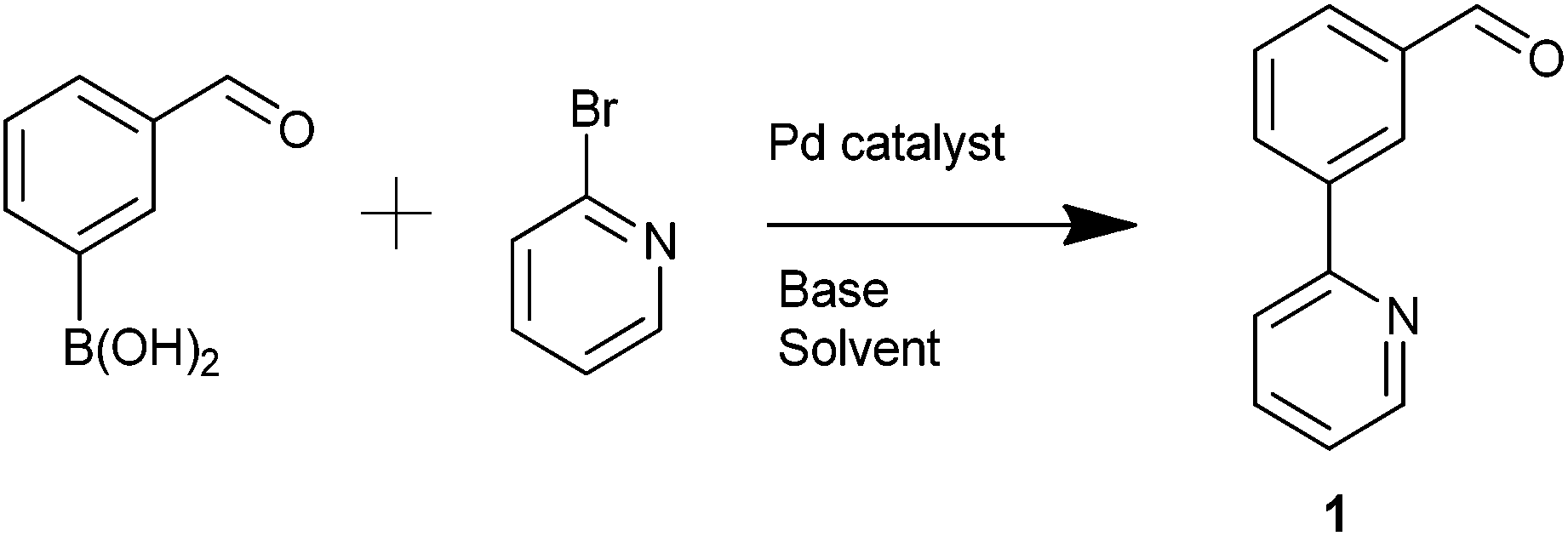
Reductive amination of 1 with HNMe2·HCl was attempted in order to synthesise the requisite unsymmetrical NCN′ ligand with the dimethyl amine and pyridine groups ideally placed to promote C–H activation towards the corresponding palladacycle. Initially sodium triacetoxyborohydride31 proved unsuccessful although the use of titanium isopropoxide/sodium borohydride32 furnished the product 2a in 81% yield (Scheme 4). Next, we attempted reductive aminations with NEt2H using both of the above conditions but to no avail.
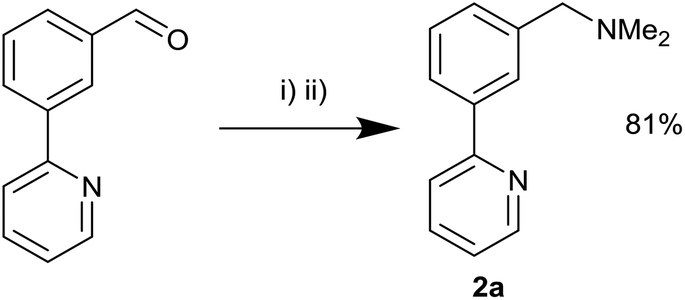 | ||
| Scheme 4 Reductive amination procedure. (i) Ti(OiPr)4, EtOH, NEt3, NHMe2·HCl. (ii) NaBH4. | ||
Given that the reductive amination procedure proved ineffectual, an alternative synthesis was devised. In previous work,29 we have shown the synthesis of the benzylic bromide 4, via benzylic alcohol 3 (Scheme 5) can allow late stage diversification via nucleophilic substitution by sulphur nucleophiles.29 Hence, the corresponding nucleophilic displacement of 4 was undertaken with nitrogen nucleophiles, yielding NCN′ ligands 2b and 2c in excellent yield (Scheme 5).
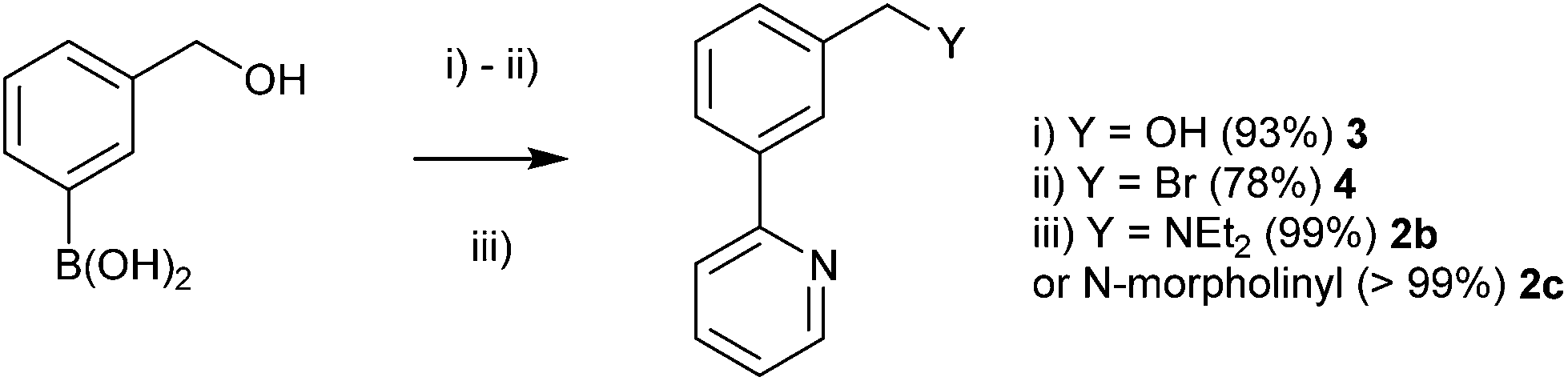 | ||
| Scheme 5 Synthesis of 4 and NCN′ ligands 2b and 2c by nucleophilic displacement with amine nucleophiles. (i) 2-bromopyridine, Pd(PPh3)4, K3PO4, toluene/H2O/EtOH. (ii) HBr, (iii) K2CO3, THF, amine. | ||
NCN′ palladacycle synthesis
Ligands 2a–c were then selected for C–H activation towards the palladacycle products. Refluxing the ligand in AcOH in the presence of Pd(OAc)2, followed by salt metathesis yielded the desired monomeric palladacycles 5a–c (Scheme 6).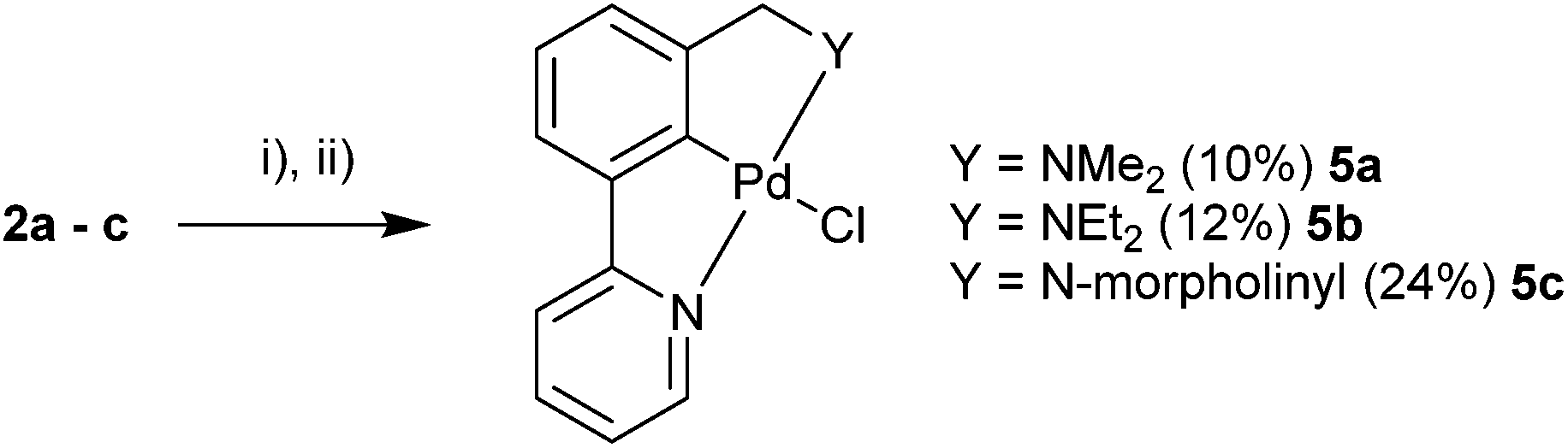 | ||
| Scheme 6 C–H activation of unsymmetrical NCN′ ligands. (i) Pd(OAc)2, acetic acid. (ii) LiCl for 5a and NaCl for 5b and 5c, acetonitrile, H2O. | ||
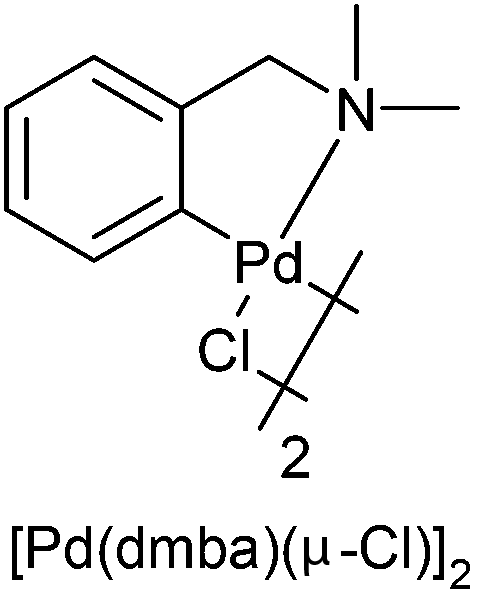
The yields for forming these palladacycles are disappointing, due to significant formation of Pd black, and therefore an optimisation study for the synthesis of 5c was attempted (Table 2). Changing the solvent to MeOH (entry 2), the palladium source to in situ generated Pd(MeCN)4(BF4)2 (entry 3),33 and a transcyclopalladation (entry 4)34 using Pd2(dmba)2Cl2 (Table 2) were attempted. It was found that indeed, the initial Pd(OAc)2 in AcOH method proved to be the most effective for palladation (entry 1).
|
|
|||
|---|---|---|---|
| Entry | Palladium source | Solvent | Yield (%) |
| 1 | Pd(OAc) 2 | AcOH | 24 |
| 2 | Pd(OAc)2 | MeOH | 2 |
| 3 | Pd(MeCN)4(BF4)2 | MeCN | 12 |
| 4 | [Pd(dmba)(μ-Cl)]2 | Toluene | — |
However, literature yields for NCN pincers are often low, with several examples provided using Pd(OAc)2 as the palladium source (Table 3), showing the wide range of yields achieved for the key C–H bond activation step from the corresponding NCN pincer ligands. Shaw's earlier work, in contrast, showed yields for PCP pincers of around 75%.2
In order to delve into such poor yields, we looked at the stability of 5c in refluxing d4 acetic acid over time via1H NMR, which showed no degradation of the palladacycle over time, under the conditions used for C–H activation. We also contemplated an oxidative addition of a Pd(0) salt to a bromide,38 although attempts to ortho-brominate 2c were unsuccessful.
PCN palladacycle synthesis
The synthetic route to unsymmetrical NCN′ pincer palladacycles has been modified to provide a route to unsymmetrical PCN pincer palladacycles. The synthesis of phosphinite palladacycles has been shown by Eberhard et al.39 from a benzyl alcohol using ClPR2, and therefore the benzyl alcohol 3 was used as the starting point for synthesis of a PCN pincer palladacycle. Due to the air sensitivity of the PCN ligand, after reaction with ClPPh2, C–H bond activation using Pd(OAc)2 in AcOH was performed in situ (Scheme 8). This synthesis yields a mixed 5-,6-membered palladacycle, and for direct comparison with our 5-,5-membered SCN,29 and NCN′ pincer palladacycles, a phenolic alcohol was synthesised (Scheme 7). Using the phenolic (6) and benzylic alcohols (3), the synthesis of PCN pincer palladacycles was undertaken (Scheme 8). The PCN pincer palladacycle structures were also confirmed using X-ray crystallography (Fig. 5). | ||
| Scheme 7 Phenol-pyridine synthesis. | ||
 | ||
| Scheme 8 Synthesis of PCN pincer palladacycles. (i) NEt3, DMAP, Et2O 0 °C. (ii) ClPPh2, rt. (iii) Pd(OAc)2, AcOH. (iv) NaCl, H2O/MeCN. 7an = 1, 7bn = 0. | ||
Growth of crystals of 5a–c and 7a and 7b by slow evaporation of CH2Cl2 from a saturated solution allowed their structures to be determined by X-ray diffraction (Fig. 4 and 5). All structures were as expected, with the metal in a distorted square planar arrangement.
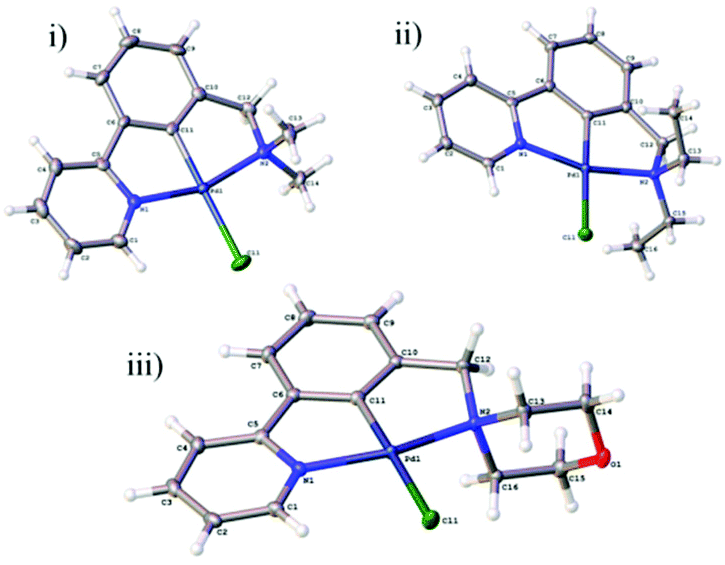 | ||
| Fig. 4 NCN′ pincer palladacycle crystal structures of (i) 5a, (ii) 5b, (iii) 5c. Structures 5a and 5b have 2 molecules in the asymmetric unit, with only one shown for clarity. | ||
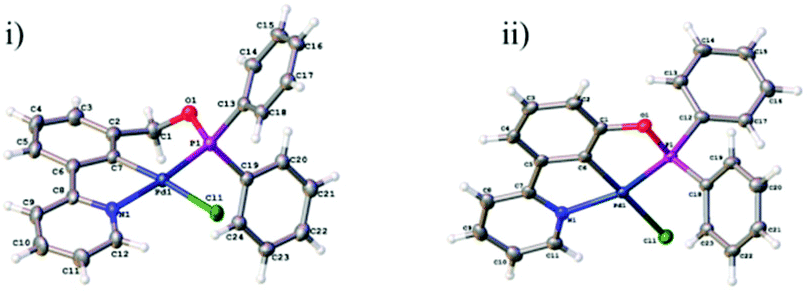 | ||
| Fig. 5 PCN pincer palladacycle crystal structures of (i) 7a, and (ii) 7b. | ||
Compound 5a crystallises in the monoclinic Pc space group with two independent molecules in the asymmetric unit. These are subject to π–π interactions (benzene centroid-pyridine centroid distances: 3.564 Å and 3.715 Å, offsets: 1.301 Å and 1.386 Å respectively). These pairs of molecules form layers with a ‘herringbone’ arrangement propagating along the crystallographic b and c axes.
The structure 5b crystallises in the monoclinic P21/c space group with two independent molecules in the asymmetric unit. The structure is formed from corrugated layers of molecules propagating along the crystallographic a and b axes with each alternate layer consisting of the same crystallographically-independent molecules.
The structure 5c crystallises in the monoclinic P21/n space group. The structure consists of π–π stacks (benzene centroid–pyridine centroid distances: 3.616 Å and 3.705 Å, offsets: 1.319 Å and 1.617 Å respectively) along the crystallographic c axis formed of alternating molecules related by inversion. There are weak C–H⋯O (C2⋯O1 = 3.458(3) Å and C8⋯O1 = 3.371 (3) Å) interactions between the stacks which lead to the formation of corrugated layers along the a and b axes.
Structure 7a crystallises in the monoclinic P21/n space group. The structure consists of columns of molecules close-packed along the crystallographic 21 screw axis parallel to the b axis with π–π interactions to neighbouring columns via the phenyl pyridine moieties (centroid–centroid distance: 3.894 Å, offset: 1.322 Å).
Structure 7b crystallises in the lower symmetry triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The structure is comprised of close-packed discrete layers parallel to the ac plane. These layers in turn are made up of close-packed ‘chains’ of C–H⋯O (C19⋯O1 = 3.405(2) Å) dimers with π–π interactions between neighbouring dimers via the phenyl pyridine moieties (centroid–centroid distance: 3.653 Å, offset: 1.098 Å). Structures 5a–c and 7a–b were given CCDC numbers 1038592, 1038593, 1038594, 1476634 and 1476635 respectively.
space group. The structure is comprised of close-packed discrete layers parallel to the ac plane. These layers in turn are made up of close-packed ‘chains’ of C–H⋯O (C19⋯O1 = 3.405(2) Å) dimers with π–π interactions between neighbouring dimers via the phenyl pyridine moieties (centroid–centroid distance: 3.653 Å, offset: 1.098 Å). Structures 5a–c and 7a–b were given CCDC numbers 1038592, 1038593, 1038594, 1476634 and 1476635 respectively.
Catalytic investigation
Next, 5c, 7a and 7b were tested in the catalytic aldol condensation (Scheme 2) and compared with a previously published SCN pincer palladacycle (8) by our group (Fig. 6)29 along with commercially available PdCl2(dtbpf) and Pd(OAc)2. The results are presented in Table 4, alongside the published data by Szabó and co-workers for symmetrical PCP and SCS pincer palladacycles, as well an unsymmetrical PCS pincer palladacycle (Scheme 2).27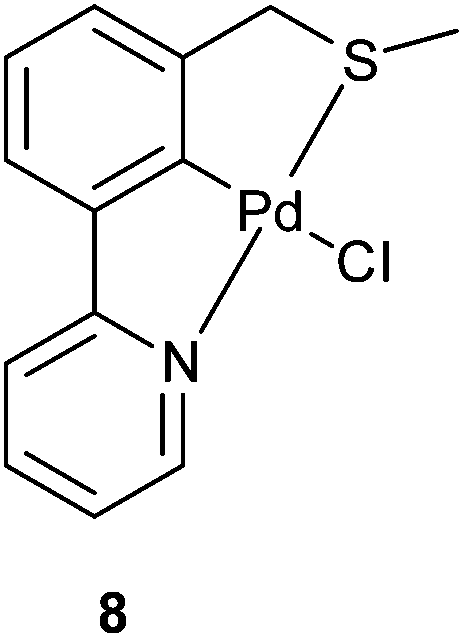 | ||
| Fig. 6 Previously published SCN pincer palladacycle by our group. | ||
|
|
|||
|---|---|---|---|
| Entry | Catalyst | trans/cis ratioa/% | Yield/% |
| a Performed in duplicate, average of 2 runs. trans/cis ratio determined by crude 1H NMR of the resulting mixture. Reaction conditions: methylisocyanoacetate (1 eq.), benzaldehyde (1 eq.), iPr2EtN (0.1 eq.), [Pd] (1 mol%), DCM (5 cm3), 24 h, rt. b Scheme 2. | |||
| 1 | 7a | 85/15 | 99 |
| 227 | PCPb | 82/18 | |
| 3 | 7b | 73/27 | Quantitative |
| 427 | SCSb | 59/41 | |
| 5 | 5c | 59/41 | 93 |
| 6 | Pd(OAc)2 | 58/42 | Quantitative |
| 7 | 8 | 57/43 | 99 |
| 827 | PCSb | 57/43 | |
| 9 | PdCl2(dtbpf) | 56/44 | Quantitative |
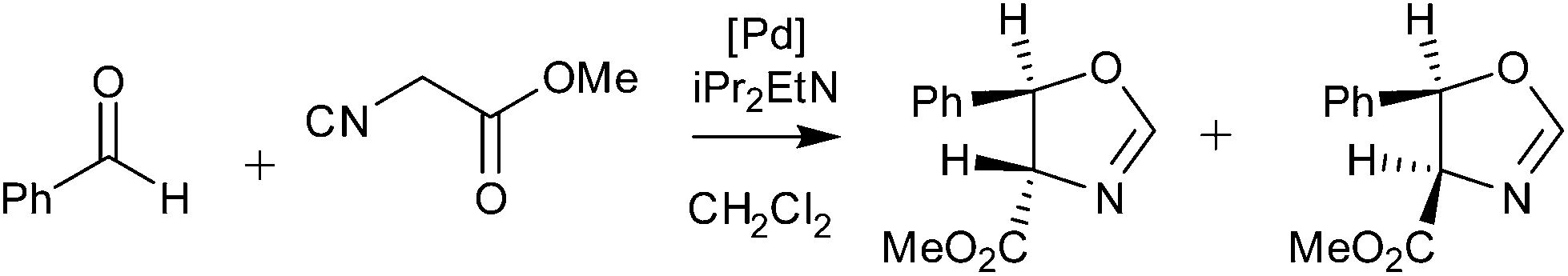
The results presented in Table 4 show that the family of palladacycles synthesised by our group, with SCN, NCN′ and PCN examples, provides the opportunity to fine tune catalytic activity, with the choice of donor group influencing the stereochemical outcomes of the aldol condensation. The novel PCN pincer palladacycle 7a provides the highest trans selectivity of all catalysts tested, and slightly more than the literature PCP example, whereas the SCN and NCN′ provide a greater proportion of the cis product.27
In addition, catalysis of the coupling of a vinylepoxide and phenylboronic acid was tested, using the NCN′, SCN and PCN pincer palladacycles, and compared to literature SeCSe results shown in Fig. 7,28 and our new results using the symmetrical SCS palladacycle (Scheme 2). The coupling and results are presented in Table 5.
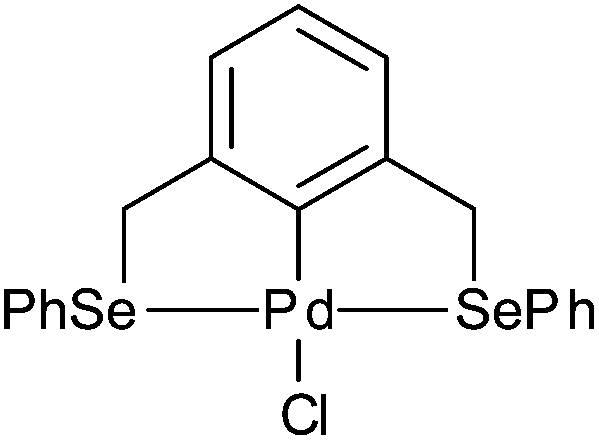 | ||
| Fig. 7 Symmetrical SeCSe pincer palladacycle. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Lineara/% | Brancheda/% | Yield/% |
|
a Performed in duplicate, average of two runs. Linear/branched ratio determined by crude 1H NMR of the resulting mixture. Reaction conditions: 2-vinyl oxirane (1 eq.), PhB(OH)2 (1.2 eq.), Cs2CO3 (2 eq.), [Pd] (4 mol%), 10:1 THF:H2O (3 cm3), 24 h rt.
|
||||
| 128 | SeCSe | 92 | 8 | 94 |
| 2 | SCS | 78 | 22 | >99 |
| 3 | 7a | 66 | 34 | 66 |
| 4 | 7b | 64 | 36 | 96 |
| 5 | 8 | 64 | 36 | 96 |
| 6 | 5c | 60 | 40 | 84 |
The results presented in Table 5 show that the symmetrical pincer palladacycles, SeCSe and SCS achieve the greatest linear selectivity (>78%), whereas the unsymmetrical pincer palladacycles achieve a higher proportion of the branched product (>34%). Clearly the presence of unsymmetrical pincer palladacycles is having an effect of the stereochemical outcome of the reaction, increasing the proportion of the branched product.
Conclusions
The simple synthesis of three NCN′ ligands has been shown. Their C–H activation yielded three new NCN′ pincer palladacycles. Similarly, two novel PCN pincer palladacycles were synthesised, with 5-,5-, and 5-,6-membered rings and all palladacycles were characterised by X-ray crystallography.The catalytic applications of the NCN′, SCN and PCN pincer palladacycles, along with several other examples were considered in two Pd(II)-mediated reactions, an aldol condensation and vinyl epoxide coupling. It was found that in the aldol condensation, the ability to fine tune the stereochemical outcome of the reaction is possible, by varying the donor atoms in the palladacycle catalyst. It was also shown in the vinyl epoxide coupling that the unsymmetrical pincer palladacycles achieve different product ratios than the symmetrical palladacycles tested, demonstrating the potential benefits of unsymmetrical palladacycles in catalytic applications.
Further work is being undertaken towards varying the families of unsymmetrical palladacycles, supported by DFT investigations into their bonding properties and reactivity. Moreover, it is hoped that such simple synthetic routes to unsymmetrical pincer ligands may encourage others to employ these in other areas of transition metal chemistry and catalysis.
Experimental
General details
Solvents and chemicals were purchased from commercial suppliers and used without further purification, with reactions taking place open to atmosphere and moisture.Instrumentation
1H and 13C spectra were recorded on either a Varian 400 or 500 MHz spectrometer. HRMS were obtained on an ESI mass spectrometer using a Bruker Daltonics Apex III, with source Apollo ESI, using methanol as the spray. Flash chromatography was performed on an automated ISCO RF75. GC measurements were obtained using a Perkin Elmer Autosystem XL Gas Chromatograph, utilizing a flame ionization detector, and a Supelco MDN-5S 30 m × 0.25 mm × 0.25 μm column, with a He mobile phase. Elemental analyses were run by the London Metropolitan University Elemental Analysis Service. Crystal structures were obtained by the UK National Crystallography Service at the University of Southampton.40:3 CH2Cl2:ethyl acetate) yielding 402 mg of the expected product 1 as a yellow oil in 74% yield. 1H NMR (500 MHz, chloroform-d) δ 10.13 (s, 1H), 8.74 (d, J = 4.8 Hz, 1H), 8.52 (s, 1H), 8.30 (d, J = 7.7 Hz, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.82–7.79 (m, 2H), 7.65 (dd, J = 7.7, 7.7 Hz, 1H), 7.32–7.28 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 192.2, 155.9, 149.9, 140.3, 137.0, 136.9, 132.7, 129.7, 129.5, 128.4, 122.8, 120.6. HRMS. Calcd for [C12H9NO + H]+ 184.0757. Found 184.0761.
:1 CH2Cl2:ethyl acetate) yielding 59 mg of the expected product 5b as a yellow solid in 12% yield. 1H NMR (500 MHz, chloroform-d) δ 9.06 (d, J = 5.5 Hz, 1H), 7.80 (ddd, J = 7.8, 7.8, 1.6 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.17 (ddd, J = 7.6, 5.5, 1.3 Hz, 1H), 7.06 (dd, J = 7.6, 7.6 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 4.16 (s, 2H), 3.51–3.44 (m, 2H), 2.85–2.78 (m, 2H), 1.61 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, chloroform-d) δ 165.0, 163.4, 152.2, 148.1, 142.4, 138.5, 124.2, 122.7, 122.4, 120.8, 118.4, 67.2, 58.1 (2C), 14.1 (2C). HRMS. Calcd for [C16H19N2Pd]+ 345.0578. Found 345.0564. Anal. Calcd for C16H19N2PdCl: C, 50.41; H, 5.02; N, 7.35. Found: C, 50.34; H, 4.92; N, 7.46.
:3 CH2Cl2:ethyl acetate) yielding 103 mg of the expected product 5c as a yellow solid in 24% yield. 1H NMR (500 MHz, chloroform-d) δ 9.16 (d, J = 5.6 Hz, 1H), 7.80 (dd, J = 7.8, 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.17 (dd, J = 7.6, 5.6 Hz, 1H), 7.09 (dd, J = 7.6, 7.6 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 4.38 (s, 2H), 4.30–4.28 (m, 2H), 4.10–4.06 (m, 2H), 3.90–3.86 (m, 2H), 3.01–2.97 (m, 2H). 13C NMR (100 MHz, chloroform-d) δ 165.1, 163.6, 152.4, 144.5, 142.4, 138.7, 124.8, 123.6, 122.6, 121.2, 118.5, 69.9, 62.8 (2C), 59.64 (2C). HRMS. Calcd for [C16H17N2OPd]+ 359.0370. Found 359.0359. Anal. Calcd for C16H17N2OPdCl: C, 48.63; H, 4.34; N, 7.09. Found: C, 48.59; H, 4.42; N, 6.97.
:2 CH2Cl2:EtOAC) yielding 250 mg of the expected product 6 as a clear liquid in 73% yield. 1H NMR (500 MHz, chloroform-d) δ (ppm): 9.51 (d, 1H), 8.63 (d, J = 4.4 Hz, 1H), 7.86–7.83 (m, 2H), 7.51 (s, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.34–7.32 (m, 1H), 7.24 (dd, J = 7.9, 7.9 Hz, 1H), 6.83 (d, J = 7.9 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ (ppm): 158.2, 156.5, 149.9, 140.5, 137.6, 130.2, 123.0, 120.6, 117.7, 116.5, 113.8. HRMS. Calcd for [C11H9NO + H]+ 172.0757. Found 172.0755.
:3 CH2Cl2:EtOAc) yielding 383 mg of the expected product (7a) as a yellow solid in 29% yield. 1H NMR (500 MHz, chloroform-d) δ (ppm): 9.38 (s, 1H), 8.08–8.03 (m, 4H), 7.88–7.79 (m, 2H), 7.63 (d, J = 7.6 Hz, 1H), 7.52–7.44 (m, 6H), 7.30 (d, J = 6.6 Hz, 1H), 7.19 (dd, J = 7.6, 7.6 Hz, 1H), 7.03 (d, J = 6.6 Hz, 1H), 4.83 (d, J = 19.6 Hz, 2H). 13C NMR (126 MHz, chloroform-d) δ (ppm): 163.3, 151.9, 149.4, 148.2, 139.2 (2C), 136.5, 133.2 (d, 2JPC = 13.4 Hz), 133.0 (d, 1JPC = 59.8 Hz, 2C), 131.7 (d, 3JPC = 2.4 Hz), 128.4 (d, 3JPC = 2.0 Hz, 4C), 128.3 (d, 2JPC = 11.6 Hz, 4C), 125.4, 124.1, 122.5 (d, 3JPC = 3.4 Hz), 118.5 (d, 3JPC = 1.9 Hz), 72.1 (d, 2JPC = 2.8 Hz). 31P NMR (162 MHz, chloroform-d) δ (ppm): 123.27. HRMS. Calcd for [C24H19NOPPd]+ 474.0239. Found 474.0239. Anal. Calcd for C24H19NOPPdCl: C, 56.49; H, 3.75; N, 2.75. Found: C, 56.48; H, 3.82; N, 2.83.
Aldol condensation reaction
Under an argon atmosphere, the palladium catalyst (0.01 eq.) was dissolved in DCM. The methylisocyanoacetate (1 eq.), benzaldehyde (1 eq.) and DIPEA (0.1 eq.) were added to the reaction vessel. The reaction was stirred at rt for 24 h, then the reaction diluted with DCM (10 cm3), washed with H2O (10 cm3), and the organic layer collected using a hydrophobic frit. The solvent was removed in vacuo, and the mixture of products was obtained as a brown oil. 1H NMR (500 MHz, chloroform-d) δ 7.47–7.27 (m, 12H), 5.75 (d, J = 11.1 Hz, 1H, cis), 5.70 (d, J = 7.8 Hz, 1H, trans), 5.10 (dd, 11.1, 2.0 Hz, 1H, cis), 4.65 (dd, 7.8, 2.2 Hz, 1H, trans), 3.85 (s, 3H, trans), 3.21 (s, 3H, cis).Vinyl epoxide coupling
The palladium catalyst (4 mol%), phenylboronic acid (1.2 eq.) and Cs2CO3 (2 eq.) were dissolved in 10:1 THF:H2O (3 cm3). The 2-vinyloxirane was then added (1 eq.), and the reaction was stirred at rt for 24 h. The solvent was removed in vacuo, dissolved in DCM (5 cm3), washed with H2O (5 cm3), and separated using a hydrophobic frit. The solvent was then removed in vacuo yielding the mixture of linear and branched products. 1H NMR (500 MHz, chloroform-d) δ 7.40–7.20 (m, 10H), 6.03 (ddd, J = 18.0, 10.7, 7.3 Hz, 1H, branched), 5.91–5.85 (m, 1H, linear), 5.77–5.69 (m, 1H, linear), 5.24–5.19 (m, 2H, branched), 4.13 (s, 2H, linear), 3.88–3.81 (m, 2H, branched), 3.55 (dt, 7.3, 7.3 Hz, 1H, branched), 3.40 (d, 6.7 Hz, 2H, linear), 1.66 (s, 1H, branched), 1.45 (s, 1H, linear). 13C NMR (100 MHz, chloroform-d) δ 140.7 (branched), 140.0 (linear), 138.3 (branched), 131.5 (linear), 130.3 (linear), 128.7 (branched), 128.5 (linear), 128.4 (linear), 126.9 (branched), 126.1 (linear), 117.0 (branched), 66.1 (branched), 63.5 (linear), 52.5 (branched), 38.6 (linear).
Acknowledgements
We would like to thank the University of Sussex for a studentship (GWR), Christopher Dadswell for the support and use of the GC equipment, and Johnson Matthey for the loan of palladium salts. We would also like to thank Dr Alaa Abdul-Sada at the University of Sussex and the EPSRC Mass Spectroscopy Facility (University of Swansea) for mass spectrometry services.Notes and references
- A. C. Cope and R. W. Siekman, J. Am. Chem. Soc., 1965, 87, 3272–3273 CrossRef CAS.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844–1848 CrossRef CAS.
- M. Beller, H. Fischer, W. A. Herrmann, K. Öfele and C. Brossmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1848–1849 CrossRef CAS.
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS.
- Palladacycles, ed. J. Dupont and M. Pfeffer, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- R. B. Bedford, Chem. Commun., 2003, 1787–1796 RSC.
- Pincer and Pincer-Type Complexes, ed. K. J. Szabó and O. F. Wendt, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014 Search PubMed.
- D. Morales-Morales, Rev. Soc. Quim. Mex, 2004, 48, 338–346 CAS.
- The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, 2007 Search PubMed.
- R. Ratti, Can. Chem. Trans., 2014, 2, 467–488 Search PubMed.
- W.-H. Zhang, S. W. Chien and T. S. A. Hor, Coord. Chem. Rev., 2011, 255, 1991–2024 CrossRef CAS.
- D. M. Grove, G. van Koten, J. N. Louwen, J. G. Noites, A. L. Spek and H. J. C. Ubbels, J. Am. Chem. Soc., 1982, 104, 6609–6616 CrossRef CAS.
- G. Ebeling, M. R. Meneghetti, F. Rominger and J. Dupont, Organometallics, 2002, 21, 3221–3227 CrossRef CAS.
- I. Moreno, R. SanMartin, B. Ines, M. T. Herrero and E. Domínguez, Curr. Org. Chem., 2009, 13, 878–895 CrossRef CAS.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS.
- H. Blaser, W. Brieden, B. Pugin, F. Spindler, M. Studer and A. Togni, Top. Catal., 2002, 19, 3–16 CrossRef CAS.
- A. Togni, C. Breutel, A. Schnyder, F. Spindler, H. Landert and A. Tijani, J. Am. Chem. Soc., 1994, 116, 4062–4066 CrossRef CAS.
- P. Steenwinkel, R. A. Gossage, T. Maunula, D. M. Grove and G. van Koten, Chem. – Eur. J., 1998, 4, 763–768 CrossRef CAS.
- A. Fleckhaus, A. H. Mousa, N. S. Lawal, N. K. Kazemifar and O. F. Wendt, Organometallics, 2015, 34, 1627–1634 CrossRef CAS.
- S. M. Khake, V. Soni, R. G. Gonnade and B. Punji, Dalton Trans., 2014, 43, 16084–16096 RSC.
- M. R. Eberhard, S. Matsukawa, Y. Yamamoto and C. M. Jensen, J. Organomet. Chem., 2003, 687, 185–189 CrossRef CAS.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed.
- R. B. Bedford, C. S. J. Cazin, M. B. Hursthouse, M. E. Light and V. J. M. Scordia, Dalton Trans., 2004, 3864–3868 RSC.
- M. Gagliardo, N. Selander, N. C. Mehendale, G. van Koten, R. J. M. Klein Gebbink and K. J. Szabó, Chem. – Eur. J., 2008, 14, 4800–4809 CrossRef CAS PubMed.
- J. Kjellgren, J. Aydin, O. A. Wallner, I. V. Saltanova and K. J. Szabó, Chem. – Eur. J., 2005, 11, 5260–5268 CrossRef CAS PubMed.
- G. W. Roffe, S. Boonseng, C. B. Baltus, S. J. Coles, I. J. Day, R. N. Jones, N. J. Press, M. Ruiz, G. J. Tizzard, H. Cox and J. Spencer, R. Soc. Open Sci., 2016, 3, 150656 CrossRef PubMed.
- T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff, R. D. Shah and A. F. Abdel-Magid, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- R. J. Mattson, K. M. Pham, D. J. Leuck and K. A. Cowen, J. Org. Chem., 1990, 55, 2552–2554 CrossRef CAS.
- S. J. Loeb, G. K. H. Shimizu and J. A. Wisner, Organometallics, 1998, 17, 2324–2327 CrossRef CAS.
- J. Aydin, N. Selander and K. J. Szabó, Tetrahedron Lett., 2006, 47, 8999–9001 CrossRef CAS.
- X.-Q. Hao, Y.-N. Wang, J.-R. Liu, K.-L. Wang, J.-F. Gong and M.-P. Song, J. Organomet. Chem., 2010, 695, 82–89 CrossRef CAS.
- J.-L. Niu, X.-Q. Hao, J.-F. Gong and M.-P. Song, Dalton Trans., 2011, 40, 5135–5150 RSC.
- J. S. Fossey and C. J. Richards, Organometallics, 2004, 23, 367–373 CrossRef CAS.
- H. P. Dijkstra, M. Q. Slagt, A. McDonald, C. A. Kruithof, R. Kreiter, A. M. Mills, M. Lutz, A. L. Spek, W. Klopper, G. P. M. van Klink and G. van Koten, Eur. J. Inorg. Chem., 2003, 830–838 CrossRef CAS.
- M. R. Eberhard, S. Matsukawa, Y. Yamamoto and C. M. Jensen, J. Organomet. Chem., 2003, 687, 185–189 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Dedicated to Professor Barry M. Trost for his many contributions to palladium chemistry. |
| ‡ Electronic supplementary information (ESI) available: Copy of spectra and crystal structure data. CCDC 1038592–1038594, 1476634 and 1476635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00198j |
| This journal is © the Partner Organisations 2016 |