 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon–sulfur composites for Li–S batteries: status and prospects
Da-Wei
Wang
ab,
Qingcong
Zeng
a,
Guangmin
Zhou
c,
Lichang
Yin
c,
Feng
Li
c,
Hui-Ming
Cheng
c,
Ian R.
Gentle
*a and
Gao Qing Max
Lu
*b
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Brisbane, Qld 4072, Australia. E-mail: i.gentle@uq.edu.au; d.wang6@uq.edu.au
bARC Centre of Excellence for Functional Nanomaterials, Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Brisbane, QLD 4072, Australia. E-mail: m.lu@uq.edu.au; d.wang6@uq.edu.au
cShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang 110016, China. E-mail: cheng@imr.ac.cn; fli@imr.ac.cn
First published on 23rd April 2013
Abstract
We review the development of carbon–sulfur composites and the application for Li–S batteries. Discussions are devoted to the synthesis approach of the various carbon–sulfur composites, the structural transformation of sulfur, the carbon–sulfur interaction and the impacts on electrochemical performances. Perspectives are summarized regarding the synthesis chemistry, electrochemistry and industrial production with particular emphasis on the structural optimization of carbon–sulfur composites.
 Da-Wei Wang | Da-Wei Wang completed his Bachelor studies at Northwestern Polytechnic University (China) in 2003 and his Ph.D. at the Institute of Metal Research, Chinese Academy of Sciences in 2009. He started postdoctoral research in collaboration with Professor Max Lu and Professor Ian Gentle at the University of Queensland (UQ) in 2009. His research interests include carbon materials and their energy applications. He received a 2012 UQ Foundation Research Excellence Award for his research to develop advanced lithium batteries. He has published 36 peer-reviewed journal papers with an h-index of 19, and total citations of over 1700. |
 Feng Li | Feng Li is a professor of the Institute of Metal Research, Chinese Academy of Sciences (IMR, CAS). He received his Ph.D. in Materials Science at IMR, CAS in 2001 supervised by Prof. Hui-Ming Cheng. He used to work at The University of Queensland, Australia and MIT, USA. He mainly works on the novel carbon based materials for lithium ion batteries, lithium sulfur battery and supercapacitors. He has published about 140 papers on peer-reviewed journals such as Angew. Chemie, Energy Environ. Sci., Adv. Mater., Adv. Funct. Mater., ACS Nano, etc. with more than 6000 citations and an h-index of 30. |
 Hui-Ming Cheng | Hui-Ming Cheng is Professor and Director of Advanced Carbons Division of Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences. He worked at AIST and Nagasaki University, Japan, and MIT, USA. His research mainly focuses on carbon nanotubes, graphene, energy storage materials, photocatalytic materials, and high-performance bulk carbon materials. He has published over 350 peer-reviewed papers. He has been Editor of Carbon since 2000 and the Editor-in-Chief of New Carbon Materials since 1998, and co-chaired the World Conference on Carbon in 2002 (Beijing) and 2011 (Shanghai). He has given more than 60 plenary/keynote/invited talks at international conferences and symposia. |
 Ian R. Gentle | Ian R. Gentle is a graduate of the University of Sydney with BSc (Hons) and PhD in Physical Chemistry. He has been at the University of Queensland since 1993 and is now Professor and Associate Dean Research in the Faculty of Science. He has held a number of leadership positions at UQ including Director of the Brisbane Surface Analysis Facility, Deputy Director of the Centre for Microscopy and Microanalysis and Program Leader in the ARC Centre of Excellence for Functional Nanomaterials. Most recently he held the position of Head of Science at the Australian Synchrotron (2008–2010). He has published around 100 journal articles in the fields of materials science, thin films, organic electronics and biological surfaces. He has also been on a number of boards and is currently the Chair of the Australian Nanotechnology Alliance. |
 Gao Qing Max Lu | G. Q. Max Lu received his PhD in Chemical Engineering from University of Queensland, Australia. He is now Deputy Vice-Chancellor (Research) of UQ. He has received numerous prestigious awards including the Federation Fellowship (twice). He is an elected Fellow of ATSE, Fellow of Institution of Chemical Engineers, and Fellow of AAS. He is the Foundation Director for the ARC Centre of Excellence for Functional Nanomaterials. Professor Lu is ISI Highly Cited Author in Materials Science with 17 |
Introduction
Carbon materials are commonly used electrode materials for their diversity, robust chemistry, conductivity, stability and abundance. Such materials, including activated carbon, nanoporous carbon, carbon nanotubes, graphene sheets and many other forms, have been widely used in supercapacitors,1–8 lithium ion batteries (anode materials),6,9–13 and lithium-air/oxygen batteries (gas diffusion cathodes).14 Carbon materials have excellent conductivity and good mechanical resilience, and therefore have shown remarkable usefulness in facilitating the performance of insulating or semiconducting cathode materials (mainly lithium-transition metal oxides).10–13A recent boom in carbon-related energy research has been triggered by the emerging focus on Li–S batteries.15 Li–S batteries utilize a lithium metal anode and a sulfur cathode. The multi-electron-transfer cathode reaction of S8 + 16 Li+ + 16 e− ↔ 8 Li2S offers an extremely high theoretical capacity of 1672 mA h g−1, while the lithium anode provides a theoretical capacity of 3842 mA h g−1. The average potential of a Li–S cell is 2.15 V with respect to Li0/Li+, which is relatively low compared to graphite–LiMO2 batteries (>3 V). The specific energy of Li–S battery is still very large, however, the medium voltage is offset by the very high capacity and reaches a theoretical value of 2567 W h kg−1. The use of lithium metal in a Li–S cell is far from optimal, as it is potentially risky due to dendrite formation.16 Many efforts have been devoted to lithium-metal free sulfur-based batteries.17–20 Unfortunately, the lithium metal anode and the lithium sulfide anode are both moisture sensitive. Large scale fabrication of battery anodes from these materials must be performed in a dry and inert environment, and this could potentially impact the industrial production of Li–S batteries.
The other obstacle that stands in the way of the large-scale uptake of Li–S technology is relevant to the sulfur cathode. Sulfur is a promising cathode material due to its advantages such as low cost, nontoxicity and virtually unlimited supply. However, several practical problems have delayed widespread application of sulfur. Sulfur itself is insulating (5 × 10−30 S cm−1 at 25 °C) and is thus unusable as an electrode material. This fact necessitates the use of carbon as a conducting additive in the sulfur cathode of Li–S batteries. The redox chemistry of sulfur in the cathode relies on a solid (cyclo-S8)–liquid (chain-polysulfides (PS, S4–82−))–solid (Li2S2/Li2S) reaction with a systematic decrease in the ion chain length. The reduction of S8 forms S82− at 2.39 V vs. Li0/Li+, which is successively reduced to S62−at 2.37 V vs. Li0/Li+ and then to S42− at 2.24 V vs. Li0/Li+.21 Because the PS ions are soluble, the reaction is fast (Fig. 1, first plateau). This process corresponds to a theoretical capacity of 418 mA h g−1. Continuing discharge will reduce the PS ion to Li2S2/Li2S which are insoluble. Due to the much slower reaction kinetics at the second plateau and the tail, which correspond to solid state reactions, the theoretical value usually cannot be achieved and a lower capacity of 1256 mA h g−1 is more realistic (corresponding to 1.5 e S−1).
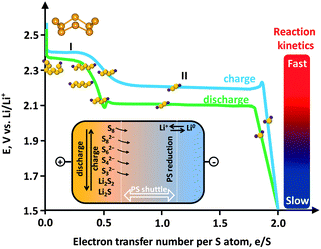 | ||
| Fig. 1 Electrochemistry of sulfur showing an ideal charge–discharge profile. Inset: polysulfide (PS) shuttle. | ||
A critical issue associated with the PS ions (S4–82−) is their solubility in an electrolyte. The dissolved PS ions easily diffuse from the cathode to anode driven by a concentration gradient (Fig. 1, inset). In fact, even sulfur can be weakly dissolved in an aprotic electrolyte.22 Reduction of the dissolved PS is as yet unclear, but is possible that it occurs through (1) ion chain scission, (2) disproportionation, and (3) reduction by the Li anode.22 The insoluble Li2S2/Li2S deposits on the Li anode can react with PS ions, yielding soluble medium-chain ions, which diffuse back to the sulfur cathode. The whole process is known as the “PS shuttle mechanism”. This shuttle phenomenon results in the following drawbacks: (1) the active mass loss from the cathode, (2) reduction of Coulombic efficiency, and (3) capacity decay upon cycling. Additionally the insoluble Li2S2/Li2S can also deposit on the cathode and is insulating.23 This makes the cathode electrochemically inaccessible due to the hindered ion and electron transport. A further issue associated with the sulfur cathode is the volume variation. The density of sulfur is 2.03 g cm−3 while that of Li2S is 1.67 g cm−3, which results in an 80% volume expansion. The current limitations of sulfur cathodes and the already proposed strategies to address these issues are summarized in Table 1. Many recent studies on carbon–sulfur composites are summarized in Table 2, where the synthesis method and the performance evaluation, typically the cyclic stability, are compared.
| Limitations | Improvements |
|---|---|
| Insulating | Conducting additives |
| PS shuttle | (i) Physical confinement (ii) chemical adsorption (iii) electrolyte design (iv) separator design |
| Volume change | (i) Porous texture (ii) buffering additives |
| Classification | Characteristics | Sulfur loading method | S percentage (by weight) | Cycle performance (1C = 1675 mA g−1) | Electrolyte/voltage window |
|---|---|---|---|---|---|
| Microporous carbon | |||||
| Sucrose-derived hydrothermal carbon nanosphere36 | Sulfur loaded in 0.7 nm micropores | Sulfur melt adsorption (150–160 °C) + vaporizing (280–300 °C) | 42% | 650 mA h g−1/400 mA g−1/500th cycle | 1 M LiPF6 PC-EC-DEC,1.0–3.0 V |
| D-Glucose-derived carbon coating on CNT38 | Sulfur loaded in 0.6 nm micropores | Sulfur melt adsorption | 40.2% | 1142 mA h g−1/0.1C/200th cycle | 1 M LiPF6 in EC/DC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in wt%),1.0–3.0 V :1 in wt%),1.0–3.0 V |
| Phenolic resin-derived porous carbon37 | Sulfur loaded in <1 nm micropores | Sulfur melt adsorption + extraction process | 16% | 200 mA h g−1/3000 mA g−1/800th cycle | 1 M LiPF6 in (EC/DMC/EMC, 1:1:1 vol.), 1.5–2.8 V |
| Mesoporous carbon | |||||
| Sucrose-derived carbon with sodium silicate as template27 | 10–30 nm | Sulfur melt adsorption | 20% | 500 mA h g−1/50 mA g−1/40th cycle | 1 M LiTFSI in EMITFSI |
| PPy-derived carbon with colloidal silica as template31 | 7–22 nm | Sulfur melt adsorption | 83.2% | 613 mA h g−1/0.1C/50th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.0–3.0 V |
| Ordered mesoporous carbon (CMK-3)28 | 3–4 nm | Sulfur melt adsorption | 70% | 1100 mA h g−1/168 mA g−1/20th cycle | 1.2 M LiPF6 in ethyl methyl sulphone |
| Phenol formaldehyde-derived carbon with TEOS and F127 as templates30 | 2.0 nm and 5.6 nm | Sulfur melt adsorption | 40% | 345 mA h g−1/1C/100th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| Phenol formaldehyde-derived carbon with TEOS and F127 as templates34 | 2.7 nm | Sulfur melt adsorption | 50% | 730 mA h g−1/1C/100th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| Hierarchical porous carbon | |||||
| Sucrose-derived carbon with silica and copolymer latex as dual-template40 | Macropore size-300 nm | Sulfur melt adsorption | 50% | 884 mA h g−1/0.1C/50th cycle | 1 M LiTFSI/DOL-DME (1:1), 0.1 M LiNO3, 1.0–3.0 V |
| Hollow carbon sphere | |||||
| Petroleum pitch with silica as template42 | Diameter 200 nm | Vapour phase infusion | 70% | 974 mA h g−1/0.5C/100th cycle | 1 M LiTFSI in tetraglyme, 1.7–3.1 V |
| Glucose derived carbon with SnO2 sphere as template43 | Diameter 300 nm | 400 °C heat treatment | 64% | 690 mA h g−1/0.1C/100th cycle | 1 M LiTFSI in tetraglyme, 1.5–3 V |
| CNT/CNF | |||||
| Carbon nanotube array49 | CNT diameter from 7–30 nm | Solvent evaporation | 63% | 900 mA h g−1/C/13/40th cycle | 1 M LiTFSI/DOL-DME (1:1), 0.25 M LiNO3, 1.0–3.0 V |
| Disordered carbon nanotubes47 | CNT diameter 200 nm | High temperature (500 °C) treatment | 40% | 700 mA h g−1/200 mA g−1/100th cycle | 1 M LiTFSI in TEGDME, 1.5–3 V |
| Styrene derived carbon hollow fiber with AAO as template50 | Hollow fiber | Sulfur melt adsorption | 75% | 730 mA h g−1/0.2C/150th cycle | 1 M LiTFSI/DOL-DME (1:1), 0.1 M LiNO3, 1.7–2.6 V |
| Graphene | |||||
| PEG modified GO-carbon black54 | Emulsion wrap 1 μm sulfur particles | Emulsion: Na2S2O3 + Triton-X100 + HCl | 70% (before annealing) | 520 mA h g−1/0.2C/100th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.7–2.6 V |
| KOH activated graphene64 | Amorphous sulfur mesoporous graphene composites | Sulfur melt adsorption | 67% | 1007 mA h g−1/0.2C/60th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.0–3.0 V |
| Graphene sheets reduced by Na2S63 | Sulfur–graphene composite | Hydrothermal NaS + NaSO3 | 75.2% | 662 mA h g−1/1000 mA g−1/100th cycle | 1 M LiCF3SO3/DOL-DME (1:1), 0.2% wt LiNO3, 1.7–2.5 V |
| Raw graphene61 | Amorphous sulfur–graphene composite | Sulfur melt adsorption (in air) | 44.5% | 819 mA h g−1/0.05C/100th cycle | 1 M LiTFSI/DOL-DME (v/v 2:8), 1.0–3.0 V |
| Ethanol derived porous graphene52 | Amorphous sulfur–graphene composite | Sulphur vaporizing | 22% | 600 mA h g−1/50 mA g−1/40th cycle | 1 M LiTFSI in PEGDME, 1.5–3.0 V |
| Reduced graphene oxide62 | Uniformly dispersed on graphene | SO32− + 2S2− + 6H+ | 63.6% | 440 mA h g−1/1250 mA g−1/500th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| Graphene55 | Graphene enveloped 1 μm sulfur particles | NaSx + H+ | 87% | 500 mA h g−1/334 mA g−1/50th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| GO with epoxy and hydroxyl groups66 | Binding between sulfur thin film and graphene | NaSx + H+ and sulfur melt adsorption | 66% | 954 mA h g−1/0.1C/50th cycle | 1 mol kg−1 LiTFSI in PYR14TFSI/PEGDME (1:1, by weight), 1.0–3.0 V |
| Graphene–CNT hybrid65 | 5 nm single walled CNT | Sulfur melt adsorption | 60% | 530 mA h g−1/1C/100th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| Nafion coated graphene57 | Sandwich type graphene–sulfur composites | Sulfur melt adsorption | 71.8% | 800 mA h g−1/0.1C/50th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.0–3.0 V |
| Flexible carbon support | |||||
| Activated carbon fiber cloth74 | Binder-free cathode | Sulfur melt adsorption | 33% | 800 mA h g−1/150 mA g−1/80th cycle | 10 wt% LiTFSI/DOL-DME (1:1) with 2 wt% LiNO3, 1.7–2.48V |
| MWCNT membrane76 | Self weaving | Emulsion: Na2S2O3 + Triton-X100 + HCl | 60% | 915 mA h g−1/1C/100th cycle | 1.85 M LiTFSI/DOL-DME (1:1) with 0.1 M LiNO3, 1.5–2.8 V |
| Graphene membrane77 | Self-supporting | Na2S2O3 + HCl | 67% | 600 mA h g−1/0.1C/100th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.0–3.0 V |
| Disordered CNT membrane75 | Binder free | In situ carbothermal reduction of sulphate | 23% | 635 mA h g−1/1500 mA g−1/100th cycle | 1.0 M LiTFSI/DOL-DME (1:1) with 0.1 wt% LiNO3, 1.5–2.8 V |
| Other carbon materials | |||||
| Nitrogen doped carbon35 | NH3 treated mesoporous carbon | Sulfur melt adsorption | 24% | 573 mA h g−1/0.05C/20th cycle | 0.5M LiTFSI/[MPPY] [TFSI], 1.0–3.0 V |
| Expanded graphite60 | / | Sulfur melt adsorption | 60% | 880 mA h g−1/280 mA g−1/70th cycle | 1 M LiTFSI/DOL-DME (1:1), 1.5–3.0 V |
| Carbon black45 | / | Sulfur precipitation | 64.7% | 832 mA h g−1/0.1C/50th cycle | 1 M LiTFSI/DOL-DME (v/v 4:1), 1.0–3.0 V |
Sulfur forms
Thermal annealing of sulfur with carbon is still the most general synthesis technique for the carbon–sulfur composite so far, and is also the best choice for industrial production. In this context, the thermal behaviour of sulfur is of interest. Elemental sulfur has a variety of crystalline and molecular/polymeric forms. The identification of these allotropic forms has been the subject of study over decades, and is summarized in Fig. 2.24–26 Ordinary sulfur (α-S), i.e. the rhombic stacking of S8, is the stable room-temperature form. It slowly converts to monoclinic sulfur (β-S) from 95.5 °C. Both rhombic and monoclinic sulfur are soluble in CS2. The melting temperature of monoclinic sulfur is 118.7 °C and the boiling temperature of sulfur is 444.6 °C. Liquid sulfur is predominantly S8 at the melting point, where cyclo-S8 and catena-S8 co-exist. S8 rings undergo thermal scission to form linear sulfenyl diradicals at 159 °C. Between 159 °C and 444.6 °C, sulfur first polymerizes and then depolymerizes, accompanied with a viscosity change which reaches a maximum at 186 to 188 °C. S8 is predominant at 444.6 °C, but is dissociated into short chain sulfur at higher temperatures (S6, S4: vapour at 600 to 800 °C, S2: vapour above 850 °C, and S: vapour above 1800 °C).26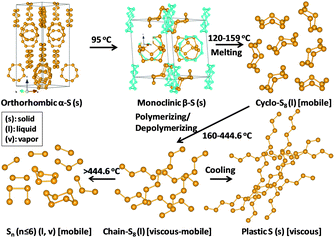 | ||
| Fig. 2 Structure transformation of sulfur. | ||
Carbon–sulfur composites
Mesoporous carbon–sulfur composites
Wang and co-workers reported the first use of a worm-like mesoporous carbon as a host material for sulfur.27 In their approach, sucrose was chosen as the carbon source and sodium silicate as the silica template source. The mixture of sulfur and mesoporous carbon was heated at 200 °C for 6 h to let the sulfur melt diffuse into the mesopores of the carbon host, and then the temperature was raised to and held at 300 °C for another 3 h to vaporize the superficial sulfur outside the carbon particles.27 An ionic liquid, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMITFSI), was used as the electrolyte solvent, in which lithium bistrifluoromethanesulfonimidate (LiTFSI) was dissolved to form a 1 M LiTFSI–EMITFSI electrolyte. Their results showed much improved cycling stability of the mesoporous carbon–sulfur composite. Two reasons were proposed to explain the better performance: (1) the physical adsorption of polysulfides by the mesopores (10–30 nm), and (2) the uniform distribution of sulfur in the mesopores, which assists with electron conduction by the composite cathode. A result noticed in Wang's work was that the stability of the same material in an organic electrolyte was not as good as that in an ionic liquid electrolyte, and the cause was suggested to be the reduced solubility of PS in the ionic liquid. Meanwhile, the reduction potentials from sulfur to PS and to Li2S2/Li2S were found to depend on the electrolyte, which may imply a PS solubility-related mechanism.The manifested advantages of mesoporous carbon were later highlighted by Ji and co-workers.28 The major progress relied on the optimal use of the ordered mesoporous carbon (CMK-3) synthesized by using the SBA-15 silica template. This CMK-3 carbon possessed uniform and narrow mesopores (3 nm) and large pore volume (2.1 cm3 g−1) (Fig. 3a). The sulfur content in the CMK-3-sulfur composite can be as high as 70 wt%. This value is lower than the theoretical value (79 wt%) because it is good for buffering the volume change during lithiation and delithiation of sulfur confined in the mesopores.28 As a result of the uniform distribution of sulfur in the mesopores, the conductivity of the composite is comparable to that of CMK-3 within errors (0.21 S cm−1vs. 0.2 S cm−1, ±0.02 S cm−1). This gave rise to a very high Coulombic efficiency (99.94%) in the first discharge–charge cycle, which indicated an extremely low fraction of PS dissolved in the electrolyte. A typical discharge–charge profile of the CMK-3–S composite shows the well resolved plateaus corresponding to the redox chemistry of sulfur, PS and lithium sulfides (Fig. 3b). It is notable that an organic electrolyte was used in this study. Ji et al. also demonstrated the enhanced stability of a PEG-coated CMK-3–sulfur composite,28 introducing the concept of carbon–sulfur–polymer ternary composites for Li–S batteries.
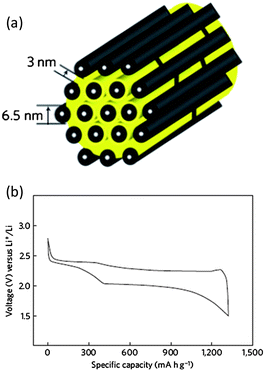 | ||
| Fig. 3 Structure scheme of the CMK-3–sulfur composite.28 Reproduced from ref. 28. Copyright 2009 Nature Publishing Group. | ||
Many other types of mesoporous carbon–sulfur composites have subsequently been developed and examined for their efficacy in promoting sulfur stability in Li–S batteries.29–34 Bimodal mesoporous carbon with 2.0 nm and 5.6 nm mesopores was used with the intention of regulating ion conduction in the mesoporous framework for high-rate performance.30 The small mesopores were located in the walls of the large mesopores without harming the structural integrity of the mesoporous framework. By screening over various mesoporous carbons with different mesopore sizes and pore volumes, Li et al. concluded that (1) large pore volume is good for increasing sulfur percentage and higher capacity of the composite (total mass of carbon and sulfur), (2) empty pore space, i.e. partial sulfur loading, is crucial for facile supply of lithium ions and adsorption of PS, and (3) polymer coating of mesoporous carbon is important for better cathode performance.31 It is important to emphasize that the empty space in C–S composites is necessary for buffering the volume change during lithiation–delithiation of the cathode. By using a post-activation treatment, Liang et al. showed the generation of micropores on the walls of mesoporous carbon.32 These micropores were expected to accommodate sulfur while the mesopores can transfer lithium ions.32 However, the cycling stability of this carbon–sulfur composite was not as good as the CMK-3–sulfur composite. The relatively larger mesopores (7.3 nm) as well as the destroyed pore walls may be the causes. Morphological control over the mesoporous carbon–sulfur composites has also been studied, where spherical particles were used.33,34 Interestingly, the introduction of nitrogen as a dopant in mesoporous carbon appeared to be beneficial for performance enhancement.35
Microporous carbon–sulfur composites
Zhang and co-workers reported the exceptional stability of sulfur confined in microporous carbon spheres.36 The carbon spheres were synthesized by refluxing sucrose (5 wt% solution) in a 6 M sulfuric acid at 120 °C for 10 h and carbonizing the black product at 1000 °C for 2 h under flowing Ar gas. The mixture of microporous carbon and sulfur was heated at 149 °C for 6 h and then the temperature raised to 300 °C and kept for 2 h to vaporize the excess sulfur. The microporous carbon spheres ranged in size from 200 to 300 nm. The micropore size was about 0.7 nm and no mesopores were detected. The sulfur loading percentage was seen to be important for high capacity. The total pore volume (0.474 cm3 g−1) allowed a theoretical sulfur loading of 49.5 wt%. A loading content of 51 wt% showed much smaller capacity (∼300 mA h g−1) than a 42 wt% loading (∼900 mA h g−1). This is likely related to the issues of lower conductivity and severer volume expansion at high sulfur loading. This composite was stable for 500 cycles, which was related to the strong adsorption of PS by the narrow micropores.36 Zhang et al. observed lithiation potential hysteresis during the discharge process which reflects the additional energy required to overcome the adsorption energy of the short-chain sulfur in micropores (Fig. 4).36 Because of the strong confinement effect, low molecular sulfur in micropores can be only fully discharged at a potential of 1 V vs. Li/Li+, compared with 1.5 V vs. Li/Li+ for sulfur in mesopores. Similar cathode behaviour of sulfur in microporous carbon was reported by Wang et al.37 and Xin et al.38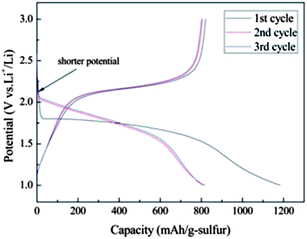 | ||
| Fig. 4 Discharge and charge profiles of the microporous carbon–sulfur composite with 42 wt% sulfur at 400 mA g−1.36 Reproduced from ref. 36. Copyright 2010 Royal Society of Chemistry. | ||
Wang et al. designed a two-step adsorption–extraction method to differentiate the differing cathode properties of sulfur confined in micropores and large pores.37 Their work clearly demonstrated the excellent efficacy of micropores in enhancing the stability of sulfur (up to 800 cycles at high rate).37 They also observed the higher binding energy of Li–S bond confined in micropores, which is likely because of the strong adsorption.37 They proposed a desolvation effect to interpret the unusual stability of sulfur in microporous carbon.37 Since the observation of ion desolvation in carbide-derived carbon, it has been widely acknowledged that solvated ions tend to be desolvated in micropores with size close to ion size.39 As generally accepted, the sulfur reduction is a solid (S)–liquid (PS ions)–solid (Li2S2/Li2S) process, and the mesopores stabilize sulfur through the adsorption of dissolved PS ions (Fig. 5, left). According to the ion-desolvation theory, the PS ions in micropores which are devoid of solvent react with desolvated ions (Fig. 5, right). This suggests a quasi-solid-state reaction of the sulfur under solvent-deficient conditions. The low Li+ conductivity in S and solid sulfides could also explain the retarded lithiation of sulfur in micropores. Xin et al. fabricated microporous carbon nanofibers in which carbon nanotubes were the backbone.38 Much improved cathode stability of sulfur was also observed, as well as potential hysteresis.38 These three works36–38 shared the same properties of microporous carbon–sulfur composites: (1) lithiation potential hysteresis and (2) excellent stability. Xin and co-workers considered the lithiation of small sulfur molecules.38 On account of the tiny 0.5–0.6 nm micropores, Xin et al. proposed that cyclo-S5–8 molecules with at least two dimensions larger than 0.5 nm cannot exist inside, while small S2–4 molecules with at least one dimension less than 0.5 nm can be hosted.38 However, characterization of the molecular structure of amorphous sulfur confined in micropores is still a great challenge.
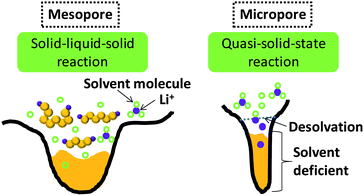 | ||
| Fig. 5 Illustration of the different lithiation mechanisms of sulfur confined in mesopores and micropores. | ||
Hierarchical porous carbon–sulfur composites
Recent efforts have been devoted to designing hierarchical porous carbon with the aim of combining the advantages of microporous texture in regards to stability with large pores for rapid ion transport.40,41 An ordered hierarchical porous carbon was fabricated by a dual-template self-assembly approach.40 Typically, 350 nm colloidal polymer particles were self-assembled with 9 nm silica spheres to form a hierarchical periodic template. Sucrose was infiltrated as the carbon source together with sulfuric acid as the carbonization catalyst. The product has a pore volume of 1.4 cm3 g−1, bimodal macropores (120 nm, 300 nm), mesopores (10 nm) and micropores. With a 50 wt% loading of sulfur, the composite could be cycled for 50 times at 167.5 mA g−1 with a capacity around 800 mA h g−1. Fish scales were used as an alternative bio-derived carbon source to produce hierarchical porous carbon.41 The natural hierarchical texture of the fish scale was inherited in the final carbon product giving rise to the hierarchical porous texture. This bio-derived carbon has a very high specific surface area (2441 m2 g−1) and a good pore volume (1.69 cm3 g−1). Benefiting from the hierarchical porosity, the initial discharge capacity of sulfur in this carbon material can be as high as 1039 mA h g−1 at 1675 mA g−1 and remain as high as 1023 mA h g−1 after 70 cycles.41Hollow carbon–sulfur composites
Jayaprakash et al. reported the synthesis of hollow carbon capsules with a mesoporous shell and their use as a sulfur host (Fig. 6a).42 In this design, the hollow carbon was synthesized using a silica template and petroleum pitch as the carbon source. The resulting product has a 3 nm average pore size and can accommodate up to 70 wt% sulfur. The vapour-phase infusion technique allowed the encapsulation of sulfur in the interior and the porous shell of the carbon capsule, which sequesters the active mass. Several advantages of this hollow carbon host were suggested: (1) maximizing the amount of sulfur sequestered by the capsules, (2) minimizing PS dissolution and shuttling, (3) preserving fast transport of lithium ions, and (4) good conductivity.42 Because of these attributes, the as-derived carbon–sulfur composite can be cycled at a low rate (850 mA g−1) over 100 cycles with a high capacity of ca. 1000 mA h g−1.42 In another approach, complex double-shelled hollow carbon spheres were synthesized using hollow SnO2 spheres as a template.43 Glucose was uniformly coated on the interior and exterior surfaces of the template and the complex hollow carbon was derived thereafter (Fig. 6b).43 The double-shelled structure was designed to host higher amounts of sulfur, suppress the outward diffusion of dissolved PS and buffer the volume change upon cycling.43 A satisfactory capacity of around 500 mA h g−1 was obtained at a current density of 850 mA g−1.43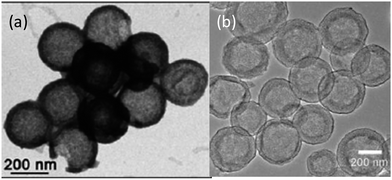 | ||
| Fig. 6 TEM images of (a) a hollow carbon capsule42 and (b) a double-shell hollow carbon sphere.43 Reproduced from ref. 42 and 43. Copyright 2011, 2012 Wiley-VCH. | ||
Carbon black–sulfur composites
Carbon black is a commercial conducting additive used widely in the battery industry. Synthesis of carbon–sulfur composites from this abundant commercial material is of crucial economic value. Wang and co-workers reported the synthesis of core–shell carbon–sulfur nanocomposite through an aqueous solution route.44 Controlled direct deposition of sulfur from the following reaction:| SO2 + 2S2− + 4H+ → 3S + 2H2O | (1) |
 | ||
| Fig. 7 (a) TEM image of the core–shell carbon–sulfur composite44 and (b) SEM image of the homogeneous carbon–sulfur composite.45 Reproduced from ref. 44 and 45. Copyright 2010, 2012 Elsevier. | ||
Carbon nanotube/fibre–sulfur composites
Sulfur-coating strategies similar to those above have been used for the synthesis of sulfur-coated carbon nanotube (CNT) nanocomposites.46 It was suggested that the low surface tension of sulfur plays an important role in wetting of the exterior surface of CNTs.46 The mixture of the CNTs and sulfur was first ball milled and then annealed at 155 °C for 24 h. The CNT–S composite showed improved capacity and stability compared to a sulfur–carbon black composite and the sulfur–CNT mixture without the coating treatment.46 Disordered CNTs (DCNTs) with less graphitic tube walls and bamboo-like hollow voids have also been used as sulfur hosts.47 The sulfur was introduced in a vapour form in a vacuum at 160, 300, and 500 °C. The thermal stability of the DCNT–S composite increased as the annealing temperature increased, and the loss due to sulfur sublimation reduced, which suggested enhanced absorption and the possibility of sulfur–carbon bonding or new compounds being produced as indicated by unknown XRD peaks.47 However, the reduction of PS ions at the first plateau gradually vanished from 160 °C to 500 °C, which was accompanied by a remarkable potential hysteresis similar to that observed in microporous carbon–sulfur composites (Fig. 4 and 8).36–38,47 Assemblies of CNTs in the forms of microspheres48 or an aligned forest49 were studied.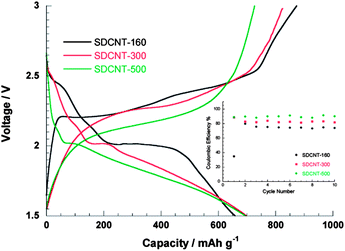 | ||
| Fig. 8 Discharge–charge profiles of the second cycles and (inset) Coulombic efficiency under 10 mA g−1 for DCNT-S composites synthesized at 160, 300 and 500 °C.47 Reproduced from ref. 47. Copyright 2011 American Chemical Society. | ||
Hollow carbon nanofibers (CNFs) were reported as a desirable host for a sulfur cathode mainly because of the limited diffusion pathways of PS ions (Fig. 9).50 This hollow design was believed to contribute the following ideal characteristics: (1) a closed structure for efficient PS containment, (2) limited surface area for sulfur–electrolyte contact, (3) sufficient space to accommodate sulfur volumetric expansion/shrinkage, and (4) a short electron and Li ion transport pathway.50 Ji et al. reported sulfur deposition onto PMMA-templated porous CNFs from a polysulfide solution.51 The reaction formula is as follows:
| Sx2− + 2H+ → (x − 1)S + H2S | (2) |
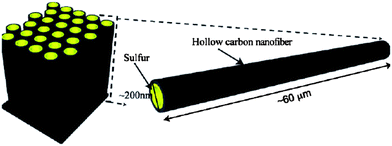 | ||
| Fig. 9 Schematic of the trapped sulfur inside vertically aligned hollow carbon nanofibers.50 Reproduced from ref. 50. Copyright 2011 American Chemical Society. | ||
The as-obtained porous CNF–S composite was further annealed to ensure that the exterior sulfur can diffuse into the interior pores of the nanofibers.51
Graphene sheet–sulfur composites
The feasibility of graphene sheets for stabilizing sulfur cathodes as well as the use of conducting additives were first reported by researchers using solvothermal-derived porous graphene sheets.52 Successive optimization of the synthesis methods resulted in diverse configurations of the graphene–sulfur composites: graphene-wrapped sulfur particles,53–56 sandwich-type composites,57–60 and sulfur coated graphene sheets.61,62 Disproportionation of thiosulfate as follows:| S2O32− + 2H+ → SO2 + S + H2O | (3) |
 | ||
| Fig. 10 (a) Graphene-sheet wrapped sulfur particles,54 Reproduced from ref. 54. Copyright 2011 American Chemical Society. (b) Sandwich-type graphene–sulfur composite.57 Reproduced from ref. 57. Copyright 2010 Royal Society of Chemistry. | ||
Graphene sheets are advantageous for wrapping sulfur because of their large lateral size, good conductivity and the flexible structure. Under ideal conditions, however, graphene sheets conduct ions along the lateral direction making ion conduction across the sheets very difficult. To overcome this problem, as well as to provide more sites for sulfur storage, activated graphene sheets have been synthesized and used as the sulfur hosts.64 KOH activation produced 3.8 nm mesopores on the graphene sheets which is large enough to transport Li ions while being small enough to hold PS ions.64 Compared with pristine graphene sheets, the activation was clearly good for rate performance and stability.64 Graphene-based hybrid materials are of special interest in energy storage. A unique graphene–single walled CNT (SWCNT) hybrid structure was developed by Zhao and co-workers.65 The graphene constituent in the hybrid formed a hexagonal hollow container for sulfur while the exterior SWCNT forests provided electron conduction pathways as well as conferring a mesoporous texture to adsorb PS ions (Fig. 11). The highly graphitic nature of this hybrid is of significance for the excellent high-rate performance.65
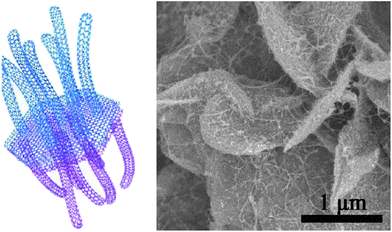 | ||
| Fig. 11 (Left) illustration of the graphene–SWCNT hybrid as a sulfur host and (right) SEM image of the hybrid-sulfur composite.65 Reproduced from ref. 65. Copyright 2012 American Chemical Society. | ||
Apart from the diverse range of synthesis chemistry and approaches for producing graphene–sulfur composites, the interaction between graphene and sulfur has also been of interest. Residual oxygen functional groups attached on graphene sheets were found to enhance the adsorption of sulfur and PS ions, both of which contribute greatly to the composite stability.66,67
Polyacrylonitrile-derived carbon–sulfur composites
Polyacrylonitrile (PAN) was first used for the preparation of carbon–sulfur composites more than a decade ago.68 A simple process was adopted by directly heating the mixture of PAN powder and sublimed sulfur at 280 to 300 °C in an Ar gas environment for 6 h. Since elemental sulfur is an effective dehydrogenating reagent, the ratio of H to C atoms in the final product (1:3) was lower than that in pristine PAN (1:1) (Fig. 12a).68 The effective dehydrogenation of PAN by sulfur gave rise to a conducting main chain. NMR results suggested the formation of a carbon double bond in the final product and the cyclization of the –CN group, which was highly thermally stable. As such, this PAN-derived carbon–sulfur composite had very high thermal stability with 70 wt% of its original weight remaining even above 600 °C. It was also noticed that no C–S bonds were formed at 280–300 °C reaction temperature and sulfur was found to be in an elemental state according to XPS. With a high amount of sulfur, even XRD can reveal the existence of elemental sulfur, suggesting a mixture of crystalline and amorphous sulfur in the final composite.69 This work also implied that by doping the PAN-derived carbon with PF6− ions the cycling properties can be considerably improved.68 To interpret the ultra-stable property of this special composite, Wang et al. proposed complex bonding between sulfur and nitrogen atoms (or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds), which could affect the reaction of sulfur with lithium. It was expected that the discharge potential hysteresis is related to the additional energy necessary to dissociate sulfur from the complex bond (Fig. 12b).69
N double bonds), which could affect the reaction of sulfur with lithium. It was expected that the discharge potential hysteresis is related to the additional energy necessary to dissociate sulfur from the complex bond (Fig. 12b).69
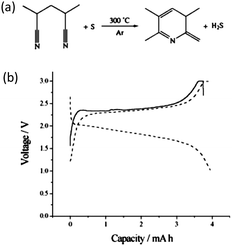 | ||
| Fig. 12 (a) Reaction mechanism during heating sulfur with PAN and (b) discharge–charge curves of a PAN-derived carbon–sulfur composite.69 Reproduced from ref. 69. Copyright 2003 Wiley-VCH. | ||
In order to improve the rate capability and utilization ratio of sulfur in PAN-derived composites, carbon nanotubes70 and graphene sheets71 have been used as electrically conducting backbones to support the composite. Several aspects of the nanocarbon–PAN–sulfur composites have been observed: (1) the intimate contact of the PAN–sulfur composite with nanocarbon surfaces ensures low interface resistance, (2) the encapsulation of sulfur by PAN-derived carbon is a good basis for high capacity and good stability, (3) the supporting backbone can hinder the agglomeration of the PAN–sulfur composite particles which may occur upon volume change of sulfur during extended cycles, and (4) the PAN–sulfur composites can help prevent the restacking of graphene sheets or carbon nanotubes and thus keep a large cathode area for reducing interface impedance.70,71 Another way to improve the reaction kinetics of PAN-derived carbon–sulfur composites is to use higher synthesis temperatures. Fanous and co-workers were able to show that the rate performance and stability of this kind of composite can be improved by increasing the synthesis temperature from 330 °C to 550 °C, but unfortunately at the cost of lower capacity.72 Taking advantage of the lithium–nitrile interaction, Guo et al. succeeded in developing a lithiated carbon–sulfur composite from PAN which represents an advanced strategy for new types of Li–S batteries.73
Flexible carbon–sulfur composites
In an innovative approach, activated carbon fibre (ACF) cloth has been recently demonstrated as a binder-free cathode support for sulfur.74 This ACF cloth is monolithic and holds the sulfur in the micropores. The structural integrity of the monolithic ACF cloth is sufficient to withstand the volume variation during lithiation and delithiation. Meanwhile, the abundant micropores that can accommodate sulfur promised excellent cathode stability.74 Zhou et al. developed a highly conducting carbon–sulfur nanotube membrane with good mechanical resilience.75 This self-supporting composite electrode can tolerate a 10 MPa stress with a 9% strain over 12000 times while maintaining unchanged conductivity (800 S cm−1) (Fig. 13a).75 The unique mechanical–electrical properties arise from the molecular level mixing of carbon with sulfur where the sulfur is intercalated between the graphitic local domains, i.e. residing in the tube walls (Fig. 13b).75 A special carbothermal route is the key to obtain this structure, where the reaction is as follows:| SO42− + 3C → S + 2CO + CO2 | (4) |
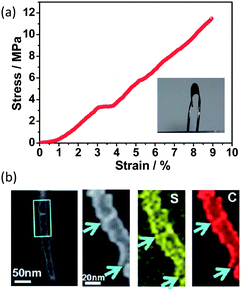 | ||
| Fig. 13 (a) Stress–strain curve of a flexible carbon–sulfur nanotube membrane cathode. Inset shows a bent S-CNT membrane. (b) STEM analysis of a single carbon–sulfur nanotube.75 Reproduced from ref. 75. Copyright 2012 Royal Society of Chemistry. | ||
This nanotubular carbon–sulfur composite has a high surface area and porous texture which is difficult to obtain through infiltration methods. The richness of the void spaces is advantageous for its capabilities of (1) compensation for volume change, (2) rapid ion transport, (3) reduced resistance polarization, and (4) adsorption of dissolved PS. Sulfur-coated carbon nanotubes76 and graphene sheets77 have also been assembled into papers by directional filtration methods for use as flexible cathodes.
Summary and outlook
Tremendous progress in carbon–sulfur composites has been achieved in recent years. Substantial attention has been paid to the morphology, chemistry and electrochemistry of carbon–sulfur composites. In the future the adsorption, intercalation, and functionalization of carbon materials still require further investigation in terms of the cathode stabilization through the confinement and physical–chemical attraction of polysulfide. For cyclic stability of sulfur, micropore adsorption or possible intercalation between graphene layers have been shown to lead to the best results (Table 2), while surface functionalization can potentially improve this further. In terms of reaction kinetics, however, mesopores, or loose confinement, e.g. graphene wrapping, are beneficial. The large pore volumes of mesoporous carbon and graphene sheets are also advantageous for high loading of sulfur that gives high capacity of the C–S composite. One also needs to bear in mind that partially filled pores can compensate for the volume change of sulfur–lithium sulfides; thus the optimal loading of sulfur is a balance between the desire for maximum capacity and the need to allow for the volume change to ensure stability. Hollow carbons with rigid shells and large internal voids are desirable for this purpose. Similarly, graphene sheets with good flexibility are excellent for buffering volume change.A summary of the efficacy of the different kinds of carbons for stability, capacity and conductivity is given in Table 3. It is clear that no one carbon form alone can meet the comprehensive performance criteria, but an effective combination of these materials is necessary to gain the best advantage from the properties of the individual building blocks.
| Carbon Form | Stability | Conductivity | Volume change |
|---|---|---|---|
| Microporous carbon | Good | Normal | Normal–good |
| Mesoporous carbon | Normal–good | Normal | Normal–good |
| Graphene sheets | Normal–good | Good | Good |
| Carbon nanotubes | Normal–good | Good | Normal |
| PAN carbon | Good | Normal | Normal |
Several critical factors are suggested here for the rational design of advanced carbon–sulfur composites:
(1) Pore size: micropores/small mesopores for strongly confining polysulfides.
(2) Large pore volume for maximum sulfur loading: carbon is not an active cathode material in Li–S batteries and its weight ratio is required to be as low as possible without harming the overall performance of sulfur–carbon cathodes. However, the very low conductivity of the composite when sulfur content increases is a critical problem to be solved.
(3) Graphitization level: a preferential content of graphitic carbon to facilitate electron conduction to the insulating sulfur/lithium sulfides.
(4) Electrolyte impregnation and lithium ion migration: a short pathway to preserve fast migration of lithium ions from the bulk electrolyte to active sulfur and the release of lithium ions from lithium sulfides. Voids are required in the final carbon–sulfur composites.
(5) Flexible or rigid carbon scaffold: volume change occurs during the discharge or charge of sulfur–carbon cathodes; the carbon host should buffer the stress-induced strain and survive over extended cycles.
(6) Low cost production and easy scale-up: templated ordered mesoporous carbon and CNTs/nanofibers are highly effective due to their periodic structure but are unlikely to be produced at an industrial scale for Li–S batteries owing to their high cost and unsatisfactory performance–cost ratio; novel continuous synthesis techniques, inclusive of but not limited to atomization carbonization and hydrothermal carbonization, are promising.
(7) An advanced technique to form carbon–sulfur composites: common methods are impregnation with the sulfur melt or sulfur organic solution, disproportionate reaction and in-situ encapsulation, and vapour diffusion. These are post-carbon-synthesis and complicate the industrial processing. A one-step method for fabrication of sulfur–carbon composites is necessary.
We emphasize here that the correlations between the carbon structure (porosity, surface chemistry, graphitic degree) and the sulfur structure need to be comprehensively studied and optimized (Fig. 14). Facile and low cost material fabrication techniques are also desired. An optimal carbon–sulfur composite will benefit from a combination of fundamental insights and an advanced synthesis approach. We also acknowledge that the sulfur cathode problem is not the only issue that impedes Li–S technology. Problems with the lithium anode, the electrolyte as well as engineering difficulties in fabricating lithium metal batteries are all great challenges to be faced, but recent dramatic progress in carbon–sulfur composites is likely to form the basis for future commercialization of Li–S batteries.
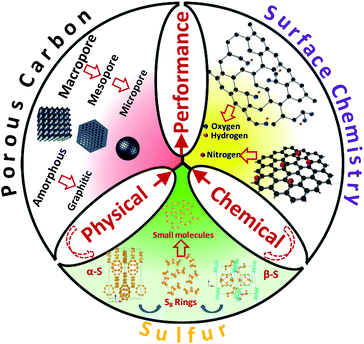 | ||
| Fig. 14 Correlations between the carbon structure and the sulfur structure for performance optimization. | ||
Acknowledgements
We acknowledge the financial supports from the University of Queensland, Foundation Research Excellence Awards for D. W. and from the National Science Foundation of China (no. 51172239).Notes and references
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774 RSC.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480 CrossRef CAS.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880 CrossRef CAS.
- H. Nishihara and T. Kyotani, Adv. Mater., 2012, 24, 4473 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11 CrossRef CAS.
- D. S. Su and R. Schlogl, ChemSusChem, 2010, 3, 136 CrossRef CAS.
- Y. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695 CrossRef CAS.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938 RSC.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1 CrossRef CAS.
- X. L. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS.
- K. Cai, M.-K. Song, E. J. Cairns and Y. Zhang, Nano Lett., 2012, 12, 6474 CrossRef CAS.
- T. Takeuchi, H. Sakaebe, H. Kageyama, H. Senoh, T. Sakai and K. Tatsumi, J. Power Sources, 2010, 195, 2928 CrossRef CAS.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486 CrossRef CAS.
- J. Hassoun and B. Scrosati, Angew. Chem., Int. Ed., 2010, 49, 2371 CrossRef CAS.
- K. Kumaresan, Y. Mikhaylik and R. E. White, J. Electrochem. Soc., 2008, 155, A576 CrossRef CAS.
- V. S. Kolosnitsyn and E. V. Karaseva, Russ. J. Electrochem., 2008, 44, 506 CrossRef CAS.
- Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387 CrossRef CAS.
- B. Meyer, Chem. Rev., 1976, 76, 367 CrossRef CAS.
- B. Meyer, Chem. Rev., 1964, 64, 429 CrossRef CAS.
- W. A. Pryor, Mechanisms of sulfur reactions, McGRAW-Hill Book Company, 1962 Search PubMed.
- J. Wang, S. Y. Chew, Z. W. Zhao, S. Ashraf, D. Wexler, J. Chen, S. H. Ng, S. L. Chou and H. K. Liu, Carbon, 2008, 46, 229 CrossRef CAS.
- X. L. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS.
- S. R. Chen, Y. P. Zhai, G. L. Xu, Y. X. Jiang, D. Y. Zhao, J. T. Li, L. Huang and S. G. Sun, Electrochim. Acta, 2011, 56, 9549 CrossRef CAS.
- G. He, X. L. Ji and L. Nazar, Energy Environ. Sci., 2011, 4, 2878 CAS.
- X. L. Li, Y. L. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. M. Nie, J. Mietek, J. G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603 RSC.
- C. D. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724 CrossRef CAS.
- X. A. Liang, Z. Y. Wen, Y. Liu, H. Zhang, L. Z. Huang and J. Jin, J. Power Sources, 2011, 196, 3655 CrossRef CAS.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591 CrossRef CAS.
- X.-G. Sun, X. Wang, R. T. Mayes and S. Dai, ChemSusChem, 2012, 5, 2079 CrossRef CAS.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531 CAS.
- D.-W. Wang, G. Zhou, F. Li, K.-H. Wu, G. Q. Lu, H.-M. Cheng and I. R. Gentle, Phys. Chem. Chem. Phys., 2012, 14, 8703 RSC.
- S. Xin, L. Gu, N.-H. Zhao, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 18510 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760 CrossRef CAS.
- B. Ding, C. Yuan, L. Shen, G. Xu, P. Nie and X. Zhang, Chem.–Eur. J., 2013, 19, 1013 CrossRef CAS.
- S. Zhao, C. Li, W. Wang, H. Zhang, M. Gao, X. Xiong, A. Wang, K. Yuan, Y. Huang and F. Wang, J. Mater. Chem. A, 2013, 1, 3334 CAS.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904 CrossRef CAS.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 9592 CrossRef CAS.
- C. Wang, J. J. Chen, Y. N. Shi, M. S. Zheng and Q. F. Dong, Electrochim. Acta, 2010, 55, 7010 CrossRef CAS.
- K. Li, B. Wang, D. Su, J. Park, H. Ahn and G. Wang, J. Power Sources, 2012, 202, 389 CrossRef CAS.
- L. X. Yuan, H. P. Yuan, X. P. Qiu, L. Q. Chen and W. T. Zhu, J. Power Sources, 2009, 189, 1141 CrossRef CAS.
- J. C. Guo, Y. H. Xu and C. S. Wang, Nano Lett., 2011, 11, 4288 CrossRef CAS.
- J.-J. Chen, Q. Zhang, Y.-N. Shi, L.-L. Qin, Y. Cao, M.-S. Zheng and Q.-F. Dong, Phys. Chem. Chem. Phys., 2012, 14, 5376 RSC.
- S. Dorfler, M. Hagen, H. Althues, J. Tubke, S. Kaskel and M. J. Hoffmann, Chem. Commun., 2012, 48, 4097 RSC.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462 CrossRef CAS.
- L. W. Ji, M. M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. G. Zhang, Energy Environ. Sci., 2011, 4, 5053 CAS.
- J. Z. Wang, L. Lu, M. Choucair, J. A. Stride, X. Xu and H. K. Liu, J. Power Sources, 2011, 196, 7030 CrossRef CAS.
- F.-f. Zhang, X.-b. Zhang, Y.-h. Dong and L.-m. Wang, J. Mater. Chem., 2012, 22, 11452 RSC.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644 CrossRef CAS.
- S. Evers and L. F. Nazar, Chem. Commun., 2012, 48, 1233 RSC.
- M.-S. Park, J.-S. Yu, K. J. Kim, G. Jeong, J.-H. Kim, Y.-N. Jo, U. Hwang, S. Kang, T. Woo and Y.-J. Kim, Phys. Chem. Chem. Phys., 2012, 14, 6796 RSC.
- Y. Cao, X. Li, I. A. Aksay, J. Lemmon, Z. Nie, Z. Yang and J. Liu, Phys. Chem. Chem. Phys., 2011, 13, 7660 RSC.
- N. W. Li, M. B. Zheng, H. L. Lu, Z. B. Hu, C. F. Shen, X. F. Chang, G. B. Ji, J. M. Cao and Y. Shi, Chem. Commun., 2012, 48, 4106 RSC.
- S. Li, M. Xie, J. B. Liu, H. Wang and H. Yan, Electrochem. Solid-State Lett., 2011, 14, A105 CrossRef CAS.
- Y. X. Wang, L. Huang, L. C. Sun, S. Y. Xie, G. L. Xu, S. R. Chen, Y. F. Xu, J. T. Li, S. L. Chou, S. X. Dou and S. G. Sun, J. Mater. Chem., 2012, 22, 4744 RSC.
- B. Wang, K. Li, D. Su, H. Ahn and G. Wang, Chem.–Asian J., 2012, 7, 1637 CrossRef CAS.
- H. Sun, G.-L. Xu, Y.-F. Xu, S.-G. Sun, X. Zhang, Y. Qiu and S. Yang, Nano Res., 2012, 5, 726 CrossRef CAS.
- Z.-K. Wei, J.-J. Chen, L.-L. Qin, A.-W. Nemage, M.-S. Zheng and Q.-F. Dong, J. Electrochem. Soc., 2012, 159, A1236 CrossRef CAS.
- B. Ding, C. Yuan, L. Shen, G. Xu, P. Nie, Q. Lai and X. Zhang, J. Mater. Chem. A, 2013, 1, 1096 CAS.
- M.-Q. Zhao, X.-F. Liu, Q. Zhang, G.-L. Tian, J.-Q. Huang, W. Zhu and F. Wei, ACS Nano, 2012, 6, 10759 CAS.
- L. W. Ji, M. M. Rao, H. M. Zheng, L. Zhang, Y. C. Li, W. H. Duan, J. H. Guo, E. J. Cairns and Y. G. Zhang, J. Am. Chem. Soc., 2011, 133, 18522 CrossRef CAS.
- L. Zhang, L. W. Ji, P. A. Glans, Y. G. Zhang, J. F. Zhu and J. H. Guo, Phys. Chem. Chem. Phys., 2012, 14, 13670 RSC.
- J. L. Wang, J. Yang, J. Y. Xie and N. X. Xu, Adv. Mater., 2002, 14, 963 CAS.
- J. L. Wang, J. Yang, C. R. Wan, K. Du, J. Y. Xie and N. X. Xu, Adv. Funct. Mater., 2003, 13, 487 CrossRef CAS.
- L. C. Yin, J. L. Wang, J. Yang and Y. N. Nuli, J. Mater. Chem., 2011, 21, 6807 RSC.
- L. C. Yin, J. L. Wang, F. J. Lin, J. Yang and Y. Nuli, Energy Environ. Sci., 2012, 5, 6966 CAS.
- J. Fanous, M. Wegner, J. Grimminger, M. Rolff, M. B. M. Spera, M. Tenzer and M. R. Buchmeiser, J. Mater. Chem., 2012, 22, 23240 RSC.
- J. Guo, Z. Yang, Y. Yu, H. D. Abruña and L. A. Archer, J. Am. Chem. Soc., 2012, 135, 763 CrossRef.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Adv. Mater., 2011, 23, 5641 CrossRef CAS.
- G. Zhou, D.-W. Wang, F. Li, P.-X. Hou, L. Yin, C. Liu, G. Q. Lu, I. R. Gentle and H.-M. Cheng, Energy Environ. Sci., 2012, 5, 8901 CAS.
- Y.-S. Su, Y. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 14495 RSC.
- J. Jin, Z. Wen, G. Ma, Y. Lu, Y. Cui, M. Wu, X. Liang and X. Wu, RSC Adv., 2013, 3, 2558 RSC.
| This journal is © The Royal Society of Chemistry 2013 |