DOI:
10.1039/C4RA01199F
(Paper)
RSC Adv., 2014,
4, 23730-23739
Molecular dynamics study on the interaction between doxorubicin and hydrophobically modified chitosan oligosaccharide†
Received
11th February 2014
, Accepted 15th April 2014
First published on 15th April 2014
Abstract
Doxorubicin (DOX) is a broad spectrum anti-tumor anthracycline antibiotic used in cancer chemotherapy, but it has certain limitations in its therapeutic effects due to non-specific targeting. Amphiphilic polymeric micelle drug delivery systems could help to improve the activity and selectivity of DOX against tumor cells. In this study, molecular dynamics simulations were performed to investigate the interaction between DOX and ten hydrophobic acid modified chitosan oligosaccharides (COS). The π–π interactions in the systems with aromaticity have been found to contribute to a great part of the van der Waals interactions and play a significant role in the DOX loading process. The encapsulation of DOX by long-chain fatty acid-grafted COS mainly depends on a high binding strength and sandwiched configuration, where the hydrophobic interactions are essential to the encapsulation process. The solvent structure around DOX and the grafted COS was found to have a relationship with the way DOX and drug carrier bind to each other. Moreover, the results derived by our computational model were compared to the experimental data obtained in our lab and the data available in the literature. It was found that the interaction strength between DOX and hydrophobically modified COS has a strong correlation with the experimental quantities, like encapsulation efficiency and drug loading rate.
1 Introduction
Doxorubicin (DOX), as shown in Fig. 1(a), is a broad spectrum anti-tumor anthracycline antibiotic used in cancer chemotherapy. It belongs to the class of cycle-phase nonspecific drugs which can kill tumor cells with a variety of cell cycles.1 It can inhibit both cellular DNA and RNA synthesis and has effects on a variety of tumors.2 However, as DOX doesn't have the capability of identifying tumor cells from normal cells, it has great toxicity to normal cells while killing tumor cells.3 That's the reason why it is not the first choice of anticancer drug. Amphiphilic polymeric micelles can form core–shell structures in aqueous solution by self-assembling. The hydrophobic blocks constitute the hydrophobic core and the hydrophilic blocks constitute the hydrophilic shell.4 Hydrophobic drugs can be encapsulated in the hydrophobic core through physical or chemical interactions.5 The study by Hu et al. has shown that the compatibility between the hydrophobic drugs and the hydrophobic blocks is the main factor that affects the drug loading ability.6 Better compatibility between the hydrophobic drugs and the hydrophobic blocks leads to increased drug loading and a slower drug release rate. In recent years, plenty of amphiphilic polymeric micelle drug delivery systems have been designed and developed to improve the activity and selectivity of DOX against tumor cells, to extend its circulation time in vivo and to reduce its side effects, thus improving the therapeutic effect.7–11
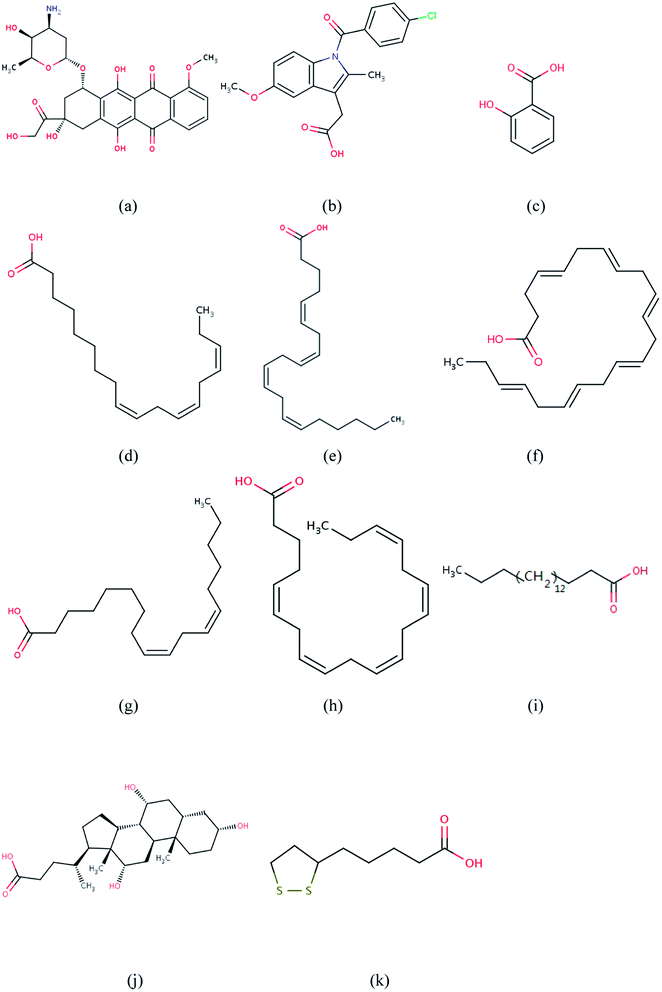 |
| | Fig. 1 Molecular structures of: (a) DOX; (b) IMN; (c) SAL; (d) LNL; (e) ACD; (f) HXA; (g) EIC; (h) EPA; (i) STE; (j) CHD; (k) LPA. | |
Chitin is a long-chain polymer of N-acetylglucosamine (a derivative of glucose) formed via covalent β-1,4 linkages. It can be found in many places and the total biomass of chitin in the natural world is second only to that of cellulose. Chitosan is a linear polysaccharide which can be obtained by the deacetylation of chitin. The structure of chitosan is very similar to that of cellulose. In the C2 position of the glucose residues in chitosan there is an amino group instead of the hydroxyl group in the same position in cellulose. However, the hydroxyl group is neutral, while the amino group is alkaline. Compared to its parent chitin, the solubility of chitosan in water is improved, but it is still very limited. Chitosan oligosaccharide (COS), the oligomer of chitosan, is the degradation product of chitosan, and it is currently the only known alkaline oligosaccharide. The solubility of COS is greatly improved compared to that of chitosan. Its good biocompatibility and biodegradability make it a good candidate for drug delivery in the biomedical field. Many studies have shown that COS has anti-tumor, anti-mutagenic and anti-oxidation effects. In addition, COS can lower blood sugar and blood pressure levels, regulate blood lipids and protect the liver. Moreover, it also has the effects of improving immunity and being anti-bacterial, anti-viral, etc.12 Because of these excellent properties of COS, its hydrophobically modified amphiphilic polymeric micelle and application to drug delivery and gene delivery have received more and more attention in recent years.11,13–16
Molecular dynamics (MD) simulation is a powerful tool which computes the motions of individual molecules and obtains detailed information on interesting systems and phenomena. During the simulation, the equation of the motion of the whole system is numerically and iteratively integrated, and the position and momentum of every molecule or atom are reserved. Therefore, MD simulation helps us to better understand the atomic-level structure and dynamics information that are difficult to observe in experiments.17 In recent years, MD simulation has been widely used in drug delivery systems to study the properties and interaction of molecules.18 It is especially good at handling issues which are difficult to investigate in laboratory experiments for drug delivery.19 In this study, MD simulation was used to investigate the interactions between the drug DOX and ten different hydrophobically modified COSs, as well as the mechanisms of drug loading in the COS systems. Besides, the ways that factors such as the functional groups and the hydrophobic properties of the hydrophobically modified COSs influence the drug loading process were analyzed, and the contributions of the van der Waals and electrostatic interactions to the drug loading process were discussed. Moreover, the results derived by the computational model were compared to the experimental data obtained in our lab and the data available in the literature. At the end of this paper, the common features of the ideal grafts are summarized, and this may help in the understanding and design of the ideal molecules for efficient and controlled DOX delivery systems.
2 Computational methods
In order to understand the mechanism of the interaction between DOX and hydrophobically modified COS, ten different acids (as shown in Table 1) were selected to modify the hydrophobicity of COS. These acids include indomethacin (IMN) and salicylic acid (SAL), which have aromatic rings; alpha-linolenic acid (LNL), arachidonic acid (ACD), docosa-4,7,10,13,16,19-hexaenoic acid (HXA), linoleic acid (EIC), icosapent (EPA) and stearic acid (STE), which are long-chain fatty acids and have high hydrophobicity; and cholic acid (CHD) and lipoic acid (LPA), both of which have a cyclic ring structure but don't have aromaticity. Their molecular structures are shown in Fig. 1. In all of the MD simulations in this study, the initial structures of these acids were taken from the Protein Data Bank (PDB), and their ligand IDs are shown in Table 1. The initial structure of the COS hexamer was taken from Glycam Biomolecule Builder (designed for carbohydrates and related molecules). The energy minimization of the COS hexamer was performed using the GLYCAM06 parameters.20 After that, the carboxyl groups of the ten acids were conjugated to the amino groups of the COS hexamer. Then the structures of all the hydrophobically modified molecules were geometrically optimized by the Gaussian 03 package,21 using the HF/6-31 G basis set.22 The atomic charges of the modified COS hexamers were also calculated by using the HF/6-31 G method. All of the MD simulations were executed by Gromacs 4.5.2.23 The force field parameters of all of the organic molecules (including DOX) were taken from the general amber force field (GAFF) of the Antechamber package, which contains the parameters of most organic and drug molecules constituted of C, H, O, N, S, P and halogens.24
Table 1 The names, ligand IDs and system labels of ten acids
| Name |
Ligand ID |
System |
| Indomethacin |
IMN |
COS/IMN |
| Salicylic acid |
SAL |
COS/SAL |
| Alpha-linolenic acid |
LNL |
COS/LNL |
| Arachidonic acid |
ACD |
COS/ACD |
| Docosa-4,7,10,13,16,19-hexaenoic acid |
HXA |
COS/HXA |
| Linoleic acid |
EIC |
COS/EIC |
| Icosapent |
EPA |
COS/EPA |
| Stearic acid |
STE |
COS/STE |
| Cholic acid |
CHD |
COS/CHD |
| Lipoic acid |
LPA |
COS/LPA |
The grafted COS chain and a DOX molecule were put into a cube with a box size of 5.0 nm. The center of mass of the complex was placed in the center of the box. Then, 3200 TIP3P25 water molecules were added into each box. Therefore, there were ten systems in our MD simulations (the system labels are shown in Table 1). These systems were energy minimized with both the steepest descent and conjugate gradient algorithms (the maximum number of steps and the energy step size of the two kinds of energy minimization were set to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 0.01 nm). Afterwards, 200 ps of NVT equilibrium and 300 ps of NPT equilibrium were conducted sequentially. After all of these pretreatments, the MD simulation was carried out in the NPT ensemble. The time step was set to 1 fs in all of the MD simulations. The temperature and pressure were controlled at 300 K and 1 bar by V-rescale26 and Parrinello–Rahman27 methods, respectively. Periodic boundary conditions were used in all dimensions and the long-range electrostatic interactions were evaluated by the Particle Mesh Ewald (PME) method.28 The intercept of PME and the cutoff of the non-bonded interaction were both 1.0 nm. The method of LINCS29 was used to constrain all of the bonds. The total simulation time was 50 ns and the last 30 ns trajectory was used to analyze the data. The total interaction energy between the drug and carriers in all of the systems in the MD simulations is defined by eqn (1):
000 and 0.01 nm). Afterwards, 200 ps of NVT equilibrium and 300 ps of NPT equilibrium were conducted sequentially. After all of these pretreatments, the MD simulation was carried out in the NPT ensemble. The time step was set to 1 fs in all of the MD simulations. The temperature and pressure were controlled at 300 K and 1 bar by V-rescale26 and Parrinello–Rahman27 methods, respectively. Periodic boundary conditions were used in all dimensions and the long-range electrostatic interactions were evaluated by the Particle Mesh Ewald (PME) method.28 The intercept of PME and the cutoff of the non-bonded interaction were both 1.0 nm. The method of LINCS29 was used to constrain all of the bonds. The total simulation time was 50 ns and the last 30 ns trajectory was used to analyze the data. The total interaction energy between the drug and carriers in all of the systems in the MD simulations is defined by eqn (1):
| | |
Eint = Ecomplex − Edrug − Ecarrier
| (1) |
where
Eint stands for the total (non-bonded) interaction energy between the drug and the carrier,
Ecomplex,
Edrug and
Ecarrier are the potential energies of the drug–carrier complex, drug, and carrier, respectively. Previous studies have shown that the interaction energy could qualitatively estimate the binding strengths of complex systems.
30,31
3 Results and discussion
3.1 π–π interactions
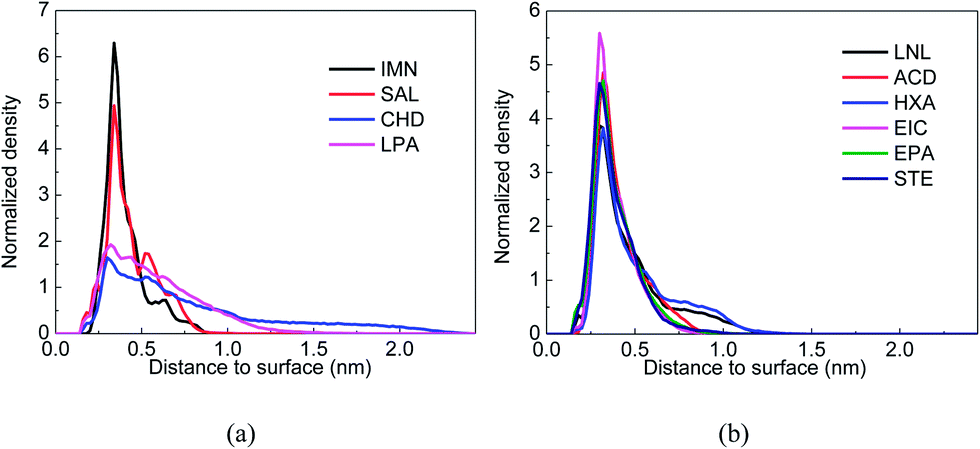
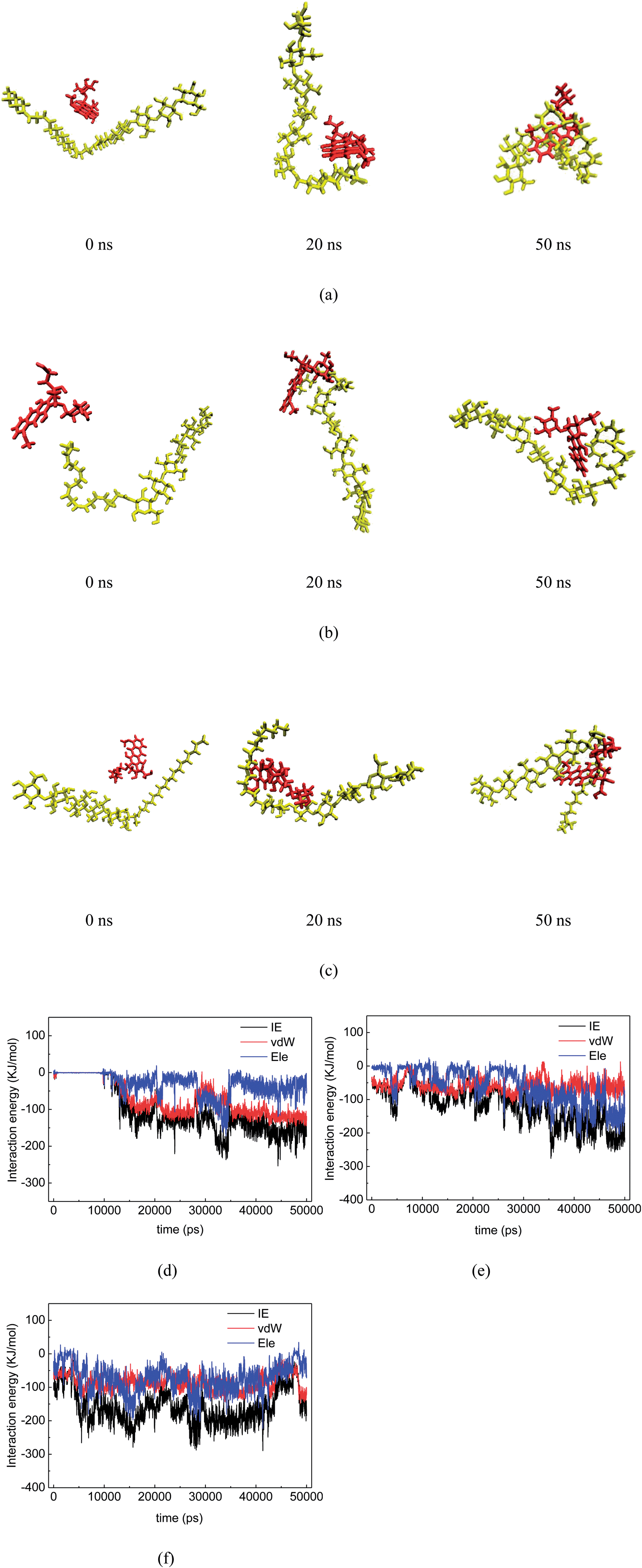
The configurations and relative positions of the drug and carriers in the simulation trajectory were carefully checked. One distinguishable phenomenon is the relatively short distance and parallel orientation between the aromatic rings in both DOX and IMM, as well as in DOX and SAL. Fig. 2(a) and (b) show snapshots of the COS/IMN and COS/SAL systems in the last frame of each MD simulation. In terms of the trajectories of both the COS/IMN and COS/SAL systems, the aromatic rings in DOX and the carrier always adopted a parallel configuration. The distance between the parallel aromatic rings in these two simulations was around 0.34 nm, and this is the typical distance between two conjugated systems with π–π interactions.33 Therefore, it strongly indicated that π–π interactions were present in these two systems. To estimate the binding strength of the drug to the carrier, Fig. 2(c) and (d) also show the interaction energies between the drug and carrier in the two systems, changing with the simulation time. The fluctuation in the interaction energies of these two systems is relatively small in the last 30 ns. This indicates that both of these systems achieved a metastable state. The averaged total interaction energies of the COS/IMN and COS/SAL systems in the last 30 ns are −100.06 and −139.34 kJ mol−1, respectively, as shown in Table 2. It is difficult to directly estimate the contribution of the π–π interactions to the total interaction energy, however, they were factored into the vdW interactions in the force field. The vdW interaction energies in these two systems are −93.95 and −59.85 kJ mol−1. This difference is probably due to the fact that in IMN there are two aromatic rings, involving a five-membered heterocycle, while in SAL there is only one phenyl ring. Therefore, IMN could form much stronger π–π interactions with DOX than SAL. The contribution of the vdW interactions to the total interaction between the drug DOX and IMN is around 94%, which also indicates the important role of the π–π interactions. Fig. 3 shows the normalized density of DOX around the drug carriers’ surfaces, which indicates the location of the drug around the carriers as well as the binding strength of the drug to the carriers. From Fig. 3(a), it was found that the peaks in the DOX density distributions around the drug carriers in both the COS/IMN and COS/SAL systems are much higher and sharper than those in the COS/CHD and COS/LPA systems. This proves that the DOX molecules in both the COS/IMN and COS/SAL systems are much closer to the hydrophobically modified COS chains, and the affinity of the drug for IMN and SAL is higher than in the other two systems. Both of the peaks are located around 0.34 nm away from the carriers’ surfaces, which is mainly attributed to the short π–π interaction distances. From the above analysis, it could be inferred that the π–π interactions in the systems of COS/IMN and COS/SAL play a significant role in the drug loading process.
 |
| | Fig. 2 (a) Snapshots of the IMN-grafted COS chain (yellow) and DOX (red), and (b) the SAL-grafted COS chain (yellow) and DOX (red) after 50 ns MD simulations. The aromatic rings in both the drug and carrier which formed π–π stacking are shown in blue, and the water molecules were omitted for clarity; (c) the total interaction energies (IEs), van der Waals interaction energies and electrostatic interaction energies between the drug and carrier in the COS/IMN and (d) COS/SAL systems. Visualization was carried out by the VMD 1.9 package.32 | |
Table 2 The number of hydrogen bonds, and the electrostatic interaction energies (Ele), vdW interaction energies (vdW) and total interaction energies (IEs) in all of the systems. Data were taken from the last 30 ns trajectories of the total 50 ns in the MD simulations
| System |
No. of H-bonds (drug–carrier) |
No. of H-bonds (drug–drug) |
No. of H-bonds (carrier–carrier) |
Ele (kJ mol−1) |
vdW (kJ mol−1) |
IE (kJ mol−1) |
| COS/IMN |
0.00 |
2.00 |
5.03 |
−6.11 |
−93.95 |
−100.06 |
| COS/SAL |
1.82 |
2.00 |
5.80 |
−79.49 |
−59.85 |
−139.34 |
| COS/LNL |
1.03 |
2.00 |
4.56 |
−56.23 |
−60.84 |
−117.07 |
| COS/ACD |
0.11 |
2.00 |
5.27 |
−8.32 |
−59.73 |
−68.05 |
| COS/HXA |
0.28 |
2.00 |
4.55 |
−17.86 |
−50.31 |
−68.17 |
| COS/EIC |
0.46 |
2.00 |
4.38 |
−44.07 |
−100.03 |
−144.10 |
| COS/EPA |
1.79 |
2.00 |
4.51 |
−85.75 |
−63.75 |
−149.50 |
| COS/STE |
1.68 |
2.00 |
5.60 |
−74.45 |
−89.64 |
−164.09 |
| COS/CHD |
0.66 |
2.00 |
4.55 |
−29.16 |
−23.85 |
−53.01 |
| COS/LPA |
0.40 |
2.00 |
5.36 |
−27.79 |
−70.80 |
−98.59 |
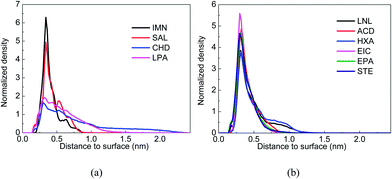 |
| | Fig. 3 Normalized density of the drug DOX around carriers’ surfaces: (a) COS/IMN (black), COS/SAL (red), COS/CHD (blue) and COS/LPA (pink); and (b) COS/LNL (black), COS/ACD (red), COS/HXA (blue), COS/EIC (pink), COS/EPA (green) and COS/STE (dark blue). | |
3.2 Single chain encapsulation
During the simulations, the phenomenon of long-chain fatty acid-grafted COS chains encapsulating DOX was observed. For example, at the initial time of the simulations, DOX and the hydrophobically modified COS chain were separated in the COS/EIC, COS/EPA and COS/STE systems, as shown in Fig. 4 (snapshot at 0 ns). Then, DOX moves closer to the hydrophobic group of the long-chain fatty acid (Fig. 4, snapshot at 20 ns), accompanied by the bending of the flexible long-chain fatty acid chain. Eventually, DOX was sandwiched between the end of the long-chain fatty acid and the COS at the end of the simulation (Fig. 4, snapshot at 50 ns). As the DOX molecule is highly hydrophobic, the sandwiched configurations buried the hydrophobic parts of the drug-carrier complexes and maximized the hydrophobic interaction between the drug and the carriers. From Fig. 3(b), it could be found that the heights and widths of the normalized densities of DOX around the carriers’ surfaces in these three systems were very close. Compared to the COS/IMN system, the heights of the density peaks in these three systems were lower, while the height of the density peak in the COS/SAL system was larger than those in the COS/EPA and COS/STE systems. However, the widths of the density peaks in these three long-chain fatty acid grafted COS systems were larger, indicating that the hydrophobic interactions in the COS/EPA and COS/STE systems were not site-specific, while the π–π interactions in the COS/IMN and COS/SAL systems were highly dependent on the interaction sites and restricted conformations. Fig. 4(c) and (d) show the total interaction energies between the drug and the carriers in the COS/EIC, COS/EPA and COS/STE systems with respect to the simulation time. Since the long chain fatty acids of these three were extremely flexible, the fluctuation in the interaction energies was high during the simulations. However, the averaged total interaction energies (over the last 30 ns trajectory) between the drug and the carriers in these three systems were −144.10 kJ mol−1, −149.50 kJ mol−1 and −164.09 kJ mol−1, respectively. They were the highest three of all of the systems, as shown in Table 2. The main reason for the high fluctuation in the energies may due to the fact that DOX was sandwiched between both the hydrophobic and hydrophilic ends and it interacted with both ends. The electrostatic interaction energies in these systems were relatively smoother than the vdW interactions, and the total interaction generally followed the trend in the fluctuation of the vdW interactions. This confirms the high flexibility of the EIC-, EPA- and STE-modified COS and indicates that the hydrophobic interactions contribute a large part to the total interaction. Previous theoretical methods have been widely used to study the kinetics of random copolymers,34 but in the MD simulation the fluctuation of the chain conformation may lead to insufficient sampling of the system. However, the high density of the drug DOX around the carriers, as well as the strong interaction energies, could still be used to qualitatively estimate the binding affinity of DOX for EIC-, EPA- and STE-modified COS. The encapsulation of DOX by a single chain mainly depends on the high binding strength and sandwiched configuration, where the hydrophobic interactions probably play an important role.
 |
| | Fig. 4 (a) Snapshots of the EIC-grafted COS chain (yellow) and DOX (red) at 0 ns, 20 ns and 50 ns in simulations; (b) snapshots of the EPA-grafted COS chain (yellow) and DOX (red) at 0 ns, 20 ns and 50 ns in simulations; (c) snapshots of the STE-grafted COS chain (yellow) and DOX (red) at 0 ns, 20 ns and 50 ns in simulations. The total interaction energies, van der Waals interaction energies and electrostatic interaction energies between the drug and the carrier in the (d) COS/EIC, (e) COS/EPA and (f) COS/STE systems. | |
3.3 More systems
In this study, a total of six long-chain fatty acid systems were selected to hydrophobically modify the COS chain. Among them, three systems (COS/EIC, COS/EPA and COS/STE) have been observed to display the phenomenon of single chain encapsulation while the other three long-chain fatty acid-modified COS systems did not exhibit this phenomenon. However, these long-chain fatty acids are very flexible and the encapsulation of the drug is a dynamic process which may occur beyond the simulation time. It is difficult to judge if the other three (LNL, ACD and HXA) fatty acid-modified COSs could encapsulate the drug DOX, on the basis of the limited simulation time. In Fig. 5(a)–(c), the interaction energies between drug and the carriers in these three systems, with respect to the simulation time, are plotted. It could be found that the averaged total interaction energies between the drug and the carriers of these systems are much smaller than those in the COS/EIC, COS/EPA and COS/STE systems. The main reason is that they did not form the sandwiched configurations of the complexes, thus the hydrophobic interactions were much weaker than those in the COS/EIC, COS/EPA and COS/STE systems.
 |
| | Fig. 5 Total interaction energies, van der Waals interaction energies and electrostatic interaction energies between the drug and the carriers in the (a) COS/LNL, (b) COS/ACD, (c) COS/HXA, (d) COS/CHD and (e) COS/LPA systems. | |
In addition to the six long-chain fatty acids and the two acids which have aromatic rings, the COS/CHD and COS/LPA systems were also checked. The changes in the interaction energies between the drug and the carriers of these two systems are shown in Fig. 5(d) and (e). The fluctuations in the interaction energies were large for these two systems, which indicates the poor stability of the drug–carrier complexes. From Fig. 3(a) it can be found that in these two systems the density distributions of DOX in the region near to the carriers, are the smallest out of all of the ten systems, which implies the poor binding affinity of DOX for CHD- and LPA-modified COS.
3.4 Solvent effect
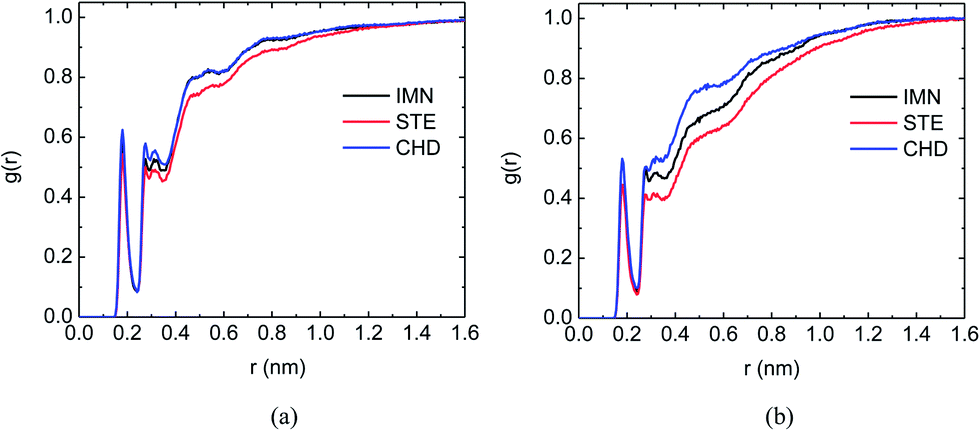
Since the COS chain was hydrophobically modified by ten different acids, the water structure around the carrier may change and have a certain influence on the DOX binding process. We have checked the radial distribution function (RDF) of oxygen (in the modified COS chain)–oxygen (in water) and oxygen (in DOX)–oxygen (in water) for all ten systems. In general, the systems with long-chain fatty acid-grafted COS encapsulating DOX have the lowest RDFs around both the drug carrier and DOX (e.g. the COS/STE system), as shown in Fig. S1 and S2.† This is because the strong hydrophobic interactions between DOX and the hydrophobic long-chain could drive a great number of water molecules out of the DOX–carrier interface. The hydrophobic interactions in fatty acid-grafted COS systems were not site-specific, while the π–π interactions in the COS/IMN and COS/SAL systems were highly dependent on the interaction site, therefore the number of water molecules driven out by the π–π interactions is less than that of the water molecules driven out by the hydrophobic interactions, and the RDFs in the COS/IMN and COS/SAL systems are higher than those in the systems having single chain encapsulation. In the COS/CHD and COS/LPA systems, due to the loose binding of DOX to the drug carrier, the RDFs in both systems are the highest. We summarized the RDFs in three typical systems (COS/IMN, COS/STE and COS/CHD) discussed above and plotted them in Fig. 6. It indicates the correlation between the solvent structures and the way that DOX and the drug carriers bind to each other (as well as the interaction energies).
 |
| | Fig. 6 (a) Radial distribution functions of oxygen (in the COS/IMN chain)–oxygen (in water) (black), oxygen (in the COS/STE chain)–oxygen (in water) (red) and oxygen (in the COS/CHD chain)–oxygen (in water) (blue). (b) Radial distribution functions of oxygen (in DOX)–oxygen (in water) in the systems of COS/IMN (black), COS/STE (red) and COS/CHD (blue). | |
3.5 Hydrogen bond analysis
As the hydrogen bond between the drug and the carrier has been found to have a great influence on the drug loading rate,18 the inter-molecular hydrogen bonds formed between DOX and the hydrophobically modified COS in all of the systems have been checked and are listed in Table 2. In addition, the intra-molecular (DOX–DOX, carrier–carrier) hydrogen bonds are also shown in Table 2. The criterion of hydrogen bonding for the donor–acceptor cutoff distance is 0.35 nm, and the cutoff angle of hydrogen-donor–acceptor is 30° (including 0°). Here the OH and NH groups are donors, and the acceptors could be O and N. From the analysis of the simulation trajectories, it was found that if DOX was away from the hydrophilic COS end and close to the hydrophobic end, the electrostatic interactions were generally small (e.g., COS/ACD, COS/IMN, etc.). If DOX was located close to the hydrophilic COS end, which increases the possibility of forming inter-molecular hydrogen bonds, the electrostatic interactions increased significantly (e.g., COS/EPA and COS/STE). Moreover, the electrostatic interaction between the drug and the carrier has a very strong correlation to the number of inter-molecular hydrogen bonds. Fig. 7 shows the linear fit of the electrostatic interaction energies and the number of hydrogen bonds between DOX and the carrier. A linear equation was obtained: y = −38.86x − 9.09 (R2 = 0.9265). In the AMBER force field there is no special term that is specific to hydrogen bonds. The hydrogen bond energy still arises from the dipole–dipole interaction of the donor and acceptor groups and is added to the electrostatic potential. This is the reason that the electrostatic interaction energy increases with the increasing of number of hydrogen bonds. In general, the strengths of the hydrogen bonds formed by OH and NH (donor) with O and N (acceptor) range from 5 to 30 kJ mol−1. The fitted equation implies that the effect of adding a hydrogen bond to the electrostatic interaction between DOX and the carrier is around 39 kJ mol−1. This is not surprising, since the formation of a hydrogen bond decreases the distance between DOX and the carrier, and both DOX and the carrier can adjust their conformation to make the polar groups complementary to each other. Therefore, the increasing electrostatic interaction energy between DOX and the carrier is not only attributed to the energy of forming hydrogen bonds, but also to the increasing electrostatic interaction between other polar groups with a shorter distance between DOX and the carrier. One has to notice that this empirical equation, based on the statistic of ten hydrophobically modified COSs, has certain limitations to its application to other systems. However, one could use it to estimate the strength of the electrostatic interaction between DOX and hydrophobically modified COS, on the basis of the number of hydrogen bonds formed. Moreover, this equation could be more accurate, based on more testing of other hydrophobically modified COS systems.
 |
| | Fig. 7 Linear relationship between the electrostatic interaction energies and the number of hydrogen bonds between DOX and the carriers, with y = −38.86x − 9.09 and R2 = 0.9265. | |
Due to the relatively rigid conformation of DOX, the hydroxyl groups and carbonyl oxygens in the conjugated aromatic ring in DOX form two stable intra-molecular hydrogen bonds (O–H⋯O) in all of the systems. However, the number of carrier–carrier intra-molecular hydrogen bonds varies due to the flexibility of the hydrophobically modified COS and the way that DOX and the carriers bind to each other (discussed in Sections 3.1–3.3).
3.6 Comparison to the experimental data
To verify our computational model, the simulation results were compared to the experimental data obtained in our lab (for details, see ESI†) and the data available in the literature. Table 3 lists the micelle size, encapsulation efficiency (EE) and drug loading (DL) rate in the experiments, as well as the interaction energies in our calculations, for each of the COS/IMN, COS/EIC and COS/CHD systems. The experimental data for the cholic acid-modified COS and DOX system is not available in the literature, so we chose the available experimental data of the deoxycholic acid (DXC)-modified COS and DOX system instead. Here the encapsulation efficiency and drug loading rate are defined as eqn (2) and (3):| | |
DL% = [We/(We + Wc)] × 100%
| (3) |
where We is the amount of the drug encapsulated in the micelle, Wt is the total amount of the drug added initially, and Wc is the amount of the drug carrier. Although in our simulations only one molecule of DOX and one single chain of hydrophobically modified COS were considered, the drug loading rates in the experiments for these systems, listed in Table 3, qualitatively follow the trend of the interaction strengths between DOX and the carriers. For example, the COS/EIC system has the strongest interaction in the computational model and this system has largest drug loading rate of 15.17% in the experiments. The encapsulation efficiencies of these systems follow a similar trend, but the COS/IMN system has a slightly higher value than the COS/EIC system. This may be due to the fact that the COS/IMN system in the experiments has a bigger micelle size (shown in Table 3) and more volume in which to encapsulate the drug molecules. One has to notice that in the experiments many properties like structure, chain length and concentration etc. of the drug carrier could affect the encapsulation efficiency and drug loading rate. Therefore, it is difficult to directly compare the interaction strength between the drug and the carrier in the simulation and the quantities (encapsulation efficiency, drug loading rate etc.) in the experiments, but they have strong correlations as discussed above. In this study, we focused on the simple model of one chain of hydrophobically modified COS and the drug, and this model gives a reasonable estimation of the interaction strength (between the drug and the carriers) which has a strong correlation with some quantities in the experiments. Recently, great progress has been achieved in the field of nanoparticles interacting with biological systems.36 A better understanding of the structures, self-assembly and thermodynamics of the targeting nanoparticles could greatly improve the design of the drug delivery system. Our future work will focus on the structural, thermodynamic and dynamic properties of the nanoparticle systems consisting of the drug DOX and longer chains of hydrophobically modified COS in a series of concentrations.
Table 3 The micelle size, encapsulation efficiency and drug loading rate in the experiments, as well as the interaction energies in our simulations, for each of the COS/IMN, COS/EIC and COS/CHD (DXC) systems
| System |
Micelle size (nm) |
EE (%) |
DL (%) |
IE (kJ mol−1) |
| For more details of the synthesis and characterization of the COS/IMN system, please see the ESI.† |
| COS/EIC35 |
205.7 ± 2.8 |
75.21 ± 2.26 |
15.17 ± 0.14 |
−144.10 |
| COS/IMNa |
345.1 ± 0.2 |
81.58 ± 0.86 |
7.76 ± 0.4 |
−100.06 |
| COS/CHD (DXC)7 |
270.5 ± 24.5 |
27.5 |
4.6 |
−53.01 |
4 Conclusion
In this study, an MD simulation was used to investigate the interactions between the drug DOX and ten hydrophobically modified COSs, as well as the mechanism of DOX loading in the hydrophobically modified COS system. The way that aromaticity and the hydrophobic properties of the hydrophobically modified COS influence the interaction strengths between DOX and the carriers as well as the DOX loading process were analyzed and discussed. The π–π interactions in the systems with aromaticity (COS/IMN and COS/SAL) contribute a big part of the van der Waals interactions and play a significant role in the DOX loading process. The encapsulation of DOX by long-chain fatty acid-grafted COS chains mainly depends on a high binding strength and sandwiched configuration, where the hydrophobic interactions play an important role. The solvent structure around DOX and the grafted COS was found to have a relationship with the way DOX and the drug carrier bind to each other. It was also found that the electrostatic interaction between the drug and the carrier has a linear relationship to the number of hydrogen bonds. Moreover, the results derived by our computational model were compared to the available experimental data. It was found that the interaction strength between DOX and the hydrophobically modified COS has a strong correlation to the experimental quantities (encapsulation efficiency and drug loading rate). From our computational model, two types of ideal hydrophobic block were suggested for the hydrophobic modification of COS for DOX delivery in the experimental evaluation. The first one includes the long-chain fatty acids like STE and EPA. Similar to hydrophobic polymers, the experimental conditions for getting long-chain fatty acid-grafted COSs are simple and easy to achieve. The second one includes acids with aromaticity like SAL (often seen in non-steroidal anti-inflammatory drugs, such as Montelukast, Bexarotene, Bezafibrate, Carprofen, etc.). Molecules which have both strong hydrophobicity and aromaticity may form more stable core–shell structures with DOX and have high drug loading rates. In conclusion, this work may help in the understanding and design of the ideal molecules for efficient and controlled DOX delivery systems. Our future work will direct more effort to studying the structural and thermodynamic properties of the systems consisting of the drug DOX and longer chains of hydrophobically modified COS in a series of concentrations. The dynamics of the self-assembly of hydrophobically modified COS and the effect of the micelle size on the DOX encapsulation efficiency and loading rate will be studied.
Acknowledgements
The authors acknowledge the financial support of National Natural Science Foundation of China (nos 30973683, 21273200 and J1210042) and Zhejiang Provincial Natural Science Foundation (LY14H300006 and LY14B030008).
References
- W. H. Mondesire, W. Jian, H. X. Zhang, J. Ensor, M. C. Hung, G. B. Mills and F. Meric-Bernstam, Clin. Cancer Res., 2004, 10, 7031–7042 CrossRef CAS PubMed.
- R. L. Momparler, M. Karon, S. E. Siegel and F. Avila, Cancer Res., 1976, 36, 2891–2895 CAS.
- S. W. Wang, E. A. Konorev, S. Kotamraju, J. Joseph, S. Kalivendi and B. Kalyanaraman, J. Biol. Chem., 2004, 279, 25535–25543 CrossRef CAS PubMed.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- A. Harada, H. Togawa and K. Kataoka, Eur. J. Pharm. Sci., 2001, 13, 35–42 CrossRef CAS.
- Y. Hu, X. Q. Jiang, Y. Ding, L. Y. Zhang, C. Z. Yang, J. F. Zhang, J. N. Chen and Y. H. Yang, Biomaterials, 2003, 24, 2395–2404 CrossRef CAS.
- K. Y. Lee, J. H. Kim, L. C. Kwon and S. Y. Jeong, Colloid Polym. Sci., 2000, 278, 1216–1219 CAS.
- J. H. Park, S. Kwon, M. Lee, H. Chung, J. H. Kim, Y. S. Kim, R. W. Park, I. S. Kim, S. B. Seo, I. C. Kwon and S. Y. Jeong, Biomaterials, 2006, 27, 119–126 CrossRef CAS PubMed.
- J. Zhang, X. G. Chen, Y. Y. Li and C. S. Liu, Nanomedicine, 2007, 3, 258–265 CrossRef CAS PubMed.
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1997, 48, 195–201 CrossRef CAS.
- Y. Z. Du, L. Wang, H. Yuan, X. H. Wei and F. Q. Hu, Colloids Surf., B, 2009, 69, 257–263 CrossRef CAS PubMed.
- V. Dodane and V. D. Vilivalam, Pharm. Sci. Technol. Today, 1998, 1, 246–253 CrossRef CAS.
- F. Q. Hu, X. L. Wu, Y. Z. Du, J. You and H. Yuan, Eur. J. Pharm. Biopharm., 2008, 69, 117–125 CrossRef CAS PubMed.
- F. Q. Hu, M. D. Zhao, H. Yuan, J. You, Y. Z. Du and S. Zeng, Int. J. Pharm., 2006, 315, 158–166 CrossRef CAS PubMed.
- S. Y. Chae, S. Son, M. Lee, M. K. Jang and J. W. Nah, J. Controlled Release, 2005, 109, 330–344 CrossRef CAS PubMed.
- S. Son, S. Y. Chae, C. Choi, M. Y. Kim, V. G. Ngugen, J. K. Kweon, M. K. Jang and J. W. Nah, Macromol. Res., 2004, 12, 573–580 CrossRef CAS.
- E. R. Lindahl, Methods Mol. Biol., 2008, 443, 3–23 CAS.
- M. Subashini, P. V. Devarajan, G. S. Sonavane and M. Doble, J. Mol. Model., 2011, 17, 1141–1147 CrossRef CAS PubMed.
- Y. Y. Li and T. J. Hou, Curr. Med. Chem., 2010, 17, 4482–4491 CrossRef CAS.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. Gonzalez-Outeirino, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian03, Revision E.01, Gaussian, Inc., Pittsburgh, PA, 2007 Search PubMed.
- A. Almond, A. Brass and J. K. Sheehan, J. Phys. Chem. B, 2000, 104, 5634–5640 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. J. Parrinello, Chem. Phys., 2007, 126, 14101–14107 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- J. Zhang, J. Z. Lou, S. Ilias, P. Krishnamachari and J. Yan, Polymer, 2008, 49, 2381–2386 CrossRef CAS PubMed.
- J. W. Shen, T. Wu, Q. Wang and H. H. Pan, Biomaterials, 2008, 29, 513–532 CrossRef CAS PubMed.
- J. W. Shen, T. Wu, Q. Wang, Y. Kang and X. Chen, ChemPhysChem, 2009, 10, 1260–1269 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. G. Timoshenko, Y. A. Kuznetsov and K. A. Dawson, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 54, 4071–4086 CrossRef CAS.
- Y. Z. Du, L. Wang, H. Yuan, X. H. Wei and F. Q. Hu, Colloids Surf., B, 2009, 69, 257–263 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01199f |
| ‡ Peng Shan and Jia-Wei Shen contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.